Mathematical Modeling and Analysis of Crosstalk between MAPK Pathway and Smad-Dependent TGF-β Signal Transduction



Abstract
:1. Introduction
2. Materials and Methods
2.1. Model of the TGF-β/Smad Pathway
2.2. Model of the MAPK Pathway
2.3. Interaction Mechanisms Reported in the Literature
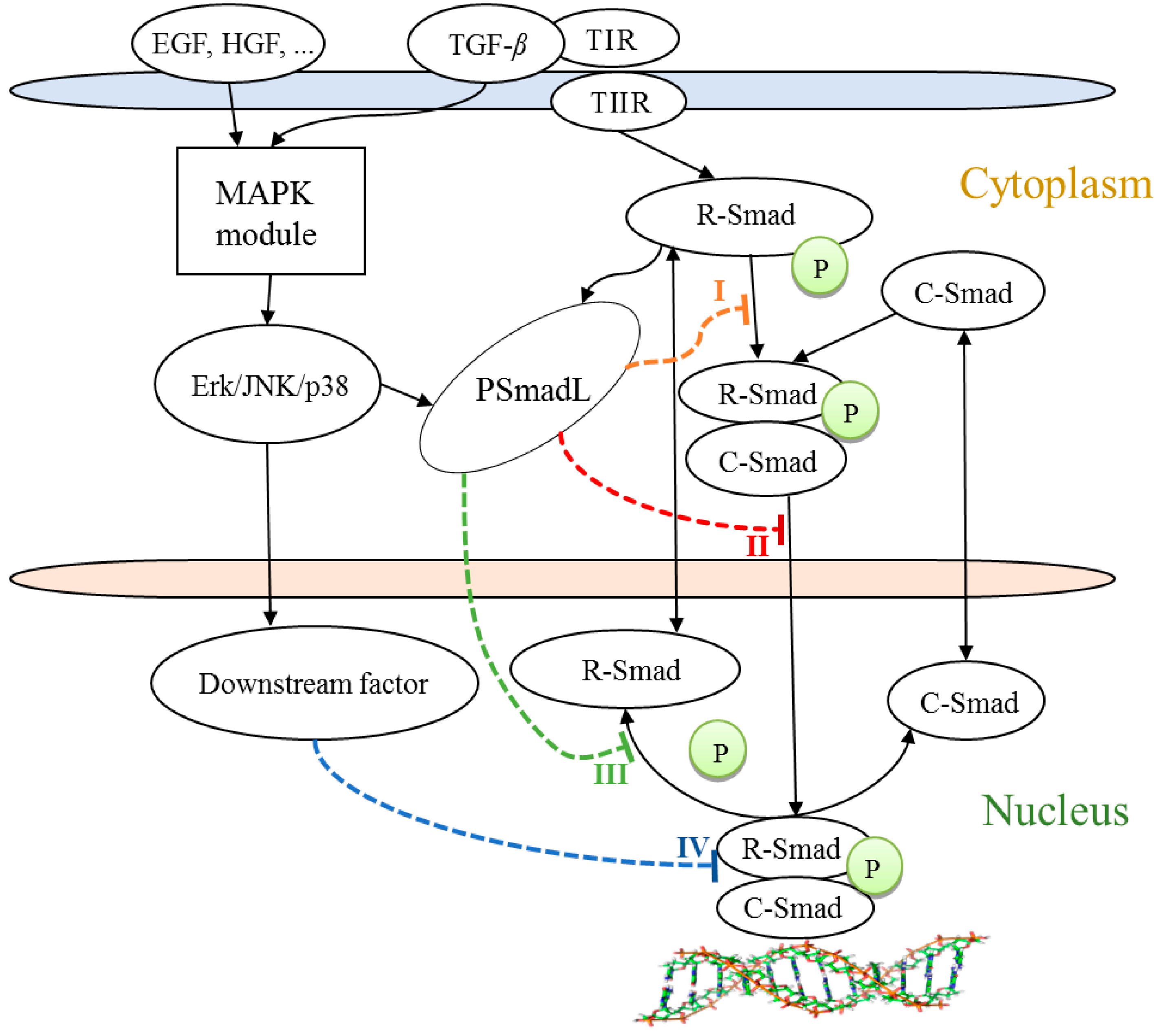
- Activated Erk, p38 or JNK regulates the translocation of the Smad complex from the cytoplasm to the nucleus [10].
- Activated Erk, p38 or JNK phosphorylates R-Smad in the linker region. This phosphorylated Smad (PSmadL) translocates into the nucleus and then inhibits the dephosphorylation, which is linked to the dissociation of the nuclear Smad complex [12].
- The nuclear transcription factors of the MAPK pathway, c-Fos and c-Jun, bind to the nuclear Smad complex to regulate its transcription activity [9].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Literatures | I | II | III | IV | Details | Assumed Mechanism |
|---|---|---|---|---|---|---|
| Hough et al. [1] | √ | Experimental paper, PSmad2/3 level and gene expression were enhanced in mesenchymal cells | PSmadL stabilizes R-Smad phosphorylation | |||
| Derynck et al. [2] | √ | √ | √ | Review paper, Transcription activity was inhibited in hyperactive Ras cells and was enhanced in normal cells | MAPK pathway changes R-Smad phosphorylation, association with Smad4 and nuclear translocation | |
| Javelaud et al. [8] | * | * | * | √ | Review paper, Smad signaling was enhanced or inhibited | PSmadL inhibits or enhances nuclear translocation |
| Inoki et al. [9] | * | * | * | √ | Experimental paper, gene expression was enhanced in mesangial cells | MAPKs regulate the nuclear translocation |
| Guo et al. [10] | * | * | * | √ | Review paper: Smad signaling was enhanced or inhibited | MAPKs phosphorylate R-Smad to control their intracellular redistribution |
