
Modeling Biofilms: From Genes to Communities


Department of Mathematical Sciences, Montana State University, Bozeman, MT 59717, USA
Processes 2017, 5(1), 5; https://doi.org/10.3390/pr5010005
Submission received: 8 October 2016
/
Revised: 5 January 2017
/
Accepted: 17 January 2017
/
Published: 23 January 2017
(This article belongs to the Special Issue Microbial Community Modeling: Prediction of Microbial Interactions and Community Dynamics)
Abstract
:Biofilms are spatially-structured communities of different microbes, which have a huge impact on both ecosystems and human life. Mathematical models are powerful tools for understanding the function and evolution of biofilms as diverse communities. In this article, we give a review of some recently-developed models focusing on the interactions of different species within a biofilm, the evolution of biofilm due to genetic and environmental causes and factors that affect the structure of a biofilm.
1. Introduction
Despite the common view of microbes in their free state, pure culture planktonic growth is rarely how microbes exist in nature. Instead, most microbial species in nature live in the form of biofilms, which are described as a multicellular consortium of microbial cells that are attached to a surface and encased in a self-secreted, extracellular polymeric matrix [1,2]. Biofilms are usually heterogeneous in both their spatial structures and component species and interact with the surrounding environment in a complicated way.
Microbial biofilms are ubiquitous in both natural and industrial settings, and bacteria living in a biofilm often behave very differently from their planktonic counterpart. Bacteria inside a biofilm are usually more resistant to antimicrobial agents [3] and usually possess big competitive advantage over bacteria growing in suspension. This means that it is often difficult to remove biofilms efficiently.
Biofilms can cause many severe problems, such as chronic infections, food contamination and equipment damage due to bio-fouling. Biofilms can also be used for good and constructive purposes, such as waste water treatment, heavy metal removal from hazardous waste sites, biofuel production and microbial fuel cells. From a neutral point of view, since much of the microbial biomass appears in the form of biofilm and due to their ability to produce and consume organic materials, the biofilm communities also have a big impact on the global ecosystem and geochemical system.
In order to promote good biofilms and prevent bad biofilms, it is important to understand the mechanisms for biofilm formation, growth and its removal. The development of a biofilm is a complicated process affected by many biological, physical and chemical factors, and understanding it requires both experimental and modeling efforts. Experiments provide directly-measured qualitative or quantitative data of biofilm properties that are of interest, such as cell counts, cell viability, biofilm morphology and EPS structure, nutrient profile, as well as genetic information. A mathematical model translates the conceptual understanding of the biofilm system into mathematical terms, usually by combining the important processes involved, but omitting the less important ones, and the solutions (either analytical or numerical) are obtained by using available mathematical or statistical tools. Since a model can connect different processes and assess their relative importance, modeling results can help us to understand the biofilm system, facilitate experimental design and make predictions that can be tested by experiments. In this sense, progresses made in experimental and modeling research always promote the development of each other.
Mathematical modeling of biofilm started in the 1970s with models studying substrate utilization and mass transport in a homogeneous slab of biofilm [4,5]. In the 1980s, models including multispecies and the non-uniform distribution of different biomass types started to emerge [6,7], but they were still primarily for one space dimension and steady-state growth dynamics. Starting in the 1990s and up to today, aided by the fast advancement in computing power and better understanding of biofilms through experimental data, multidimensional, multispecies, multisubstrate models are being developed to incorporate realistic biofilm morphology, biofilm mechanics, interactions between biofilm and the environment and interactions between different species within a biofilm, as well as various time scales involved in biofilm-related processes [8,9,10,11,12].
There is a rich literature on the review of mathematical models for biofilms. A few representative ones are listed below.
The IWAtask group gives an excellent review of mathematical modeling of biofilms [13]. The book explains the basic steps in creating a mathematical model, emphasizes that the “golden rule” of modeling is that “a model should be as simple as possible, and only as complex as needed” and presents the model derivation based on mass conservation in detail. Models are classified into analytical models (A), Pseudo-Analytic models (PA), Numerical one-dimensional (N1) and multi-dimensional Numerical models (N2 and N3). The features, definitions and equations, as well as the application of each type of model are discussed. The performances of all models for solving three characteristic benchmark problems are compared, which help identify the trade-offs inherent to using different types of models. The book also points out the significance of the definitions and units of model parameters.
Klapper and Dockery [14] discuss how macroscale physical factors might influence the composition, structure and function of ecosystems within microbial communities from the modeling perspective and emphasizes that despite its difficulty and complexity, it is important to include the physical, chemical and biological processes at a variety of time and length scales in the model to fully understand the physiology and ecology of the microbial communities. Specific modeling aspects discussed include quorum sensing, growth, mechanics and antimicrobial tolerance mechanisms.
Wang and Zhang [15] give a chronological review of some biofilm models developed from the 1980s to the early 2000s. Based on their dimensionality, the way in which diffusion is treated and the complexity in terms of the incorporation of the physics, chemistry and biological effects, models are classified into four main categories: one-dimensional continuum models, diffusion-limited aggregate models, continuum-discrete diffusion models and biofilm-fluid coupled models.
In recent years, fast advancement in experimental technologies, such as microscopy and high-throughput sequencing, has provided an abundance of data at both the genetic and community level and helped researchers to understand biofilms much better. In particular, it is a common belief that biofilms should be viewed as spatially-structured communities of microbes, and the structure and function of the communities are determined by both the surrounding environment and the local interactions between different species within biofilms via complex metabolic networks [16,17,18,19,20]. To understand biofilms as a diverse community and the evolution of different species and its genetic causes, mathematical modeling again serves as a powerful tool. Experimental data with a high level of detail provide both opportunities and challenges for researchers working on biofilm modeling. On the one hand, there are more data available to improve conceptual understanding of biofilm and to compare with model predictions; on the other hand, more sophisticated models are demanded in order to accommodate the data. Song et al. [21] give a methodological review on mathematical modeling of microbial community dynamics. Widder et al. [22] address the challenges in building predictive models for understanding the function and dynamics of Microbial Communities (MCs). Several specific examples where model-experiment integration has already resulted in important insights into MC function and structure are discussed. These include inferring species interactions from proximal data, predicting species interactions using stoichiometric models and kinetic models for community dynamics. The conclusion is that addressing this challenge requires close coordination of experimental data collection and method development with mathematical model building.
In this article, we review some recent mathematical models that focus on studying biofilms as a diverse community. The rest of the article is organized as follows. Section 2 discusses the genetic basis for biofilm development based on experimental results and its mathematical modeling with emphasis on the models based on the idea of Quorum Sensing (QS); Section 3 discusses models based on Flux Balance Analysis (FBA) and stoichiometry; Section 4 discusses models based on statistical inference; Section 5 discusses models with novel growth kinetics and the ability to resolve the complex spatial structure of biofilm. Table 1 gives a brief overview of the models discussed in the article.
2. Modeling the Genetic Basis of Biofilm Development
Costerton et al. [3] pointed out that biofilms consist of microcolonies on a surface and that within these microcolonies, the bacteria have developed into organized communities with functional heterogeneity. Clinical characteristics of biofilm infections are discussed, and multiple mechanisms of biofilm resistance to antimicrobial agents are proposed. Furthermore, P. aeruginosa and the chronic lung infections it causes in most patients afflicted with the recessive genetic disease Cystic Fibrosis (CF) are used as a model to reveal information about the molecular and genetic basis of biofilm development. There is evidence [23,24] showing that during the attachment phase of biofilm development, the transcription of specific genes (such as the genes required for the synthesis of the extracellular polysaccharide) is activated. Research on quorum sensing in Gram-negative bacteria [25,26] has shown that acyl homoserine lactone signals are produced by individual bacterial cells. At a critical cell density, these signals can accumulate and trigger the expression of specific sets of genes. Detachment and dispersal of planktonic cells from biofilms could also have a genetic basis. It has been suggested that increased expression of the alginate lyase in the mucoid strain of P. aeruginosa led to alginate degradation and increased cell detachment [27,28]. Antibiotic therapy in patients colonized with P. aeruginosa often gives a measure of relief from symptoms, but fails to cure the basic ongoing infection [29,30]. One interpretation of this is that the antibiotics act on the planktonic cells that are shed by the biofilms, but cannot eliminate the antibiotic-resistant sessile biofilm communities, and the microcolonies of sessile bacteria in the lung act as niduses for the spread of the infection [31,32]. The conclusion from [3] is that the effective control of biofilm infections will require a concerted effort to develop therapeutic agents that target the biofilm phenotype and community signaling-based agents that prevent the formation, or promote the detachment, of biofilms.
