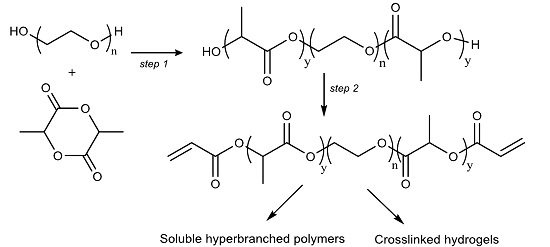
1. Introduction
Hydrogels are cross-linked polymer networks that have a high affinity for water but do not dissolve in it, thus retaining their three dimensional structures [
1]. Hydrogels have very wide applicability in the biomedical and pharmaceutical fields including tissue engineering [
2,
3,
4], diagnostics, and drug delivery [
5,
6]. Hydrogels are biocompatible because they cause minimal tissue irritation with little cell adherence when in contact with the extracellular matrix due to their hydrophilicity and soft nature [
7]. In tissue engineering and regenerative medicine, there are many advantages if hydrogels can form in situ; for example, an injectable macromer system can occupy an irregular defect and then crosslink to attain a permanent feature. This would be desirable, particularly, if cells or molecular signals could be incorporated in the injection mixture [
8]. Solidification of materials in situ can then be achieved by either physical or chemical means [
9,
10].
Hydrogels based on poly (ethylene glycol) (PEG) and polyester block co-polymers are especially useful as many are known to be temperature-sensitive in aqueous solutions as they undergo a phase transition from the sol to the gel state at higher temperatures [
11,
12,
13]. PEG is a synthetic polymer and is soluble in water, non-toxic, and can be eliminated from the body depending on its size. Incorporation of lactate segments into the PEG polymer backbone can introduce biodegradability thus facilitating degradation of the materials in vivo [
14,
15]. Various polyesters have been used to form the hydrophobic blocks to PEG based polymers, including poly(lactide) (PLA) [
16], poly(ε-caprolactone) (PCL) [
17], and poly(glycolic-co-lactic acid) (PLGA) [
18], via ring opening polymerisation of lactide, ε-caprolactone, or glycolide with PEG, respectively.
Co-polymers based on PEG and PLA are of interest because of their biocompatibility and biodegradability [
19]. PLA–PEG block co-polymers have been synthesized by various means including the ring opening polymerisation of lactide in the presence of PEG. Deng et al. first reported the use of ring opening catalysts in the co-polymerisation of
d,
l-lactide with PEG [
20]. The use of stannous 2-ethylhexanoate as a catalyst at 180 °C through bulk polymerisation was first reported by Zhu [
21]. These co-polymers have a tri-block nature. Such co-polymers using PEG with different chain lengths have been reported and used to prepare biodegradable hydrogels. It appears that PEG-PLA tri-block co-polymers with a short domain of PEG at molecular weights below 6000 g·mol
−1 are rarely studied. Co-polymers with varying segment lengths and distributions have been synthesized and it is found that their properties were influenced by these variations [
22]. Lee et al. prepared P
LLA–PEG multi-block co-polymers [
12] with long blocks of PEG (
Mn = 2000–10,000 g·mol
−1) and P
LLA (
Mn = 2000–4500 g·mol
−1). Luo made P
LLA–PEG multi-block co-polymers with shorter PEG (
Mn = 600, 2000 g·mol
−1) and PLA (
Mn = 1000, 2000, 3000 g·mol
−1) [
23]. To enable P
DLLA-PEG-P
DLLA co-polymers to undergo free radical polymerisation, they were further functionalized with vinyl groups at the terminal ends. Sawhney and Hubbell first described the synthesis of synthetic co-polymers which are hydrolytically degradable and cross-linkable consisting of a hydrophilic PEG central domain which was then co-polymerised with poly(lactic acid) (PLA) and end-capped with acrylate groups [
14]. Since then a variety of such macromers have been created in which PEG is modified with various hydrolytically degradable ester moieties and terminal acrylate/methacrylate groups [
24]. Varying the PEG molecular weight was found to influence the degree of gel swelling and other mechanical properties including degradation. Hubbell further prepared hydrogels by crosslinking acrylated multi-arm PEGs with thiol compounds via a Michael-type addition [
25].
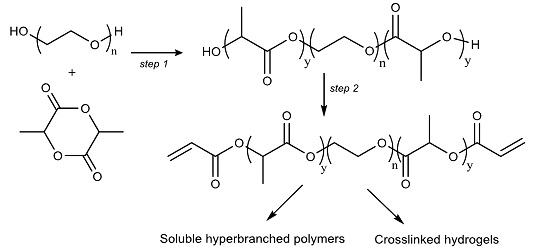
In this work, we prepared tri-block P
DLLA-PEG-P
DLLA diacrylate macromers with a relatively short PEG length (
Mn = 1000 g·mol
−1), which were used in further polymerisation with or without other PEG based co-monomers via free radical polymerisation to prepare both soluble hyperbranched polymers and crosslinked biodegradable hydrogels. Generally, other reported syntheses have been performed using relatively large PLA fractions to primarily change the hydrophobic/hydrophilic balance [
26]. This study also gives details of the swelling behavior of the prepared hydrogels. The swelling curves show a reduction in the swelling ratio due to degradation after maximum swelling is achieved and is seen to commence first in the material with the highest lactoyl content. The degradation is hypothesized to be a result of hydrolysis of the lactoyl domains in the material. The low critical solution temperatures (LCSTs) of these co-polymers and its derived macromer materials can be tailored to be on the order of physiological temperatures.
The polymer samples (linear block and hyperbranched) obtained were characterised by
1H NMR, Gel Permeation Chromatography (GPC), and FTIR. Moreoever, the diacryl-P
DLLA-PEG-P
DLLA marcromers were utilized to prepare crosslinked biodegradable hydrogels. The experiments demonstrated the challenges of working with these materials due to the unstable nature of ester functional groups within the P
DLLA-PEG-P
DLLA co-polymers and the macromers. Photo-polymerisation and the Michael addition reaction were used for the preparation of crosslinked hydrogels. To the best of our knowledge, this is the first report on the study of such macromers with PEG 1000 (g·mol
−1) as the core and short poly-lactoyl terminal domains conferring on the co-polymer an amphiphilic character of varying degree. Soluble hyperbranched polymers and crosslinked hydrogels prepared from the diacryl-P
DLLA-PEG-P
DLLA macromers can be used as nanocarriers or depot systems for drug delivery [
27] and tissue engineering applications [
28].
