RNA-Seq as an Emerging Tool for Marine Dinoflagellate Transcriptome Analysis: Process and Challenges


1
School of Bioscience and Biotechnology, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, Bangi 43600, Selangor, Malaysia
2
School of Environmental and Natural Resources Sciences, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, Bangi 43600, Selangor, Malaysia
3
Institute of Systems Biology, Universiti Kebangsaan Malaysia, Bangi 43600, Selangor, Malaysia
*
Author to whom correspondence should be addressed.
Processes 2018, 6(1), 5; https://doi.org/10.3390/pr6010005
Submission received: 16 November 2017
/
Revised: 28 December 2017
/
Accepted: 5 January 2018
/
Published: 11 January 2018
Abstract
:Dinoflagellates are the large group of marine phytoplankton with primary studies interest regarding their symbiosis with coral reef and the abilities to form harmful algae blooms (HABs). Toxin produced by dinoflagellates during events of HABs cause severe negative impact both in the economy and health sector. However, attempts to understand the dinoflagellates genomic features are hindered by their complex genome organization. Transcriptomics have been employed to understand dinoflagellates genome structure, profile genes and gene expression. RNA-seq is one of the latest methods for transcriptomics study. This method is capable of profiling the dinoflagellates transcriptomes and has several advantages, including highly sensitive, cost effective and deeper sequence coverage. Thus, in this review paper, the current workflow of dinoflagellates RNA-seq starts with the extraction of high quality RNA and is followed by cDNA sequencing using the next-generation sequencing platform, dinoflagellates transcriptome assembly and computational analysis will be discussed. Certain consideration needs will be highlighted such as difficulty in dinoflagellates sequence annotation, post-transcriptional activity and the effect of RNA pooling when using RNA-seq.
1. Introduction
Dinoflagellates are a marine unicellular eukaryotic organism and these organisms are one of the major components of marine phytoplankton besides diatoms [1]. Dinoflagellates have been studied extensively for their role in maintaining coral reefs and their toxin production especially during the event of Harmful algae blooms (HABs) in the ocean [2,3,4]. HABs caused by dinoflagellates has been associated with water discoloration and accumulation of toxins through the marine food chain, gave a severe negative impact toward economic and health [5,6]. However, their exceptionally large genome size (3–245 Gb) and permanently condensed chromosomes hinder the genomic approach to unravel dinoflagellates genome complexity especially in regards to their toxin production [7]. Nevertheless, transcriptomic is deemed as a practical alternative for genome survey of non-model organism such as dinoflagellates [8].
Transcriptomic is a reference to the total RNA presence and quantity in cells including messenger RNA (mRNA), small RNA (smRNA), ribosomal RNA (rRNA), transfer RNA (tRNA) and non-coding RNA [9]. Unlike genome, which is relatively stable and unchanged, transcriptomes tend to change under different environmental conditions and growth stages [10]. Thus, transcriptomic acts as a powerful tool to unravel the complexity of dinoflagellates physiology and gene regulation. Transcriptomic will be able to identify and characterize the total RNA expressed in the cells during a particular growth stage or time, to quantify differently expressed genes and to determine functional structure of the gene [11]. The advancement of the Next-Generation Sequencing (NGS) technologies surely provide a huge impact on the profiling of the dinoflagellates transcriptome (Table 1).
RNA-seq is one of the transcriptomic methods to sequence the entire transcriptome in the cell at high resolution with a low cost. This method has been employed extensively for both eukaryotic and prokaryotic organisms [10,22,23,24]. Thus, in this review, we provide an overview of the process and technical aspect of dinoflagellates transcriptomics technology using RNA-seq and highlight the considerations and challenges using RNA-seq.
2. Dinoflagellates Transcriptomics: A Brief History
In the early stages of the dinoflagellates transcriptomics study, the differential-display reverse transcription-PCR (DDRT-PCR) was used to identify the over-expressed or under-expressed genes in dinoflagellates [25,26,27]. In general, DDRT-PCR requires a large amount of RNA, cannot recognize under or over-expressed genes accurately and can only compare two types of population simultaneously [28]. Due to these issues, this approach was not popular among scientists and the microarrays have remained the most popular approach for the transcriptome profiling for almost a decade (Table 2).
Microarray technology has the ability to simultaneously detect tens of thousands of transcriptomes and has led to important advances in tackling a wide range of biological problems, including the identification of genes that are differentially expressed between different environmental conditions and growth phases [36,42]. Despite this, microarray technology has several limitations, for example the background hybridization limits the accuracy of the expression measurements, particularly for transcripts present in low abundance. Microarray also has limitations in capturing only those genes for which probes are designed, thus, probe design is crucial for microarray approach and the target sequence is needed prior to designing the probe [43].
Another method which has been applied for dinoflagellates transcriptomics is Serial Analysis of Gene Expression (SAGE). This method enables the large-scale analysis of transcriptome without prior knowledge of the genes thus making it the superior method, compared to microarray [44]. Coyne et al. [37] developed a novel approach to facilitate the construction of SAGE libraries for dinoflagellates which require a lower amount of RNA to identify genes expressed during toxic zoospores stages of Pfiesteria shumwayae. Apparently, the SAGE method did not get too much attention and this method was soon replaced by Massively Parallel Signature Sequencing (MPSS) which covers a deeper sequencing depth and more sensitive to low abundance genes compared to SAGE [2,38]. Utilization of the MPSS method has proven useful for Alexandrium spp. transcriptome profiling and its regulation [2,38].