Monds and O’Toole [33] give a critical review of the causal basis of biofilm formation and its molecular underpinnings. It discusses the concept of biofilm formation as a developmental process by evaluating experimental data and concludes that the developmental model of biofilm formation must be approached as a model in need of further validation, rather than coveted as a robust platform on which to base scientific inference. Here, the definition of developmental process is a “series of stable and meta-stable changes in the form and function of a cell, where those changes are part of the normal life cycle of the cell” according to [34]. Furthermore, the molecular requirement implicit in a developmental process is that a series of hierarchically-ordered genetic elements control temporal transition through the developmental pathway in response to specific cues. The review starts with the origin of the developmental model of biofilm formation as an analogy with Myxococcus xanthus fruiting-body formation [35], then provides some unequivocal support for the developmental model, including both structural transitions occurring during biofilm formation [36] and various phenotypes with biofilm-specific properties, such as increased antibiotic tolerance [37]. After that, two specific case studies are presented as evidence that bacteria have evolved genetic pathways that serve to directly link environmental cues to the regulation of stage-specific transitions in biofilm formation. One case is that low extracellular phosphate blocks microcolony formation by Pseudomonas fluorescens [38,39]; the other case is intracellular iron as a signal for biofilm maturation [40,41]. However, there is still lack of success in uncovering comprehensive genetic programs specific for the regulation of biofilm development [42,43]. Moreover, the development model also requires that groups of biofilm pathways are connected in a hierarchically-ordered genetic network, and there are causal links between form and function. Folkesson et al. [44] provide valuable evidence in this direction by showing that Escherichia coli biofilms formed by F-plasmid-containing cells were more structured and had increased tolerance to colistin relative to cells without the F-plasmids, but it is still not possible to say that pathways controlling the structural development of an E. coli biofilm are directly coupled with pathways for the formation of a subpopulation of cells with increased tolerance to colistin. Next, two examples are used to demonstrate biofilms as multicellular organisms with functional differentiation and alternative cell fates. The first one by Klausen et al. [45] investigated differential roles for motile and non-motile subpopulations in determining the topology (mushroom-like or flat) of P. aeruginosa biofilms. Burrows [46] gave a thorough review on the twitching motility of P. aeruginosa through Type IV pili (T4P) structure and function. The second one [47] examined the spatiotemporal patterns of cell-specific gene expression in B. subtilis biofilms and indicated that different cell types vary in abundance and location in the biofilm over time. Finally, an alternative experimental model for biofilm formation based on the ecological adaptation of individuals was proposed by Klausen et al. [48]. In this experiment, deterministic responses are integrated with stochastic interactions with the environment to shape biofilm form and function, where biofilm evolution has been driven by the selection for individual competitiveness in complex and dynamic environments.
Many models have been proposed to describe the genetic processes that regulate biofilm development, and a few representative ones are discussed below.
Reed et al. [49] proposed the gene-centric approach for integrating environmental genomics and biogeochemical models. In this model, the production rate or j-th gene is given by:
where is gene abundance (genes per unit volume), is the thermodynamic potential factor accounting for the chemical energy available to drive the metabolism, is the specific growth rate, is the concentration of a reactant or nutrient s, is the half-saturation constant of the reactant or nutrient s, is the concentration of inhibitor x and is the half-saturation constant of inhibitor x. Furthermore, metabolic plasticity, whereby growth via one metabolism can lead to the propagation of functional genes associated with other metabolisms, is incorporated into the model by introducing the following governing equation for the gene abundance:
where is the number of the i-th gene per unit mass of cells that contains this gene, is a probabilistic measure of the co-occurrence of genes i and j within a genome and λ is the mortality rate constant of a gene. Equations (1)–(2) describing the microbial community are coupled to chemical dynamics by usual reaction equations. There are several advantages of this model: most of its parameters are either directly measurable (, ) or easily obtained by calculation (); numerical solutions from the model give gene abundances and chemical concentrations that allow direct comparisons between model predictions and experimental results; and the metabolic plasticity could be important for understanding the complex microbial community dynamics. Zhang et al. [50] developed a theory for the analysis and prediction of the spatial and temporal patterns of gene and protein expression within microbial biofilms based on similar ideas. The theory integrates the phenomena of solute reaction and diffusion, microbial growth, mRNA or protein synthesis, biomass advection and gene transcript or protein turnover. Case studies illustrate the capacity of the theory to simulate heterogeneous spatial patterns and predict microbial activities in biofilms that are qualitatively different from those of planktonic cells.
Genetic changes via horizontal gene transfer often make taxonomic distinction among species obscure; thus, sometimes, it is convenient to characterize the dynamics of microbial communities by different traits. Trait-based models were developed based on this idea and applied for analyzing the diversity of ecosystems. Shipley et al. [51] developed the maximum entropy (MaxEnt) model, and Laughlin et al. [52] developed the the Traitspace model; both have been applied to predict the relative abundance of species for plant communities. Both approaches are based on statistical methods and are composed of three key elements: an underlying trait distribution, a performance filter defining the fitness of traits in different environments and a projection of the performance filter along some environmental gradient. The objective of the modeling is to estimate the relative abundance of the some species in a given environment by incorporating information about individual-level functional traits. The MaxEnt model tends to overestimate the relative abundance of species since it maximizes the evenness of their distribution. On the other hand, the Traitspace model tends to underestimate the relative abundance of species since it is based on Bayesian theory and predicts a low probability of abundances for functional groups that do not pass through environmental filters. Though the trait-based models have not be widely adopted for modeling of the biofilm yet, it is certainly promising to apply the methodology to study biofilm as a microbial community. For example, Lennon et al. [53] have found that certain traits are related to the biofilm-producing capability of strains and identified functional groups of microorganisms that will help predict the structure and functioning of microbial communities under contrasting soil moisture regimes. Furthermore, current biofilm models can provide accurate predictions of the environmental gradients inside the biofilm, which can be used as the input of the trait-based models.
Quorum sensing is the regulation of gene expression in response to fluctuations in cell-population density [54,55,56]. Quorum sensing bacteria produce and release chemical signal molecules called autoinducers, and the concentrations of autoinducers increase as the cell density increases. Once the concentration of an autoinducer reaches a threshold, an alteration in gene expression is triggered. Recent research on many different bacterial species has shown that quorum sensing systems play an important role in regulating the expression of genes involved in biofilm formation, biofilm maturation, biofilm dispersal and detachment [57,58,59]. Naturally, modeling of QS is an important part of the general effort in modeling the genetic processes involved in biofilm development. Ward [60] gives a good review of early mathematical modeling of QS.
The modeling of QS starts with a circuit describing the gene regulation involved in the QS system, which is usually given by a schematic diagram showing all of the genes, autoinducers and the corresponding positive and negative interactions. For example, the las and rhl systems in P. aeruginosa [61] are extensively studied. James et al. [62] and Dockery and Keener [63] pioneered the work on modeling QS at the molecular level. These early QS models use a system of coupled Ordinary Differential Equations (ODEs) to describe the dynamics of the intracellular concentrations of genes (or proteins), autoinducers and substrates, where the reaction kinetics are carefully designed to reflect the interactions within the QS system. Due to their simple forms, these models can be investigated by both numerical and analytical tools, and results suggest that QS works as a biochemical switch between two stable steady states of the system, one with low levels of autoinducer and one with high levels of autoinducer.
The signal production in QS in biofilm can be affected by many physical, chemical and biological factors [64]. Examples include diffusion of nutrients and QS molecules inside the biofilm and mass transfer affected by the hydrodynamics of the bulk fluid and biofilm structure. For example, Kirisits et al. [65] studied the influence of the hydrodynamic environment on QS in a P. aeruginosa biofilm and concluded that the amount of biofilm biomass required for full QS induction of the population increases as the flow rate increases.