2. Results and Discussion
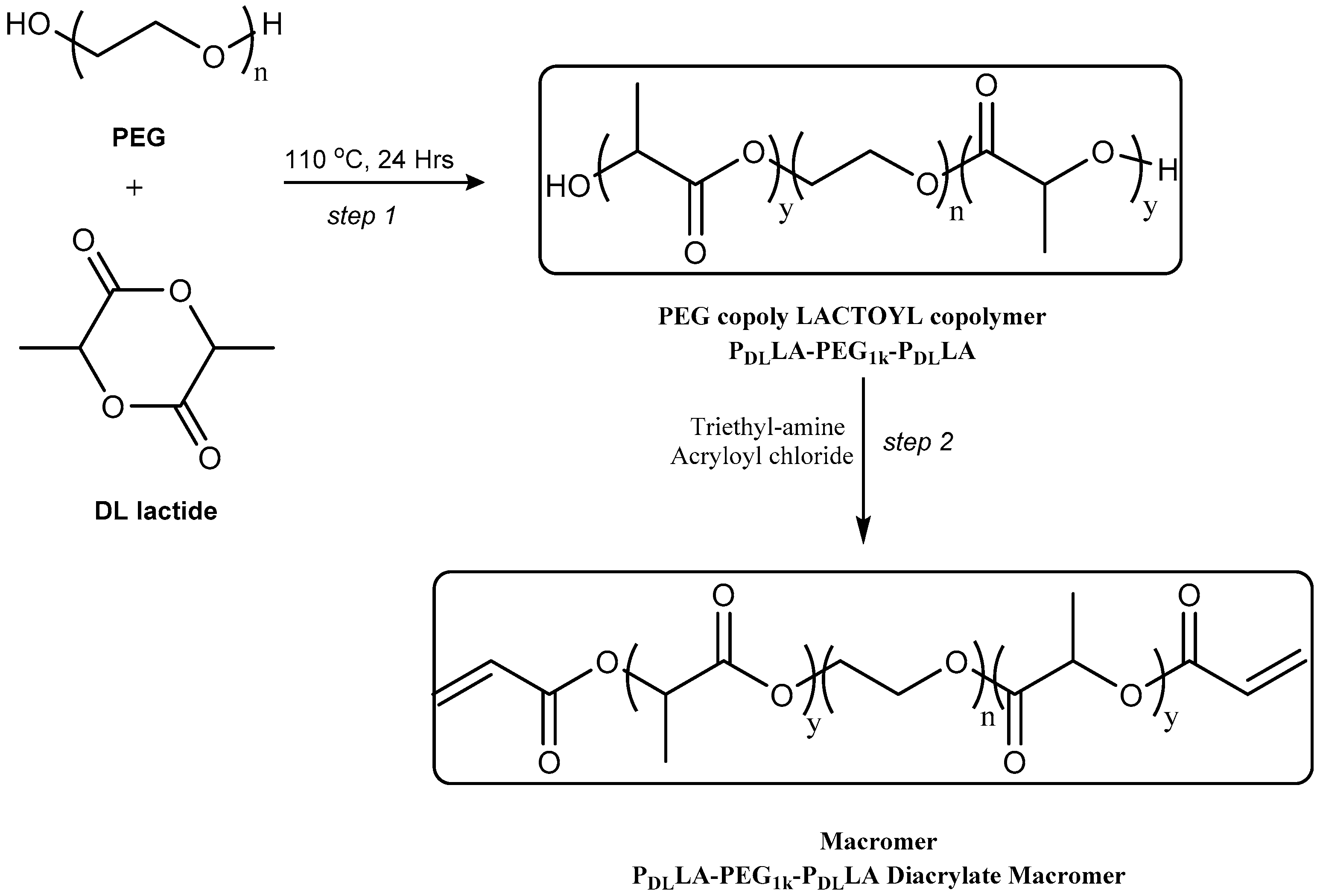
In this study, the molecular weight of the hydrophilic PEG was chosen as 1000 g·mol
−1, to which hydrophobic lactoyl terminal domains were added to both ends of the PEG chain. Diacryl-P
DLLA-PEG
1k-P
DLLA macromers were prepared in two steps (
Scheme 1). The first step was the co-polymerisation of PEG and
d,
l-Lactide. The co-polymer was subsequently acrylated using acryloyl chloride to produce the macromer (
Figure S1 in the Supplementary Materials). An alternative method was also attempted by using a one-pot two-stage procedure, in which the P
DLLA-PEG
1k-P
DLLA co-polymer was formed as an intermediate and the second acrylation step, without separation and purification of the P
DLLA-PEG
1k-P
DLLA co-polymer from the reaction mixture, was then performed. Both methods demonstrated a similar yield and controllability, thus the one-pot method was adopted (
Table 1).
Step 1: Synthesis of PDLLA-PEG1k-PDLLA co-polymers. Telechelic P
DLLA-PEG
1k-P
DLLA co-polymers were synthesised via a ring opening polymerisation (ROP) of the cyclic-diester monomer
d,
l-lactide. The ROP was initiated by the α and ω hydroxyl terminal groups of the PEG catalysed by stannous 2-ethylhexanoate and consisted of the step-wise addition of the lactide. It was important to eliminate any water from the reaction mixture to avoid hydrolysis of the products. PEG and
d,
l-lactide were dried as described in the experimental section. A range of P
DLLA-PEG
1k-P
DLLA co-polymers were prepared by varying the ratio of
d,
l-lactide to PEG (
Table 1).
The degree of polymerisation was determined by
1H NMR (see
Figures S2 and S3 in the Supplementary Materials) and it was found to be dependent on the feed molar ratio of the reactants, i.e., the PEG and the
d,
l-lactide (see
Table 1). Typically, the PEG signal –CH
2–CH
2–O– was found at δ = 3.65, and the methyl signal for the lactoyl group was found at δ = 1.6, thus for
Table 1 entry 6 the integrations were found to be 1.48 and 1.0, respectively. Each lactoyl methyl residue contains three protons and if we assume equi-molar addition at both ends of the PEG block, the total number of protons for the lactoyl signal is equal to 6
m where
m is the number of lactoyl residues on each end of the co-polymer. Note that PEG
Mn = 1000 g·mol
−1 consists of, on average,
n = 22.32 repeating residues (–O–CH
2–CH
2) with an additional H and an –OH at each terminal of the linear molecule, i.e., an additional 18 g·mol
−1. Therefore, the number of lactoyl units in the co-polymer,
m, can be calculated by Equation (1):
The above co-polymer was named 1KL10, i.e., 10 lactoyl residues on each end of the PEG domain. The PEG/Lactoyl ratios in the co-polymers were generally found to be lower than the ratio in the feed. This was in part due to the loss of any residual water from the pre-weighed PEG. Also the conversion of lactide was not complete as unreacted lactide vaporised on the cooler parts of the reaction vessel and thus was effectively removed from the reaction mixture and subsequently removed during purification. If PEG of higher molecular weight is used, the relative hydroxyl content will decline as there are two moles of hydroxyl groups per mole of linear PEG. The present study used PEG of molecular weight of 1000 (g·mol
−1) which is relatively low compared to most other studies involving this reaction. Thus, with increasing molar mass of PEG for a given weight of PEG there are fewer hydroxyl end groups, which function as initiation sites with the consequent lowering of the conversion ratio of the lactide under the same reaction conditions. The experimental entries show that longer reaction time reduced the lactoyl content in the co-polymer. For example, comparing entries 6 and 7 (in
Table 1), the longer reaction time resulted in lower lactoyl content despite otherwise similar conditions. This may be result of cleavage of the lactoyl chain under these conditions. It should also be noticed from
Table 1 that the co-polymer yield was variable and often low, as is illustrated in entries 5 and 6 (
Table 1), with a great disparity in yield between the two similar products. This was possibly due to the failure to obtain optimal precipitation during the extraction step when a fine suspension resulted which could not be isolated by either filtration or centrifugation. Moreover, P
DLLA-PEG
1k-P
DLLA co-polymers demonstrated poor water solubility (
Table 1) due to the incorporation of hydrophobic PLA with hydrophilic PEG. As the mass fraction of the PLA in the co-polymer increases, its water solubility decreases. We can see (
Table 1) that the co-polymer 1KL3 was soluble whereas those with higher lactide content were insoluble. These results are in agreement with the findings of Sawhney [
14]. In general, a high PEG/PLA ratio and a low molar mass confer water solubility. After synthesis via ROP, the P
DLLA-PEG
1k-P
DLLA products were recovered, either by dissolution in dichloromethane and precipitation in anhydrous ether which was then dried under reduced pressure after filtration, or by dissolving the co-polymer mixture in ice-cold water and then gradually increasing its temperature above to its cloud point at about 52 °C at which the co-polymer precipitated out of the solution.
Step 2: Diacryl-PDLLA-PEG1k-PDLLA Macromers. The P
DLLA-PEG
1k-P
DLLA co-polymer, with both α and ω hydroxyl end-groups, was end-capped with acrylate groups which could then be further used for cross linking. The degree of acrylation was determined by
1H NMR (see
Figure S3 in Supplementary Materials). Vinyl proton NMR signals typically appeared at δ = 5.9, 6.2, and 6.4 ppm. Thus, for entry 3 (
Table 2), where the integration of the acrylate signal is 0.04 and for the methyl is 1, the total number of vinyl protons per mole of the macromer was determined as
x where
As the vinyl groups each comprise three protons at each end of the macromer chain (i.e., a total of six per molecule), the given the macromer is 59.5% acrylated on average. This would translate to a value for the maximum molecular weight for the di-acrylated macromer of Mn = 2549 g·mol−1 (i.e., for each di-acrylated molecule).
