Subunit-Specific Role of NF-κB in Cancer
by
 ,
,
Barbara Kaltschmidt
1, 
Johannes F. W. Greiner
2,
Hussamadin M. Kadhim
2 and
Christian Kaltschmidt
2,* 1
AG Molecular Neurobiology, University of Bielefeld, 33615 Bielefeld, Germany
2
Department of Cell Biology, University of Bielefeld, 33615 Bielefeld, Germany
*
Author to whom correspondence should be addressed.
Biomedicines 2018, 6(2), 44; https://doi.org/10.3390/biomedicines6020044
Submission received: 1 March 2018
/
Revised: 11 April 2018
/
Accepted: 12 April 2018
/
Published: 17 April 2018
(This article belongs to the Special Issue Roles of NF-κB in Cancer and Their Therapeutic Approaches)
Abstract
:The transcription factor NF-κB is a key player in inflammation, cancer development, and progression. NF-κB stimulates cell proliferation, prevents apoptosis, and could promote tumor angiogenesis as well as metastasis. Extending the commonly accepted role of NF-κB in cancer formation and progression, different NF-κB subunits have been shown to be active and of particular importance in distinct types of cancer. Here, we summarize overexpression data of the NF-κB subunits RELA, RELB, and c-REL (referring to the v-REL, which is the oncogene of Reticuloendotheliosis virus strain T) as well as of their upstream kinase inhibitor, namely inhibitor of κB kinases (IKK), in different human cancers, assessed by database mining. These data argue against a universal mechanism of cancer-mediated activation of NF-κB, and suggest a much more elaborated mode of NF-κB regulation, indicating a tumor type-specific upregulation of the NF-κB subunits. We further discuss recent findings showing the diverse roles of NF-κB signaling in cancer development and metastasis in a subunit-specific manner, emphasizing their specific transcriptional activity and the role of autoregulation. While non-canonical NF-κB RELB signaling is described to be mostly present in hematological cancers, solid cancers reveal constitutive canonical NF-κB RELA or c-REL activity. Providing a linkage to cancer therapy, we discuss the recently described pivotal role of NF-κB c-REL in regulating cancer-targeting immune responses. In addition, current strategies and ongoing clinical trials are summarized, which utilize genome editing or drugs to inhibit the NF-κB subunits for cancer treatment.
Keywords:
NF-κB; RELA; cREL; RELB; tumor; cancer; transformation; inflammation; gene expression; tumor necrosis factor; Treg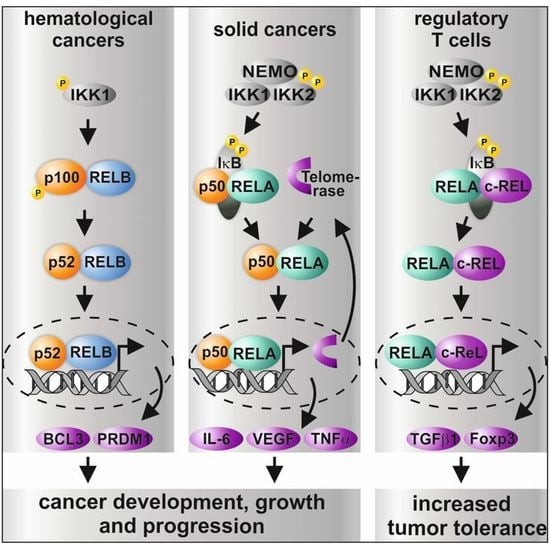
1. The NF-κB Family—An Introduction
The transcription factor nuclear factor “kappa-light-chain-enhancer” of activated B-cells (NF-κB) [1,2] plays a key role in a broad range of cellular processes like cell growth, apoptosis, inflammation, learning, and memory as well as immunity [3,4]. The transcription factor is ubiquitously expressed and responds to diverse stimuli, particularly including infectious agents, cytokines, or growth factors [5,6]. According to its various cellular functions, deregulation of NF-κB signaling is strongly associated with cancer formation and progression [7,8].
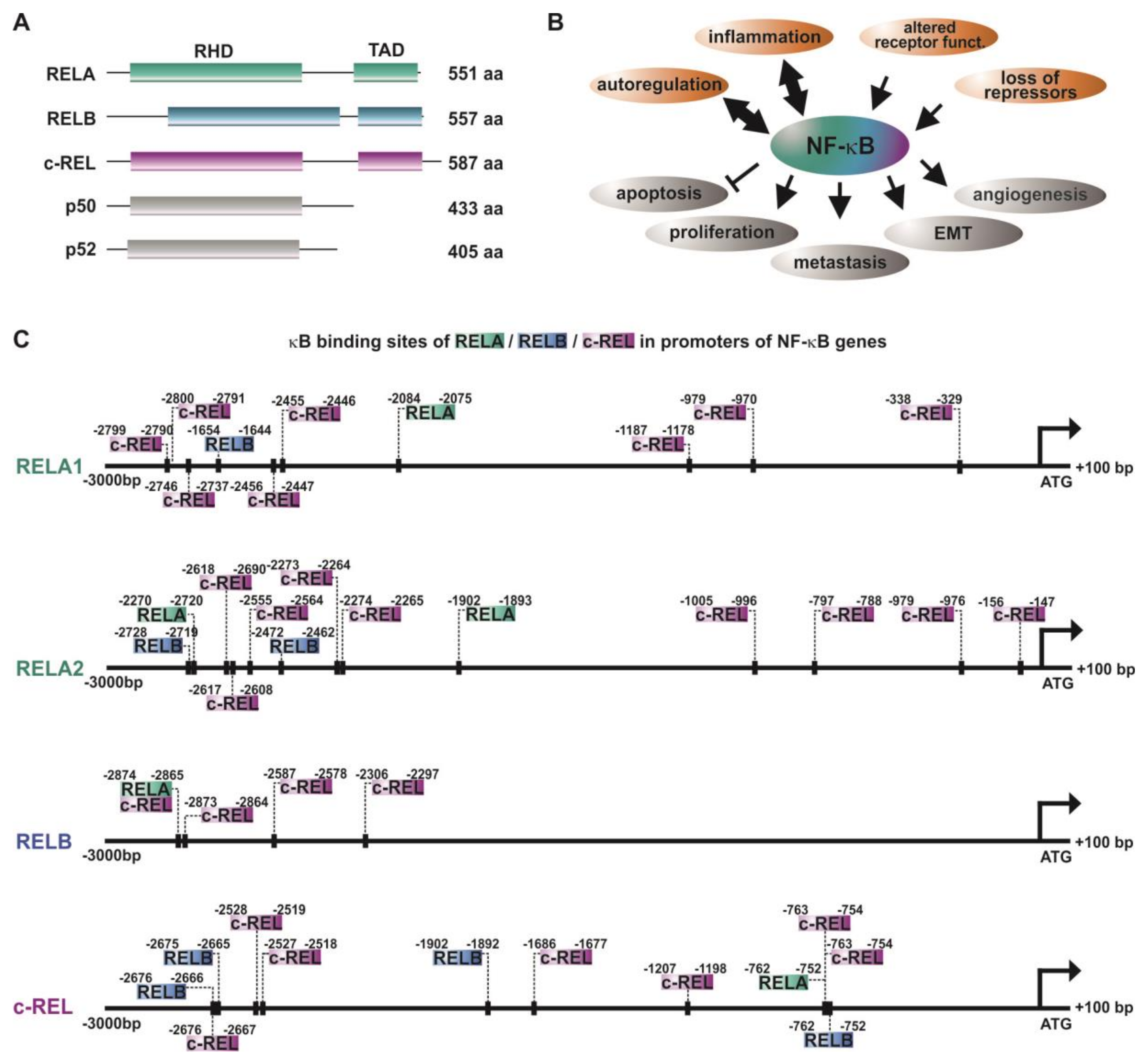
The NF-κB family is composed of five subunits, namely, RELA (p65), RELB, c-REL, p50, and p52 (Figure 1A), all comprising a conserved REL homology domain (RHD) near the N-terminus. This domain is crucial for DNA binding (N-terminal part of RHD), dimerization of NF-κB family members, as well as interaction with the inhibitors of κB (IκBs) (C-terminal part of RHD). Via the RHD, NF-κB family members can form homo- or heterodimers, like p50/RELAp65, RELB/p50, p52/c-REL, or RELA/RELA. In addition, the subunits RELA, RELB, and c-REL contain a C-terminal transactivation domain (TAD) [9,10].