These more advanced QS models use either the continuum approach with Partial Differential Equations (PDEs) or the individual-based approach, both capable of capturing the spatial structure of the biofilm and its interaction with the surrounding environment, to study the effect of QS on either the biofilm structure or interactions among species within a biofilm. A continuum model involving QS will be discussed in Section 5, and here, we describe the work of Nadell et al. [66], which implemented detailed simulations using individual-based modeling methods [67,68,69] to investigate evolutionary competitions between strains that differ in their polymer production and quorum-sensing phenotypes. It is known that EPS secretion in the process of biofilm formation is under quorum-sensing control in a number of bacterial model systems in very different ways. For example, P. aeruginosa activates EPS production at high cell density [70]. In contrast, V. cholerae initiates EPS secretion after attaching to a surface and losing flagellar activity, but halts EPS secretion once it reaches its high cell density quorum-sensing threshold [71]. The model presented in [66] focuses on three strains with the following behavior: (1) no polymer secretion and no quorum sensing (); (2) constitutive polymer secretion and no quorum sensing (); and (3) polymer secretion under negative quorum-sensing control, such that EPS secretion stops at high cell density (). Cells consume substrate according to their strain-specific metabolism kinetics and produce additional biomass; all cells secrete an autoinducer without cost and at a constant rate, and cells synthesize EPS only when the local autoinducer concentration is below the quorum-sensing threshold concentration, which is represented by a single dimensionless parameter. Results from Nadell et al. [66] suggest that cells have a competitive advantage over , but only for a limited time window. In contrast, cells suffer an initial disadvantage due to a lower growth rate when competing with cells, then rapidly ascend to a majority in the biofilm and remain there indefinitely. In addition, the strain can invade populations composed mostly of either or cells, but not vice versa. The importance of the work in [66] is that it provides an evolutionary model that can be used to make predictions on the evolution of specific biological outcomes based on the biological constraints, and these predictions can be tested by experiments. In particular, it predicts that pathogenic strains, such as V. cholerae, selected for rapid colonization of, and efficient dispersal from, human hosts or other temporary environments, will exhibit negative quorum-sensing-regulated EPS production. In contrast, upregulation of EPS secretion at high cell density, which focuses resource investment into sustained local competitive ability, is more likely to be favored for organisms occupying specific niches long term, such as P. aeruginosa in chronic infections.
Mathematical tools used for modeling genetic processes related to biofilm include differential equations, statistical methods and individual-based approaches. These models usually enjoy success, but face challenges at the same time. For example, it is challenging to apply the gene-centric model to complex ecological systems since it is not easy to obtain associations between functional genes and reactions. Among many models for studying QS in biofilm systems, most of them focus on upregulation and downregulation of certain genes, and only a few emphasize the effect of QS on biofilm structure, function and its interaction with the environment, which leaves much room for model improvement.
3. Models Based on Flux Balance Analysis and Stoichiometry
Interactions of different species within a biofilm are closely related to the substrate consumption and metabolite exchange, and mathematical models based on FBA are excellent tools for predicting these interactions.
Early work in this direction involves the synthesis of metabolic pathways. Seressiotis and Bailey [72] developed a computer software system for metabolic pathway synthesis, which can be used to identify biochemical pathways, to predict on a qualitative basis the effects of adding or deleting enzymatic activities to or from the cellular environment, to classify pathways with respect to cellular objectives and to extract information about metabolic regulation. Mavrovouniotis et al. [73] extended the work in [72] by including stoichiometric constraints. Schilling et al. [74] gave a review on the development of computer-aided algorithms for the synthesis of metabolic pathways and explained the important algebraic concepts used in pathway analysis, such as null space and convex cone.
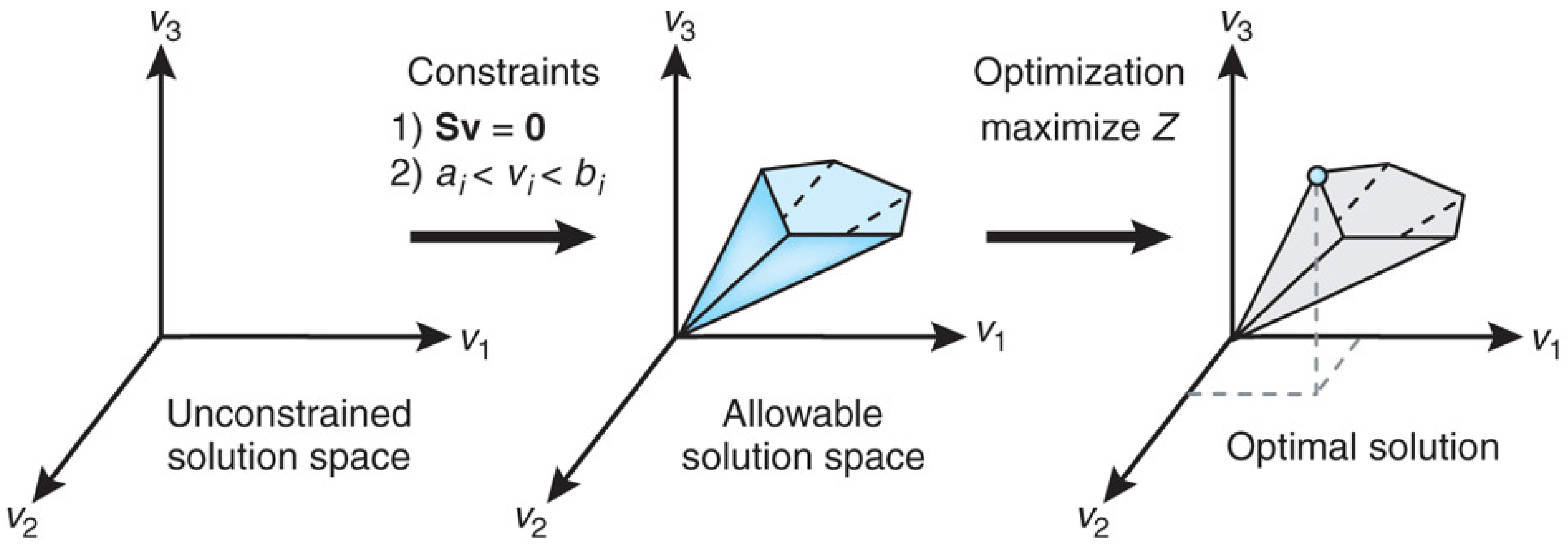
Orth et al. [75] covered the theoretical basis of FBA and provide several practical examples and a software toolbox for performing the calculations. Figure 1 from [75] explains the conceptual basis of FBA as a constraint optimization problem. In FBA, metabolic reactions are represented as a stoichiometric matrix () of size . Each row of represents one unique compound (for a system with m compounds), and each column represents one reaction (n reactions). The entry of is the stoichiometric coefficient denoting the number of moles of the i-th compound formed in the j-th reaction. The coefficient is positive if the metabolite is produced, negative if the metabolite is consumed and zero if the metabolite does not participate in a particular reaction. The flux through all of the reactions in a network is represented by the vector of length n, and the concentrations of all metabolites are represented by the vector of length m. The system of mass balance equations at steady state () gives a set of equality constraints . Each reaction also has upper and lower bounds, which gives a set of inequality constraints on the flux components, namely . FBA seeks to maximize or minimize an objective function , which can be any linear combination of fluxes, where is a vector of weights indicating how much each reaction (such as the biomass reaction when simulating maximum growth) contributes to the objective function. The constraint optimization problem is usually solved by linear programming. It is important to note that the stoichiometric matrix can be directly constructed from knowledge of an organism’s metabolic genotype, which in turn can be efficiently determined from the results of genome annotation.
Models based on FBA are used to study the role of interspecies exchange of metabolites in determining the spatiotemporal dynamics of microbial communities. Harcombe et al. [76] developed a model that integrates dynamic Flux Balance Analysis (dFBA) [77] with diffusion on a lattice and applied it to engineered communities. Simulations from the model predict the species ratio to which a two-species (E. coli/S. enterica) mutualistic consortium converges, the equilibrium composition of an engineered three-member (E. coli/S. enterica/M. extorquens AM1) community and the beneficial effect of a competitor in spatially-structured mutualism. All predictions are confirmed by experimental results. The strength of the model is highlighted by the fact that it requires very few free parameters and no a priori assumptions on whether or how species would interact.
Phalak et al. [78] developed a model to investigate the multispecies metabolism of a biofilm consortium comprised of two common chronic wound isolates: the aerobe P. aeruginosa and the facultative anaerobe S. aureus. The model combines genome-scale metabolic reconstructions for growth rates via FBA and partial differential equations for metabolite diffusion and provides both temporal and spatial predictions with genome-scale resolution. In particular, the two-species system was predicted to support a maximum biofilm thickness much greater than P. aeruginosa alone, but slightly less than S. aureus alone, suggesting an antagonistic metabolic effect of P. aeruginosa on S. aureus.
Sigurdsson et al. [79] used a systems biology approach to identify candidate drug targets for biofilm-associated P. aeruginosa. This study employed the published reconstruction of P. aeruginosa iMO1056 [80] and used FBA to simulate different medium and oxygen conditions. The effect of single and double gene deletion on bacterial growth in planktonic and biofilm-like environmental conditions was investigated. Condition-dependent genes were found that could be used to slow growth specifically in biofilm-associated P. aeruginosa. In particular, eight gene pairs were found to be synthetically lethal in oxygen-limited environments, and these gene sets may serve as metabolic drug targets to combat biofilm-associated P. aeruginosa. Results from [79] show that FBA can be used to determine key metabolic differences between planktonic and biofilm colonies and shed light on searching for novel drug targets.