The quantity of DCM solvent, i.e., the concentration of the reactants is critical to the rate of acrylation. Too much DCM will re-dissolve the triethylamine hydrochloride salt. It was noted that the precipitate of the salt correlated with high acrylation. It is presumed that if the solvent for the acrylation is in excess, this reduces the rate of the acrylation reaction by reducing the concentration of the reactants. Thus a minimal amount of solvent is used, sufficient for dissolving the co-polymer.
For the macromer 1KL3 (
Table 2, entries 11 and 13), it can be seen that the greater degree of acrylation occurs for entry 13 (80.3%) with a lower acryloyl chloride ratio of 1:1.2:2.5 compared to entry 11 (14.9%) synthesised with a ratio 1:4:4. This situation is reversed with macromer 1KL8 with its relatively higher lactoyl content. Thus, for entries 5 and 10, the entry 5 (acrylation 33.1%) was synthesised with a lower acryloyl chloride ratio of 1:1.2:2.5 (cf. 75.7% for entry 10). However, excessive ratios should be avoided because of the possibility of causing cleavage of the polymer chain especially as the reaction is exothermic and because of the need to maintain the reaction at 0 °C at least at the early stage. To ensure that the highest possible conversion to acrylate was achieved, the reaction was allowed to run for 24 h in accordance with the procedure outlined by Zhang et al. [
29]. The ice temperature affected the product quality and if not kept low, a deep yellow colour resulted from the exothermic reaction between the acryloyl chloride and the diol groups of the co-polymer.
The P
DLLA-PEG
1k-P
DLLA Co-Polymer and Diacryl-P
DLLA-PEG
1k-P
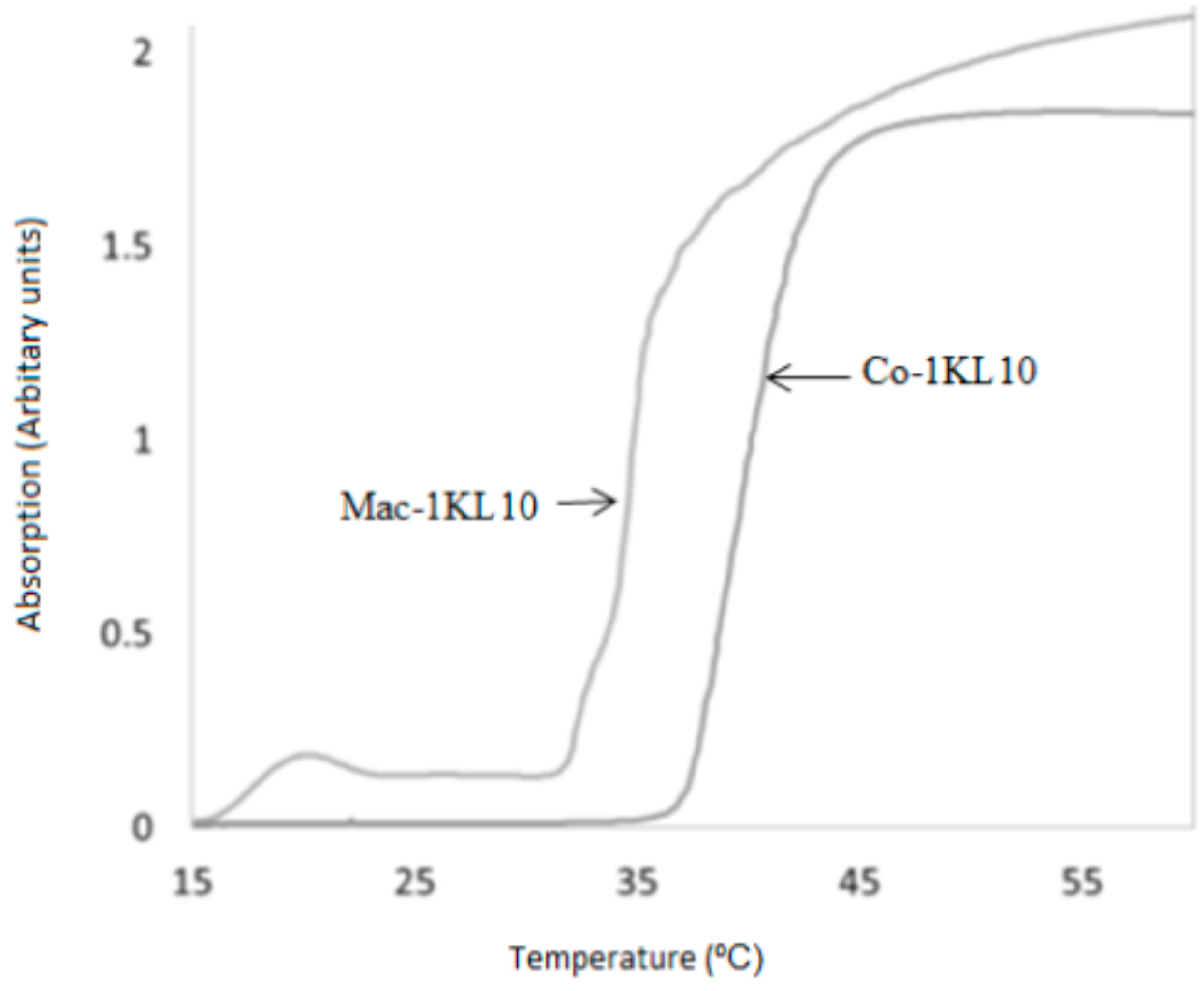
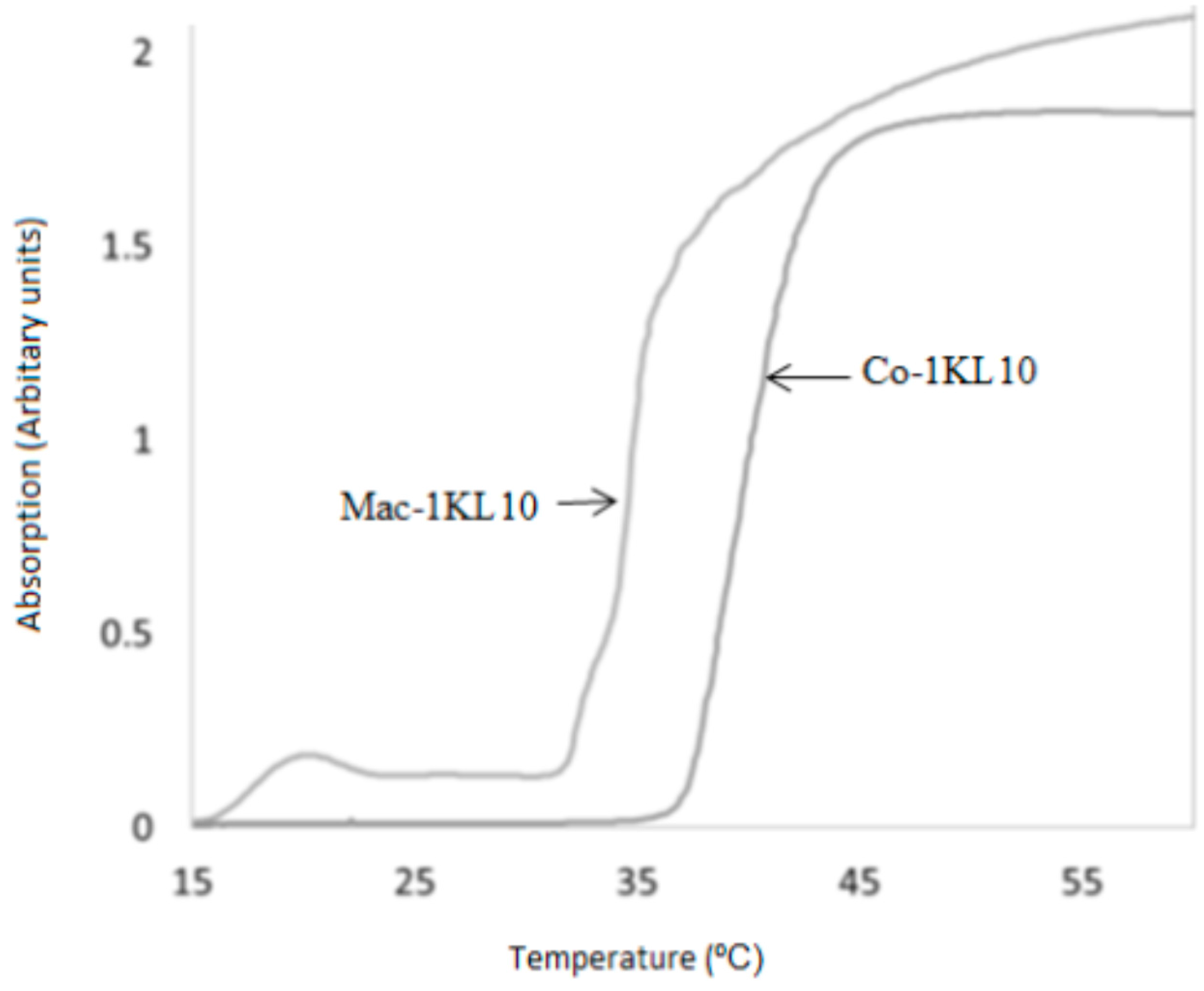
DLLA Macromer showed thermoresponsive behaviors. Varying the ratios of the hydrophilic/hydrophobic domains within the co-polymer results in changing the phase transition temperature in the aqueous solution; the more hydrophilic the co-polymer, the higher the LCST. The LCST for macromer 1KL10 (Mac-1KL10) and co-polymer 1KL10 (Co-1KL10), respectively, were determined. The Mac-1KL10 and the Co-1KL10 were dissolved in deionized water, and the change in absorbency with increasing temperature was measured at a wavelength of 550 nm using UV-Vis spectrophotometry (
Figure 1). The LCST was found to be around 34 °C and 40 °C, respectively. These findings are consistent with LCSTs determined by visual observation when the temperatures for this transition were found to be 27 °C and 32 °C, respectively.