Inactive NF-κB dimers are localized within the cytoplasm, since the NLS (nuclear localization signal) within the RHD is masked by IκBs. During canonical NF-κB signaling, binding of ligands such as cytokines, growth factors, or lipopolysaccharides to their respective receptors (see below, Section 2) leads to the phosphorylation of the IκB kinase (IKK) complex comprised of IKK1/IKK2 (IKKα/IKKβ) and NEMO (NF-κB essential modulator). Phosphorylated IKKs, in particular IKK2, in turn phosphorylate IκBα, which subsequently undergoes proteasome-mediated degradation via polyubiquitinylation. Degradation of IκBα leads to demasking of the nuclear translocation site of the NF-κB p50/RELA heterodimer. In turn, translocation into the nucleus occurs. This results in the expression of NF-κB-target genes via binding to the respective target sites [4,9]. On the contrary, non-canonical NF-κB signaling induced by distinct members of the tumor necrosis factor (TNF) family like lymphotoxin-β relies on the phosphorylation of IKK1 via NIK (NF-κB-inducing kinase). IKK1 mediates the phosphorylation of p100, associated to RELB, inducing the proteasomal processing of p100 to p52 [14]. The p52/RELB heterodimer is able to enter the nucleus and activate specific target genes via binding to selective κB sites. Both the canonical and the non-canonical pathway have been described to be closely linked to cancer formation and progression [15] (Figure 1B, see also Section 2). In addition, atypical NF-κB pathways, as in the case of epidermal growth factor receptor (EGFR) tyrosine kinase-dependent NF-κB activation, were likewise described to promote cancer [16].
2. NF-κB in Inflammation and Cancer
In response to physical or physiological stress, injury, or infection, inflammation takes place as a key defense process of innate immunity aiming to restore the physiological situation. NF-κB is broadly described to be one of the key transcription factors regarding pro-inflammatory signaling, particularly activated by the presence of pro-inflammatory cytokines (like TNFα or IL-1), lipopolysaccharides (LPS) of the bacterial cell wall [17], or viral and bacterial nucleic acids [18]. Recognition of cytokines or LPS species is mediated by the respective receptors, such as TNF receptors or Toll-like microbial pattern recognition receptors (TLRs). As described above, binding of such ligands to their respective receptors leads to canonical NF-κB signaling, ultimately resulting in the translocation of released NF-κB p50/RELA into the nucleus and binding onto κB elements located in distinct target genes. Among the broad range of target genes of NF-κB, the most prominent ones in terms of inflammation are also pro-inflammatory cytokines, such as TNFα [19,20], IL-1 [21], and T cell regulatory ones, such as IL-2 [22] (proliferation) or IL-8 [23] (recruitment). The resulting feed-forward loops of NF-κB-activation, particularly in the case of TNFα, make NF-κB a booster of pro-inflammatory signaling, which augments the inflammation. In the case of cancer, these signaling cascades and the resulting production of pro-inflammatory cytokines likewise recruit cytotoxic immune cells targeting and eliminating the transformed cells [24]. However, the presence of active NF-κB in cancer is a double-edged sword. Although being a mediator of immune responses eliminating cancer cells, NF-κB was observed to be constitutively active in many types of cancer arising from a prolonged chronic inflammatory microenvironment or induced by various oncogenic mutations [8,25]. In a seminal review, Baud and Karin listed 11 types of blood-born cancers (including frequent ones such as acute myeloid leukemia (AML)) and 23 solid tumors (including frequent ones such colon cancer), which showed activated NF-κB signaling [26]. By way of example, elevated NF-κB activity resulting in the accumulation of pro-inflammatory cytokines in the tumor was reported to directly contribute to a pro-tumorigenic microenvironment in colon cancer [27]. Despite this close relation between inflammatory NF-κB signaling and cancer, NF-κB directly mediates vital tumor-promoting mechanisms. NF-κB activity was shown to stimulate cell proliferation, prevent apoptosis, and promote tumor angiogenesis, epithelial-to-mesenchymal transition, invasiveness, as well as metastasis [8,28,29] (Figure 1B). For further details, see a recent review by Taniguchi and Karin [30]. Extending this commonly accepted role of NF-κB in cancer formation and progression, different NF-κB subunits have been shown to be active and of particular importance in distinct types of cancers [11]. In the following, we will discuss the current literature depicting the roles of different NF-κB subunits, their autoregulation, and specific transcriptional activity in cancer and outline how particular subunits and upstream kinases contribute to cancer progression.
3. Autoregulation of NF-κB—A Potential Driver on the Road to Cancer Development?
In addition to the canonical, non-canonical, and atypical activation of NF-κB (see also Section 1), NF-κB RELA, RELB, and c-REL have been described to be activated by autoregulation [31,32,33,34]. Accordingly, the promoter analysis of NF-κB-subunits performed in the present study depicted the presence of various κB-binding sites for RELA, RELB, and c-REL (Figure 1C). While RELA and c-REL promoters contain binding sites for all three transactivating subunits RELA, RELB, and c-REL, the promoter of RELB showed only binding sites for cREL and RELA/c-REL in its proximal region. Next to the transactivating subunits, p50 and p52 are likewise known to be autoregulated [35,36]. As depicted in Table 1, several tumor types show various levels of overexpression of the NF-κB-transactivating subunits. A mechanistic reason for this observation might be a feed-forward autoregulation. In this line, a broad range of different κB binding sites within the NF-κB promoters shown here suggest NF-κB feed-forward loops to act as boosters of vital tumor-promoting mechanisms, like cell proliferation, angiogenesis, invasiveness, and metastasis. In addition, these autoregulatory mechanisms may at least in part account for the constitutive activity of NF-κB observed in a broad range of cancers [25,26].
4. Activity of Distinct NF-κB Upstream Kinases in Cancer
To investigate the role of the upstream regulators of NF-κB-signaling IKK1 and IKK2 in human cancers, we applied database mining using COSMIC to determine their levels of overexpression (Table 2) [37,38].