The application of models based on FBA in studying biofilm as a microbial community is very promising, but also challenging. In particular, efficient and standardized methods are necessary for generating reliable stoichiometric models when a large number of species is involved. Furthermore, it is important to develop mathematical tools that can effectively incorporate omics-based metabolic pathway information into kinetic functions, which can be used directly in kinetic growth models. The cybernetic approach developed by Song et al. sheds some light on future research in this direction, and a review of this approach is given by [81].
4. Models Based on Statistical Inference
Species within a biofilm rarely live in isolation; instead, they often coexist and have complex interactions that affect the community structure and function [82,83,84]. The types of interactions include win-win (mutualism), win-zero (commensalism), win-lose (predation , parasitism), zero-lose (amensalism) and lose-lose (competition). Community-wide information on microbial interactions can be obtained using statistical inference based on correlations between taxon abundances from high-throughput sequence data [85,86].
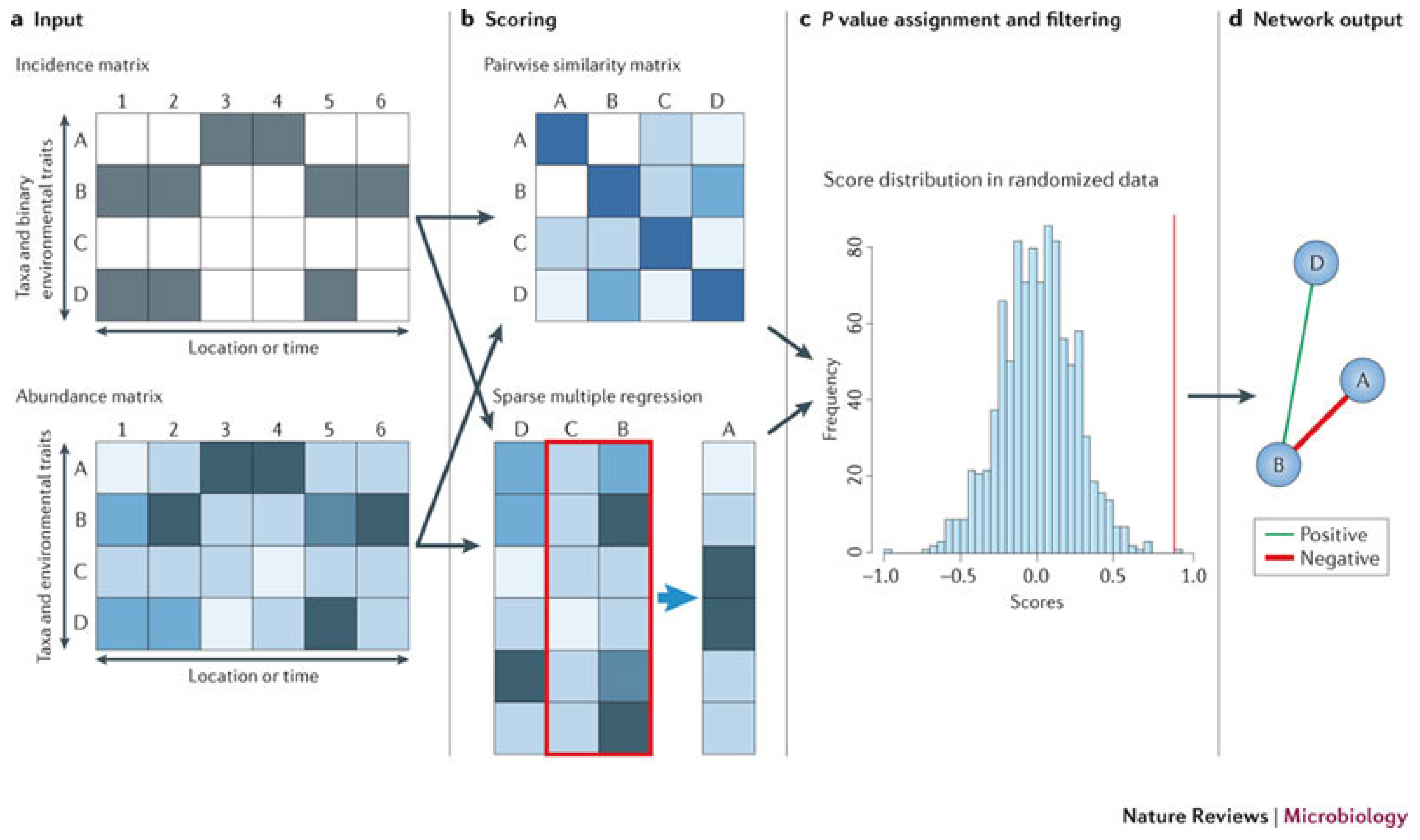
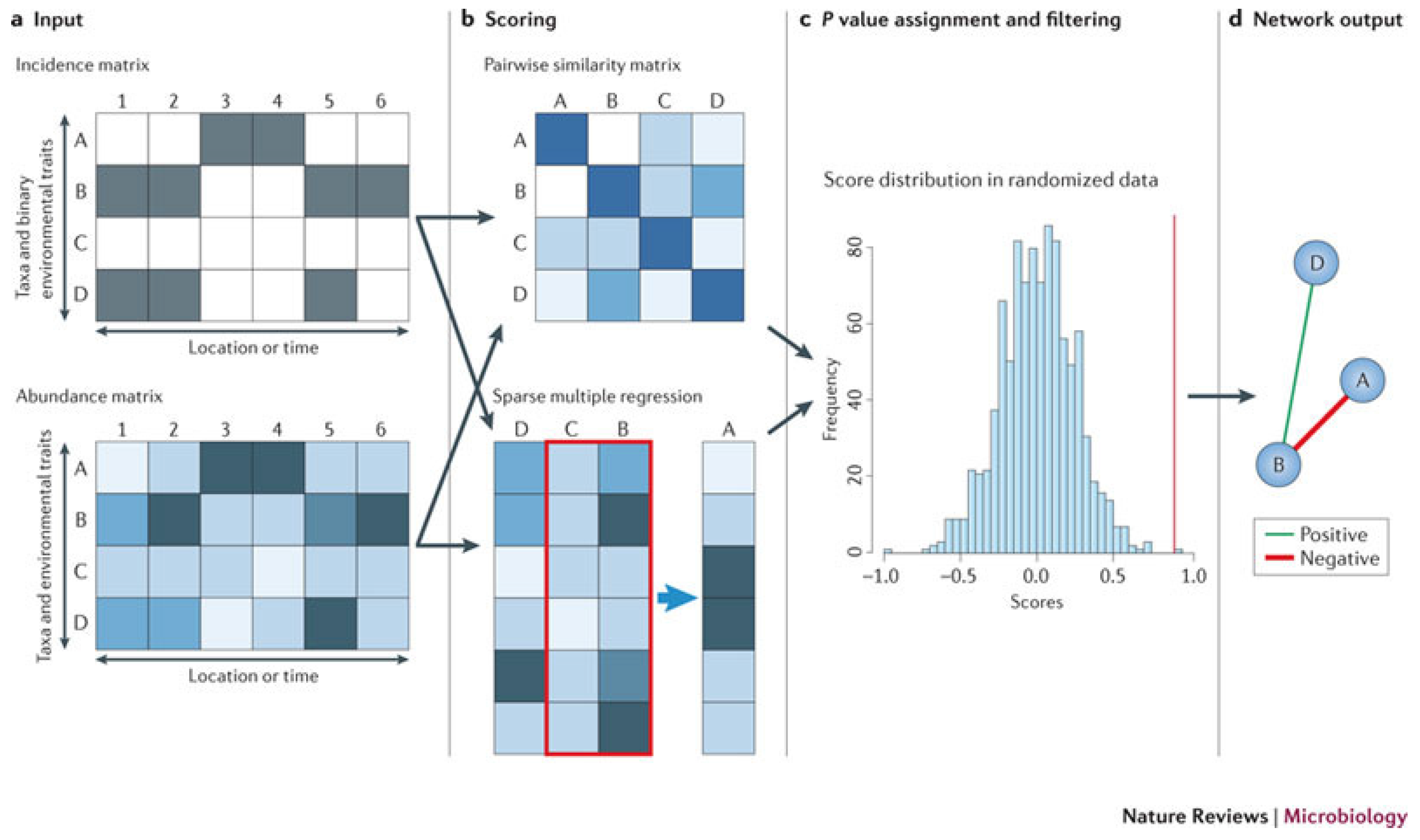
Faust and Raes [87] reviewed strategies to construct community models from abundance data and use the models to predict the outcome of community alterations and the effects of perturbations. The prediction of microbial association networks from abundance data is known as the network inference problem [88]. The network inference methods can be classified into two categories: the similarity-based methods, which predict pairwise relationships, and the regression- and rule-based methods, which predict complex relationships. Figure 2 from [87] explains the principle of similarity- and regression-based network inference. Network inference starts from an incidence or an abundance matrix. Pairwise scores between two taxa are then computed using a suitable similarity or distance measure, and relationships involving more than two taxa are detected by either multiple regression or association rule mining [89]. Then, a random score distribution is generated by repeating the scoring step, and the P-value is computed to measure the significance of the predicted relationship. Finally, taxon pairs with P-values below a given threshold are visualized as a network. Inferred networks can be considered as static models of microbial communities, which describes the community status at a particular time. However, time series data obtained by network inference methods can provide important input (such as growth rates or interaction strengths) for dynamic models of microbial interactions; see [90] for modeling of cheese fermentation community interactions with generalized Lotka–Volterra equations. Network inference has several strengths. It is generic; it can integrate different data types; and it can identify community properties that are encoded in the network structure. However, network inference also suffers from several pitfalls, such as normalization, similarity measure biases, the choice of appropriate null models and multiple testing issues. Despite these pitfalls, network inference is a versatile tool for studying microbial interactions, can be used to build dynamic models that can predict community stability, alternative stable states and microbial succession and ultimately shed light on the manipulation of microbial communities to enhance the abundances of beneficial species and to suppress harmful ones.