Moreover, the FTIR spectra for the co-polymer and its macromer (see
Figure S4 in the Supplementary Materials) show strong absorption at 3510 cm
−1 for the PEG precursor due to the terminal hydroxyl group and this signal is reduced due to acrylation although not eliminated. A strong absorption at 1756 cm
−1 for the 1KL3 confirms the presence of the ester due to the lactoyl moieties. A weak signal for the –C=C– in the region 1680–1640 cm
−1 can also be seen.
3. Preparation of Soluble Hyperbranched Polymers from Free Radical Polymerisation (FRP) of Diacryl-PDLLA-PEG1k-PDLLA Macromer in the Presence and Absence of Co-Monomer PEGMEMA
To prepare branched polymers, free radical polymerisations were performed using the diacryl-P
DLLA-PEG
1k-P
DLLA macromer alone (homo-polymerisation, entries 1 and 2 in
Table 3) and also with another co-monomer, i.e., PEGMEMA (co-polymerisation, entries 3–5 in
Table 3).
Polymerisation was found to occur in both DMF and chloroform (entries 1 and 2 in
Table 3). However, the conversion was higher in DMF and the PDI was given as 1.03 which indicates a controlled polymerisation mechanism under the reaction conditions used. GPC revealed that higher molecular weights were obtained in chloroform but the conversion was lower given that the temperature and reaction time were comparable. This difference may reflect the difference in solubility and polarity between the two solvents as it is observed that the chloroform was found to be a better solvent but the DMF yielded a slightly translucent solution. The co-polymerisations with PEGMEMA (entries 3–5 in
Table 3) were observed, evidenced by the GPC data, however, the molecular weights obtained were not as high as for the homo-polymerisations of the macromer (see entries 2 and 3) when the reactions were undertaken in the same solvent and under similar experimental conditions. If the concentration was increased, this caused a lower molecular weight product to be synthesised despite doubling the reaction time (see entries 4 and 5 in
Table 3). It was also noted that all polymerisations were to a greater or lesser degree conducted under heterogeneous conditions. Entries 4 and 5 in
Table 3 were conducted in THF and the monomer to solvent ratios was generally lower, reflecting better solubility than those entries using DMF and chloroform.
The polydispersity (PDI) of the branched polymers obtained from free radical polymerisation of the diacryl-P
DLLA-PEG
1k-P
DLLA macromer with or without PEGMEMA were generally fairly narrow (less than 2, see
Table 3). This may be due to the fact that the GPC samples were obtained upon gelation and, therefore, represent a soluble extract of the reaction mixture.
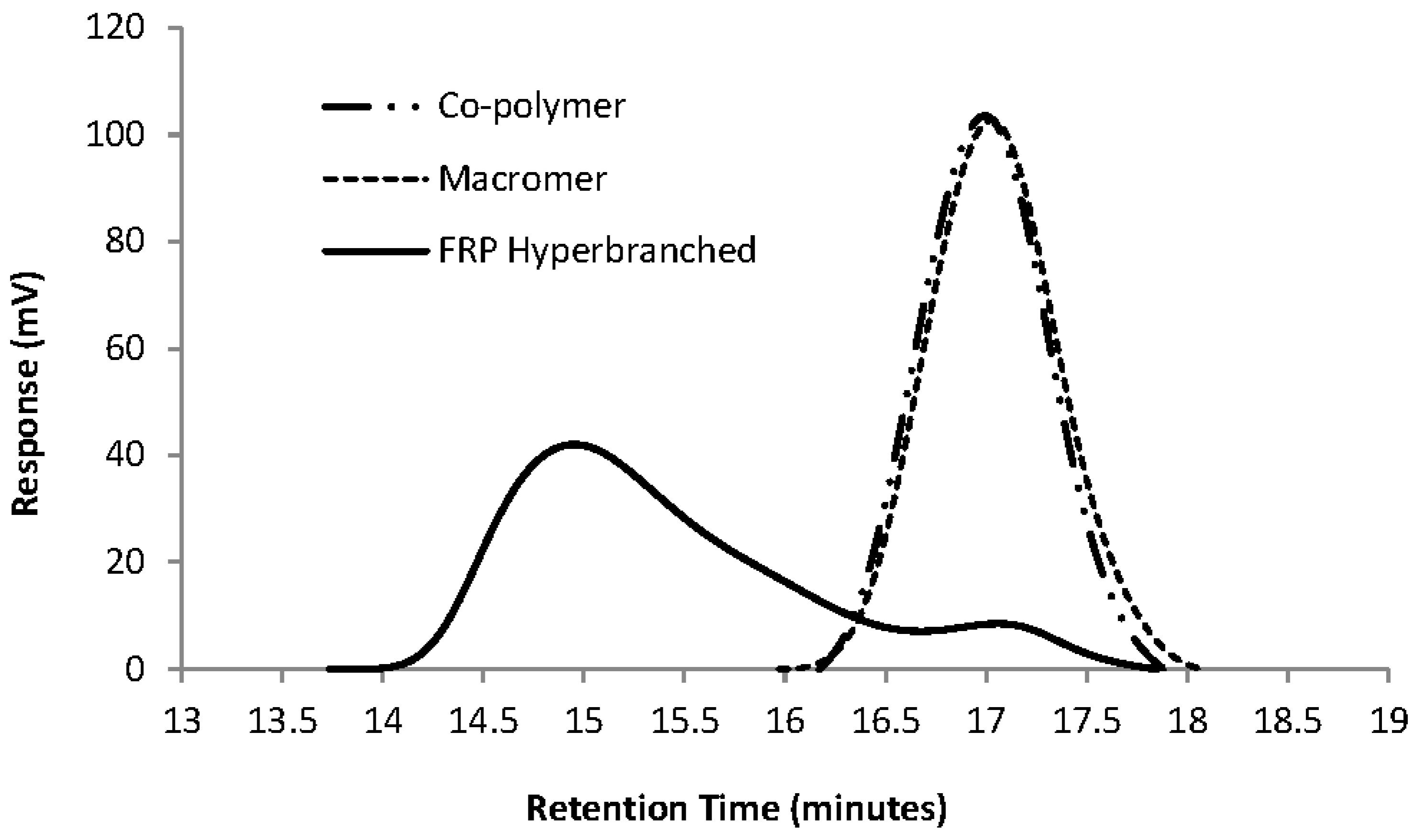
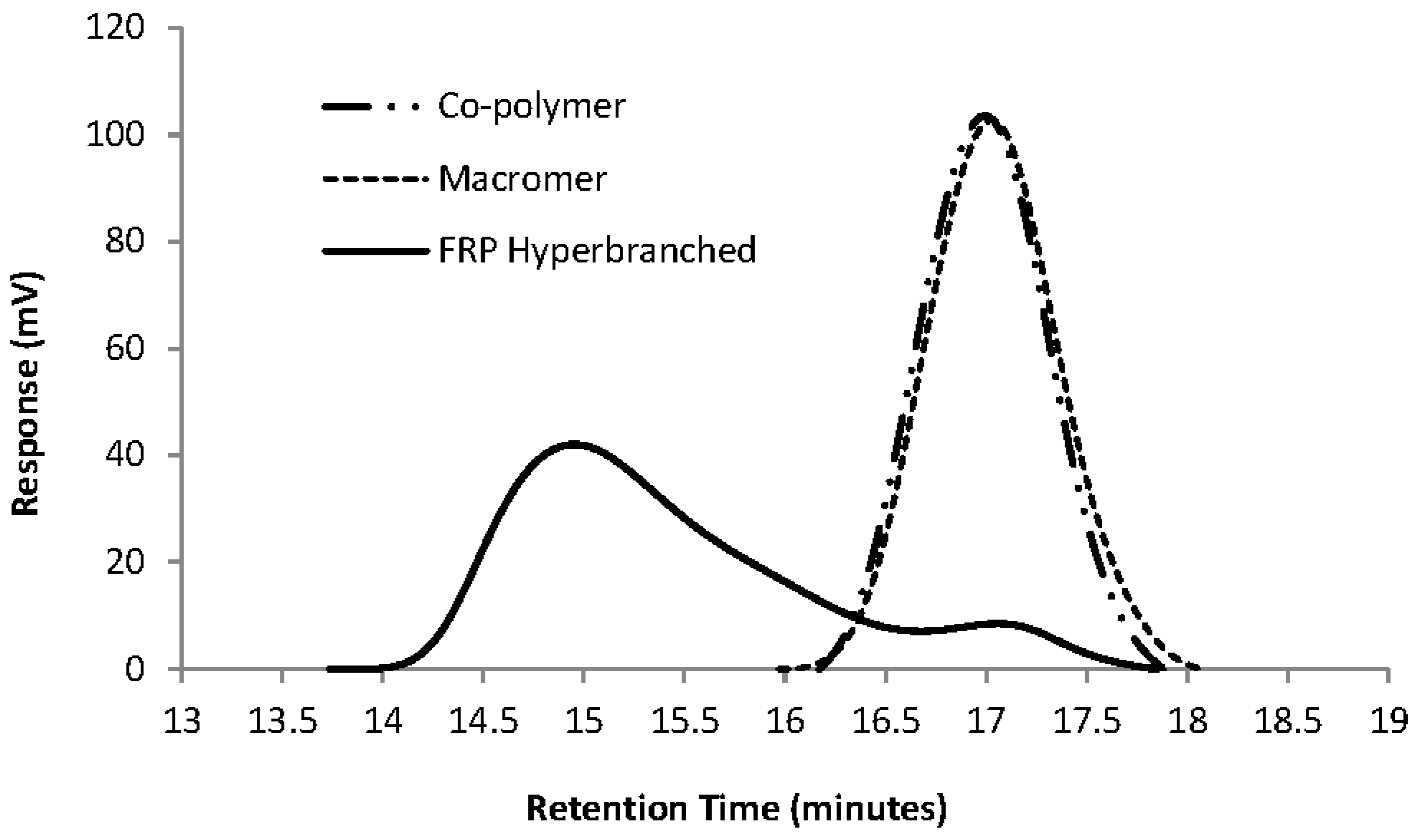
Figure 2 gives a GPC overlay of the FRP homo-polymerisation of Mac-1KL3 (entry 2 in
Table 3) as a comparison with its precursor, the co-polymer 1KL3 (Co-1KL3).
These experimental results show that soluble (most likely hyperbranched) polymers can be synthesized by free radical polymerisation of the diacryl-P
DLLA-PEG
1k-P