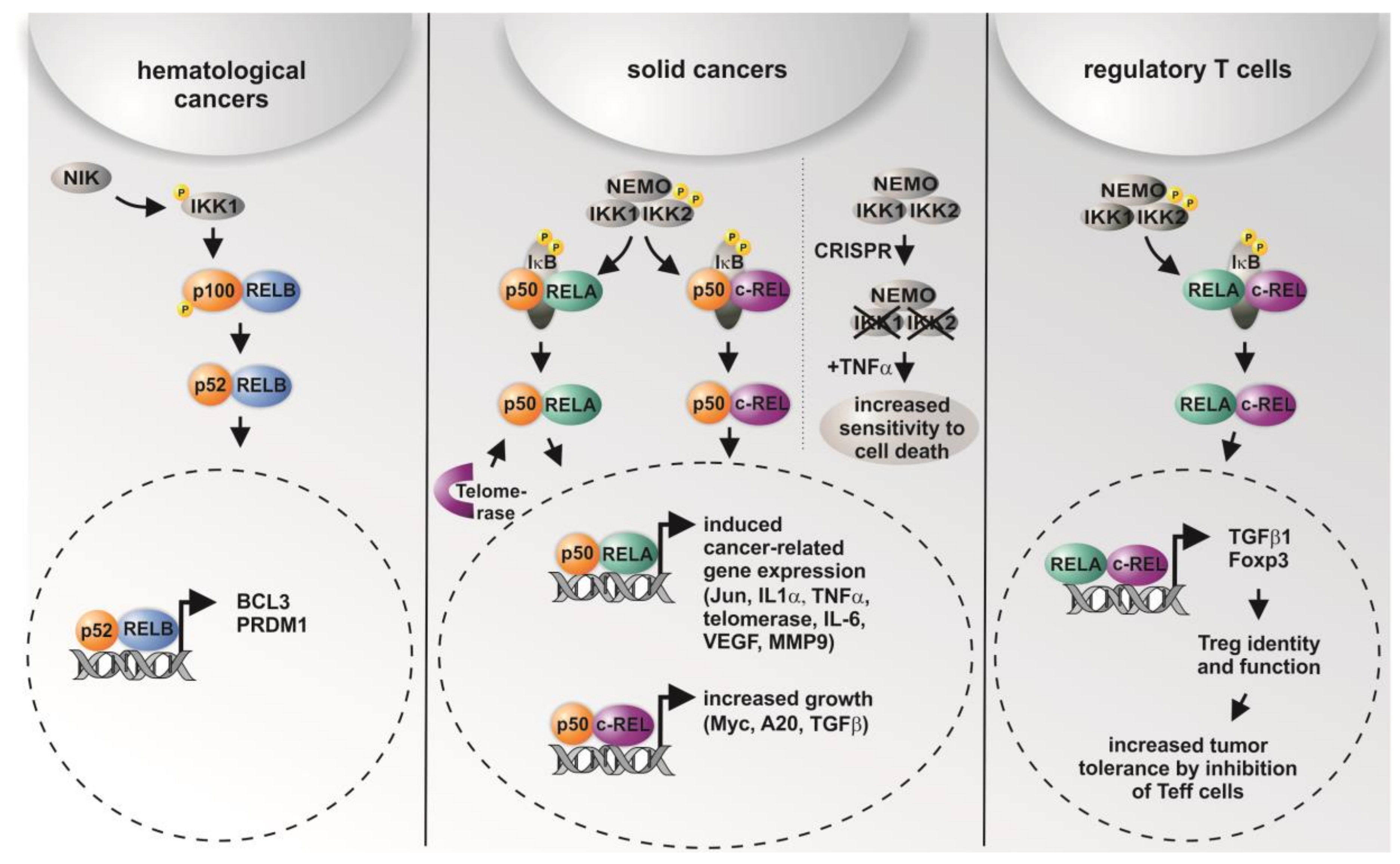
Here, IKK1 and IKK2 showed distinct levels of overexpression in different types of cancer, with IKK2 being overexpressed in cancers arising in the large intestine, the oesophagus, and the lung (Table 2). Accordingly, data from a lung cancer mouse model indicated that tumor cell proliferation was significantly impaired after deletion of IKK2 [39]. Interestingly, IKK-mediated phosphorylation of IκB was shown to mainly depend on the IKK2 catalytic subunit of the IKK complex in mice [40], particularly in terms of prevention of apoptosis [41]. On the contrary, we recently observed TNF-α-mediated cell death only in human cells lacking IKK1 and IKK2 and not in single CRISPR/Cas-mediated IKK knockouts, suggesting that both IKK1 and IKK2 are required for functional TNF-signaling [38] (Figure 2). However, knockout of IKK2 was shown to be associated with about a one-third reduced number of tumors in a colitis-associated cancer model. Surprisingly, deletion of IKK2 in enterocytes led to an increased expression of COX-2, IL-6, and MIP-2, whereas TNF-α, IL-1, and ICAM were not affected. In the myeloid compartment, the number of tumors per mouse was reduced by about 50% after deletion of IKK2 [42]. Constitutive IKK2 activation in intestinal epithelial cells was further demonstrated to induce intestinal tumors in mice [43]. These findings are in accordance with the profound overexpression of IKK2 observed in cancers of the large intestine (Table 2) [38]. On the functional level, IKK2 was shown to directly promote the development of lung cancer in an inflammation-dependent manner triggered by tobacco smoke, which was abrogated by ablation of IKK2 in myeloid cells [44]. Applying a model of breast cancer progression, Huber and colleagues showed IKK2-dependent activation of NF-κB to be essential for epithelial-to-mesenchymal transition and metastasis [45]. Furthermore, the activation of NF-κB by overexpression of constitutively active IKK-2 in prostate cancer cell lines promoted the growth of prostate cancer cells in bone [46]. Accordingly, IKK1 activated by cytokines was shown to control prostate cancer metastasis, with the amount of active nuclear IKK1 correlating with metastatic progression of mouse and human prostate cancer [47].
Next to IKKs, downstream signaling of IκBs is likewise associated to cancer development and progression. Pikarsky and colleagues reported a super-repressor of IκB in hepatocytes to act as a tumor promoter in inflammation-induced liver cancer [48]. Furthermore, in Hodgkin’s disease, a hematologic malignancy, the overexpression of a truncated form of IκB is linked to constitutive NF-κB (p50/RELA) activity [49]. In addition, we observed reduced cell growth and a retarded G1/S transition in human cercival cancer cells, accompanied by an increase in cyclin D1-dependent kinase activity after overexpression of IκBα. We further demonstrated a crosstalk of IκBα overexpression with cell cycle checkpoints via a reduction of transcription factor p53 and elevation of p21WAF [50].
5. Differential Roles of NF-κB Subunits in Cancer
To provide an overview on the occurrences of distinct NF-κB subunits in cancer subtypes, we assessed the overexpression of the NF-κB subunits RELA, RELB, and c-REL in human cancers by database mining, using COSMIC (Table 1) [12,37].