| Hayashida et al. [11] | √ | √ | Experimental paper, PSmad2/3 level was enhanced in human mesangial cells | MAPKs increase the R-Smad phosphorylation and its association with Smad4 | ||
| Funaba et al. [12] | √ | Experimental paper, transcription activity was enhanced in mink lung epithelial cells and COS7 cells | MAPKs increase the half-life of Smad2 and the amount of Smad complex | |||
| Burch et al. [13] | * | Experimental paper, transcription activity was enhanced in vascular smooth-muscle cells | PSmadL accumulates in nucleus and upregulates transcription | |||
| Kretzschmar et al. [14] | √ | Experimental paper, nuclear PSmad2/3 level and transcription activity was inhibited in oncogenic Ras mammary and lung epithelial cells | PSmadL attenuates R-Smad nuclear accumulation | |||
| Chapnick et al. [19] | √ | √ | Review paper, Smad signaling was inhibited in cancer cells | PSmadL attenuates R-Smad phosphorylation | ||
| Zhang [25] | √ | √ | Review paper, Smad signaling was inhibited or enhanced | PSmadL attenuates R-Smad to inhibit its association with Smad4 | ||
| Blanchette et al. [27] | * | * | * | √ | Experimental paper, nuclear Smad2 level and gene expression were enhanced in mammalian fur cell | MAPKs increase nuclear translocation |
| Massagué [31] | √ | * | * | √ | Review paper, Smad signaling was inhibited by Erk in I, IV and enhanced by p38 and JNK in IV | MAPKs attenuates Smad accumulation in nucleus |
2.4. Global Sensitivity Analysis


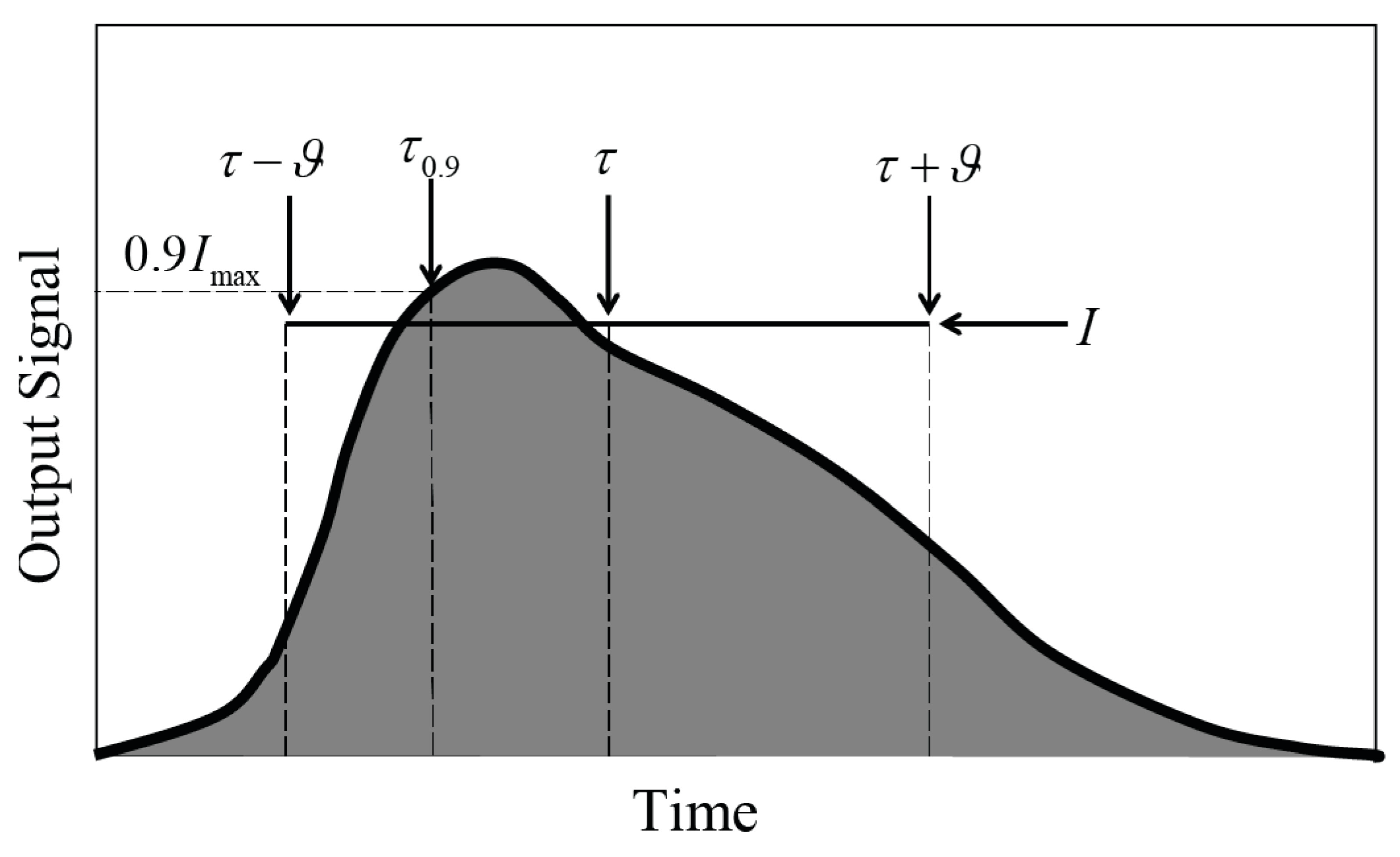
2.5. Measures for Evaluating Output Signals




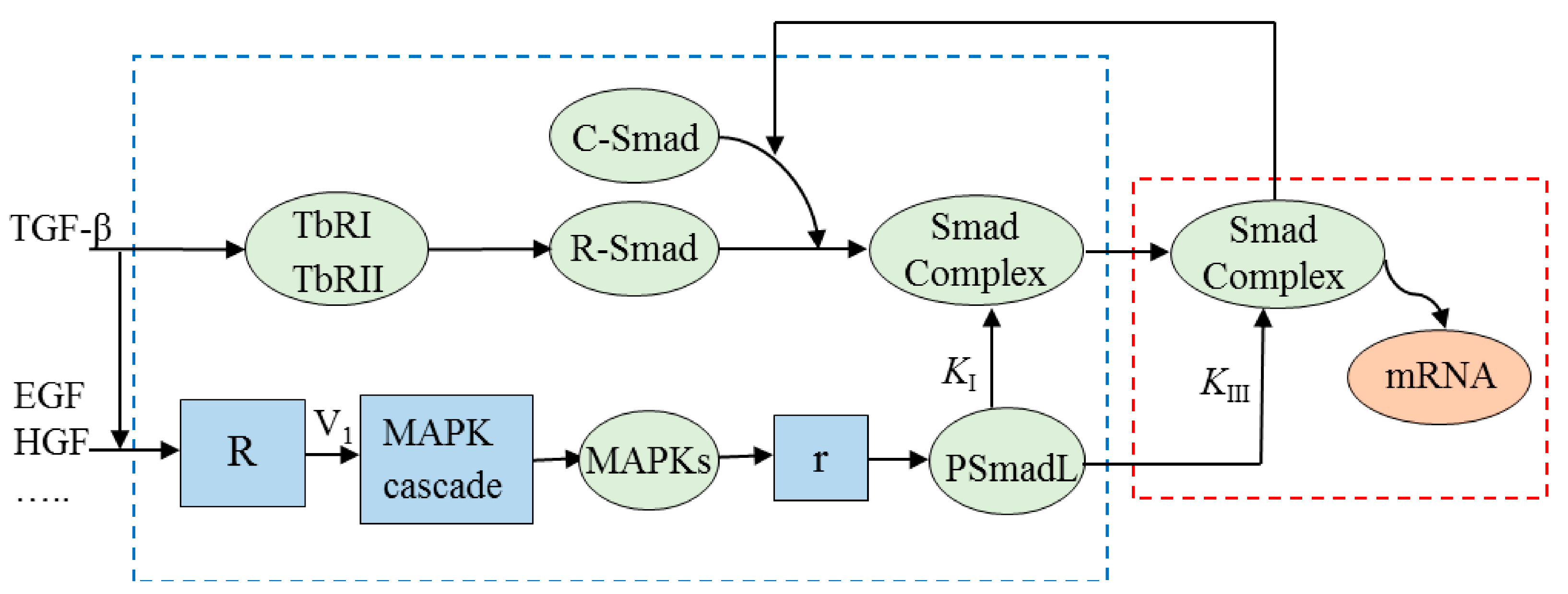
3. Model Development
3.1. Potential Crosstalk Mechanisms
3.2. Modeling Potential Crosstalk Mechanisms


 is the original dephosphorylation rate, equal to 0.1174 min−1 [6].