DLLA macromer at a relatively low yield (less than 30%), before gelation occurs. However, it was found to be challenging using the diacryl-P
DLLA-PEG
1k-P
DLLA macromer as a multifunctional monomer to prepare soluble hyperbranched polymers by radical polymerisations, including controlled radical polymerisation approaches such as atom transfer radical polymerisation and reversible addition-fragmentation chain transfer polymerisation. This is because the diacryl-P
DLLA-PEG
1k-P
DLLA macromer is unstable, thus the polymerisation conditions should be carefully considered and chosen, including reaction temperature, initiators, and solvents. Nevertheless, the soluble hyperbranched polymers prepared from the diacryl-P
DLLA-PEG
1k-P
DLLA macromers could have potential as nanocarriers to deliver drugs for nanomedicine applications or could be used to fabricate hydrogels for regenerative medicine applications [
28].
3.1. Synthesis of Chemical Crosslinked Hydrogels from Diacryl-PDLLA-PEG1k-PDLLA Macromers via Michael-Addition Reaction
The diacrylate macromer with its vinyl functionality is able to undergo a Michael addition type reaction when it is used with a suitable reagent containing thiol functional groups. The macromer should be capable of a homo-polymerisation when used on its own or possibly in the presence of another vinyl functional monomer such as the PEGMEMA monomer, where co-polymerisation should occur. Therefore, a number of reactions were performed (
Table 4). It can be seen that the homo-polymerisation of the macromer occurred in all the solvents used, i.e., water, DMSO, and PBS (entries 1–3 and 6). However, the reactions with PEGMEMA were unsuccessful (entries 4, 7, and 8). The Michael addition requires the presence of a base to act as a catalyst. The basic conditions were supplied by the use of PBS buffer (pH 7.4), triethylamine, or sodium hydroxide. The reactions with the macromer only resulted in a white gel indicating that the reaction occurred. Co-polymerisation with PEGMEMA was also attempted but did not yield any evidence of reaction other than in the solvent THF. This could be explained by the lower reactivity of the methacrylate groups in PEGMEMA with thiol groups for undertaking the Michael addition reaction, compared to the acrylate groups in diacryl-P
DLLA-PEG
1k-P
DLLA macromers.
3.2. Photocrosslinked Hydrogels from Macromers: Synthesis, Swelling, and Degradation
The diacryl-P
DLLA-PEG
1k-P
DLLA macromer can be used to prepare hydrogels as either a homo-polymer or as a co-polymer, with for example PEGMEMA, which is also water soluble. With this in mind, a swelling study was undertaken on the hydrogels prepared from the photo-polymerisation of macromer 1KL3 and on hydrogels prepared from the co-polymerisation of macromer 1KL11 and PEGMEMA (
Table 5).
Photo-polymerisation was conducted at 25 °C by irradiation with UV light. A 1% aqueous solution of the initiator Irgacure 2959 was prepared, and the macromer was then dissolved at different ratios of Macromer/PEGMEMA to the concentrations shown. The reaction mixture was then subjected to UV exposure. After a given exposure time the resulting solids were gently washed with de-ionised water and dried in a vacuum oven.
