ADAR Mediated RNA Editing Modulates MicroRNA Targeting in Human Breast Cancer

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. NGS Sequencing of MCF-7 and MDA-MB-231
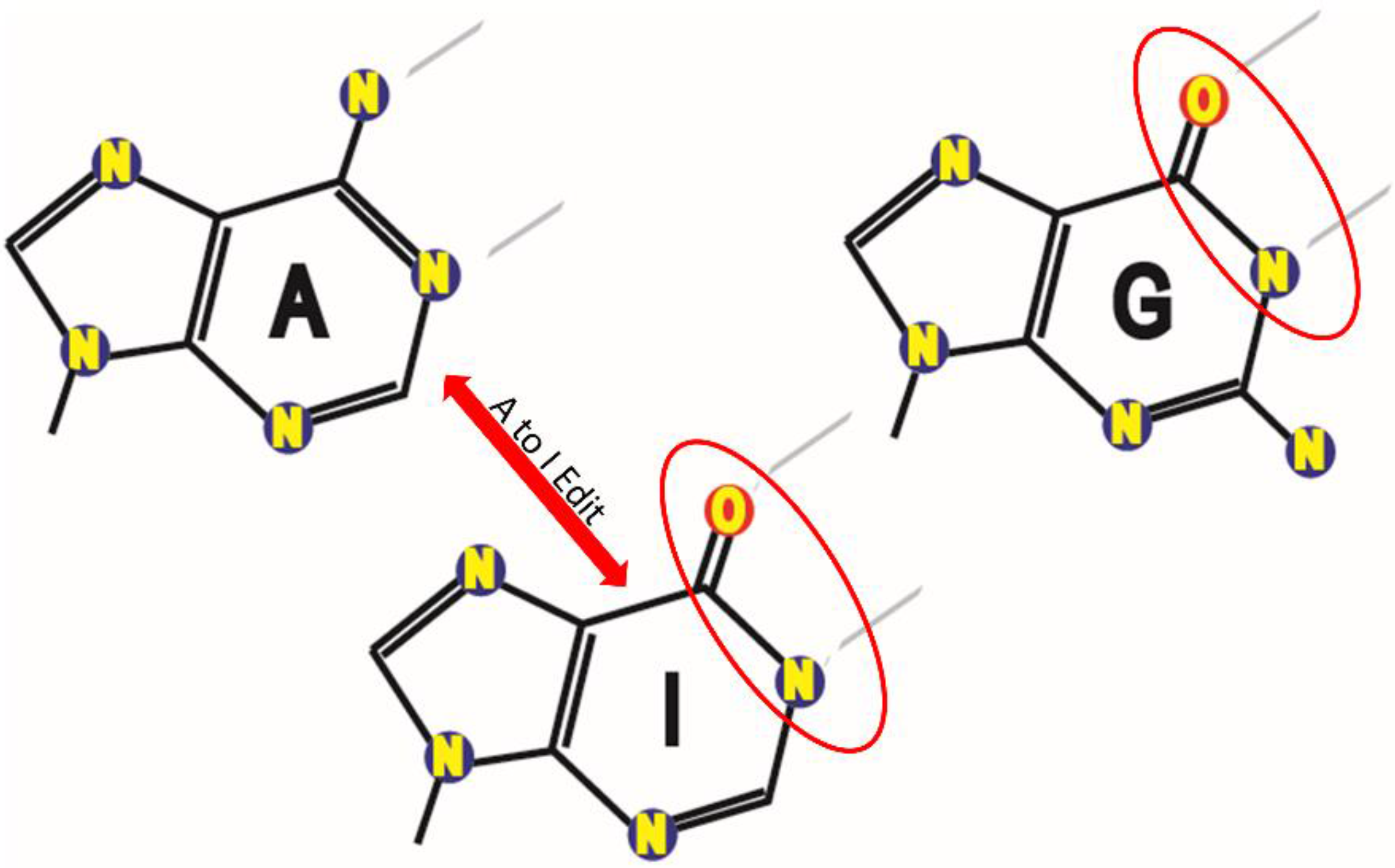
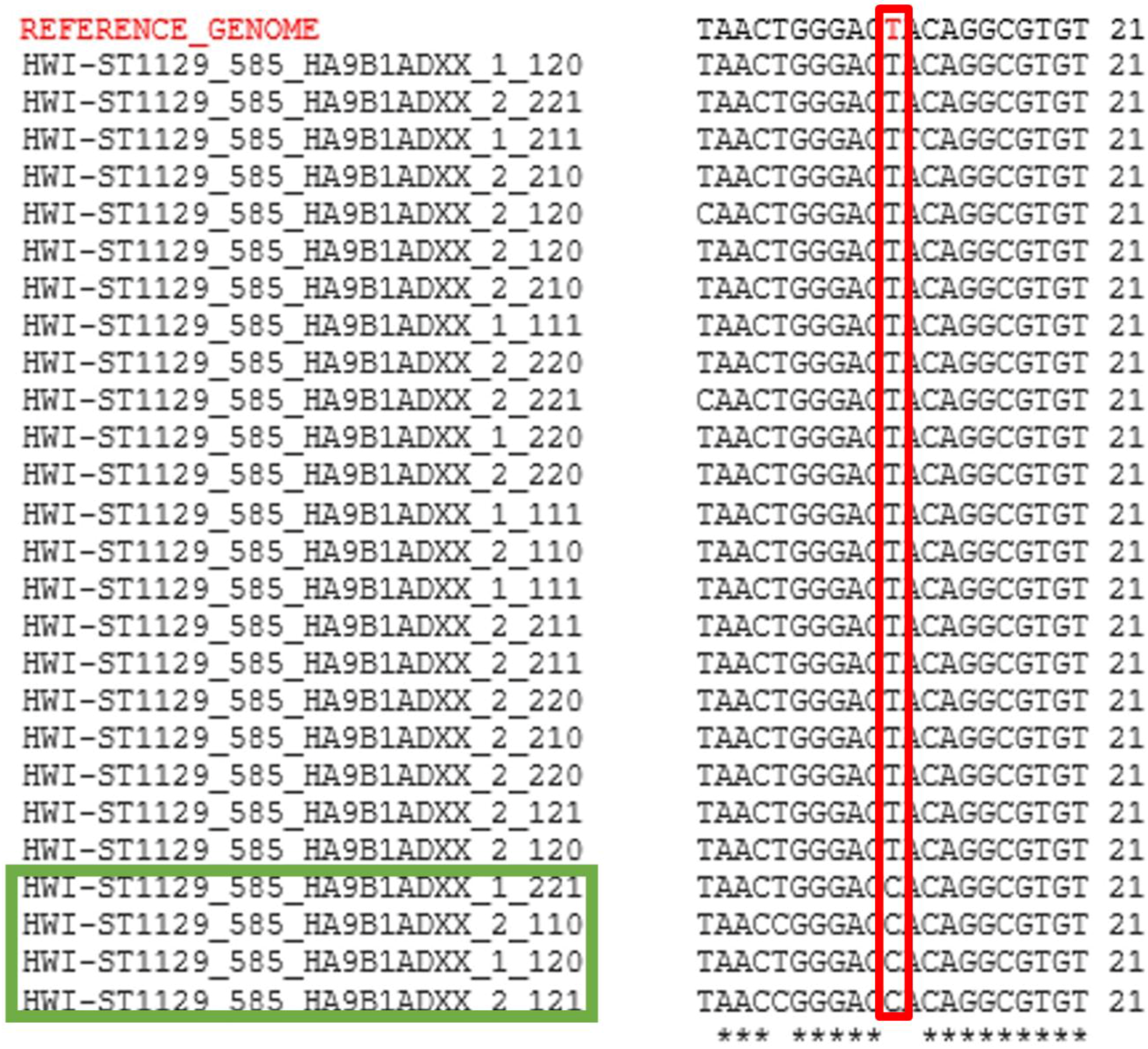
2.2. Identification of A-to-G Edits in Breast Cancer Cells
2.3. Computational Identification of MiRNAs Biased towards Editing
2.4. Small RNA-Seq Analysis
2.5. Cell Growth Assay
2.6. Western Blot Analysis
3. Results
3.1. Identification of RNA Edit Sites
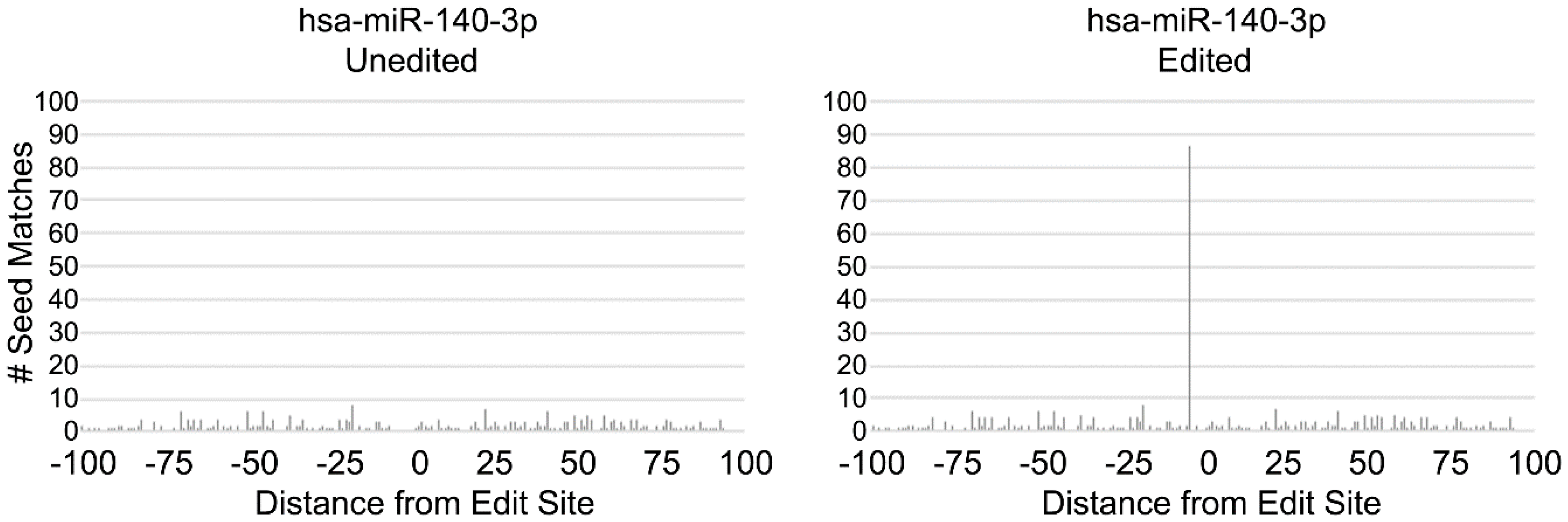
3.2. MiRNAs Biased towards Editing
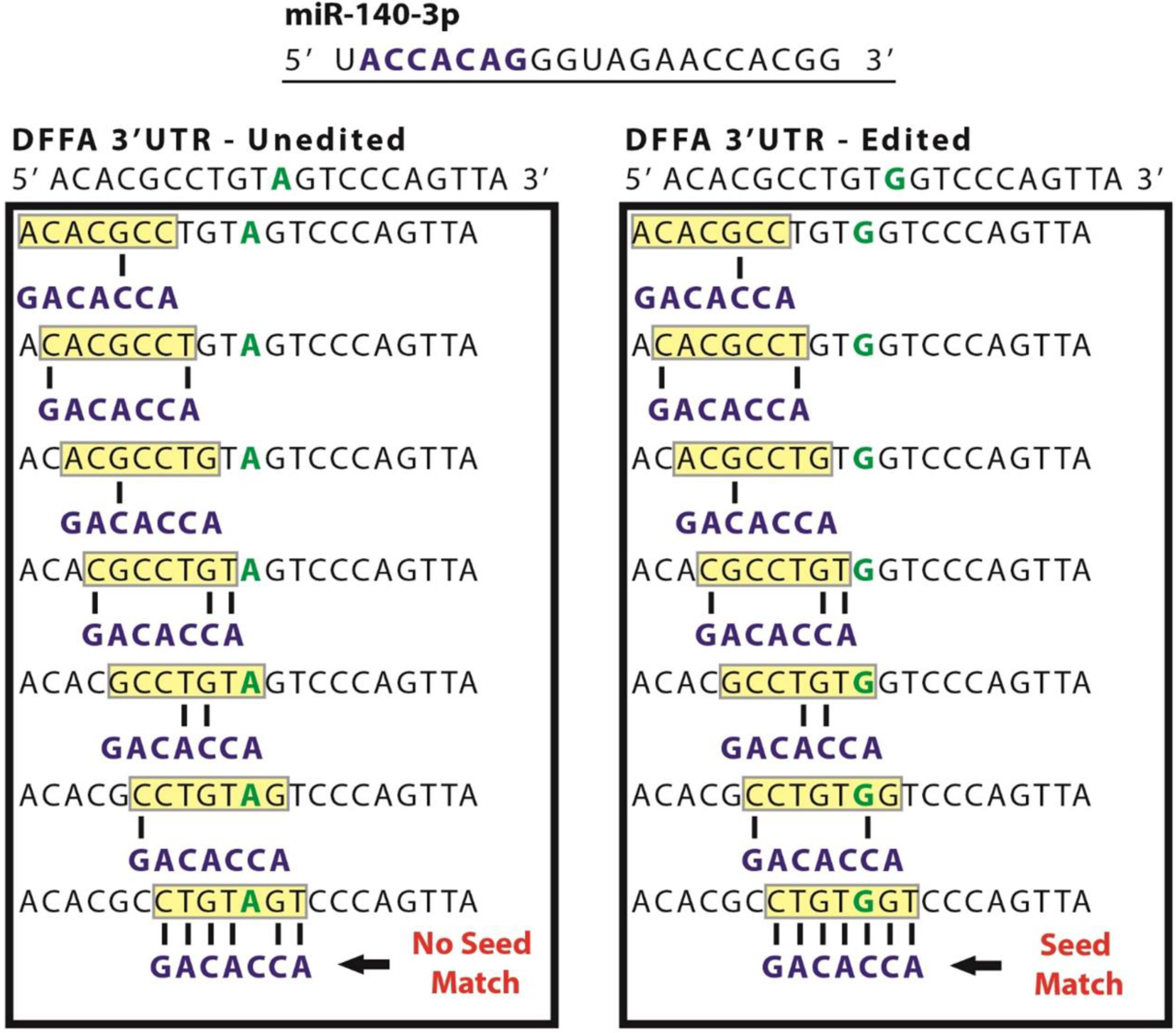
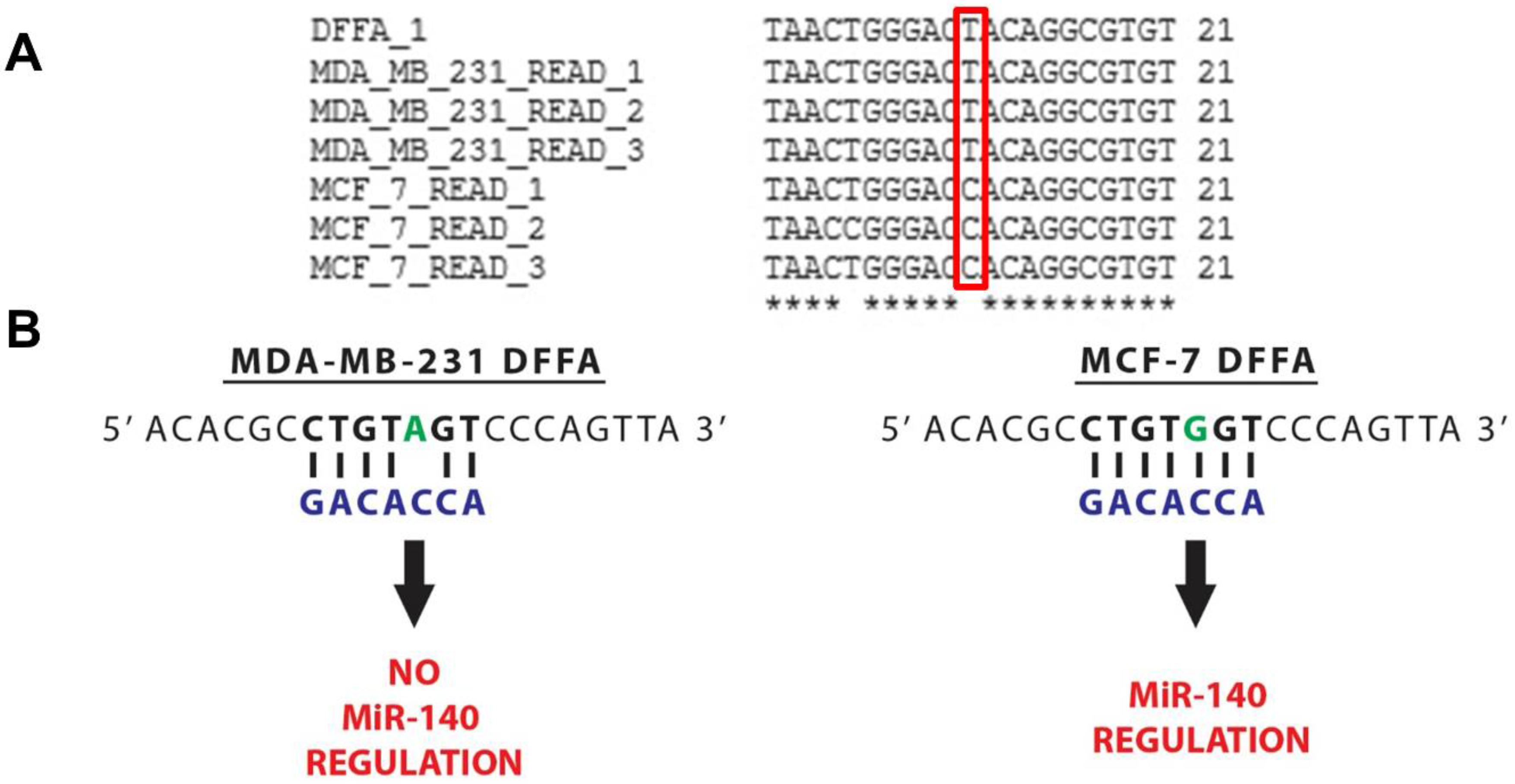
3.3. MiR-140 Is Able to Target DFFA in MCF-7 but not MDA-MB-231
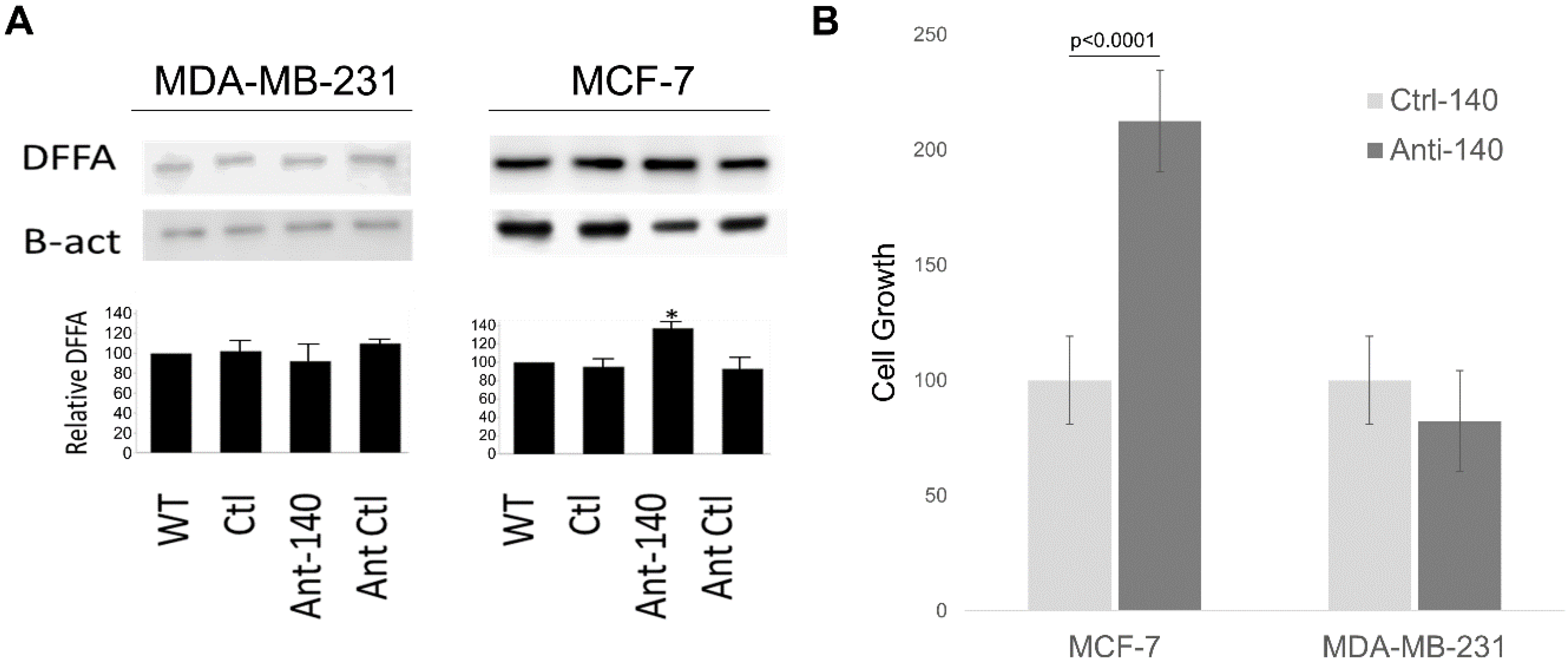
3.4. Inhibiting miR-140-3p Increases DFFA Expression in MCF-7
3.5. Inhibiting miR-140-3p Increases MCF-7 Cellular Proliferation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Levanon, E.Y. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Guo, J. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Li, J.B. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 2009, 324, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Williams, B.; Wold, B.J.; Mortazavi, A. RNA editing in the human ENCODE RNA-seq data. Genome Res. 2012, 22, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Bahn, J.H. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 2012, 22, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Maas, S. Genome-wide evaluation and discovery of vertebrate A-to-I RNA editing sites. Biochem. Biophys. Res. Commun. 2011, 412, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Carmichael, G.G. Gene regulation by SINES and inosines: Biological consequences of A-to-I editing of Alu element inverted repeats. Cell Cycle 2008, 7, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.D.; Kim, T.T.; Walsh, T.; Kobayashi, Y.; Matise, T.C.; Buyske, S.; Gabriel, A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004, 14, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Seeburg, P.H. A-to-I editing: New and old sites, functions and speculations. Neuron 2002, 35, 17–20. [Google Scholar] [CrossRef]
- Rieder, L.E.; Reenan, R.A. The intricate relationship between RNA structure, editing, and splicing. Semin. Cell Dev. Biol. 2012, 23, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.R.; Qiao, J.J.; Chan, T.H.; Zhu, Y.H.; Li, F.F.; Liu, H.; Chen, L. Adenosine-to-inosine RNA editing mediated by ADARs in esophageal squamous cell carcinoma. Cancer Res. 2014, 74, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Crews, L.A.; Barrett, C.L.; Chun, H.J.; Court, A.C.; Isquith, J.M.; Zipeto, M.A.; Dao, K.H.T. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Y.; Lin, C.H.; Chan, T.H.; Chow, R.K.; Song, Y.; Qi, L. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med. 2013, 19, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.A. Deletion of the RNA-editing enzyme ADAR1 causes regression of established chronic myelogenous leukemia in mice. Int. J. Cancer 2013, 132, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, Y. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J. Clin. Investig. 2012, 122, 4059–4076. [Google Scholar] [CrossRef] [PubMed]
- Paz, N. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007, 17, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; Grocock, R.J.; van Dongen, S.; Enright, A.J.; Dicks, E.; Futreal, P.A.; Stratton, M.R. RNA editing of human microRNAs. Genome Biol. 2006, 7, R27. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005, 6, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Zamore, P.D. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002, 297, 2056–2060. [Google Scholar]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Maegdefessel, L. The emerging role of microRNAs in