is the original dephosphorylation rate, equal to 0.1174 min−1 [6].3.3. Integrating the Individual Components of the Model

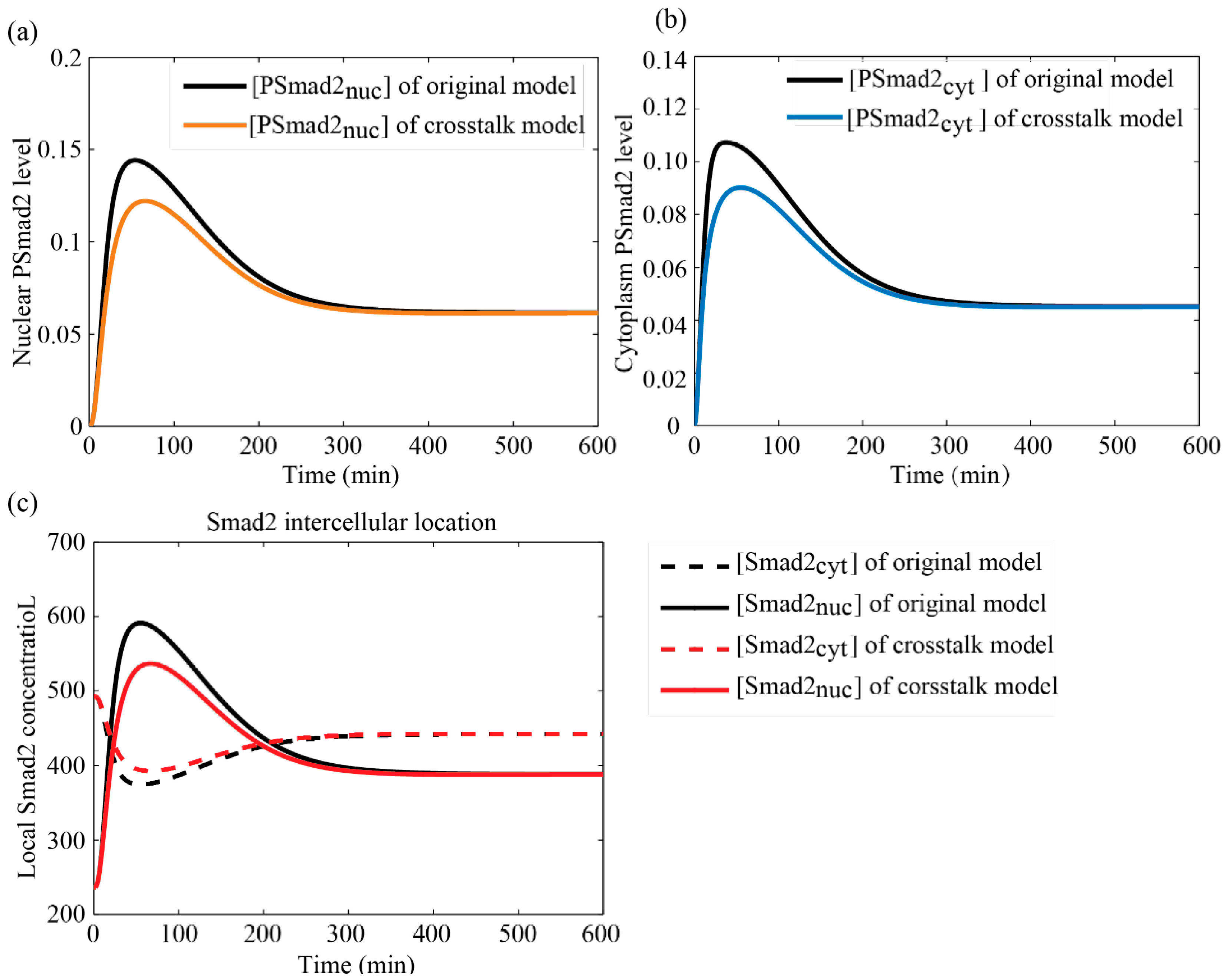
3.4. Simulation Results
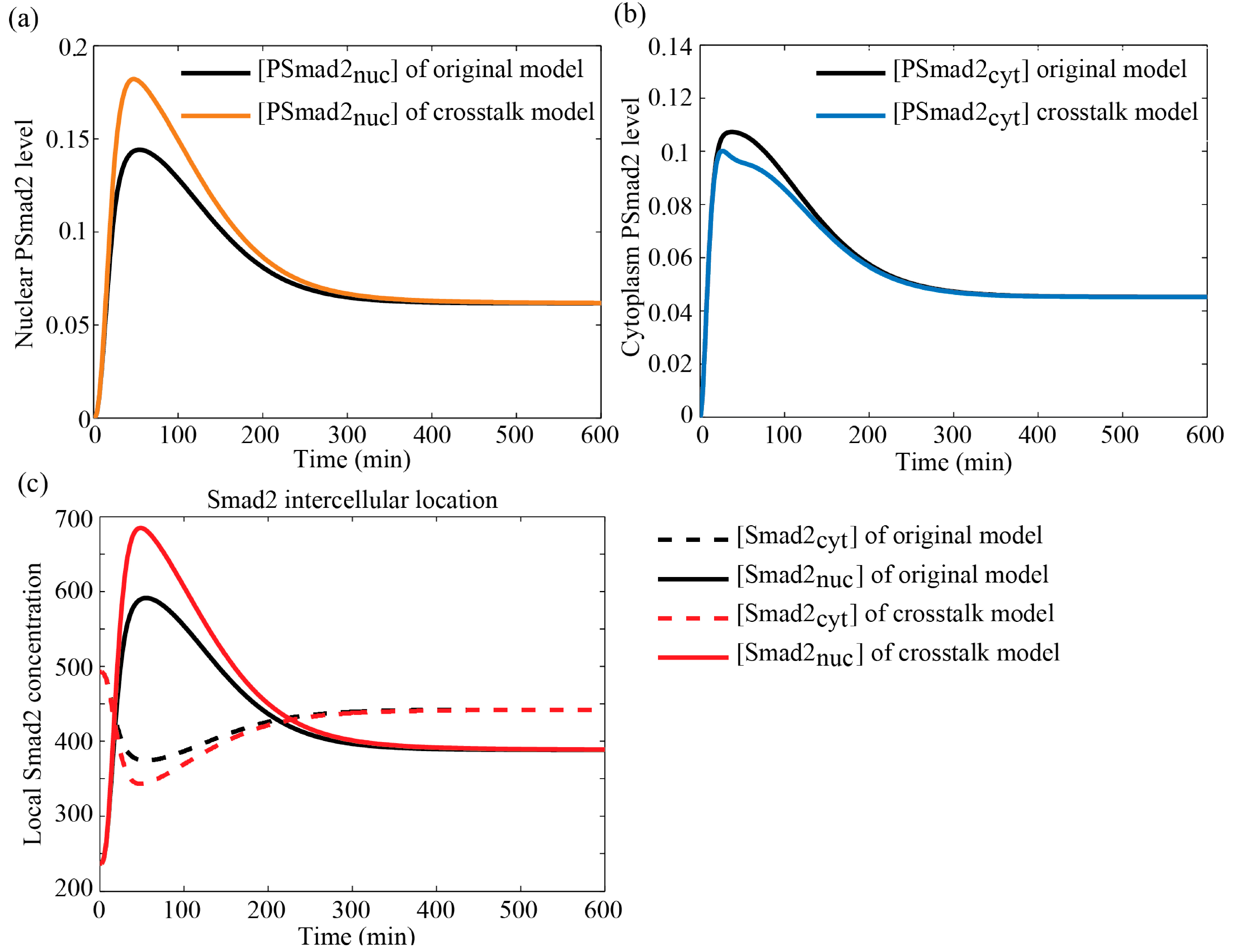
3.4.1. Comparisons between Crosstalk Model and Original Model


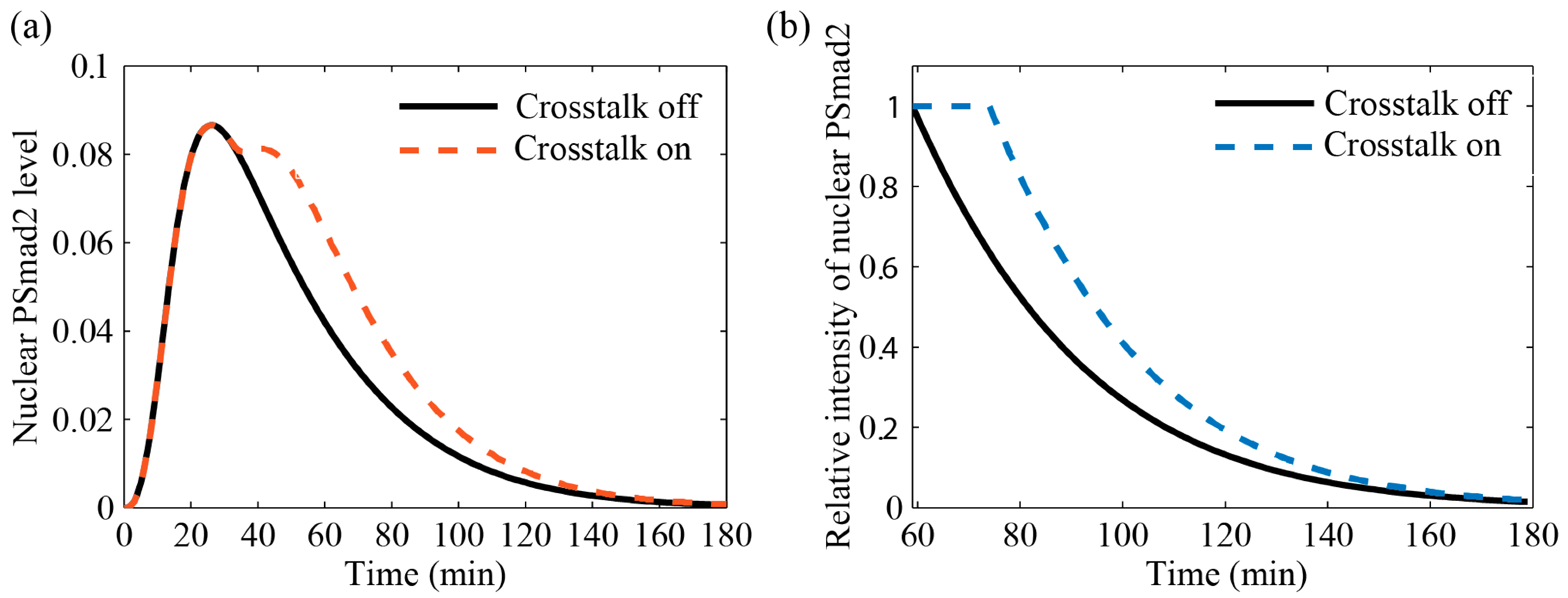
3.4.2. Comparison with Literature Data

4. Results and Discussion
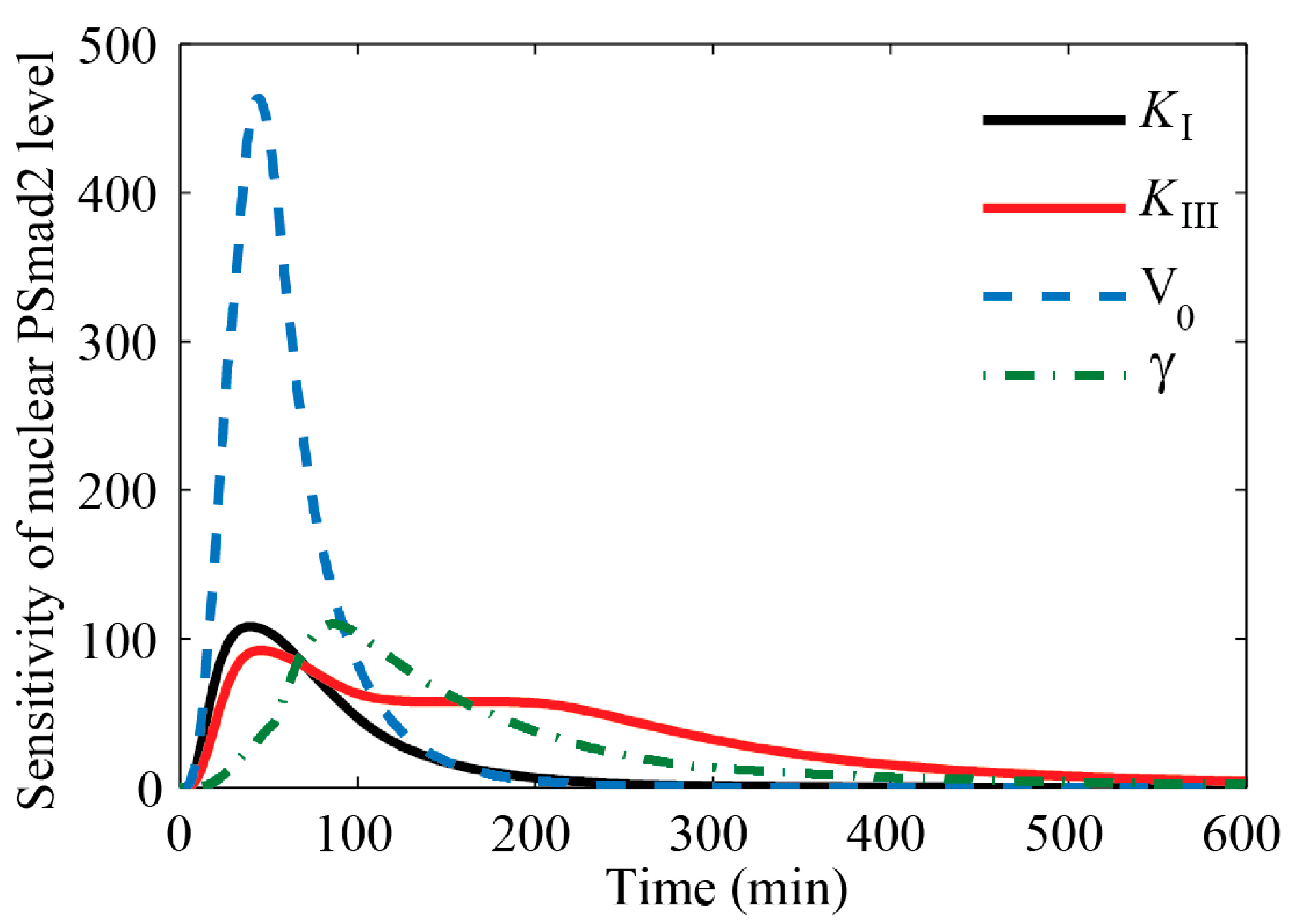
4.1. Sensitivity Analysis of Key Parameters

4.2. Balance between Two Competing Effects Defines the Specific Cell Responses
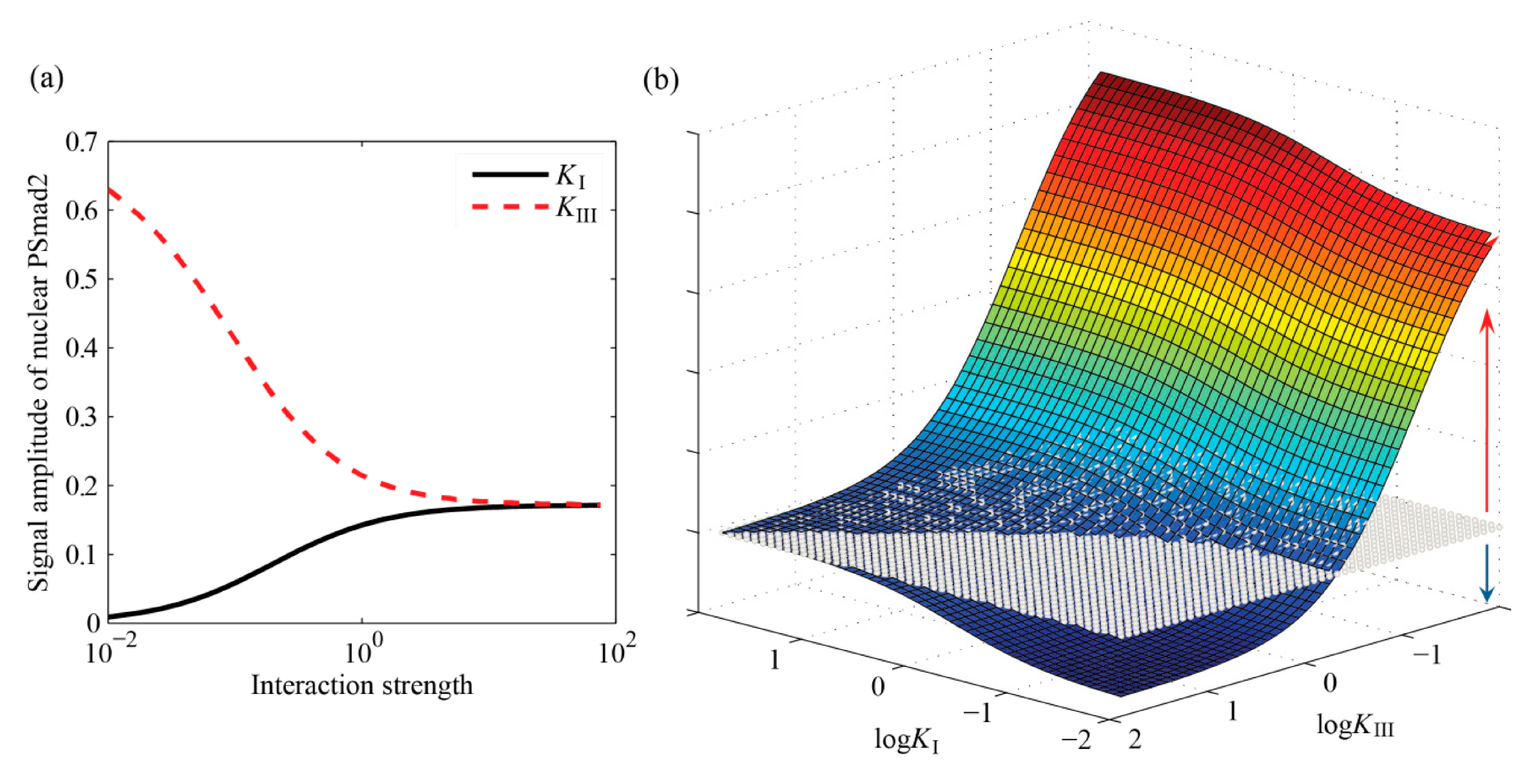
4.3. Effects of Interaction Parameters on Cell Responses
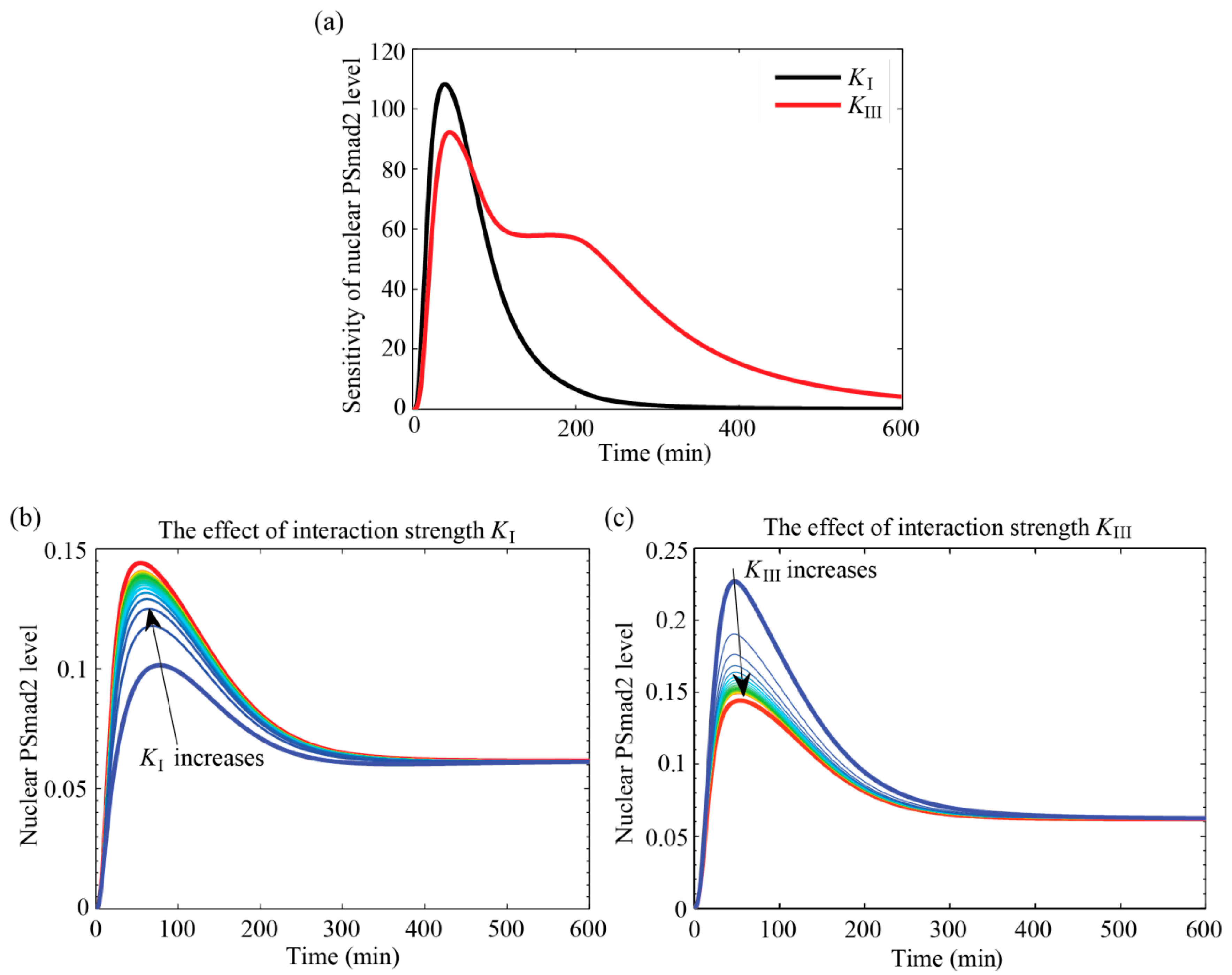
4.3.1. Interaction Parameters Are Identifiable and have Distinct Effects on the Outputs

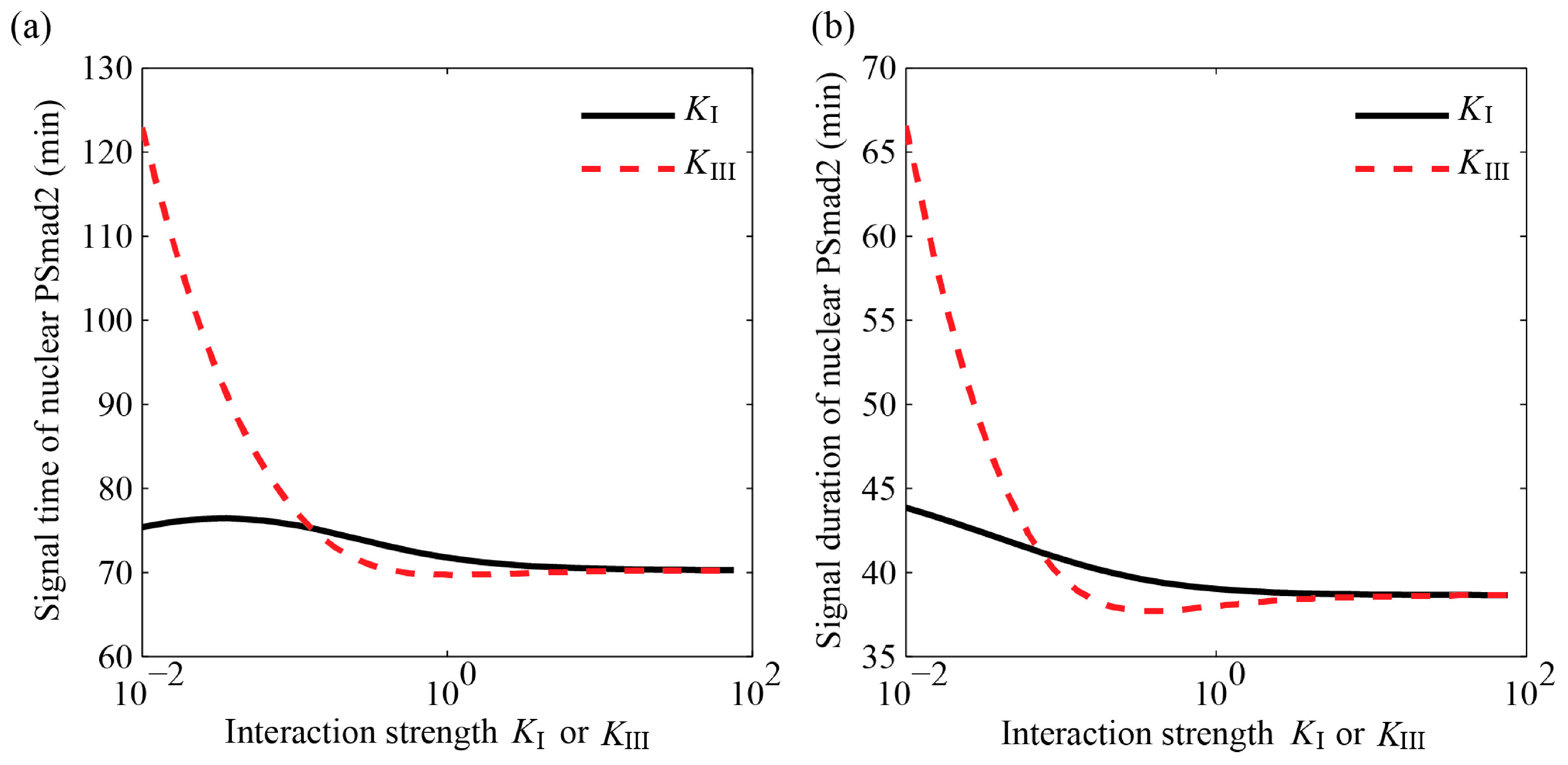
4.3.2. MAPK Pathway Extends the TGF-β/Smad Signal Transduction Duration, Which Is More Strongly Affected by Values of KIII than KI

4.3.3. Specific Outcomes Can Be Described through Appropriate Choices for KI and KIII

5. Conclusions
Acknowledgments
Author Contributions
Appendix

























| Parameter | Biological Meaning | Value |
|---|---|---|
| vT1R | type I receptor production rate constant | 0.0103 nM·min−1 |
| vT2R | type II receptor production rate constant | 0.02869 nM·min−1 |
| kiEE | internalization rate constant of receptor from cell surface to early endosome | 0.33 min−1 |
| krEE | recycling rate constant of receptor from early endosome to cell surface | 0.033 min−1 |
| kicave | internalization rate constant of receptor from cell surface to caveolar lipid-raft | 0.33 min−1 |
| krcave | recycling rate constant of receptor from caveolar lipid-raft to cell surface | 0.03742 min−1 |
| kcd | constitutive degradation rate constant for ligand-receptor complex in early endosome | 0.005 min−1 |
| kLCR | ligand-receptor complex formation rate constant from TGF-β and receptors | 2197 N·m2·min−1 |