Currently, the RNA-seq is a new option for studying the whole genome transcriptome profile of an organism. RNA-Seq is a direct sequencing of transcripts by high-throughput sequencing technologies using next-generation sequencing (NGS). The RNA-seq approach does not depend on knowledge of the target sequence prior to probe selection and can avoid the related biases introduced during hybridization of microarrays [43]. RNA-seq is also capable of measuring the abundance of expressed transcripts for both low-levels and high-levels transcripts with a greater sensitivity, compared to the microarray method [11]. The high-throughput NGS analysis plays a major role in molecular biology field to measure the activity of thousands of genes in parallel. Changes in gene expression due to different environmental conditions or growth stages can be examined through absolute quantification of transcripts. Although the RNA-seq method is still new in transcriptome analysis, it has considerable advantages in application of gene expression profiling, alternative splicing, SNP discovery, mapping and quantification of transcriptome compared to other transcriptome profiling methods [45]. We strongly believe that RNA-seq strategy will revolutionize dinoflagellates transcriptome analysis as the entire transcriptome can be sequenced in a high-throughput manner with low cost, high sensitivity and without introducing any bias like other transcriptomic methods.
3. Strategies for Dinoflagellates RNA-Seq
3.1. Isolation of RNA
For efficient and high-throughput transriptome profiling, the most important operation is to obtain high quality RNA samples. The first step of RNA extraction is to release all the RNA by effective disruption of the tough dinoflagellate cell wall without degrading the RNA in the cell (Figure 1). This step is often very challenging due to the fact that dinoflagellates are known to have thick cell wall. Available commercial lysis buffers are known inefficiently to lyse dinoflagellates cells completely, therefore are unable to prevent the RNA degradation. It is essential to use extra technique to disrupt the cells or tissue of dinoflagellates to obtain maximum yield and high quality of the RNA.
Several cell disruption methods have been tested (bead beating, grinding using a micropestle and sonication) against dinoflagellates Alexandrium catenella [46]. Cell disruption using micropestle were reported the most efficient method to disrupt the dinoflagellates cells. Bead beating and sonication for more than 20 s always lead to degradation of the RNA due to the combination of the RNA shearing and warming of the sample, which would activate RNases. Rosic and Hoegh-Guldberg [47] performed a few RNA extraction protocols to evaluate the most efficient method for extracting the RNA for dinoflagellates. Several methods were tested and interestingly a modified RNA extraction procedure that combines two existing commercial kits, Trizol and Qiagen RNeasy kits were demonstrated to recover high-quality RNA under specific homogenization conditions.
Next, the quality of the obtained RNA is tested on denaturing formaldehyde gels for the presence of intact 18S and 28S rRNA bands. Presence of smearing on the gel indicates that the obtained RNA was degraded and not suitable for sequencing [24]. The most recent protocol utilizing Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) electropherogram which defines the RNA quality using RNA Integrity Number (RIN) calculated by the machine through generated electropherogram and RIN. Values higher than 6 represent RNA with preserved integrity [24,47]. However, several studies have suggested that researchers should not blindly trust the RIN number as a presence of chloroplast as rRNA from dinoflagellates may interfere with RIN calculations. The generated electropherogram should be interpreted manually for the presence of a clear peak of 18S/28S rRNA and a flat inter-region between those regions represent a good quality of RNA.
3.2. Preparation of cDNA Library
Currently, RNA-Seq approach relies on the transforming of RNA into cDNA molecules using random hexamers or oligo(dT) primers before sequencing, as RNA cannot be sequenced directly [48]. It is possible for researchers to directly sequence total cellular RNA, but the RNA sequences will likely be dominated by non-mRNA sequences as more than 80% of total cellular transcriptome composed of the ribosomal RNA (rRNA) [49]. Therefore, for high-throughput sequencing it is generally necessary to remove rRNA from the total RNA.
The next step for the construction of a cDNA library is the RNA fragmentation process. This process is important to fragmentize the long RNA into short sizes of fragments, according to the NGS sequencing platform. In most cases, RNA is fragmented prior to the reverse transcription to cDNA molecules allowing a more uniform fragment across the transcript and allowing for a more comprehensive analysis of transcriptome, but resulting in depleted transcript ends [10]. However, when using the Roche 454 platform, the fragmentation is done after the reverse transcription to cDNA molecules, but rendering the sequencing depth lower than Illumina or SOLiD platform [24].
3.3. High-Throughput Sequencing
From the cDNA obtained, each cDNA fragment, with or without amplification, is then sequenced in a high-throughput manner to obtain short sequences from one end (single-end sequencing) or both ends (pair-end sequencing) using the NGS platform [11]. Current sequencing step for dinoflagellates RNA relies on the widely used NGS platform such as Illumina and Roche 454 [15,50]. Each sequencing platform has their own advantages and disadvantages thus a careful consideration needs to be addressed according to the experiment goals. In general, both of the platforms have substantial variances in read length and sequencing procedures. For instance, Roche 454 offers longer reads up to 500 nt which significantly will reduce the effort to assemble the transcripts ambiguously [51]. On the other hand, Illumina provides equivalent or better base-call error, frameshift frequency and contig length assemblies than Roche 454 [51]. Thus, to have low sequencing error rate and greater sequencing depth, Illumina seems to be the best choice as this platform offer <1% mismatch error and high sequencing depth that will facilitate detection of low expressed genes [51]. Detailed comparison and technical aspect of each NGS platform for RNA-seq including methodological procedure and sequencing chemistry will not be discussed here as it has already been discussed in a few review articles [52,53,54]. Another option for RNA-seq is using two or more platforms for high-throughput sequencing. For example, the longer reads of Roche 454 datasets can be used as a scaffold to connect the short reads of SOLiD datasets [55].