Faust et al. [91] applied an ensemble method based on multiple similarity measures [92] in combination with Generalized Boosted Linear Models (GBLMs) [93] to taxonomic marker (16S rRNA gene) profiles of the Human Microbiome Project (HMP) cohort [94], resulting in a global network of 3005 significant co-occurrence and co-exclusion relationships between 197 clades occurring throughout the human microbiome. Analysis of the network revealed strong organization of the human microbiota into body area niches, mostly among closely-related individual body sites representing microbial habitats. For example, Fusobacterium species can bridge organisms in the development and maturation of oral biofilms by co-aggregation through physical contact, allowing a more complex use of resources, such as sugars and proteins. The approach in [91] provides a starting point for future mechanistic studies of the microbial ecology of the human microbiome.
Widder et al. [95] investigated the effect of features inherent to fluvial networks on the structure and function of biofilm communities in these ecosystems by combining co-occurrence analyses of biofilms based on pyrosequencing profiling and a probabilistic hydrological model. Co-occurrence networks were constructed using 454 pyrosequencing data of the 16S rRNA gene from benthic biofilms from 114 streams of the pre-alpine Ybbs catchment. Results suggested that hydrological disturbance and metacommunity dynamics affect the co-occurrence patterns of benthic biofilm communities in fluvial networks. In particular, the removal of gatekeepers disproportionately contributed to network fragmentation. This study provides a linkage between the biofilm communities and flow dynamics across fluvial networks, which are important for understanding the whole ecosystem processes.
Although network inference methods can uncover previously-unknown interactions, they still require validation by experimental data, which could be challenging. Some of these interactions could be indirect, such as bacteria modifying their environments via the secretion of metabolically-costly proteins and metabolites [96]. Since these indirect interactions usually happen on larger spatial scales than direct interactions, it is very important to develop non-invasive spatially-resolved experimental techniques to collect structure and population data.
5. Kinetic Growth Models and Spatial Heterogeneity
Kinetic models predict growth rates of different species within a biofilm based on the concentrations of growth-limiting nutrients and species-dependent parameters, such as maximum growth rate. Probably the most widely-used kinetic model is the Michaelis–Menten (or Monod) kinetic model [97] with the growth rate μ given by:
where is the maximum growth rate, S is the concentration of the growth-limiting nutrient and is the half-saturation constant. The formula given by (3) can be readily generalized to cases with more than one limiting nutrient [13] by multiplying the contribution from each nutrient together ( for the i-th nutrient), but usually, and are considered constants. Under the nutrient-saturation condition, (3) can be approximated by the zero order kinetics, where μ is independent of S (. Under the very low nutrient condition, (3) can be approximated by the first order kinetics, where μ is proportional to S (. These two approximations provide convenient mathematical bounds on the Monod kinetic forms and have the advantages of allowing analytic solutions [50] to the model equations for simple scenarios (ODE model or 1D in space).
The success of the kinetic models depends crucially both on their particular formula and parameter values. Since the formula is often empirical and the parameter values are usually measured from pure and mixed cultures growing in laboratory reactors, they may miss important factors of the growth kinetics in the biofilm community developed in the natural environment, and their application may require validation. Recently, new kinetic models have been developed to address this problem. Quéméner and Bouchez [98] and Jin et al. [99] developed kinetic models with thermodynamics included. The model proposed in [98] is based on the theory of Boltzmann statistics and builds a relationship between microbial growth rate and available energy, thus connecting microbial population dynamics to the thermodynamic driving forces of the surrounding ecosystem. The work in [99] modified the Monod kinetics by a thermodynamic potential factor, which accounts for the chemical energy available from the reaction (acetate oxidation and sulfate reduction in this case) and evaluated the feasibility of applying experimentally-obtained parameters to the natural environment. The results suggest that some parameters, such as maximum growth rate, can be applied directly to the environment; but others, such as half-saturation constants, should be determined using data from the environment of interest. Bonachela et al. [100] relaxed the requirement that the maximum nutrient uptake rate be a constant; instead, the maximum uptake rate was assumed to increase monotonically as the external nutrient concentration decreases. The model predicts larger uptake and growth rates than the standard Monod kinetic, which explains the ability of marine microbes to persist under extreme nutrient limitation.
Biofilms usually have a complex spatial structure, which contributes to their distinctive properties, such as strong antibiotic resistance and diverse population (niches for different species). Mathematical models designated for characterizing the spatial structure of biofilms include Individual-based Models (IbMs) [8,67], continuum models [101,102] and hybrid models [9], which combine both. For IbMs, biofilms are represented as a collection of individual microbes (usually hard spheres) whose growth and movement are determined by a set of rules depending on the local environment, such as the availability of nutrients and space. Results from IbMs usually provide more detailed information and are generally considered superior for studying interactions at the microbe level. Continuum models represent the biomass by functions depending continuously on time and space, and these functions are governed by differential equations derived by using physical, chemical and biological principles. Continuum models allow both numerical solutions obtained using readily-available numerical methods and theoretical analysis of the qualitative behavior of the solutions, such as stability [103], and they are often considered more applicable at larger spatial scales.
Recently, models incorporating novel features have been successfully applied to study biofilms as a microbial community. Below, we discuss an individual-based model and a continuum model, respectively.
Storck et al. [104] developed an individual-based, mass-spring modeling framework to study the effect of cell properties on the structure of biofilms. In this model, cells are represented by a collection of particles connected by springs, which allows variable morphology (e.g., cocci, bacilli and filaments). Three types of structures are considered: the primary structure, which defines the shape of individual cells; the secondary structure, which defines microbial assemblies related by filial links between immediate siblings; and the tertiary structure, which defines non-filial cell-cell and cell-substratum links, such as sticking and anchoring connections. Forces acted on the cells include both elastic force from the springs they are attached to and DLVOforce (combination of the van der Waals and electrostatic force). Simulation results of the growth of rod-shaped cells on a planar surface suggest that the biofilm may grow as a monolayer if there are no anchoring and filial links; the biofilm can be much thicker and much less spread if there are cell-substratum anchoring; and with filial links, but no anchoring to the substratum, a biofilm with an irregular shape (less circularity) is more likely to develop. Simulation results of the activated sludge floc structure suggest that in the floc with filament branching, the filaments are shorter than that of a floc made of straight filaments growing at a similar rate, therefore resulting in greater floc density and attenuating the bulking tendency of filamentous sludge. Furthermore, simulated flocs with spherical floc formers (in contrast to rod-shaped floc formers) were less dense, since the denser packing of spherical cells in a colony leads to a smaller cluster volume, which lowers the chance to encounter a filament former. These simulations demonstrate the close relationship between the fundamental controlling mechanisms, such as the intracellular, intercellular and cell-substratum links, and the diverse biofilm structures.