| klid | ligand induced degradation rate constant for ligand-receptor complex in caveolar lipid-raft | 0.02609 min−1 |
 | constitutive degradation rate constant for type I receptor in early endosome | 0.005 min−1 |
 | constitutive degradation rate constant for type II receptor in early endosome | 0.025 min−1 |
 | nuclear import rate constant for Smad2 | 0.16 min−1 |
 | nuclear export rate constant for Smad2 | 1 min−1 |
 | nuclear import rate constant for Smad4 | 0.08 min−1 |
 | nuclear export rate constant for Smad4 | 0.5 min−1 |
| kSmads_complex | formation rate constant for the phosphorylated Smad complex | 6.85 × 10−5 n·M2·min−1 |
 | nuclear import rate constant for the phosphorylated Smad complex | 0.16 min−1 |
 | dissociation rate constant for the nuclear phosphorylated Smad complex | 0.1174 min−1 |
| VC/Vn | ratio of cytoplasmic to nuclear volume | 3 |
| V0/Vextra | ratio of cytoplasmic volume to the average extracellular medium volume per cell | 0.001 |
| V0 | the concentration of activated receptor MKKKK at t = t0 | 0.18 nM·s−1 |
| γ | the characteristic time of activated receptor MKKKK | 0.004 |
| t0 | the delayed time of receptor response to extracellular stimuli | 0 |
| KI | interaction strength | 5 |
| KIII | interaction strength | 5 |
| Variable | Biological Meaning | Initial Value (nM) |
|---|---|---|
| MKKK | MAPK kinase kinase | 100 |
| MKKKP | Singly phosphorylated MKKK | 0 |
| MKK | MAPK kinase | 300 |
| MKKP | Singly phosphorylated MKK | 0 |
| MKKPP | doubly phosphorylated MKK | 0 |
| MAPK | MAP kinase | 300 |
| MAPKP | Singly phosphorylated MAPK | 0 |
| MAPKPP | doubly phosphorylated MAPK | 0 |
| T1Rsurf | type I receptor at cell surface | 0.237 |
| T1Rcave | type I receptor in caveolar lipid-raft | 2.092 |
| T1REE | type I receptor in early endosome | 1.148 |
| T2Rsurf | type II receptor at cell surface | 0.202 |
| T2Rcave | type II receptor in caveolar lipid-raft | 1.778 |
| T2REE | type II receptor in early endosome | 1.148 |