cardiovascular disease. J. Intern. Med. 2014, 276, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.; Shi, Y. MicroRNAs: Small molecules with big roles in neurodevelopment and diseases. Exp. Neurol. 2014, 268, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.D.; Lund, A.H. MicroRNA and cancer. Mol. Oncol. 2012, 6, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Ota, H. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Chendrimada, T.P.; Shiekhattar, R.; Nishikura, K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007, 8, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Fukami, T.; Gotoh, S.; Takamiya, M.; Aoki, Y.; Nakajima, M. RNA Editing Modulates Human Hepatic Aryl Hydrocarbon Receptor Expression by Creating MicroRNA Recognition Sequence. J. Biol. Chem. 2016, 291, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, C.S.; Varelas, X.; Monti, S. Altered RNA editing in 3′ UTR perturbs microRNA-mediated regulation of oncogenes and tumor-suppressors. Sci. Rep. 2016, 6, 23226. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, R.; Stearns, T.M.; Griswold, A.L.; Mehta, A.; Czachor, A.; Fukumoto, J.; Kolliputi, N. Detection of canonical A-to-G editing events at 3′ UTRs and microRNA target sites in human lungs using next-generation sequencing. Oncotarget 2015, 6, 35726–35736. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Fukami, T.; Gotoh, S.; Nakajima, M. A-to-I RNA Editing Up-regulates Human Dihydrofolate Reductase in Breast Cancer. J. Biol. Chem. 2017, 292, 4873–4884. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Levanon, E.Y. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple Sequence Alignment Using ClustalW and ClustalX. Curr. Protocals Bioinform. 2002. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum. Mol. Genet. 2009, 18, 4801–4807. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, J.J.; Seeburg, P.H. A-to-I RNA editing: Effects on proteins key to neural excitability. Neuron 2012, 74, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, C.; Alicea, D.; Gonzalez, M.; Bykhovskaia, M.; Marie, B. Adar is essential for optimal presynaptic function. Mol. Cell. Neurosci. 2013, 52, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lopez, J.; de Jde, D.; Mazo, J. Hourca Del Reprogramming of microRNAs by adenosine-to-inosine editing and the selective elimination of edited microRNA precursors in mouse oocytes and preimplantation embryos. Nucleic Acids Res. 2013, 41, 5483–5493. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H. Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology 2003, 109, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zou, H.; Slaughter, C.; Wang, X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997, 89, 175–184. [Google Scholar] [CrossRef]
- Song, B. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene 2009, 28, 4065–4074. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, B.; Eades, G.; Zhou, Q. Roles of microRNA-140 in stem cell-associated early stage breast cancer. World J. Stem Cells 2014, 6, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Li, Q. Downregulation of miR-140 promotes cancer stem cell formation in basal-like early stage breast cancer. Oncogene 2013, 33, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T. Introducing molecular subtyping of breast cancer into the clinic. J. Clin. Oncol. 2009, 27, 1153–1154. [Google Scholar] [CrossRef] [PubMed]
- Pozo-Guisado, E.; Alvarez-Barrientos, A.; Mulero-Navarro, S.; Santiago-Josefat, B.; Fernandez-Salguero, P.M. The antiproliferative activity of resveratrol results in apoptosis in MCF-7 but not in MDA-MB-231 human breast cancer cells: Cell-specific alteration of the cell cycle. Biochem. Pharmacol. 2002, 64, 1375–1386. [Google Scholar] [CrossRef]
- Salem, O.; Erdem, N.; Jung, J.; Munstermann, E.; Worner, A.; Wilhelm, H.; Körner, C. The highly expressed 5′isomiR of hsa-miR-140-3p contributes to the tumor-suppressive effects of miR-140 by reducing breast cancer proliferation and migration. BMC Genom. 2016, 17, 566. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR | miRBase ID | Seed (RC) | Targets (Edited) | Targets (Unedited) | Expected |
|---|---|---|---|---|---|
| A | |||||
| hsa-miR-513a-5p | MIMAT0002877 | CCTGTGA | 258 | 0 | 0.63 |
| hsa-miR-450b-3p | MIMAT0004910 | GATCCCA | 252 | 4 | 0.79 |
| hsa-miR-769-3p | MIMAT0003887 | GATCCCA | 252 | 4 | 0.79 |
| hsa-miR-6089 | MIMAT0023714 | CGGCCTC | 219 | 0 | 3.83 |
| hsa-miR-4691-3p | MIMAT0019782 | GTGGCTG | 181 | 0 | 1.16 |
| hsa-miR-3189-3p | MIMAT0015071 | CCCAAGG | 140 | 5 | 0.48 |
| hsa-miR-140-3p | MIMAT0004597 | CTGTGGT | 139 | 0 | 1.11 |
| hsa-miR-3065-3p | MIMAT0015378 | GGTGCTG | 118 | 0 | 0.5 |
| hsa-miR-3940-3p | MIMAT0018356 | CCGGGCT | 111 | 0 | 0.72 |
| hsa-miR-3680-3p | MIMAT0018107 | ATGCAAA | 108 | 2 | 0.82 |
| B | |||||
| hsa-miR-5089-5p | MIMAT0021081 | AATCCCA | 0 | 644 | 21.39 |
| hsa-miR-6504-3p | MIMAT0025465 | CTGTAAT | 58 | 587 | 19.93 |
| hsa-miR-6506-5p | MIMAT0025468 | ATCCCAG | 18 | 377 | 21.57 |
| hsa-miR-619-5p | MIMAT0026622 | ATCCCAG | 18 | 377 | 21.57 |
| hsa-miR-4775 | MIMAT0019931 | AAAATTA | 0 | 351 | 19.37 |