Emerenini et al. [105] developed a continuum model that includes biofilm growth, production of quorum sensing molecules, cell dispersal triggered by quorum sensing molecules and reattachment of cells. In this model, two distinct cell types are considered: the sessile cells in the biofilm and the motile cells, which can move into and in the liquid phase. The volume fraction of sessile cells and EPS is denoted by M; the motile cell density is denoted by N; and the concentrations of growth-controlling nutrients and autoinducers are denoted by C and A, respectively. Dispersal of cells from the biofilm is controlled by the local autoinducer concentration through Hill kinetics with switching threshold τ and maximum dispersal rate . Re-attachment of cells in the biofilm is controlled by the local biofilm density M through Monod kinetics with maximum rate . The autoinducer production rate is controlled by the local autoinducer concentration, which implicitly represents the switch between down- and up-regulated cells. The governing equations for are reaction-diffusion-type equations with density-dependent (M) diffusion coefficients. Simulation results suggest that single quorum sensing-based mechanism can explain both periodic dispersal in discrete events and continuous dispersal, depending on the value of switching threshold parameter τ. For smaller values of τ, the switching threshold is reached quickly, leading to a rapid dispersal of the biomass before the biofilm can grow into a large size. After the first dispersal event, the biofilm population starts growing again, and the autoinducer concentration increases again, resulting in an almost periodic pattern of discrete dispersal events. For bigger values of τ, it takes a much longer time to reach the switching threshold, and the biofilm develops into a stronger colony before the onset of dispersal. Release of cells from the biofilm into the liquid phase appears continuous, and the biofilm population reaches a plateau. Simulation also suggests that re-attachment of dispersed cells is negligible. The study in [105] indicated that important properties, such as biofilm mass and thickness, can be modified by changing the QS threshold and dispersal rates, and the systems can change between continuous and oscillating behavior. The findings can also help to optimize treatment strategies. For example, promoting quorum sensing can enhance cell dispersal and limit biofilm thickness, which could increase the efficacy of antibiotic treatment, since planktonic cells are generally assumed to be more vulnerable to antibiotic treatment.
IbMs have the obvious strength of describing the detailed biofilm structure and interactions at the cell level, and the forces between individual particles can be derived from first principles. On the other hand, IbMs also suffer the drawback of high computational cost, especially when the goal is to model a biofilm containing a very large number of cells. Therefore, it is important to use efficient numerical methods in the implementation of IbMs, and parallel computing is often the choice [106]. The continuum models can often be analyzed using well-established differential equation theory, and there are many numerical packages available for solving the corresponding discretized system of algebraic equations. However, continuum models often depend on some empirical formula; examples include the effective diffusion coefficient of nutrients inside the biofilm and the constitutive equation for the stress-strain relation when modeling the biofilm as a viscoelastic fluid, and derivation of such a formula is often nontrivial.
6. Conclusions
We present a review of some recently-developed mathematical models that focus on studying biofilms as diverse communities. Despite obvious overlapping, these models are categorized based on their principal methodologies, such as trait-based models, QS, FBA, statistical inference and spatially-resolved models with specific growth kinetics. There are some important topics that have been left out, such as models incorporating stochasticity and evolutionary game theory.
Even though currently-available models can describe many aspects of biofilms accurately, it still remains a challenge to build models that can predict the overall behavior of biofilms as complex and evolving communities. First, time scales involved in biofilm-related processes can vary in as many as ten orders of magnitude, ranging from the fast scale for fluid dynamics to the slow scale for biofilm growth, which is an obvious challenge for modeling. To address this, it is often necessary to assume equilibrium in the fast processes or a quasi-static biofilm profile, depending on the problem of interest. Second, fast advancement in the experimental technologies has provided an abundance of omics data at both the genetic and community level, and many community-scale models have been proposed to describe the interaction between biology, chemistry and physics inside biofilms. However, there is still the lack of a systematic approach to link the observational data to the community-level understanding, namely to tie system kinetics to omics data in a tractable and general way by translating omics to rate functions at the cellular level. The method based on the FBA approach is very promising in this direction. Third, long-term challenges for modeling biofilm as an MC include the necessity to incorporated evolutionary processes, social evolution and bacterial strategies, community assembly and historical contingency, as well as the importance of spatial structure. Addressing these challenges would inevitably require an integrated approach that not only selectively combines multiple relevant models by adding their strengths, but also combines modeling effort and experimental findings.
Acknowledgments
This work was supported by NSF Award DMS-1516951.
Conflicts of Interest
The author declares no conflict of interest.
References
- Costerton, J.W. The Biofilm Primer; Springer Science & Business Media: Berlin, Germany, 2007; Volume 1. [Google Scholar]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Harremoës, P. Biofilm kinetics. In Water Pollution Microbiology; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 1978. [Google Scholar]
- Harris, N.P.; Hansford, G. A study of substrate removal in a microbial film reactor. Water Res. 1976, 10, 935–943. [Google Scholar] [CrossRef]
- Rittmann, B.E.; McCarty, P.L. Evaluation of steady-state biofilm kinetics. Biotechnol. Bioeng. 1980, 22, 2359–2373. [Google Scholar] [CrossRef]
- Wanner, O.; Gujer, W. A multispecies biofilm model. Biotechnol. Bioeng. 1986, 28, 314–328. [Google Scholar] [CrossRef] [PubMed]
- Kreft, J.U.; Booth, G.; Wimpenny, J.W.T. BacSim, a simulator for individual-based modelling of bacterial colony growth. Microbiology 1998, 144, 3275–3287. [Google Scholar] [CrossRef] [PubMed]
- Picioreanu, C.; Loosdrecht, M.C.M.; Heijnen, J.J. Mathematical modeling of biofilm structure with a hybrid differential-discrete cellular automaton approach. Biotechnol. Bioeng. 1998, 58, 101–116. [Google Scholar] [CrossRef]
- Picioreanu, C.; Kreft, J.U.; Loosdrecht, M.C. Particle-Based Multidimensional Multispecies Biofilm Model. Appl. Environ. Microbiol. 2004, 70, 3024–3040. [Google Scholar] [CrossRef] [PubMed]
- Alpkvist, E.; Picioreanu, C.; Loosdrecht, M.C.M.; Heyden, A. Three-dimensional biofilm model with individual cells and continuum EPS matrix. Biotechnol. Bioeng. 2006, 94, 961–979. [Google Scholar] [CrossRef] [PubMed]
- Alpkvist, E.; Klapper, I. Description of Mechanical Response Including Detachment Using a Novel Particle Method of Biofilm/Flow Interaction. Water Sci. Technol. 2007, 55, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Wanner, O.; Eberl, H.; Morgenroth, E.N.D.; Picioreanu, C.; Rittmann, B.; Van Loosdrecht, M. Mathematical Modeling of Biofilms; IWA Publishing: London, UK, 2006; Volume 18. [Google Scholar]
- Klapper, I.; Dockery, J. Mathematical description of microbial biofilms. SIAM Rev. 2010, 52, 221–265. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, T. Review of mathematical models for biofilms. Solid State Commun. 2010, 150, 1009–1022. [Google Scholar] [CrossRef]
- May, R.M. Species coexistence and self-organizing spatial dynamics. Nature 1994, 37, 28. [Google Scholar]
- Davey, M.E.; O’toole, G.A. Microbial biofilms: From ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 2000, 64, 847–867. [Google Scholar] [CrossRef] [PubMed]
- Tilman, D. Niche tradeoffs, neutrality, and community structure: A stochastic theory of resource competition, invasion, and community assembly. Proc. Natl. Acad. Sci. USA 2004, 101, 10854–10861. [Google Scholar] [CrossRef] [PubMed]
- De Meester, N.; Derycke, S.; Rigaux, A.; Moens, T. Active dispersal is differentially affected by inter-and intraspecific competition in closely related nematode species. Oikos 2015, 124, 561–570. [Google Scholar] [CrossRef]