| LRCsurf | ligand-receptor complex at cell surface | 0 |
| LRCcave | ligand-receptor complex in caveolar lipid-raft | 0 |
| LRCEE | ligand-receptor complex in early endosome | 0 |
| Smad2c | Smad2 in the cytoplasm | 492.61 |
| Smad2n | Smad2 in the nucleus | 236.45 |
| Smad4c | Smad4 in the cytoplasm | 1149.4 |
| Smad4n | Smad4 in the nucleus | 551.72 |
| Smads_complexc | Smad complex in the cytoplasm | 0 |
| Smads_complexn | Smad complex in the nucleus | 0 |
| TGF-β | TGF-β in the extracellular medium | 0.080 |
Conflicts of Interest
References
- Hough, C.; Radu, M.; Doré, J.J. Tgf-β induced Erk phosphorylation of smad linker region regulates smad signaling. PLoS One 2012, 7, e42513. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Kyprianou, N.; Isaacs, J.T. Expression of transforming growth factor-β in the rat ventral prostate during castration-induced programmed cell death. Mol. Endocrinol. 1989, 3, 1515–1522. [Google Scholar] [CrossRef]
- Guo, Y.; Kyprianou, N. Restoration of transforming growth factor β signaling pathway in human prostate cancer cells suppresses tumorigenicity via induction of caspase-1-mediated apoptosis. Cancer Res. 1999, 59, 1366–1371. [Google Scholar]
- Zhu, B.; Fukada, K.; Zhu, H.; Kyprianou, N. Prohibitin and cofilin are intracellular effectors of transforming growth factor β signaling in human prostate cancer cells. Cancer Res. 2006, 66, 8640–8647. [Google Scholar]
- Zi, Z.; Klipp, E. Constraint-based modeling and kinetic analysis of the Smad dependent TGF-β signaling pathway. PLoS One 2007, 2, e936. [Google Scholar] [CrossRef]
- Tian, X.-J.; Zhang, H.; Xing, J. Coupled reversible and irreversible bistable switches underlying TGFβ-induced epithelial to mesenchymal transition. Biophys. J. 2013, 105, 1079–1089. [Google Scholar] [CrossRef]
- Javelaud, D.; Mauviel, A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-β: Implications for carcinogenesis. Oncogene 2005, 24, 5742–5750. [Google Scholar] [CrossRef]