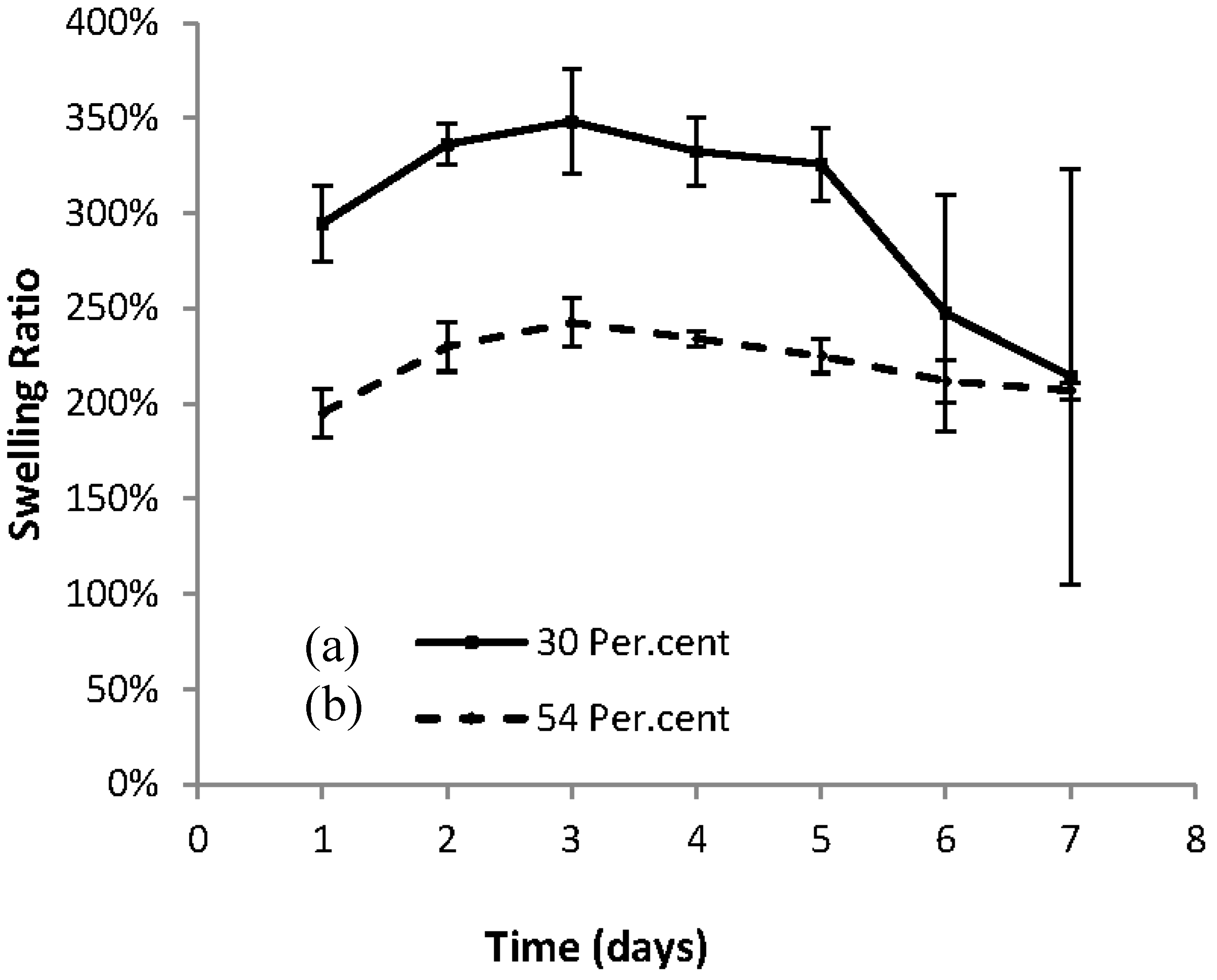
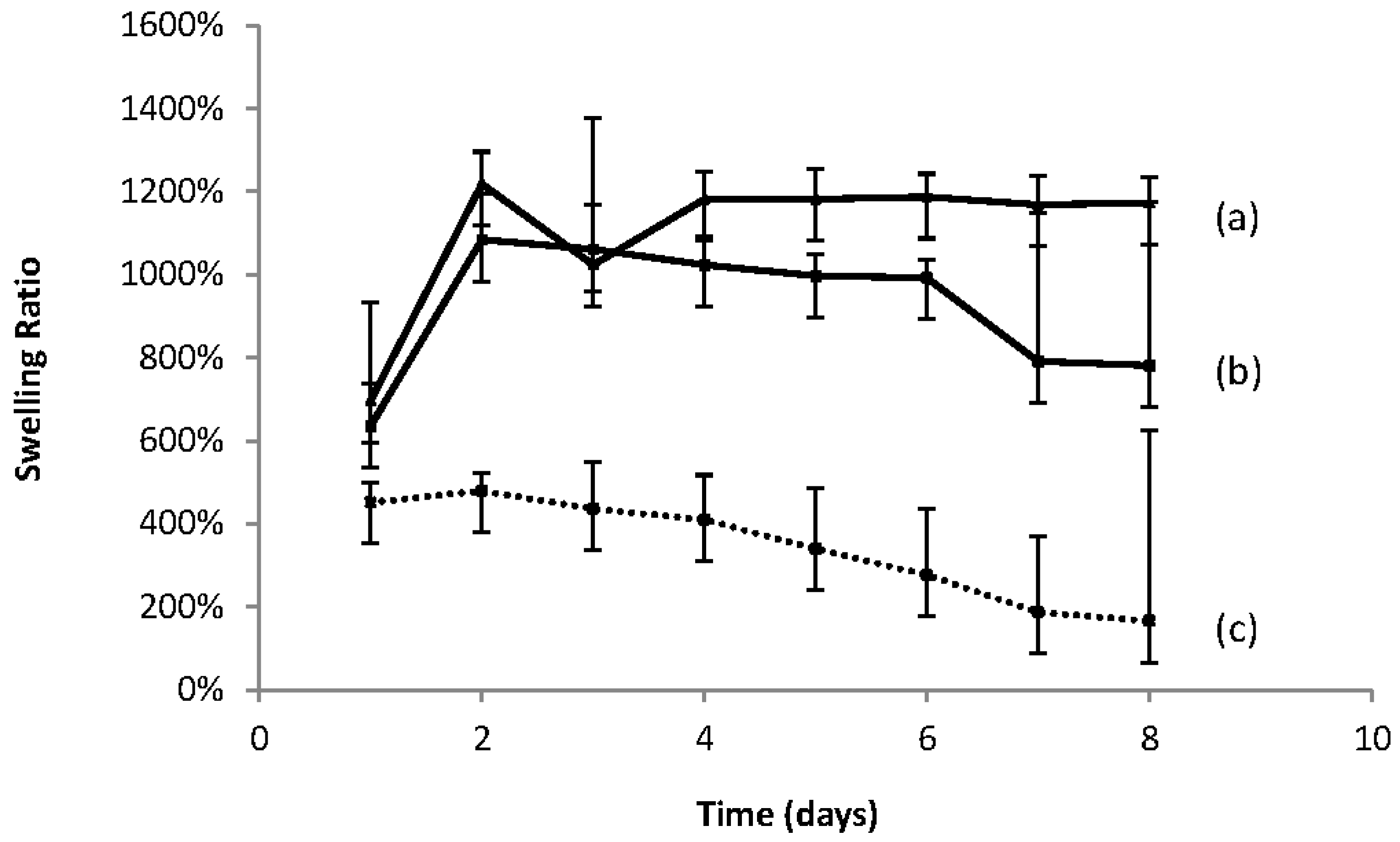
In
Figure 3, plots are shown of the Swelling Ratio vs. Time for the dried cross-linked macromer gel 1KL3 in water, and it is seen that the 30% hydrogel becomes more swollen than the 54% hydrogel. This may be a result of the hydrogel structure being more “open” as a consequence of a less crosslinked structure being formed when less macromer was used. An open structure with lower crosslinking density is likely to be more water absorbent and have a higher swelling ratio. Both curves have maximum swelling in approximately 3 days, and then they both start to degrade. This indicates that the 54% hydrogel is not capable of absorbing more water which would be the case if the material were merely slower in uptake due to its density of crosslinking. It is also observed that after being fully swollen, degradation occurs but this is more rapid for the 30% hydrogel which is again a consequence of its higher water content. The 30% hydrogel mass, with its fewer crosslinking points and consequent lower mechanical strength, is likely to disintegrate more rapidly if these links are broken as a result of hydrolysis.