In line with the concept of subunit-specific gene regulation in cancer [11], we found profound differences in the overexpression of particular NF-κB subunits in distinct types of cancer. On the contrary, gene amplification and/or mutations within the coding region were only found in neglectable amounts in the COSMIC database. For instance, RELA is most dominantly overexpressed in ovarian cancer and cancer of adrenal glands in comparison to RELB and c-REL, while the overexpression of c-REL is most abundantly found in lung cancers, compared to that of the other subunits (Table 1). In 2016, Scheidereit and coworkers reported the cell survival of Hodgkin lymphoma (HL) cells to be predominantly controlled by the non-canonical NF-κB pathway. In particular, knockdown of p52/RELB in HL cells resulted in 95% reduction of viability. Using combined ChIP-sequencing and microarray analyses, the authors further showed a low frequency of RELA bound to DNA, but a high frequency of DNA-bound p50- and p52-containing complexes, also including p50/p52 heterodimers [51]. Non-canonical NF-κB signaling was further reported to be active in 20% of the samples from 155 multiple myeloma patients. Here, constitutive activation of the non-canonical RELB/p52 pathway was associated with abnormalities like bi-allelic deletion events, mutations, and gene rearrangements in the genes NFKB1 (p50/p105) and NFKB2 (p52/p100) [52]. Furthermore ectopic expression of RELB can inhibit the growth of tumor xenografts in mice [53]. C-REL is frequently amplified in B cell lymphoma and could function as a tumor-promoting transcription factor, but c-rel-/-mice also could develop an earlier onset of B cell lymphoma [54]. In summary, non-canonical NF-κB-signaling seems to predominantly contribute to hematological cancers (Figure 2).
In contrast to its non-canonical counterpart, canonical NF-κB signaling is described to be present in solid cancers (Figure 2). Shukla and colleagues reported an increased expression of RELA and p50 in human high-grade prostate adenocarcinomas, leading to constitutive NF-κB activity. Active NF-κB p50/RELA led to increased expression of NF-κB target genes MMP9 and VEGF, commonly involved in cell migration and vascularization. Accordingly, NF-κB activity was related to tumor progression due to transcriptional regulation of these NF-κB target genes [57,59]. An increased NF-κB RELA signaling was likewise observed in tumor-initiating stem-like cells in human prostate cancer [60]. Applying a set of 1826 fully annotated prostate cancers, Gannon and colleagues showed a significant association between an increase in the nuclear frequency of NF-κB RELA and Gleason score, which is used to score prostate cancer grade, although the contribution of NF-κB RELA to a post-surgical predictive model appears modest [61]. In lung cancer, NF-κB RELA is known to be required for K-Ras-induced lung tumorigenesis, while lung tumors with RELA deletion show increased apoptosis accompanied by reduced spread and a lower grade [62]. In addition, Mukhopadhyay and colleagues showed highly increased levels of NF-κB p50 in nine of 11 non-small-cell lung carcinoma tissues [63]. KrasG12D-induced IKK2/NF-κB activation, resulting in increased expression of IL1-α and p62 and respective feed-forward loops, was demonstrated to be required for the development of pancreatic ductal adenocarcinoma [64]. In 2003, Nair and coworkers showed a constitutive activation of NF-κB RELA during human cervical cancer progression. Here, NF-κB RELA was demonstrated to be particularly activated in high-grade squamous intraepithelial lesions and squamous cell carcinomas of the human uterine cervix [65]. Interestingly, NF-κB-dependent transcription was recently shown to be directly regulated by telomerase. In particular, telomerase directly bound to the NF-κB RELA subunit, thus regulating NF-κB-dependent gene expression by binding κB sites in the promoter regions of IL-6 and TNF-α, both critical for inflammation and cancer progression [58] (Figure 2). The effect of telomerase on the strong activation of colony formation of tumor stem cells could be repressed by siRNA knockdown of RELA. Given the transcriptional regulation of telomerase by NF-κB RELA, Gosh and coworkers suggested a feed-forward regulation, linking chronic inflammation to increased activity of telomerase in human cancer [58]. Next to RELA, the NF-κB subunit c-REL was likewise shown to possess a key role in tumor formation. In 2000, Cogswell and colleagues revealed the induction of mammary tumors by c-REL expression in mouse models of breast cancer [66]. Shehata and coworkers further demonstrated a sixfold slower cell growth in cultivated cervical cancer cells after expression of the c-REL homolog Xrel3 from Xenopus laevi [67]. We recently investigated the role of c-REL in human cervical cancer cells using CRISPR/Cas9n-mediated gene editing. Knockout of c-REL resulted in significantly decreased basal expression levels of Myc, A20, and TGFβ, accompanied by a significantly reduced proliferative behavior and a significant delay in the prometaphase of mitosis (see also Figure 2 for overview). Compared to the wild type, an increased resistance against chemotherapeutic agents was observable in c-REL knockout cells [12]. Next, by directly promoting cancer cell growth and proliferation, c-REL was very recently shown to possess an important role in cancer-targeting immune responses with highly promising implications for therapeutic approaches [68]. Enabling tumor progression, activated CD4+Foxp3+ regulatory T cells (Tregs) are known to migrate to the tumor site and inhibit of CD8 effector T cells (Teffs), which are mainly responsible for anti-tumor immune responses [69]. In melanoma, large amounts of Tregs have been observed [70] and associated with impaired prognosis, while a lesser amount of Tregs was associated with increased survival in stage 4 melanoma patients [71]. In their groundbreaking study, Grindberg-Bleyer and colleagues demonstrated NF-κB cREL as the critical subunit for identity and function of activated CD4+Foxp3+ Tregs in melanoma (see also Figure 2). Notably, deletion or inhibition of c-REL, but not of RELA, in Tregs resulted in reduced melanoma growth and potentiated anti-PDL1 therapy, a ligand presented by cancer cells and dendritic cells to evade the immune system by binding to the immunosuppressive programmed death (PD) receptor on CD8+ Teff cells [68]. In the following, we will emphasize the therapeutic implications of these findings as wells current strategies to utilize genome editing or drugs for targeted deletion/inhibition of NF-κB subunits in cancer therapy.
6. Targeting NF-κB Subunits via Genome Editing or Drugs—Therapeutic Implications
Given the important roles of distinct NF-κB subunits in cancer development and progression, we aim to summarize currently used drugs targeting NF-κB subunits for cancer treatment. One drug utilized in the clinics is the NF-κB inhibitor Bortezomib [72] (developed by Millenium Pharmaceuticals (Cambridge, MA, USA) as Velcade, also known as Neomib (Getwell Pharmaceuticals, Gurgaon, Haryana, India) or Bortecad (Cadila Healthcare, Ahmedabad, Gujarat, India), a reversible 26S proteasome inhibitor of IκB-α degradation. This drug is certified in Europe as monotherapy for pre-treated adult patients with progressive multiple myeloma. Next to Bortezomib, the NF-κB inhibitor Thalidomide is also clinically applied. In 2002, Majumdar and colleagues showed Thalidomide to abrogate TNFα-dependent activation of IKKs and I-Bα [73]. First evaluated in patients with refractory multiple myeloma in the 1990s, Thalidomide is now known to cause responses in 30–50% of myeloma patients as a single agent and acts synergistically with corticosteroids and chemotherapy [74,75]. In addition, a phase III clinical trial is presently studying a combination of Aspirin (an IKK inhibitor) and Esomeprazole (a proton pump inhibitor) to prevent esophageal cancer in patients with Barrett’s metaplasia (ClinicalTrials.gov Identifier: NCT00357682). Furthermore, a phase 3 clinical trial is using high-dose ascorbic acid, a well-known NF-κB inhibitor [76], as a pharmaceutical for a combination therapy for colorectal cancer (ClinicalTrials.gov Identifier: NCT02969681). Recently, a subunit-specific inhibitor for c-REL was discovered, which might be useful for inhibiting Tregs (patent filed, IPO: WO2017058881A1). Thus, the inhibition of c-REL might be a new way to treat tumors pharmacologically. In addition to the application of NF-κB-inhibiting drugs, a recent increase in clinical studies applying CRISPR/Cas-mediated knockout strategies suggest that gene therapy might be considered in future therapeutic approaches (e.g., five clinical trials with PD-1 knockout engineered T cells; information retrieved in February 2018 from ClinicalTrials.gov).