3.4. De Novo Transcriptome Assembly
Following sequencing by the NGS platform such as Illumina and Roche 454, the sequence reads generated are often ranging from 50 to 330 bp resulting in the necessity for the reconstruction of full length transcripts using a suitable assembler program [56]. However, the reads generated contain artefacts originated from low complexity reads, adapter mismatch or even during the sequencing. There are several tools available for RNA-seq raw data trimming [57]. The generated reads are either aligned to a reference genome or transcripts. For non-model organisms like dinoflagellates, reference genomes do not exist. De novo transcriptome assembly is used to produce a genome-scale transcription map that consists of both the transcriptional structure and/or level of expression for each gene [11].
Although recently draft genomes of the coral symbionts dinoflagellates from the genus Symbiodinium were obtained and may serve as key model organism for transcriptome assembly, it was found to not be densely covered by high-quality draft genome sequences [14,18,58,59,60]. Moreover, dinoflagellates transcriptome exhibits complex alternative splicing patterns and RNA-seq samples genetically exhibit polymorphism [61,62]. Thus, the de novo transcriptome assembly approach remains the best option for dinoflagellates transcriptome assembly. Several de novo transcriptome assembly programs have been used widely in dinoflagellates transcriptomics studies such as Trinity, ABySS de novo and SOAPdenovo [15,63,64,65]. In general, these assemblers utilize the De Bruijn graph-based approach where the generated reads are broken into smaller reads called k-mers. The overlapping of these k-mers are then used to create a contig. A more comprehensive working principle of how De Bruijn graph-based approach works in de novo transcriptome assembly can be found here [66].
3.5. Sequence Annotation
Complete annotation and quantification of all genes and their isoforms across samples is crucial for understanding the biology of organism [67]. Functional annotation is a process in transcriptomics data analysis to categorize the genes presence in the data generated to their respective functional classes, which can be very useful in understanding the physiological meaning of large amounts of genes and to assess functional differences between subgroups of sequences [68]. However, to annotate each of the transcripts as completely as possible requires a powerful computational tool or else the value of generated data will be minimal. In dinoflagellates transcriptomics study, the contig usually annotated by Blast alignment against NCBI non-redundant protein (NR) and nucleotide (NT), Kyoto Encyclopedia of Genes and Genomes (KEGG), SwissProt and InterPro database [34,69,70]. Blast2go is a powerful computational tool for high-throughput functional annotation of RNA sequences and data mining based on Gene Ontology (GO), for a non-model organism with a user-friendly interface and is less labor intensive [71].
3.6. Computational Analysis for Differently Expressed Genes
The final goal of the majority dinoflagellates transcriptomics study is focusing on the targeting of differently expressed genes in the different biological conditions to get a better understanding about molecular regulation in the dinoflagellates physiology [1,34,72]. RNA-seq provides data in the form of read coverage to quantify transcript abundance presence in the sample to provide additional information such as a distribution for a particular transcript [67]. The method to analyse differently expressed genes is built based on the statistical model. Fisher’s exact test and likelihood ratio test is based on the assumption of the number of reads coming from a gene or transcript isoform following a binomial, or Poisson distribution [11]. Several studies have been done also applying Fisher’s exact test using several statistical packages such as DEGseq and IDEG6 to find the differently expressed genes in dinoflagellates during different biological conditions [2,50,63].
4. Challenges and Consideration in Dinoflagellates Transcriptome Studies Using RNA-Seq
Despite the powerful potential of the transcriptomics approach for gene expression study in dinoflagellates, there are a few limitations that have been reported in this approach. Transcriptome surveys in non-model species will be much easier if there are genomic resources available for a related species to aid in assembly and functional annotation purposes [8]. Annotation of dinoflagellates genes remains as one of the biggest challenges due to lack of genomics data for dinoflagellates [34]. Automated annotation of A. minutum contigs only yield a hit for 28% of the contigs and the annotation of Oxyrrhis marina transcriptomics data also failed to identify 76% of these contigs [34,50].
As some of differently expressed genes cannot be annotated, this is clearly one of the major limitations not just to reveal the genetic basis for any traits but also for elucidating the environmental adaptation mechanism. A study done by Gibson et al. [73] found that there is no strong correlation between either low sequence or assembly quality to these un-annotated genes in eukaryotic genomes. These un-annotated genes might be an indication for a novel pathway to yet be discovered in dinoflagellates [74]. Recently, two draft genomes of dinoflagellates have been sequenced [7,58]. Approximately 42,000 protein-coding genes were detected in the S. minimum draft genomes and this work will give hope and insight on further gene identification and transcriptome assembly in dinoflagellates.
RNA turnover also needs to be considered during the cell harvesting prior to RNA extraction especially for the gene expression studies in dinoflagellates. The extracted RNA in the ready-state abundance of mRNAs in a cell must represent an actual snapshot of the transcriptome during the desired condition. The mRNAs act as template for protein synthesis and it also provides the rates of transcription and degradation in the cells. Survey of mRNA stability in dinoflagellates using in vivo labeling method revealed quite diverse mRNA half-life in Karenia brevis ranging from 42 min to 144 h [75]. However, a recent report demonstrated that mRNA degradation may occur rapidly with some genes start to degrade as early as 2 min [76]. Utilizing RNA stabilization solution such as RNAlater and RNAlater ICE seems effective to stop the dinoflagellates gene expression without compromising RNA integrity [46].