- Ellis, C.N.; Traverse, C.C.; Mayo-Smith, L.; Buskirk, S.W.; Cooper, V.S. Character displacement and the evolution of niche complementarity in a model biofilm community. Evolution 2015, 69, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Song, H.S.; Cannon, W.R.; Beliaev, A.S.; Konopka, A. Mathematical modeling of microbial community dynamics: A methodological review. Processes 2014, 2, 711–752. [Google Scholar] [CrossRef]
- Widder, S.; Allen, R.J.; Pfeiffer, T.; Curtis, T.P.; Wiuf, C.; Sloan, W.T.; Cordero, O.X.; Brown, S.P.; Momeni, B.; Shou, W.; et al. Challenges in microbial ecology: Building predictive understanding of community function and dynamics. ISME J. 2016, 10, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.; Geesey, G. Regulation of the alginate biosynthesis gene algC in Pseudomonas aeruginosa during biofilm development in continuous culture. Appl. Environ. Microbiol. 1995, 61, 860–867. [Google Scholar] [PubMed]
- Petrova, O.E.; Sauer, K. Sticky situations: Key components that control bacterial surface attachment. J. Bacteriol. 2012, 194, 2413–2425. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, W.C.; Winans, S.C.; Greenberg, E.P. Quorum sensing in bacteria: The LuxR-LuxI family of cell density-responsive transcriptional regulators. J. Bacteriol. 1994, 176, 269. [Google Scholar] [CrossRef] [PubMed]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.; Chakrabarty, A.M. Role of alginate lyase in cell detachment of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 1994, 60, 2355–2359. [Google Scholar] [PubMed]
- Lamppa, J.W.; Griswold, K.E. Alginate lyase exhibits catalysis-independent biofilm dispersion and antibiotic synergy. Antimicrob. Agents Chemother. 2013, 57, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Stewart, P.S. Chlorine penetration into artificial biofilm is limited by a reaction-diffusion interaction. Environ. Sci. Technol. 1996, 30, 2078–2083. [Google Scholar] [CrossRef]
- Pabst, B.; Pitts, B.; Lauchnor, E.; Stewart, P.S. Gel-Entrapped Staphylococcus aureus Bacteria as Models of Biofilm Infection Exhibit Growth in Dense Aggregates, Oxygen Limitation, Antibiotic Tolerance, and Heterogeneous Gene Expression. Antimicrob. Agents Chemother. 2016, 60, 6294–6301. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Travis, S.M.; Greenberg, E.P.; Welsh, M.J. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 1996, 85, 229–236. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Alaiwa, M.H.A.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Monds, R.D.; O’Toole, G.A. The developmental model of microbial biofilms: Ten years of a paradigm up for review. Trends Microbiol. 2009, 17, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, M.; Dworkin, M. Developmental Biology of the Bacteria; Benjamin/Cummings Publishing Company: Reading, MA, USA, 1985. [Google Scholar]
- O’Toole, G.; Kaplan, H.B.; Kolter, R. Biofilm formation as microbial development. Annu. Rev. Microbiol. 2000, 54, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, P.; Sauer, K.; Davies, D.; Costerton, J.W. Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 2002, 56, 187–209. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial Biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef] [PubMed]
- Jenal, U.; Malone, J. Mechanisms of cyclic-di-GMP signaling in bacteria. Annu. Rev. Genet. 2006, 40, 385–407. [Google Scholar] [CrossRef] [PubMed]
- Römling, U.; Galperin, M.Y.; Gomelsky, M. Cyclic di-GMP: The first 25 years of a universal bacterial second messenger. Microbiol. Mol. Biol. Rev. 2013, 77, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Banin, E.; Vasil, M.L.; Greenberg, E.P. Iron and Pseudomonas aeruginosa biofilm formation. Proc. Natl. Acad. Sci. USA 2005, 102, 11076–11081. [Google Scholar] [CrossRef] [PubMed]
- Wiens, J.R.; Vasil, A.I.; Schurr, M.J.; Vasil, M.L. Iron-regulated expression of alginate production, mucoid phenotype, and biofilm formation by Pseudomonas aeruginosa. MBio 2014, 5, e01010-13. [Google Scholar] [CrossRef] [PubMed]
- Lazazzera, B.A. Lessons from DNA microarray analysis: The gene expression profile of biofilms. Curr. Opin. Microbiol. 2005, 8, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Franklin, M.J.; Williamson, K.S.; Folsom, J.P.; Boegli, L.; James, G.A. Contribution of stress responses to antibiotic tolerance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2015, 59, 3838–3847. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, A.; Haagensen, J.A.; Zampaloni, C.; Sternberg, C.; Molin, S. Biofilm induced tolerance towards antimicrobial peptides. PLoS ONE 2008, 3, e1891. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.; Heydorn, A.; Ragas, P.; Lambertsen, L.; Aaes-Jørgensen, A.; Molin, S.; Tolker-Nielsen, T. Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol. Microbiol. 2003, 48, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- Burrows, L.L. Pseudomonas aeruginosa twitching motility: Type IV pili in action. Annu. Rev. Microbiol. 2012, 66, 493–520. [Google Scholar] [CrossRef] [PubMed]
- Vlamakis, H.; Aguilar, C.; Losick, R.; Kolter, R. Control of cell fate by the formation of an architecturally complex bacterial community. Genes Dev. 2008, 22, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.; Gjermansen, M.; Kreft, J.U.; Tolker-Nielsen, T. Dynamics of development and dispersal in sessile microbial communities: Examples from Pseudomonas aeruginosa and Pseudomonas putida model biofilms. FEMS Microbiol. Lett. 2006, 261, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.C.; Algar, C.K.; Huber, J.A.; Dick, G.J. Gene-centric approach to integrating environmental genomics and biogeochemical models. Proc. Natl. Acad. Sci. USA 2014, 111, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Pabst, B.; Klapper, I.; Stewart, P.S. General theory for integrated analysis of growth, gene, and protein expression in biofilms. PLoS ONE 2013, 8, e83626. [Google Scholar] [CrossRef] [PubMed]
- Shipley, B.; Vile, D.; Garnier, É. From plant traits to plant communities: A statistical mechanistic approach to biodiversity. Science 2006, 314, 812–814. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, D.C.; Joshi, C.; Bodegom, P.M.; Bastow, Z.A.; Fulé, P.Z. A predictive model of community assembly that incorporates intraspecific trait variation. Ecol. Lett. 2012, 15, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Lennon, J.T.; Aanderud, Z.T.; Lehmkuhl, B.; Schoolmaster, D.R. Mapping the niche space of soil microorganisms using taxonomy and traits. Ecology 2012, 93, 1867–1879. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [PubMed]
- Williams, P. Quorum sensing, communication and cross-kingdom signalling in the bacterial world. Microbiology 2007, 153, 3923–3938. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.; Koh, K.; Queck, S.; Labbate, M.; Lam, K.; Kjelleberg, S. Biofilm formation and sloughing in Serratia marcescens are controlled by quorum sensing and nutrient cues. J. Bacteriol. 2005, 187, 3477–3485. [Google Scholar] [CrossRef] [PubMed]
- Arciola, C.R.; Campoccia, D.; Speziale, P.; Montanaro, L.; Costerton, J.W. Biofilm formation in Staphylococcus implant infections. A review of molecular mechanisms and implications for biofilm-resistant materials. Biomaterials 2012, 33, 5967–5982. [Google Scholar] [CrossRef] [PubMed]
- McDougald, D.; Rice, S.A.; Barraud, N.; Steinberg, P.D.; Kjelleberg, S. Should we stay or should we go: Mechanisms and ecological consequences for biofilm dispersal. Nat. Rev. Microbiol. 2012, 10, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Ward, J. Mathematical Modeling of Quorum-Sensing Control in Biofilms. In Control of Biofilm Infections by Signal Manipulation; Springer: Berlin/Heidelberg, Germany, 2008; pp. 79–108. [Google Scholar]
- Pesci, E.C.; Pearson, J.P.; Seed, P.C.; Iglewski, B.H. Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa. J. Bacteriol. 1997, 179, 3127–3132. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Nilsson, P.; James, G.; Kjelleberg, S.; Fagerström, T. Luminescence control in the marine bacterium Vibrio fischeri: An analysis of the dynamics of lux regulation. J. Mol. Biol. 2000, 296, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Dockery, J.D.; Keener, J.P. A mathematical model for quorum sensing in Pseudomonas aeruginosa. Bull. Math. Biol. 2001, 63, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Horswill, A.R.; Stoodley, P.; Stewart, P.S.; Parsek, M.R. The effect of the chemical, biological, and physical environment on quorum sensing in structured microbial communities. Anal. Bioanal. Chem. 2007, 387, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Kirisits, M.J.; Margolis, J.J.; Purevdorj-Gage, B.L.; Vaughan, B.; Chopp, D.L.; Stoodley, P.; Parsek, M.R. Influence of the hydrodynamic environment on quorum sensing in Pseudomonas aeruginosa biofilms. J. Bacteriol. 2007, 189, 8357–8360. [Google Scholar] [CrossRef] [PubMed]