7. Conclusions
Although NF-κB might be considered as a major factor in cancer development and progression, distinct NF-κB subunits seem to be active in different kinds of cancer. While non-canonical NF-κB RELB signaling is described to be mostly present in hematological cancers, solid cancers reveal canonical NF-κB RELA (p65) and/or c-REL activity. These particular subunits contribute to cancer formation and invasiveness as a result of their specific transcriptional activity, inter alia via feed-forward loops as in the case of TNFα or telomerase. Currently ongoing clinical trials target NF-κB-dependent signaling by application of drugs or CRISPR/Cas-mediated genome editing impinging on potentially NF-κB-driven processes. Thus, although the here summarized data emphasize the importance to assure subunit specificity, NF-κB seems to be a highly promising target for cancer treatment. Michael Karin and coworkers suggested over the years a universal activation of NF-κB in cancer by inflammatory cytokines [30]. It might be important to note that our analysis of the COSMIC database argues against this universal mechanism of cancer-mediated activation of NF-κB. Here, we suggest a much more elaborated mode of NF-κB regulation in terms of a tumor type-specific upregulation of the NF-κB subunits.
Acknowledgments
Studies in own lab were funded in part by the University of Bielefeld, which provides funds for open access publishing. Further funding was provided by the Thyssen-Stiftung and the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie Grant Agreement No. 766181, project “DeLIVER”, which also provides funds covering the costs to publish in open access. We thank the anonymous reviewers for valuable advice.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sen, R.; Baltimore, D. Inducibility of kappa immunoglobulin enhancer-binding protein NF-kappaB by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C. NF-kappaB in the nervous system. Cold Spring Harb. Perspect. Biol. 2009, 1, a001271. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C. NF-kappaB in long-term memory and structural plasticity in the adult mammalian brain. Front. Mol. Neurosci. 2015, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.E.; Huang, D.B.; Chen, Y.Q.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature 1998, 391, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Slotta, C.; Schluter, T.; Ruiz-Perera, L.M.; Kadhim, H.M.; Tertel, T.; Henkel, E.; Hubner, W.; Greiner, J.F.W.; Huser, T.; Kaltschmidt, B.; et al. Crispr/cas9-mediated knockout of c-rel in hela cells results in profound defects of the cell cycle. PLoS ONE 2017, 12, e0182373. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. Jaspar 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by ikkalpha of a second, evolutionary conserved, nf-kappa b signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C.; Pinciroli, P.; Valeri, B.; Ferri, R.; Ditto, A.; Umezawa, K.; Sensi, M.; Canevari, S.; Tomassetti, A. Ligand-dependent egfr activation induces the co-expression of il-6 and pai-1 via the nfkb pathway in advanced-stage epithelial ovarian cancer. Oncogene 2012, 31, 4139–4149. [Google Scholar] [CrossRef] [PubMed]
- Greiner, J.F.; Muller, J.; Zeuner, M.T.; Hauser, S.; Seidel, T.; Klenke, C.; Grunwald, L.M.; Schomann, T.; Widera, D.; Sudhoff, H.; et al. 1,8-cineol inhibits nuclear translocation of NF-kappaB p65 and NF-kappaB-dependent transcriptional activity. Biochim. Biophys. Acta 2013, 1833, 2866–2878. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Greiner, J.F.; Zeuner, M.; Brotzmann, V.; Schafermann, J.; Wieters, F.; Widera, D.; Sudhoff, H.; Kaltschmidt, B.; Kaltschmidt, C. 1,8-cineole potentiates irf3-mediated antiviral response in human stem cells and in an ex vivo model of rhinosinusitis. Clin. Sci. 2016, 130, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Shakhov, A.N.; Collart, M.A.; Vassalli, P.; Nedospasov, S.A.; Jongeneel, C.V. Kappa b-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J. Exp. Med. 1990, 171, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Collart, M.A.; Baeuerle, P.; Vassalli, P. Regulation of tumor necrosis factor alpha transcription in macrophages: Involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappaB. Mol. Cell. Biol. 1990, 10, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Prager, D. Transactivation of the interleukin-1alpha promoter by human t-cell leukemia virus type i and type ii tax proteins. Blood 1996, 87, 3410–3417. [Google Scholar] [PubMed]
- Serfling, E.; Barthelmas, R.; Pfeuffer, I.; Schenk, B.; Zarius, S.; Swoboda, R.; Mercurio, F.; Karin, M. Ubiquitous and lymphocyte-specific factors are involved in the induction of the mouse interleukin 2 gene in t lymphocytes. EMBO J. 1989, 8, 465–473. [Google Scholar] [PubMed]
- Kunsch, C.; Rosen, C.A. NF-kappaB subunit-specific regulation of the interleukin-8 promoter. Mol. Cell. Biol. 1993, 13, 6137–6146. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Bhat-Nakshatri, P.; Martin, D.A.; Goulet, R.J., Jr.; Sledge, G.W., Jr. Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol. Cell. Biol. 1997, 17, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Pettaway, C.A.; Uehara, H.; Bucana, C.D.; Fidler, I.J. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 2001, 20, 4188–4197. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018. [Google Scholar] [CrossRef]
- Hannink, M.; Temin, H.M. Structure and autoregulation of the c-rel promoter. Oncogene 1990, 5, 1843–1850. [Google Scholar] [PubMed]
- Bren, G.D.; Solan, N.J.; Miyoshi, H.; Pennington, K.N.; Pobst, L.J.; Paya, C.V. Transcription of the relb gene is regulated by NF-kappaB. Oncogene 2001, 20, 7722–7733. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Baeuerle, P.A.; Kaltschmidt, C. Cloning of the murine rela (p65 nf-kappa b) gene and comparison to the human gene reveals a distinct first intron. Gene 1996, 176, 119–124. [Google Scholar] [CrossRef]
- Capobianco, A.J.; Gilmore, T.D. Repression of the chicken c-rel promoter by vrel in chicken embryo fibroblasts is not mediated through a consensus NF-kappaB binding site. Oncogene 1991, 6, 2203–2210. [Google Scholar] [PubMed]
- Lombardi, L.; Ciana, P.; Cappellini, C.; Trecca, D.; Guerrini, L.; Migliazza, A.; Maiolo, A.T.; Neri, A. Structural and functional characterization of the promoter regions of the NFKB2 gene. Nucleic Acids Res. 1995, 23, 2328–2336. [Google Scholar] [CrossRef] [PubMed]
- Ten, R.M.; Paya, C.V.; Israel, N.; Le Bail, O.; Mattei, M.G.; Virelizier, J.L.; Kourilsky, P.; Israel, A. The characterization of the promoter of the gene encoding the p50 subunit of NF-kappaB indicates that it participates in its own regulation. EMBO J. 1992, 11, 195–203. [Google Scholar] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. Cosmic: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811.38. [Google Scholar] [CrossRef] [PubMed]