Although mRNA is eventually translated into protein, there is a poor correlation between mRNA and protein abundance and these correlations are far from having a linear and simple relationship [77,78]. Several post-transcriptional events may uncouple the correlation between mRNA and protein abundance, including the presence of a RNA secondary structure, regulatory RNA such as miRNA, codon bias, translational efficiency and protein turnover [78]. To date, the transcriptomics approach using either microarray or RNA-seq only record the levels of RNA (cDNA) present and are thus not able to detect any post transcriptional events [79]. Although until now there is no detailed study regarding correlation between mRNA and protein abundance in dinoflagellates, this limitation needs to be considered as there is strong proof for post-transcriptional gene regulation such as regulation by miRNA in dinoflagellates [4,19,24,33,80,81,82].
Proteomic and western blotting are the two most common methods to validate protein expressions in dinoflagellates that may help to detect the post transcriptional events, however both methods are hard to be applied due to the difficulty in protein identification and the availability of suitable antibodies for dinoflagellates [80,83]. Evans [84] also reported that the differently expressed genes during different experimental conditions are not correlated with the phenotypic changes of the organism. Differently expressed genes are always targeted by researchers to capture the biomarker for environmental stress conditions or even for toxin biosynthesis. However, different types of stress response may be resulting in the same kind of cellular damages [84]. Analysis of dinoflagellates transcriptomics data revealed that there is overlapping response between different types of environmental stress such as photosynthesis related genes [1,42]. Thus, a careful and detailed bioinformatics analysis is needed to elucidate specific stress related genes. Harvesting time for RNA extraction is crucial, especially for time series expression experiments. This is due to the fact that the cell will only express a small fraction of the genes that are present in their genome. Multiple harvesting time is recommended to allow one to distinguish between primary and secondary responses especially related to a stress study [85].
RNA pooling is one of the practices used by some researchers to increase the dinoflagellates RNA diversity representation without increasing the cost of sequencing and can help to reduce workload intensity in a large-scale experiment [12,34,86,87]. RNA samples from multiple experimental conditions or different growth stages are pooled together before preparation of the cDNA library to reduce the effects of biological variation and increased the diversity of RNA. Kendziorski et al. [88] demonstrated that RNA pooling is reducing the effects of biological variation only, but not the biological variation itself. This practice is often desirable when the main interest of the study is to gain vast transcriptomics data from the same individual without concerning the expression level (e.g., comparison of toxic and non-toxic dinoflagellates) as the transcriptome expression level is subjected to non-linear distortion during data normalization [34,88]. Another limitation is the fact that the information regarding the biological variation cannot be traced back and differently expressed genes from individual RNA samples cannot be detected [89]. It is crucial to check the quality of individual RNA samples before pooling the RNA as the quality of transcriptomics data will be affected by a contaminated or degraded sample [86]. The study on comparative pooled RNA in toxic versus non-toxic strains of A. minutum revealed that the putative candidates involved in toxin biosynthesis and regulation, or acclimation of PSP toxins were only present in the mRNA pools of toxic strain but some of these genes could not be annotated due to the lack of known similar sequences.
5. Conclusions
The RNA-seq method is a powerful tool for dinoflagellates transcriptome study and we have highlighted a brief overview of dinoflagellates technical aspect of transcriptome strategies using RNA-seq as well as a few considerations for this method. Application and utilization of RNA-seq strategies for dinoflagellates transcriptome analysis will benefit us with the knowledge especially regarding gene regulation, novel gene and single nucleotide polymorphism discovery in this mystery of phytoplankton, dinoflagellates.
Acknowledgments
This research was supported by MOSTI, Malaysia under the research grant 04-01-02-SF1244.
Author Contributions