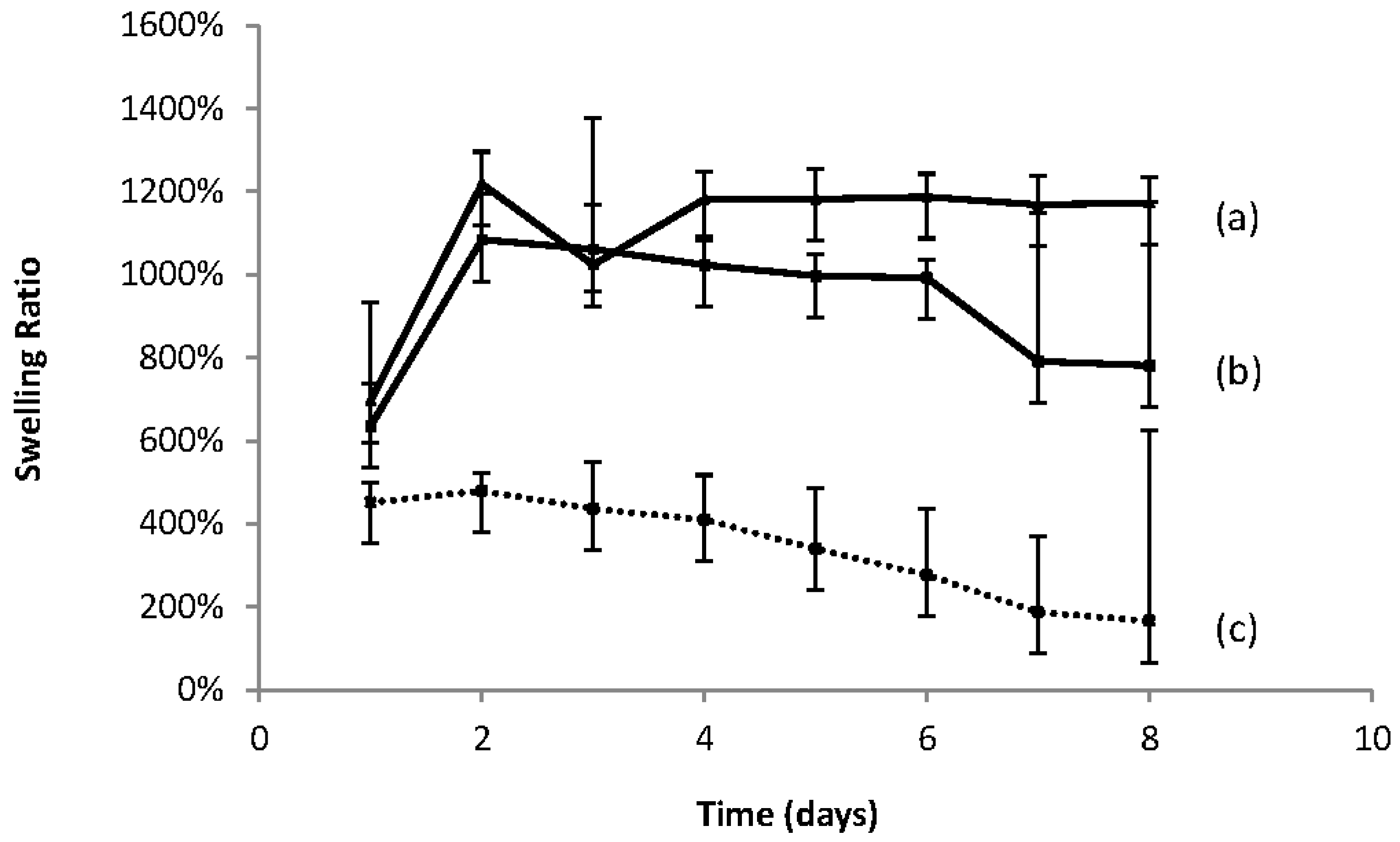
Figure 4c shows the swelling curve for a 50% concentration for the UV cross-linked gel macromer 1KL11 (entry 3 in
Table 5) which has a greater lactoyl component than that for macromer 1KL3 (entry 2 in
Table 5). It is observed that 1KL11 has a higher swelling ratio which could be due to its low crosslinking density compared to 1KL3, although the hydrogel from 1KL11 has a higher lactoyl content which generally confers greater hydrophobicity to the macromer. This explanation is given more credence by the fact that 1KL3 undergoes a faster degradation than 1KL11 (entry 3 in
Table 5) which is presumably due to the greater exposure of its lactoyl domains to water and therefore a greater likelihood of hydrolytic cleavage.
It may be seen from
Figure 4 that both the macromer and the co-polymers of PEGMEMA absorbed water to achieve swelling. Of the co-polymers,
Figure 4 shows that hydrogels with the higher PEGMEMA content (
Figure 4a) had the highest swelling ratio, followed by the other co-polymer (
Figure 4b), and finally the macromer only (
Figure 4c) exhibited the least swelling. It is thought that the lower concentration of the macromer crosslinking agent in
Figure 4a leads to a more open structure with a lower crosslinking density. This would allow for ready access of water to the hydrophilic regions of the cross linked structure, i.e., the PEG domains contained within the macromer and the PEGMEMA. This would give rise to the highest swelling ratio of the three materials. The homo-polymerised macromer (
Figure 4c) formed hydrogels with the lowest swelling ratios. This, in line with the above argument, is due to its dense crosslinked structure which hinders water access to the interior structure. Conversely, the hydrophobic regions, due to the presence of the lactoyl domains within the macromer, are found in the highest ratio in
Figure 4c and contribute to the lowest degree of swelling. The lactoyl content is reduced further in
Figure 4b and is at its lowest value in
Figure 4a, which has the highest swelling ratio.
The swelling curves also show a reduction in the swelling ratio after approximately 2 days for all three entries, indicating the hydrogels begin to degrade after the swelling reaches a maximum. The degradation is due to the hydrolysis of the lactoyl domains in the material. The macromer with the highest lactoyl content (
Figure 4c) is seen to first commence degradation after two days. The material with the lowest lactoyl content (
Figure 4a) is seen to be the last to commence degradation.
4. Experimental (Materials and Methods)
4.1. Materials
Poly(ethylene glycol) (PEG) (Mn = 1000 g·mol−1) was obtained from Polysciences Inc. (Europe GmbH, Germany). The following chemicals were purchased from Sigma Aldrich and used as received: d,l-lactide (3,6-dimethyl-1,4-dioxane-2,5-dione), stannous 2-ethylhexanoate, triethylamine, acryloyl chloride, poly(ethylene glycol) methyl ether methylacrylate (PEGMEMA, Mn = 475 g·mol−1) (containing inhibitors 100 ppm MEHQ and 200 ppm BHT), 1,1′-azobis (cyclohexanecarbonitrile) (ACHN), Irgacure 2959 (2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone), lithium bromide (LiBr), dry IR-grade potassium bromide (KBr), pentaerythritol tetrakis (3-mercaptopropionate) (QT) cross-linker, deuterated chloroform (CDCl3), dimethyl sulfoxide-d6 (DMSO-d6), tetrahydrofuran (THF), dichloromethane (DCM), toluene, and dimethylformamide (DMF).