- Slotta, C.; Storm, J.; Pfisterer, N.; Henkel, E.; Kleinwachter, S.; Pieper, M.; Ruiz-Perera, L.M.; Greiner, J.F.W.; Kaltschmidt, B.; Kaltschmidt, C. Ikk1/2 protect human cells from tnf-mediated ripk1-dependent apoptosis in an nf-kappab-independent manner. Biochim. Biophys. Acta 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yeddula, N.; Leblanc, M.; Ke, E.; Zhang, Y.; Oldfield, E.; Shaw, R.J.; Verma, I.M. Reduced cell proliferation by ikk2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 2012, 14, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Van Antwerp, D.; Mercurio, F.; Lee, K.F.; Verma, I.M. Severe liver degeneration in mice lacking the ikappab kinase 2 gene. Science 1999, 284, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.W.; Chu, W.; Hu, Y.; Delhase, M.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. The ikkbeta subunit of ikappab kinase (ikk) is essential for nuclear factor kappab activation and prevention of apoptosis. J. Exp. Med. 1999, 189, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. Ikkbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Vlantis, K.; Wullaert, A.; Sasaki, Y.; Schmidt-Supprian, M.; Rajewsky, K.; Roskams, T.; Pasparakis, M. Constitutive ikk2 activation in intestinal epithelial cells induces intestinal tumors in mice. J. Clin. Investig. 2011, 121, 2781–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering ikkbeta- and jnk1-dependent inflammation. Cancer Cell 2010, 17, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Sterling, J.A.; Edwards, J.R.; DeGraff, D.J.; Lee, C.; Park, S.I.; Matusik, R.J. Activation of nf-kappa b signaling promotes growth of prostate cancer cells in bone. PLoS ONE 2013, 8, e60983. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.L.; Tan, W.; Ricono, J.M.; Korchynskyi, O.; Zhang, M.; Gonias, S.L.; Cheresh, D.A.; Karin, M. Nuclear cytokine-activated ikkalpha controls prostate cancer metastasis by repressing maspin. Nature 2007, 446, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Brandl, C.; Florian, C.; Driemel, O.; Weber, B.H.; Morsczeck, C. Identification of neural crest-derived stem cell-like cells from the corneal limbus of juvenile mice. Exp. Eye Res. 2009, 89, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C.; Hehner, S.P.; Droge, W.; Schmitz, M.L. Repression of NF-kappaB impairs hela cell proliferation by functional interference with cell cycle checkpoint regulators. Oncogene 1999, 18, 3213–3225. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, K.A.; Kaergel, E.; Heinig, M.; Fontaine, J.F.; Patone, G.; Muro, E.M.; Mathas, S.; Hummel, M.; Andrade-Navarro, M.A.; Hubner, N.; et al. A roadmap of constitutive NF-kappaB activity in hodgkin lymphoma: Dominant roles of p50 and p52 revealed by genome-wide analyses. Genome Med. 2016, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; Van Wier, S.; Tiedemann, R.; Shi, C.X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Toma, J.G.; Akhavan, M.; Fernandes, K.J.; Barnabe-Heider, F.; Sadikot, A.; Kaplan, D.R.; Miller, F.D. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat. Cell Biol. 2001, 3, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Techawattanawisal, W.; Nakahama, K.; Komaki, M.; Abe, M.; Takagi, Y.; Morita, I. Isolation of multipotent stem cells from adult rat periodontal ligament by neurosphere-forming culture system. Biochem. Biophys. Res. Commun. 2007, 357, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Kameswaran, V.; Tone, Y.; Li, L.; Liou, H.C.; Greene, M.I.; Tone, M.; Chen, Y.H. Development of foxp3(+) regulatory T cells is driven by the c-Rel enhanceosome. Immunity 2009, 31, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Libermann, T.A.; Baltimore, D. Activation of interleukin-6 gene expression through the NF-kappaB transcription factor. Mol. Cell. Biol. 1990, 10, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; MacLennan, G.T.; Fu, P.; Patel, J.; Marengo, S.R.; Resnick, M.I.; Gupta, S. Nuclear factor-kappaB/p65 (Rel A) is constitutively activated in human prostate adenocarcinoma and correlates with disease progression. Neoplasia 2004, 6, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase directly regulates NF-kappaB-dependent transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Widera, D.; Zander, C.; Heidbreder, M.; Kasperek, Y.; Noll, T.; Seitz, O.; Saldamli, B.; Sudhoff, H.; Sader, R.; Kaltschmidt, C.; et al. Adult palatum as a novel source of neural crest-related stem cells. Stem Cells 2009, 27, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, V.K.; Studer, L.; Gerald, W.; Socci, N.D.; Scher, H.I. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat. Commun. 2011, 2, 162. [Google Scholar] [CrossRef] [PubMed]
- Gannon, P.O.; Lessard, L.; Stevens, L.M.; Forest, V.; Begin, L.R.; Minner, S.; Tennstedt, P.; Schlomm, T.; Mes-Masson, A.M.; Saad, F. Large-scale independent validation of the nuclear factor-kappaB p65 prognostic biomarker in prostate cancer. Eur. J. Cancer 2013, 49, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Ebbs, A.; Levantini, E.; Baldwin, A.S. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010, 70, 3537–3546. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, T.; Roth, J.A.; Maxwell, S.A. Altered expression of the p50 subunit of the NF-kappaB transcription factor complex in non-small cell lung carcinoma. Oncogene 1995, 11, 999–1003. [Google Scholar] [PubMed]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. Krasg12d-induced ikk2/beta/NF-kappaB activation by il-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Venkatraman, M.; Maliekal, T.T.; Nair, B.; Karunagaran, D. NF-kappaB is constitutively activated in high-grade squamous intraepithelial lesions and squamous cell carcinomas of the human uterine cervix. Oncogene 2003, 22, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, P.C.; Guttridge, D.C.; Funkhouser, W.K.; Baldwin, A.S., Jr. Selective activation of nf-kappa b subunits in human breast cancer: Potential roles for NF-kappaB2/p52 and for Bcl-3. Oncogene 2000, 19, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Shehata, M.; Shehata, F.; Pater, A. Apoptosis effects of xrel3 c-Rel/Nuclear factor-kappa B homolog in human cervical cancer cells. Cell Biol. Int. 2005, 29, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-kappaB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell 2017, 170, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory t cells in tumor immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Jandus, C.; Bioley, G.; Speiser, D.E.; Romero, P. Selective accumulation of differentiated foxp3(+) cd4 (+) T cells in metastatic tumor lesions from melanoma patients compared to peripheral blood. Cancer Immunol. Immunother. 2008, 57, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, J.M.; Gonzalez, R.; Lewis, K.D.; Robinson, W.A.; Richter, D.A.; Palmer, B.E.; Wilson, C.C.; McCarter, M.D. Increased survival from stage IV melanoma associated with fewer regulatory T cells. J. Surg. Res. 2009, 154, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Kauffman, M. Development of the proteasome inhibitor velcade (bortezomib). Cancer Investig. 2004, 22, 304–311. [Google Scholar] [CrossRef]
- Majumdar, S.; Lamothe, B.; Aggarwal, B.B. Thalidomide suppresses NF-kappa B activation induced by TNF and H2O2, but not that activated by ceramide, lipopolysaccharides, or phorbol ester. J. Immunol. 2002, 168, 2644–2651. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Mehta, J. Thalidomide in cancer. Biomed. Pharmacother. 2002, 56, 4–12. [Google Scholar] [CrossRef]
- Strasser, K.; Ludwig, H. Thalidomide treatment in multiple myeloma. Blood Rev. 2002, 16, 207–215. [Google Scholar] [CrossRef]
- Carcamo, J.M.; Pedraza, A.; Borquez-Ojeda, O.; Golde, D.W. Vitamin C suppresses TNF alpha-induced NF kappa B activation by inhibiting I kappa B alpha phosphorylation. Biochemistry 2002, 41, 12995–13002. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
NF-κB and autoregulation of NF-κB subunits in cancer. (A) Schematic view of the NF-κB-family ([11]). (B) Principal mechanisms causing overexpression/activation of NF-κB as well as the cellular effects of NF-κB acitivity leading to cancer development and progression. RHD: REL homology domain, TAD: transactivation domain. (C) The promoters of NF-κB subunits RELA, RELB, and c-REL contain various κB sites enabling autoregulation of NF-κB in cancer. Promoter analysis was done as described in [12]. Briefly, sequences of promoter regions (3000 bp downstream and 100 bp upstream of the respective ATG) of interest were taken from Eukaryotic Promoter Database (epd.vital-ti.ch) for Homo sapiens. The binding sites for the gene of interest in the chosen promoter sequence were looked up using the JASPAR Tool [13]) with a relative score threshold of 85%.