M.A.A. drafted the manuscript. A.A., G.U., H.B. edited and added valuable insights into the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cooper, J.T.; Sinclair, G.A.; Wawrik, B. Transcriptome analysis of Scrippsiella trochoidea CCMP 3099 reveals physiological changes related to nitrate depletion. Front. Microbiol. 2016, 7, 639. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Evans, A.N.; Kulis, D.M.; Hackett, J.D.; Erdner, D.L.; Anderson, D.M.; Bhattacharya, D. Transcriptome profiling of a toxic dinoflagellate reveals a gene-rich protist and a potential impact on gene expression due to bacterial presence. PLoS ONE 2010, 5, e9688. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Beszteri, S.; Tillmann, U.; Cembella, A.; John, U. Growth-and nutrient-dependent gene expression in the toxigenic marine dinoflagellate Alexandrium minutum. Harmful Algae 2011, 12, 55–69. [Google Scholar] [CrossRef] [Green Version]
- Baumgarten, S.; Bayer, T.; Aranda, M.; Liew, Y.J.; Carr, A.; Micklem, G.; Voolstra, C.R. Integrating microRNA and mRNA expression profiling in Symbiodinium microadriaticum, a dinoflagellate symbiont of reef-building corals. BMC Genom. 2013, 14, 704. [Google Scholar] [CrossRef] [PubMed]
- Perini, F.; Casabianca, A.; Battocchi, C.; Accoroni, S.; Totti, C.; Penna, A. New approach using the real-time PCR method for estimation of the toxic marine dinoflagellate Ostreopsis cf. ovata in marine environment. PLoS ONE 2011, 6, e17699. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Malkiel, E.; Katz, J.; Adolf, J.E.; Place, A.R. A dinoflagellate exploits toxins to immobilize prey prior to ingestion. Proc. Natl. Acad. Sci. USA 2010, 107, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Lin, S. Genomic understanding of dinoflagellates. Res. Microbiol. 2011, 16, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, R.; Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 2011, 107, 1. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genom. Hum. Genet. 2009, 10, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Ba, Y.; Zhuang, Q.; Zhong, G. RNA-Seq technology and its application in fish transcriptomics. OMICS J. Integr. Biol. 2014, 18, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Gen. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.; Aranda, M.; Sunagawa, S.; Yum, L.K.; DeSalvo, M.K.; Lindquist, E.; Coffroth, M.A.; Voolstra, C.R.; Medina, M. Symbiodinium transcriptomes: Genome insights into the dinoflagellate symbionts of reef-building corals. PLoS ONE 2012, 7, e35269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, S.-F.; Lin, L.; Wang, D.-Z. Comparative transcriptome analysis of a toxin-producing dinoflagellate Alexandrium catenella and its non-toxic mutant. Mar. Drugs 2014, 12, 5698–5718. [Google Scholar] [CrossRef] [PubMed]
- Gierz, S.L.; Forêt, S.; Leggat, W. Transcriptomic analysis of thermally stressed Symbiodinium reveals differential expression of stress and metabolism genes. Front. Plant Sci. 2017, 8, 271. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.E.; Pepper, A.E.; Campbell, L. De novo assembly and characterization of the transcriptome of the toxic dinoflagellate Karenia brevis. BMC Genom. 2014, 15, 888. [Google Scholar] [CrossRef] [PubMed]
- Pawlowiez, R.; Morey, J.S.; Darius, H.T.; Chinain, M.; Van Dolah, F.M. Transcriptome sequencing reveals single domain Type I-like polyketide synthases in the toxic dinoflagellate Gambierdiscus polynesiensis. Harmful Algae 2014, 36, 29–37. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.F.; Lin, L.; Wang, D.Z. Whole Transcriptomic Analysis Provides Insights into Molecular Mechanisms for Toxin Biosynthesis in a Toxic Dinoflagellate Alexandrium catenella (ACHK-T). Toxins 2017, 9, 213. [Google Scholar] [CrossRef] [PubMed]
- Rosic, N.; Ling, E.Y.S.; Chan, C.-K.K.; Lee, H.C.; Kaniewska, P.; Edwards, D.; Dove, S.; Hoegh-Guldberg, O. Unfolding the secrets of coral-Algal symbiosis. ISME J. 2015, 9, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Lin, X.; Li, L.; Li, M.; Palenik, B.; Lin, S. Transcriptomic and microRNAomic profiling reveals multi-faceted mechanisms to cope with phosphate stress in a dinoflagellate. ISME J. 2017, 11, 2209–2218. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Zhang, H.; Hannick, L.; Lin, S. Metatranscriptome profiling reveals versatile N-nutrient utilization, CO 2 limitation, oxidative stress, and active toxin production in an Alexandrium fundyense bloom. Harmful Algae 2015, 42, 60–70. [Google Scholar] [CrossRef]
- Ogura, A.; Lin, M.; Shigenobu, Y.; Fujiwara, A.; Ikeo, K.; Nagai, S. Effective gene collection from the metatranscriptome of marine microorganisms. BMC Genom. 2011, 12, S15. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Thomson, N.R. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Tjaden, B. De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.A.; Ponnala, L.; Weber, C.A. Strategies for transcriptome analysis in nonmodel plants. Am. J. Bot. 2012, 99, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Taroncher-Oldenburg, G.; Anderson, D.M. Identification and characterization of three differentially expressed genes, encoding S-adenosylhomocysteine hydrolase, methionine aminopeptidase, and a histone-like protein, in the toxic dinoflagellate Alexandrium fundyense. Appl. Environ. Microbiol. 