4.2. Synthesis of PDLLA-PEG1k-PDLLA Co-Polymers
The PEG was dried either by heating under a stream of dry nitrogen gas with stirring at 150 °C for 3 h or was dried by azeotropic distillation with toluene.
d,
l-lactide was dried either by adding it to the dry melt of the PEG and heating for 30 min under a stream of nitrogen gas at 150 °C or was recrystallized in ethyl acetate before use. A typical procedure for the synthesis of the co-polymer e.g., 1KL2.7 (entry 11 in
Table 1) is described below: a total of 30 g (30 mmol) of dried PEG was placed in a two neck flask and heated under a stream of nitrogen at 150 °C for 3 h with constant stirring to which 12.97 g (90 mmol) of
d,
l-lactide was then added and heating was continued for another 30 min under nitrogen to remove any water impurity. The stannous 2-ethylhexanoate (194 μL (0.6 mM)) was transferred to the reaction flask and the temperature reduced to 130 °C. The reaction was left to proceed for 24 h and was then terminated by turning off the heat and allowing to cool. A stock solution of stannous 2-ethylhexanoate was prepared to enable accurate delivery of the correct amount into the reaction vessel. After the reaction, the resulting co-polymer was then dissolved in dichloromethane, precipitated in anhydrous ether, then dried in a vacuum oven at room temperature. The PEG with its α,ω-dihydroxy end groups acted as a ring opening reagent to initiate the polymerisation of the
d,
l-lactide [
14]. Other co-polymers were prepared by varying the ratio of
d,
l-lactide to PEG (see
Table 1).
4.3. Synthesis of Diacryl-PDLLA-PEG1k-PDLLA Macromers
The above PDLLA-PEG1k-PDLLA co-polymers were end capped with acrylate moieties to introduce vinyl functionalities and thus form polymerisable diacryl-PDLLA-PEG1k-PDLLA macromers. In a typical procedure, a total of 12 g (8.38 mM) of PDLLA-PEG1k-PDLLA co-polymer was dissolved in 100 mL of dichloromethane in a 250 mL two neck round bottom reaction flask and cooled in an ice bath to 0 °C. Triethylamine, 2.8 mL (20.11 mM) was added to the mixture under constant stirring, and then 3.39 mL (41.90 mM) of acryloyl chloride was slowly added. The reaction mixture was maintained at 0 °C for several hours and allowed to continue at room temperature for another 12 h. The mixture was then filtered to remove the white precipitate of triethylamine hydrochloride and the macromer was separated by dropwise addition into a large excess of anhydrous diethyl ether. It was then redissolved in dichloromethane and re-precipitated out of a large excess of dry hexane and dried under vacuum at room temperature overnight. Alternatively, to separate out the macromer product, the mixture was dissolved in ice cold water (5–8 °C), and the resulting solution was then heated to 80 °C and caused the co-polymer to precipitate, thus leaving the unreacted monomers in the solution. The macromer was isolated by removing the supernatant liquid and was dried under vacuum at room temperature.
4.4. Preparation of Soluble Hyperbranched Polymers Using Diacryl-PDLLA-PEG1k-PDLLA Macromers via Free Radical Polymerisation
Hyperbranched polymers were prepared by homo-polymerisation of the diacryl-P
DLLA-PEG
1k-P
DLLA macromer and also by the co-polymerisation of this macromer with PEGMEMA via free radical polymerisation (FRP). Typically, for the homo-polymerisation of 1KL10, 1.64 g (0.714 mmol) of macromer (prepared according to entry 3 in
Table 2) was dissolved in 2 mL of DMF to which 0.004 g (0.24 wt %) of the initiator ACHN was then added. The mixture was purged with nitrogen for 15 min to remove any dissolved oxygen. The mixture was then heated to 65 °C and GPC samples were taken at suitable intervals. For the co-polymerisation of the macromer and PEGMEMA, the molar ratio of macromer to PEGMEMA was varied according to
Table 3. Samples were withdrawn at required intervals for GPC analysis.
4.5. Characterisations of PDLLA-PEG1k-PDLLA Co-Polymers, Diacryl-PDLLA-PEG1k-PDLLA Macromers, and Hyperbranched Polymers
The co-polymer and macromer structures were determined by 1H NMR spectroscopy. The spectra were obtained on a Brucker 500 MHz NMR and analysed using MestReNova-Lite software (Version 11.0). Deuterated chloroform (CDCl3) with tetramethylsilane (TMS) as an internal standard was used as the solvent. Gel permeation chromatography (GPC) was used to determine the size of the macromers and polymers as this method separates analytes on the basis of size/hydrodynamic volume. Chromatograms were recorded on a PL-GPC 50 Plus Integrated GPC/SEC System from Agilent Technologies. The number average molecular weight (Mn), the weight average molecular weight (Mw), and the polydispersity index (Mw/Mn) were determined using an RI (refractive index) detector. The columns (PLgel Mixed-C column 300 mm in length, two in series) were eluted using THF and calibrated with poly (methyl methacrylate) standard. All calibrations and analyses were performed at a flow rate of 1 mL/min at 40 °C. FTIR spectra were recorded on a PerkinElmer Spectra 100 FTIR Spectrometer. Samples were cast as thin films on sodium chloride disks from a chloroform solution or were prepared as KBr disks. Thermal responsive properties of the polymers were studied by measuring the low critical solution temperatures (LCSTs) of the polymers in aqueous solutions (0.1 w/v % concentration in de-ionised water) using a Cary 100 UV-Vis spectrophotometer. The parameters for the measurements used were the heating rate as 1 °C/min, data collection rate as 0.06 °C/point, absorbance wavelength at 550 nm, and the temperature range between 15–60 °C.
4.6. Preparation of Biodegradable Hydrogels from Linear Diacryl-PDLLA-PEG-PDLLA Macromers
Biodegradable hydrogels were prepared using the diacryl-PDLLA-PEG1k-PDLLA macromer by homo-polymerisation and co-polymerisation with PEGMEMA via the Michael addition reaction and free radical photopolymerisation.
4.6.1. Michael Addition Method
Chemically cross-linked hydrogels were prepared using diacryl-P
DLLA-PEG
1k-P
DLLA macromers and PEGMEMA by means of the Michael-addition reaction between their acrylate groups and the thiol functional groups in QT cross-linker (
Table 4). A typical homo-polymerisation involves the addition of 0.5 g (0.019 mmol) of macromer (
Mn = 2693.43 g·mol
−1) to a vial containing 500 µL of phosphate buffered solution (pH 7.4). Then 171.6 µL of QT was added in a stoichiometric molar ratio of vinyl group to thiol of 1:1. Triethylamine (TEA, 42.13 µL) was then added, and the sample was incubated at 37 °C for 2 h. Upon completion of the reaction, a transparent gel was formed. Co-polymerisation with the monomer PEGMEMA was undertaken using various ratios of macromer to PEGMEMA according to
Table 4.
4.6.2. Photo-Crosslinking Method
A 1% (
w/
v) stock solution of the photo-initiator Irgacure 2959 in de-ionised water was prepared. The homo-polymerisation of Macromer 1KL3 was performed by dissolving it in Irgacure stock solution to a final concentration of 54% and 30%. Respectively, 1 mL of each solution was placed into a small glass vial to form a layer with thickness of about 1 mm which was exposed to UV light (2.3 mW/cm
2) overnight at room temperature. Photo-polymerisation of Macromer 1KL11 and PEGMEMA was performed using the same procedures, and the ratio of macromer to PEGMEMA was varied according to
Table 5.
4.7. Swelling Studies
For the macromer 1KL3 hydrogel, two samples were prepared at 54% and 30% concentration (
Table 5), and then were tested for their swelling characteristics (
Figure 3). The dry weights of the materials were determined (
w0), and then hydrogels were formed from the co-polymers by the addition of de-ionised water and were allowed to soak at room temperature (25 °C). The excess surface water was gently removed by means of a fine pipette and gently touching the surface with tissue paper. The weight of the swollen gel was taken (
ws). The swelling ratio was defined as:
where
ws is the weight of the swollen hydrogel and
w0 is the weight of the dried hydrogel.
Readings were taken every 24 h and the results were tabulated and presented as a plot of swelling ratio vs. time in days. The tests were performed in triplicate.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}