| hsa-miR-4735-5p | MIMAT0019860 | AAATTAG | 6 | 305 | 17.31 |
| hsa-miR-6514-3p | MIMAT0025485 | ACAGGCA | 10 | 216 | 9.59 |
| hsa-miR-4794 | MIMAT0019967 | TAGCCAG | 10 | 173 | 8.05 |
| hsa-miR-664a-5p | MIMAT0005948 | TAGCCAG | 10 | 173 | 8.05 |
| hsa-miR-1273e | MIMAT0018079 | TCAAGCA | 2 | 169 | 5.22 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, J.T.; Patterson, D.G.; King, V.M.; Amin, S.V.; Polska, C.J.; Houserova, D.; Crucello, A.; Barnhill, E.C.; Miller, M.M.; Sherman, T.D.; et al. ADAR Mediated RNA Editing Modulates MicroRNA Targeting in Human Breast Cancer. Processes 2018, 6, 42. https://doi.org/10.3390/pr6050042
Roberts JT, Patterson DG, King VM, Amin SV, Polska CJ, Houserova D, Crucello A, Barnhill EC, Miller MM, Sherman TD, et al. ADAR Mediated RNA Editing Modulates MicroRNA Targeting in Human Breast Cancer. Processes. 2018; 6(5):42. https://doi.org/10.3390/pr6050042
Chicago/Turabian StyleRoberts, Justin T., Dillon G. Patterson, Valeria M. King, Shivam V. Amin, Caroline J. Polska, Dominika Houserova, Aline Crucello, Emmaline C. Barnhill, Molly M. Miller, Timothy D. Sherman, and et al. 2018. "ADAR Mediated RNA Editing Modulates MicroRNA Targeting in Human Breast Cancer" Processes 6, no. 5: 42. https://doi.org/10.3390/pr6050042