2000, 66, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Hosoi-Tanabe, S.; Tomishima, S.; Nagai, S.; Sako, Y. Identification of a gene induced in conjugation-promoted cells of toxic marine dinoflagellates Alexandrium tamarense and Alexandrium catenella using differential display analysis. FEMS Microbiol. Lett. 2015, 251, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Karako-Lampert, S.; Hershkovits, G.; Stambler, N.; Simon-Blecher, N.; Achituv, Y.; Dubinsky, Z.; Katcoff, D.J. Differential gene expression in Symbiodinium microadriaticum clade B following stress. Mar. Biotechnol. 2006, 8, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Sturtevant, J. Applications of differential-display reverse transcription-PCR to molecular pathogenesis and medical mycology. Clin. Microbiol. Rev. 2000, 13, 408–427. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Velculescu, V.E.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Serial analysis of gene expression. Science 1995, 270, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.; Johnson, M.; Bridgham, J.; Golda, G.; Lloyd, D.H.; Johnson, D.; Roth, R. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol. 2000, 18, 630. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, O.K.; Hastings, J. Novel dinoflagellate clock-related genes identified through microarray analysis. J. Phycol. 2003, 39, 519–526. [Google Scholar] [CrossRef]
- Van Dolah, F.M.; Lidie, K.B.; Morey, J.S.; Brunelle, S.A.; Ryan, J.C.; Monroe, E.A.; Haynes, B.L. Microarray Analysis of Diurnal-and Circadian-Regulated Genes in the Florida Red-Tide Dinoflagellate Karenia Brevis (Dinophyceae). J. Phycol. 2007, 43, 741–752. [Google Scholar] [CrossRef]
- Yang, I.; John, U.; Beszteri, S.; Glöckner, G.; Krock, B.; Goesmann, A.; Cembella, A.D. Comparative gene expression in toxic versus non-toxic strains of the marine dinoflagellate Alexandrium minutum. BMC Genom. 2010, 11, 248. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Selander, E.; Pavia, H.; John, U. Grazer-induced toxin formation in dinoflagellates: A transcriptomic model study. Eur. J. Phycol. 2011, 46, 66–73. [Google Scholar] [CrossRef]
- Johnson, J.G.; Janech, M.G.; Van Dolah, F.M. Caspase-like activity during aging and cell death in the toxic dinoflagellate Karenia brevis. Harmful Algae 2014, 31, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Coyne, K.J.; Burkholder, J.M.; Feldman, R.A.; Hutchins, D.A.; Cary, S.C. Modified serial analysis of gene expression method for construction of gene expression profiles of microbial eukaryotic species. Appl. Environ. Microbiol. 2004, 70, 5298–5304. [Google Scholar] [CrossRef] [PubMed]
- Erdner, D.L.; Anderson, D.M. Global transcriptional profiling of the toxic dinoflagellate Alexandrium fundyense using massively parallel signature sequencing. BMC Genom. 2016, 7, 88. [Google Scholar] [CrossRef]
- Leggat, W.; Hoegh-Guldberg, O.; Dove, S.; Yellowlees, D. Analysis of an EST library from the dinoflagellate (Symbiodinium sp.) symbiont of reef-building corals. J. Phycol. 2007, 43, 1010–1021. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Stüken, A.; Orr, R.J.S.; Kellmann, R.; Murray, S.A.; Neilan, B.A.; Jakobsen, K.S. Discovery of nuclear-encoded genes for the neurotoxin saxitoxin in dinoflagellates. PLoS ONE 2011, 6, e20096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morey, J.S.; Monroe, E.A.; Kinney, A.L.; Beal, M.; Johnson, J.G.; Hitchcock, G.L.; Van Dolah, F.M. Transcriptomic response of the red tide dinoflagellate, Karenia brevis, to nitrogen and phosphorus depletion and addition. BMC Genom. 2011, 12, 346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Wakatsuki, T.; Hada, A.; Ryo, A. Use of serial analysis of gene expression (SAGE) technology. J. Immunol. Methods 2001, 250, 45–66. [Google Scholar] [CrossRef]
- Mutz, K.O.; Heilkenbrinker, A.; Lönne, M.; Walter, J.G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Harlow, L.D.; Koutoulis, A.; Hallegraeff, G.M. A novel, simplified technique for preservation and rapid isolation of total RNA from the toxic dinoflagellate Alexandrium catenella (Dinophyceae). Phycologia 2006, 45, 311–318. [Google Scholar] [CrossRef]
- Rosic, N.N.; Hoegh-Guldberg, O. A method for extracting a high-quality RNA from Symbiodinium sp. J. Appl. Phycol. 2010, 22, 139–146. [Google Scholar] [CrossRef]