- Nadell, C.D.; Xavier, J.B.; Levin, S.A.; Foster, K.R. The evolution of quorum sensing in bacterial biofilms. PLoS Biol. 2008, 6, e14. [Google Scholar] [CrossRef] [PubMed]
- Kreft, J.U.; Picioreanu, C.; Wimpenny, J.W.; van Loosdrecht, M.C. Individual-based modelling of biofilms. Microbiology 2001, 147, 2897–2912. [Google Scholar] [CrossRef] [PubMed]
- Xavier, J.B.; Picioreanu, C.; Van Loosdrecht, M. A framework for multidimensional modelling of activity and structure of multispecies biofilms. Environ. Microbiol. 2005, 7, 1085–1103. [Google Scholar] [CrossRef] [PubMed]
- Picioreanu, C.; Van Loosdrecht, M.C.; Heijnen, J.J. Effect of diffusive and convective substrate transport on biofilm structure formation: A two-dimensional modeling study. Biotechnol. Bioeng. 2000, 69, 504–515. [Google Scholar] [CrossRef]
- Sakuragi, Y.; Kolter, R. Quorum-sensing regulation of the biofilm matrix genes (pel) of Pseudomonas aeruginosa. J. Bacteriol. 2007, 189, 5383–5386. [Google Scholar] [CrossRef] [PubMed]
- Heithoff, D.M.; Mahan, M.J. Vibrio cholerae biofilms: Stuck between a rock and a hard place. J. Bacteriol. 2004, 186, 4835–4837. [Google Scholar] [CrossRef] [PubMed]
- Seressiotis, A.; Bailey, J.E. MPS: An artificially intelligent software system for the analysis and synthesis of metabolic pathways. Biotechnol. Bioeng. 1988, 31, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Mavrovouniotis, M.L.; Stephanopoulos, G.; Stephanopoulos, G. Computer-aided synthesis of biochemical pathways. Biotechnol. Bioeng. 1990, 36, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Schilling, C.H.; Schuster, S.; Palsson, B.O.; Heinrich, R. Metabolic pathway analysis: Basic concepts and scientific applications in the post-genomic era. Biotechnol. Progress 1999, 15, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Harcombe, W.R.; Riehl, W.J.; Dukovski, I.; Granger, B.R.; Betts, A.; Lang, A.H.; Bonilla, G.; Kar, A.; Leiby, N.; Mehta, P.; et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014, 7, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Edwards, J.S.; Doyle, F.J. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Phalak, P.; Chen, J.; Carlson, R.P.; Henson, M.A. Metabolic modeling of a chronic wound biofilm consortium predicts spatial partitioning of bacterial species. BMC Syst. Biol. 2016, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, G.; Fleming, R.M.; Heinken, A.; Thiele, I. A systems biology approach to drug targets in Pseudomonas aeruginosa biofilm. PLoS ONE 2012, 7, e34337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberhardt, M.A.; Puchałka, J.; Fryer, K.E.; Dos Santos, V.A.M.; Papin, J.A. Genome-scale metabolic network analysis of the opportunistic pathogen Pseudomonas aeruginosa PAO1. J. Bacteriol. 2008, 190, 2790–2803. [Google Scholar] [CrossRef] [PubMed]
- Ramkrishna, D.; Song, H.S. Dynamic models of metabolism: Review of the cybernetic approach. AIChE J. 2012, 58, 986–997. [Google Scholar] [CrossRef]
- James, G.; Beaudette, L.; Costerton, J. Interspecies bacterial interactions in biofilms. J. Ind. Microbiol. 1995, 15, 257–262. [Google Scholar] [CrossRef]
- Hansen, S.K.; Rainey, P.B.; Haagensen, J.A.; Molin, S. Evolution of species interactions in a biofilm community. Nature 2007, 445, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Periasamy, S.; Mukherjee, M.; Xie, C.; Kjelleberg, S.; Rice, S.A. Biofilm development and enhanced stress resistance of a model, mixed-species community biofilm. ISME J. 2014, 8, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Microbial community structure and its functional implications. Nature 2009, 459, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Freilich, S.; Kreimer, A.; Meilijson, I.; Gophna, U.; Sharan, R.; Ruppin, E. The large-scale organization of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Res. 2010, 38, 3857–3868. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- De Smet, R.; Marchal, K. Advantages and limitations of current network inference methods. Nat. Rev. Microbiol. 2010, 8, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Imieliński, T.; Swami, A. Mining association rules between sets of items in large databases. In ACM Sigmod Record; ACM: New York, NY, USA, 1993; Volume 22, pp. 207–216. [Google Scholar]
- Mounier, J.; Monnet, C.; Vallaeys, T.; Arditi, R.; Sarthou, A.S.; Hélias, A.; Irlinger, F. Microbial interactions within a cheese microbial community. Appl. Environ. Microbiol. 2008, 74, 172–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitchison, J. The Statistical Analysis of Compositional Data; Chapman and Hall, Ltd.: London, UK, 1986. [Google Scholar]
- Buehlmann, P. Boosting for high-dimensional linear models. Ann. Stat. 2006, 559–583. [Google Scholar] [CrossRef]
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar] [Green Version]
- Widder, S.; Besemer, K.; Singer, G.A.; Ceola, S.; Bertuzzo, E.; Quince, C.; Sloan, W.T.; Rinaldo, A.; Battin, T.J. Fluvial network organization imprints on microbial co-occurrence networks. Proc. Natl. Acad. Sci. USA 2014, 111, 12799–12804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, L.; Viana, M.; Brown, S.P. Cooperative secretions facilitate host range expansion in bacteria. Nat. Commun. 2014, 5, 4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michealis, L.; Menten, M. The kinetics of invertase activity. Biochem. Z. 1913, 49, 333–369. [Google Scholar]
- Desmond-Le Quéméner, E.; Bouchez, T. A thermodynamic theory of microbial growth. ISME J. 2014, 8, 1747. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Roden, E.E.; Giska, J.R. Geomicrobial kinetics: Extrapolating laboratory studies to natural environments. Geomicrobiol. J. 2013, 30, 173–185. [Google Scholar] [CrossRef]
- Bonachela, J.A.; Raghib, M.; Levin, S.A. Dynamic model of flexible phytoplankton nutrient uptake. Proc. Natl. Acad. Sci. USA 2011, 108, 20633–20638. [Google Scholar] [CrossRef] [PubMed]
- Alpkvist, E.; Klapper, I. A Multidimensional multispecies continuum model for heterogenous biofilm. Bull. Math. Biol. 2007, 69, 765–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cogan, N.; Wang, Q. Phase field models for biofilms. II. 2-D numerical simulations of biofilm-flow interaction. Commun. Comput. Phys. 2008, 4, 72–101. [Google Scholar]
- Dockery, J.D.; Klapper, I. Finger formation in biofilm layers. SIAM. J. Appl. Math. 2001, 62, 853–869. [Google Scholar]
- Storck, T.; Picioreanu, C.; Virdis, B.; Batstone, D.J. Variable cell morphology approach for individual-based modeling of microbial communities. Biophys. J. 2014, 106, 2037–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerenini, B.O.; Hense, B.A.; Kuttler, C.; Eberl, H.J. A mathematical model of quorum sensing induced biofilm detachment. PLoS ONE 2015, 10, e0132385. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kahan, S.; Momeni, B. Simulating microbial community patterning using biocellion. Eng. Anal. Multicell. Sys. Methods Protoc. 2014, 1151, 233–253. [Google Scholar]
Figure 1.
The conceptual basis of FBA as constraint-based modeling. Reprinted from [75] with permission from Nature Publishing Group.
Figure 1.
The conceptual basis of FBA as constraint-based modeling. Reprinted from [75] with permission from Nature Publishing Group.

Figure 2.
Principle of similarity- and regression-based network inference. Reprinted from [87] with permission from Nature Publishing Group.
Figure 2.
Principle of similarity- and regression-based network inference. Reprinted from [87] with permission from Nature Publishing Group.

{kind=link}
{kind=link}
Table 1.
Summary of biofilm models discussed in the article. QS, Quorum Sensing; FBA, Flux Balance Analysis; IbM, Individual-based Model.
| Model Category | Specific Models/Mathematical Tools | Biofilm Aspects Modeled |
|---|---|---|
| Genetic modeling | gene-centric model trait-based model QS model | biofilm structure, genetic composition species interaction, ecosystem diversity species interaction, biofilm structure |
| FBA model | constraint optimization problem | species interaction, biofilm structure |
| Statistical inference model | similarity-based method, regression-based method | species interaction, community stability |
| Kinetic growth model | IbM continuum model | biofilm structure, interaction with environment, mechanical property |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, T. Modeling Biofilms: From Genes to Communities. Processes 2017, 5, 5. https://doi.org/10.3390/pr5010005
AMA Style
Zhang T. Modeling Biofilms: From Genes to Communities. Processes. 2017; 5(1):5. https://doi.org/10.3390/pr5010005
Chicago/Turabian StyleZhang, Tianyu. 2017. "Modeling Biofilms: From Genes to Communities" Processes 5, no. 1: 5. https://doi.org/10.3390/pr5010005
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.