- Inoki, K.; Haneda, M.; Ishida, T.; Mori, H.; Maeda, S.; Koya, D.; Sugimoto, T.; Kikkawa, R. Role of mitogen-activated protein kinases as downstream effectors of transforming growth factor-β; in mesangial cells. Kidney Int. 2000, 58, S76–S80. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.-F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 2008, 19, 71–88. [Google Scholar]
- Hayashida, T.; Decaestecker, M.; Schnaper, H.W. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-β-dependent responses in human mesangial cells. FASEB J. 2003, 17, 1576–1578. [Google Scholar]
- Funaba, M.; Zimmerman, C.M.; Mathews, L.S. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J. Biol. Chem. 2002, 277, 41361–41368. [Google Scholar]
- Burch, M.L.; Yang, S.N.; Ballinger, M.L.; Getachew, R.; Osman, N.; Little, P.J. TGF-β stimulates biglycan synthesis via p38 and ERK phosphorylation of the linker region of Smad2. Cell. Mol. Life Sci. 2010, 67, 2077–2090. [Google Scholar] [CrossRef]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massague, J. A mechanism of repression of TGF-β/Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef]
- Kretzschmar, M.; Doody, J.; Massagu, J. Opposing BMP and EGF signalling pathways converge on the TGF-β family mediator Smad1. Nature 1997, 389, 618–622. [Google Scholar] [CrossRef]
- Oft, M.; Peli, J.; Rudaz, C.; Schwarz, H.; Beug, H.; Reichman, E. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996, 10, 2462–2477. [Google Scholar] [CrossRef]
- Calonge, M.A.J.; Massagué, J. Smad4/DPC4 silencing and hyperactive Ras jointly disrupt transforming growth factor-β antiproliferative responses in colon cancer cells. J. Biol. Chem. 1999, 274, 33637–33643. [Google Scholar] [CrossRef]
- Massagué, J.; Chen, Y.-G. Controlling TGF-β signaling. Genes Dev. 2000, 14, 627–644. [Google Scholar]