- Nagaraj, S.H.; Gasser, R.B.; Ranganathan, S. A hitchhiker’s guide to expressed sequence tag (EST) analysis. Brief. Bioinform. 2007, 8, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Lundeberg, J. The plasticity of the mammalian transcriptome. Genomics 2010, 95, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lowe, C.D.; Mello, L.V.; Samatar, N.; Martin, L.E.; Montagnes, D.J.S.; Watts, P.C. The transcriptome of the novel dinoflagellate Oxyrrhis marina (Alveolata: Dinophyceae): Response to salinity examined by 454 sequencing. BMC Genom. 2011, 12, 519. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Corey, D.R. RNA sequencing: Platform selection, experimental design, and data interpretation. Nucleic Acid Ther. 2012, 22, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blythe, M.J.; Kao, D.; Malla, S.; Rowsell, J.; Wilson, R.; Evans, D.; Aboobaker, A.A. A dual platform approach to transcript discovery for the planarian Schmidtea mediterranea to establish RNAseq for stem cell and regeneration biology. PLoS ONE 2010, 5, e15617. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Gen. 2010, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Gen. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Shoguchi, E.; Shinzato, C.; Kawashima, T.; Gyoja, F.; Mungpakdee, S.; Koyanagi, R.; Hamada, M. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 2013, 23, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Cheng, S.; Song, B.; Zhong, X.; Lin, X.; Li, W.; Cai, M. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 2015, 350, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.A.; Beltran, V.H.; Hill, R.; Kjelleberg, S.; McDougald, D.; Steinberg, P.D.; Van Oppen, M.J.H. Sex, Scavengers, and Chaperones: Transcriptome Secrets of Divergent Symbiodinium Thermal Tolerances. Mol. Biol. Evol. 2016, 33, 2201–2215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hou, Y.; Miranda, L.; Campbell, D.A.; Sturm, N.R.; Gaasterland, T.; Lin, S. Spliced leader RNA trans-splicing in dinoflagellates. Proc. Natl. Acad. Sci. USA 2007, 104, 4618–4623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dungan, C.F.; Lin, S. Introns, alternative splicing, spliced leader trans-splicing and differential expression of pcna and cyclin in Perkinsus marinus. Protist 2011, 162, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sui, Z.; Chang, L.; Kang, K.; Ma, J.; Kong, F.; Zhou, W.; Wang, J.; Guo, L.; Geng, H.; et al. Transcriptome de novo assembly sequencing and analysis of the toxic dinoflagellate Alexandrium catenella using the Illumina platform. Gene 2014, 537, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Koid, A.E.; Terrado, R.; Campbell, V.; Caron, D.A.; Heidelberg, K.B. Changes in gene expression of Prymnesium parvum induced by nitrogen and phosphorus limitation. Front. Microbiol. 2015, 6, 631. [Google Scholar] [CrossRef]
- Jang, S.H.; Jeong, H.J.; Chon, J.K.; Lee, S.Y. De novo assembly and characterization of the transcriptome of the newly described dinoflagellate Ansanella granifera: Spotlight on flagellum-associated genes. Mar. Genom. 2016, 33, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Chen, Z. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wohlrab, S.; Groth, M.; Glöckner, G.; Guillou, L.; John, U. Transcriptomic profiling of Alexandrium fundyense during physical interaction with or exposure to chemical signals from the parasite Amoebophrya. Mol. Ecol. 2016, 25, 1294–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Wang, H.; Suh, Y.S.; Ki, J.-S. Transcriptomic profiles reveal the genome-wide responses of the harmful dinoflagellate Cochlodinium polykrikoides when exposed to the algicide copper sulfate. BMC Genom. 2016, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Barshis, D.J.; Ladner, J.T.; Oliver, T.A.; Palumbi, S.R. Lineage-specific transcriptional profiles of Symbiodinium spp. Unaltered by heat stress in a coral host. Mol. Biol. Evol. 2014, 31, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.K.; Smith, Z.; Fuqua, C.; Clay, K.; Colbourne, J.K. Why so many unknown genes? Partitioning orphans from a representative transcriptome of the lone star tick Amblyomma americanum. BMC Genom. 2013, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.M.; Rödelsperger, C.; Eichholz, K.; Tillmann, U.; Cembella, A.; McGaughran, A.; John, U. Transcriptomic characterisation and genomic glimps into the toxigenic dinoflagellate Azadinium spinosum, with emphasis on polykeitde synthase genes. BMC Genom. 2015, 16, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morey, J.S.; Van Dolah, F.M. Global analysis of mRNA half-lives and de novo transcription in a dinoflagellate, Karenia brevis. PLoS ONE 2013, 8, e66347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudrimont, A.; Voegeli, S.; Viloria, E.C.; Stritt, F.; Lenon, M.; Wada, T.; Becskei, A. Multiplexed gene control reveals rapid mRNA turnover. Sci. Adv. 2017, 3, e1700006. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.H. Next generation sequencing of microbial transcriptomes: Challenges and opportunities. FEMS Microbiol. Lett. 2009, 302, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, S.A.; Van Dolah, F.M. Post-transcriptional Regulation of S-Phase Genes in the Dinoflagellate, Karenia brevis. J. Eukaryot. Microbiol. 2011, 58, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Qiu, L.; Hou, Z.; Zhang, Q.; Wu, J.; Gao, Q.; Song, L. Computational identification of microRNAs from the expressed sequence tags of toxic dinoflagellate Alexandrium tamarense. Evol. Bioinform. 