Figure 1.
NF-κB and autoregulation of NF-κB subunits in cancer. (A) Schematic view of the NF-κB-family ([11]). (B) Principal mechanisms causing overexpression/activation of NF-κB as well as the cellular effects of NF-κB acitivity leading to cancer development and progression. RHD: REL homology domain, TAD: transactivation domain. (C) The promoters of NF-κB subunits RELA, RELB, and c-REL contain various κB sites enabling autoregulation of NF-κB in cancer. Promoter analysis was done as described in [12]. Briefly, sequences of promoter regions (3000 bp downstream and 100 bp upstream of the respective ATG) of interest were taken from Eukaryotic Promoter Database (epd.vital-ti.ch) for Homo sapiens. The binding sites for the gene of interest in the chosen promoter sequence were looked up using the JASPAR Tool [13]) with a relative score threshold of 85%.

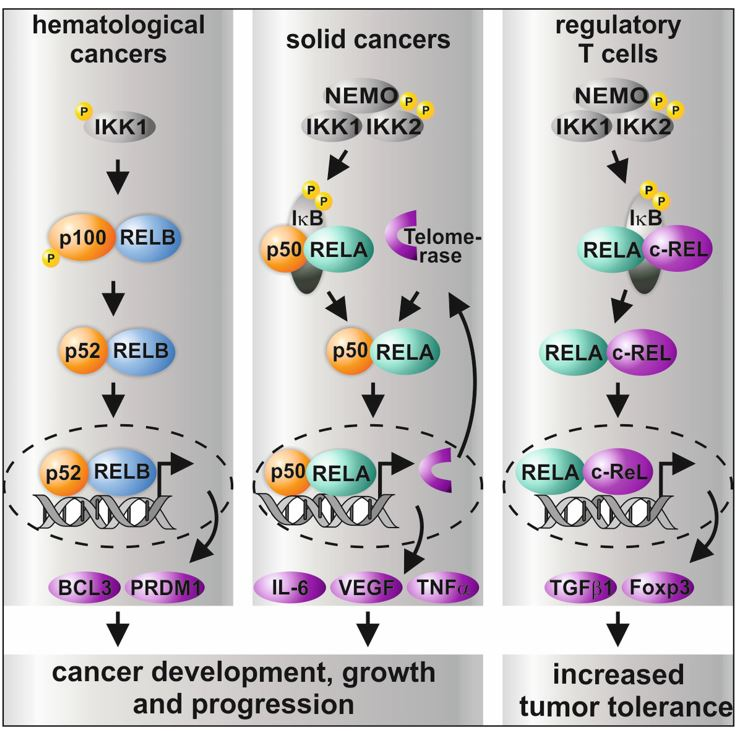
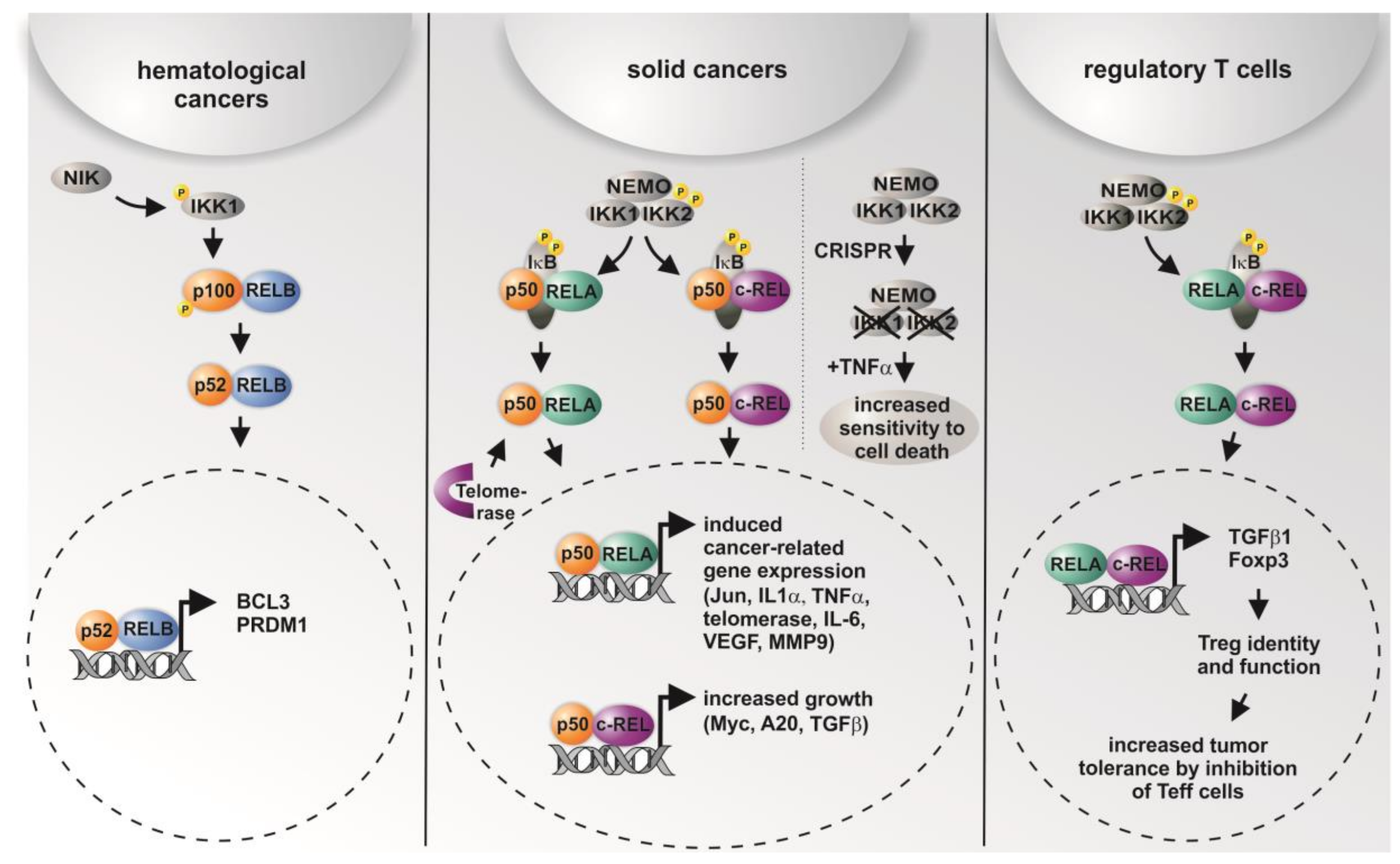
Figure 2.