- Chapnick, D.A.; Warner, L.; Bernet, J.; Rao, T.; Liu, X. Partners in crime: The TGF-β and MAPK pathways in cancer progression. Cell Biosci. 2011. [Google Scholar] [CrossRef]
- Heinrich, R.; Neel, B.G.; Rapoport, T.A. Mathematical models of protein kinase signal transduction. Mol. Cell 2002, 9, 957–970. [Google Scholar] [CrossRef]
- Saez-Rodriguez, J.; Kremling, A.; Conzelmann, H.; Bettenbrock, K. Modular analysis of signal transduction networks. Control Syst. IEEE 2004, 24, 35–52. [Google Scholar]
- Kholodenko, B.N. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur. J. Biochem. 2000, 267, 1583–1588. [Google Scholar] [CrossRef]
- Schoeberl, B.; Eichler-Jonsson, C.; Gilles, E.D.; Müller, G. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat. Biotechnol. 2002, 20, 370–375. [Google Scholar] [CrossRef]
- Hartsough, M.T.; Mulder, K.M. Transforming growth factor β activation of p44mapk in proliferating cultures of epithelial cells. J. Biol. Chem. 1995, 270, 7117–7124. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2008, 19, 128–139. [Google Scholar] [CrossRef]
- Gui, T.; Sun, Y.; Shimokado, A.; Muragaki, Y. The roles of mitogen-activated protein kinase pathways in TGF-β-induced epithelial-mesenchymal transition. J. Signal Transduct. 2012, 2012. [Google Scholar] [CrossRef]
- Blanchette, F.; Rivard, N.; Rudd, P.; Grondin, F.; Attisano, L.; Dubois, C.M. Cross-talk between the p42/p44 MAP kinase and Smad pathways in transforming growth factor β1-induced furin gene transactivation. J. Biol. Chem. 2001, 276, 33986–33994. [Google Scholar]
- Pessah, M.; Prunier, C.; Marais, J.; Ferrand, N.; Mazars, A.; Lallemand, F.; Gauthier, J.M.; Atfi, A. c-jun interacts with the corepressor TG-interacting factor (TGIF) to suppress Smad2 transcriptional activity. Proc. Natl. Acad. Sci. USA 2001, 98, 6198–6203. [Google Scholar] [CrossRef]
- Suzuki, K.; Wilkes, M.C.; Garamszegi, N.; Edens, M.; Leof, E.B. Transforming growth factor β signaling via Ras in mesenchymal cells requires p21-activated kinase 2 for extracellular signal-regulated kinase-dependent transcriptional responses. Cancer Res. 2007, 67, 3673–3682. [Google Scholar]