2013, 9, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Sui, Z.; Zhang, S.; Du, Q.; Ren, Y.; Liu, Y.; Kong, F.; Zhong, J.; Ma, Q. Identification of microRNAs in the toxigenic Dinoflagellate Alexandrium catenella by high-throughput illumina sequencing and bioinformatic analysis. PLoS ONE 2015, 10, e0138709. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z.; Zhang, H.; Zhang, Y.; Zhang, S.F. Marine dinoflagellate proteomics: Current status and future perspectives. J. Proteom. 2014, 105, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.G. Considerations for the use of transcriptomics in identifying the ‘genes that matter’ for environmental adaptation. J. Exp. Biol. 2015, 218, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.R.; Urano, K.; Delrot, S.; Pezzotti, M.; Shinozaki, K. Effects of abiotic stress on plants: A systems biology perspective. BMC Plant Biol. 2011, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Pronk, T.E.; van der Veen, J.W.; Ezendam, J.; Van Loveren, H.; Pennings, J.L. Effects of pooling RNA from samples treated with different compounds for determining class specific biomarkers and processes in toxicogenomics. Toxicol. In Vitro 2011, 25, 1841–1847. [Google Scholar] [CrossRef] [PubMed]
- Jaeckisch, N.; Yang, I.; Wohlrab, S.; Glöckner, G.; Kroymann, J.; Vogel, H.; John, U. Comparative genomic and transcriptomic characterization of the toxigenic marine dinoflagellate Alexandrium ostenfeldii. PLoS ONE 2011, 6, e28012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendziorski, C.; Irizarry, R.A.; Chen, K.S.; Haag, J.D.; Gould, M.N. On the utility of pooling biological samples in microarray experiments. Proc. Natl. Acad. Sci. USA 2005, 102, 4252–4257. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.; Schrey, A.W.; Richards, C.L. Ten years of transcriptomics in wild populations: What have we learned about their ecology and evolution? Mol. Ecol. 2015, 24, 710–725. [Google Scholar] [CrossRef] [PubMed]
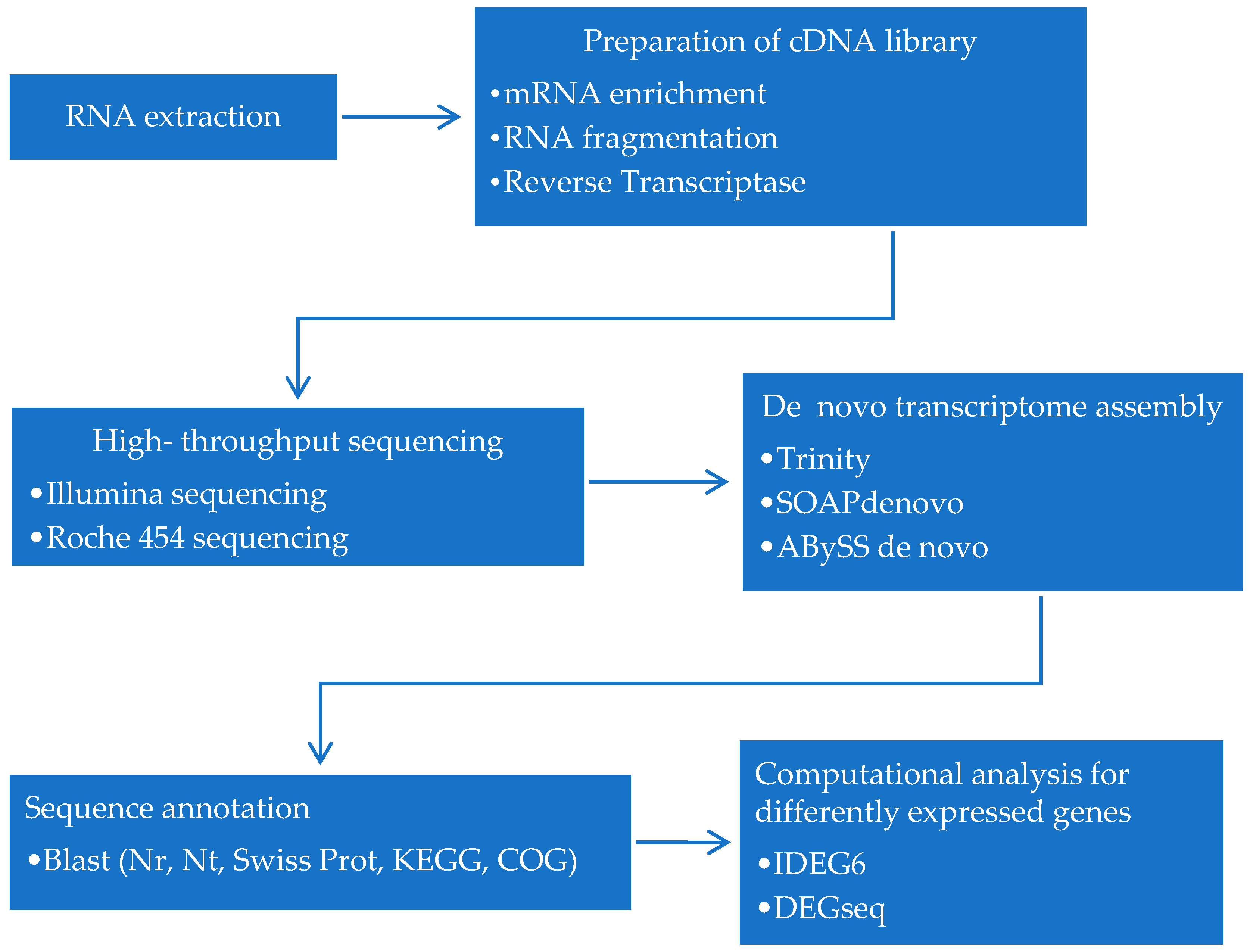
Figure 1.
Current strategies and workflow for dinoflagellates transcriptomics study. Following RNA extraction, cDNA library will be generated and then will be sequenced using NGS platform. Then, the generated reads will be assembled and annotated before computational analysis can be done.
Figure 1.
Current strategies and workflow for dinoflagellates transcriptomics study. Following RNA extraction, cDNA library will be generated and then will be sequenced using NGS platform. Then, the generated reads will be assembled and annotated before computational analysis can be done.

{kind=link}
Table 1.
Representative publication for application of RNA-seq in dinoflagellates transcriptome study.
Table 1.
Representative publication for application of RNA-seq in dinoflagellates transcriptome study.
| RNA-Seq | Major Application | Platform | References |
|---|---|---|---|
| Transcriptome sequencing | Transcriptome profiling | Roche 454 | [12] |
| Comprehensive analysis of differently gene expression | Illumina | [13,14] | |
| Single Nucleotide Polymorphisms (SNP) discovery | Illumina | [15] | |
| Discovery and characterization of novel genes | Roche 454 | [16] | |
| Isoform discovery | Illumina | [17] | |
| Pathway analysis | Illumina | [18] | |
| Small RNA sequencing | microRNA profiling | Illumina | [4,19] |
| Gene regulation | Illumina | [19] | |
| Meta-transcriptome sequencing | Analysis of community RNA pool and ecological function of gene | Roche 454 | [20] |
| Community taxonomic structure | Roche 454 | [21] |
Table 2.
A brief history in dinoflagellates transcriptome analysis.
| Year | Event | References |
|---|---|---|
| 1995 | Development of microarray method | [29] |
| 1995 | Development of SAGE method | [30] |
| 2000 | Development of MPSS method | [31] |
| 2000 | First DDRT-PCR study to discover differently expressed genes | [25] |
| 2003–2012 | Report on dinoflagellates transcriptome analysis using microarray | [32,33,34,35,36] |
| 2004 | Modified SAGE method for dinoflagellates gene expression analysis introduced | [37] |
| 2006–2010 | Report on dinoflagellates transcriptome analysis using MPSS | [2,38] |
| 2007 | Report on dinoflagellates transcriptome analysis using Expression Sequence Tags (EST) | [39] |
| 2008 | Development RNA-seq method | [40] |
| 2011–2017 | Report on dinoflagellates transcriptome analysis using RNA-seq | [19,41] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Akbar, M.A.; Ahmad, A.; Usup, G.; Bunawan, H. RNA-Seq as an Emerging Tool for Marine Dinoflagellate Transcriptome Analysis: Process and Challenges. Processes 2018, 6, 5. https://doi.org/10.3390/pr6010005
AMA Style
Akbar MA, Ahmad A, Usup G, Bunawan H. RNA-Seq as an Emerging Tool for Marine Dinoflagellate Transcriptome Analysis: Process and Challenges. Processes. 2018; 6(1):5. https://doi.org/10.3390/pr6010005
Chicago/Turabian StyleAkbar, Muhamad Afiq, Asmat Ahmad, Gires Usup, and Hamidun Bunawan. 2018. "RNA-Seq as an Emerging Tool for Marine Dinoflagellate Transcriptome Analysis: Process and Challenges" Processes 6, no. 1: 5. https://doi.org/10.3390/pr6010005
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.