Graphical overview on the differential roles of NF-κB subunits and their transcriptional activity in distinct types of cancer and in regulatory T cells. While non-canonical signaling is mostly present in hematological cancer, solid cancer shows predominantly canonical signaling via p50/RELA or p50/cREL. In addition, CRISPR/Cas-mediated double knockout (KO) of IKK1/2 was recently shown to result in increased sensitity to TNF-α-mediated cell death [38]. In regulatory T cells (Tregs), activation of RELA/cREL results in distinct target gene expression leading to active Tregs inhibiting effector T cells (Teff), which infiltrate the tumor [11,12,52,55,56,57,58].
Figure 2.
Graphical overview on the differential roles of NF-κB subunits and their transcriptional activity in distinct types of cancer and in regulatory T cells. While non-canonical signaling is mostly present in hematological cancer, solid cancer shows predominantly canonical signaling via p50/RELA or p50/cREL. In addition, CRISPR/Cas-mediated double knockout (KO) of IKK1/2 was recently shown to result in increased sensitity to TNF-α-mediated cell death [38]. In regulatory T cells (Tregs), activation of RELA/cREL results in distinct target gene expression leading to active Tregs inhibiting effector T cells (Teff), which infiltrate the tumor [11,12,52,55,56,57,58].

{kind=link}
{kind=link}
{kind=link}
Table 1.
Overexpression of NF-κB subunits in distinct human cancer tissues. COSMIC was used for database mining [37]. Parts of this table are published in part [12]). n.a: not assessed.
| Cancer Tissue | RELA | RELB | c-REL | |||
|---|---|---|---|---|---|---|
| % Overexpressed | No. Tested | % Overexpressed | No. Tested | % Overexpressed | No. Tested | |
| Ovary | 11.65 | 266 | 3.38 | 266 | 7.52 | 266 |
| Lung | 2.36 | 1019 | 4.12 | 1019 | 7.26 | 1019 |
| Urinary tract | 2.45 | 408 | 4.41 | 408 | 7.11 | 408 |
| Endometrium | 1.99 | 602 | 8.8 | 602 | 6.81 | 602 |
| Pancreas | 2.79 | 179 | 6.7 | 179 | 6.7 | 179 |
| Haematopoietic and lymphoid | 4.07 | 221 | 1.36 | 221 | 6.33 | 221 |
| Soft tissue | 3.42 | 263 | 1.9 | 263 | 6.08 | 263 |
| Cervix | 1.3 | 307 | 7.17 | 307 | 5.86 | 307 |
| Upper aerodigestive tract | 2.49 | 522 | 4.02 | 522 | 5.75 | 522 |
| Kidney | 2.83 | 600 | 4.5 | 600 | 5.5 | 600 |
| Thyroid | 1.36 | 513 | 3.7 | 513 | 5.46 | 513 |
| Large intestine | 1.87 | 610 | 5.25 | 610 | 4.92 | 610 |
| Stomach | 7.02 | 285 | 7.37 | 285 | 4.91 | 285 |
| Liver | 3.75 | 373 | 6.97 | 373 | 4.83 | 373 |
| Central nervous system(CNS) | 4.45 | 697 | 3.73 | 697 | 4.73 | 697 |
| Prostate | 4.62 | 498 | 5.02 | 498 | 4.62 | 498 |
| Breast | 4.17 | 1104 | 4.26 | 1104 | 3.71 | 1104 |
| Skin | 6.34 | 473 | 4.23 | 473 | 3.59 | 473 |
| Oesophagus | 2.4 | 125 | 2.4 | 125 | 3.2 | 125 |
| Adrenal gland | 12.66 | 79 | 5.06 | 79 | 2.53 | 79 |
| Nervous system (NS) | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| Bone | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
Table 2.
Overexpression of IκB kinases IKK1 and IKK2 in distinct human cancer tissues. COSMIC was used for database mining [37].
Table 2.
Overexpression of IκB kinases IKK1 and IKK2 in distinct human cancer tissues. COSMIC was used for database mining [37].
| Cancer Tissue | IKK1 | IKK2 | ||
|---|---|---|---|---|
| % Overexpressed | No. Tested | % Overexpressed | No. Tested | |
| Breast | 7.07 | 1104 | 9.6 | 1104 |
| Lung | 5.1 | 1019 | 7.16 | 1019 |
| Adrenal Gland | 5.06 | 79 | 1.27 | 79 |
| Endometrium | 4.98 | 602 | 13.12 | 602 |
| Oesophagus | 4.8 | 125 | 24.8 | 125 |
| Liver | 4.56 | 373 | 5.36 | 373 |
| Pancreas | 4.47 | 179 | 4.47 | 179 |
| Urinary tract | 4.41 | 408 | 4.9 | 408 |
| Stomach | 4.21 | 285 | 7.72 | 285 |
| Ovary | 4.14 | 266 | 7.52 | 266 |
| Thyroid | 4.09 | 513 | 2.34 | 513 |
| Prostate | 3.21 | 498 | 5.02 | 498 |
| Haematopoietic and lymphoid | 3.17 | 221 | 5.43 | 221 |
| Upper aerodigestive tract | 2.87 | 522 | 6.13 | 522 |
| Large intestine | 2.46 | 610 | 18.52 | 610 |
| Central nervous system(CNS) | 2.44 | 697 | 3.59 | 697 |
| Cervix | 1.95 | 307 | 5.54 | 307 |
| Soft tissue | 1.9 | 263 | 6.08 | 263 |
| Kidney | 1.83 | 600 | 3.33 | 600 |
| Skin | 1.48 | 473 | 8.25 | 473 |
| Biliary tract | n.a. | n.a. | n.a. | n.a. |
| Bone | n.a. | n.a. | n.a. | n.a. |
| Nervous system (NS) | n.a. | n.a. | n.a. | n.a. |
| Pituitary | n.a. | n.a. | n.a. | n.a. |
| Salivary gland | n.a. | n.a. | n.a. | n.a. |
| Testis | n.a. | n.a. | n.a. | n.a. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kaltschmidt, B.; Greiner, J.F.W.; Kadhim, H.M.; Kaltschmidt, C. Subunit-Specific Role of NF-κB in Cancer. Biomedicines 2018, 6, 44. https://doi.org/10.3390/biomedicines6020044
AMA Style
Kaltschmidt B, Greiner JFW, Kadhim HM, Kaltschmidt C. Subunit-Specific Role of NF-κB in Cancer. Biomedicines. 2018; 6(2):44. https://doi.org/10.3390/biomedicines6020044
Chicago/Turabian StyleKaltschmidt, Barbara, Johannes F. W. Greiner, Hussamadin M. Kadhim, and Christian Kaltschmidt. 2018. "Subunit-Specific Role of NF-κB in Cancer" Biomedicines 6, no. 2: 44. https://doi.org/10.3390/biomedicines6020044
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.