- Liberati, N.T.; Datto, M.B.; Frederick, J.P.; Shen, X.; Wong, C.; Rougier-Chapman, E.M.; Wang, X.F. Smads bind directly to the Jun family of AP-1 transcription factors. Proc. Natl. Acad. Sci. USA 1999, 96, 4844–4849. [Google Scholar]
- Massagué, J. How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Chu, Y.; Hahn, J. Parameter set selection via clustering of parameters into pairwise indistinguishable groups of parameters. Ind. Eng. Chem. Res. 2008, 48, 6000–6009. [Google Scholar] [CrossRef]
- Dai, W.; Bansal, L.; Hahn, J.; Word, D. Parameter set selection for dynamic systems under uncertainty via dynamic optimization and hierarchical clustering. AIChE J. 2014, 60, 181–192. [Google Scholar] [CrossRef]
- Morris, M.D. Factorial sampling plans for preliminary computational experiments. Technometrics 1991, 33, 161–174. [Google Scholar] [CrossRef]
- Martin, M.M.; Buckenberger, J.A.; Jiang, J.; Malana, G.E.; Knoell, D.L.; Feldman, D.S.; Elton, T.S. TGF-β1 stimulates human AT1 receptor expression in lung fibroblasts by cross talk between the Smad, p38 MAPK, JNK, and PI3K signaling pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L790–L799. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, J.; Dai, W.; Hahn, J. Mathematical Modeling and Analysis of Crosstalk between MAPK Pathway and Smad-Dependent TGF-β Signal Transduction. Processes 2014, 2, 570-595. https://doi.org/10.3390/pr2030570
Liu J, Dai W, Hahn J. Mathematical Modeling and Analysis of Crosstalk between MAPK Pathway and Smad-Dependent TGF-β Signal Transduction. Processes. 2014; 2(3):570-595. https://doi.org/10.3390/pr2030570
Chicago/Turabian StyleLiu, Ji, Wei Dai, and Juergen Hahn. 2014. "Mathematical Modeling and Analysis of Crosstalk between MAPK Pathway and Smad-Dependent TGF-β Signal Transduction" Processes 2, no. 3: 570-595. https://doi.org/10.3390/pr2030570