
Achieving a Near-Theoretical Maximum in Peak Capacity Gain for the Forensic Analysis of Ignitable Liquids Using GC×GC-TOFMS


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumental Conditions
2.3. Data Processing
3. Results and Discussion
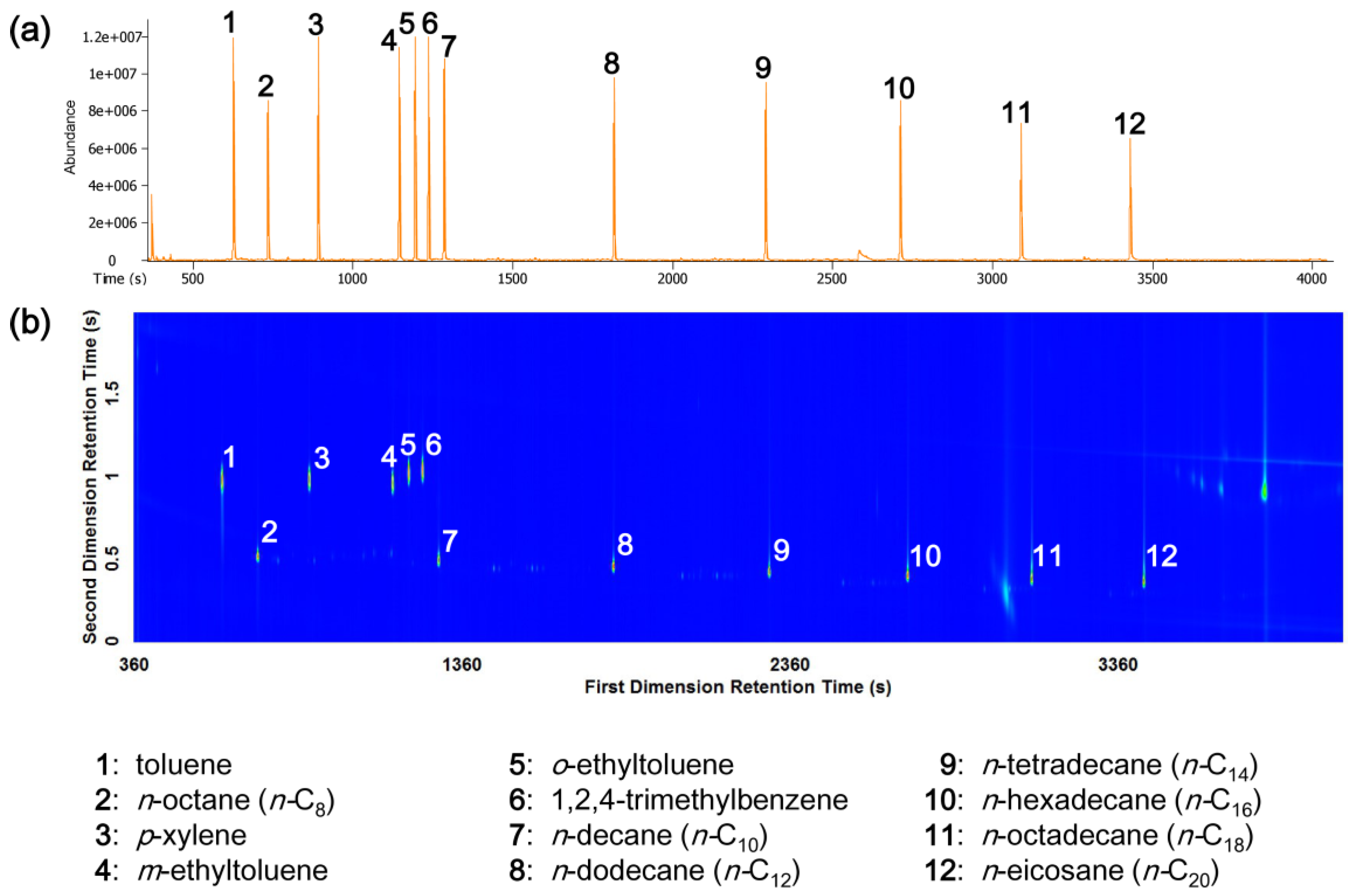
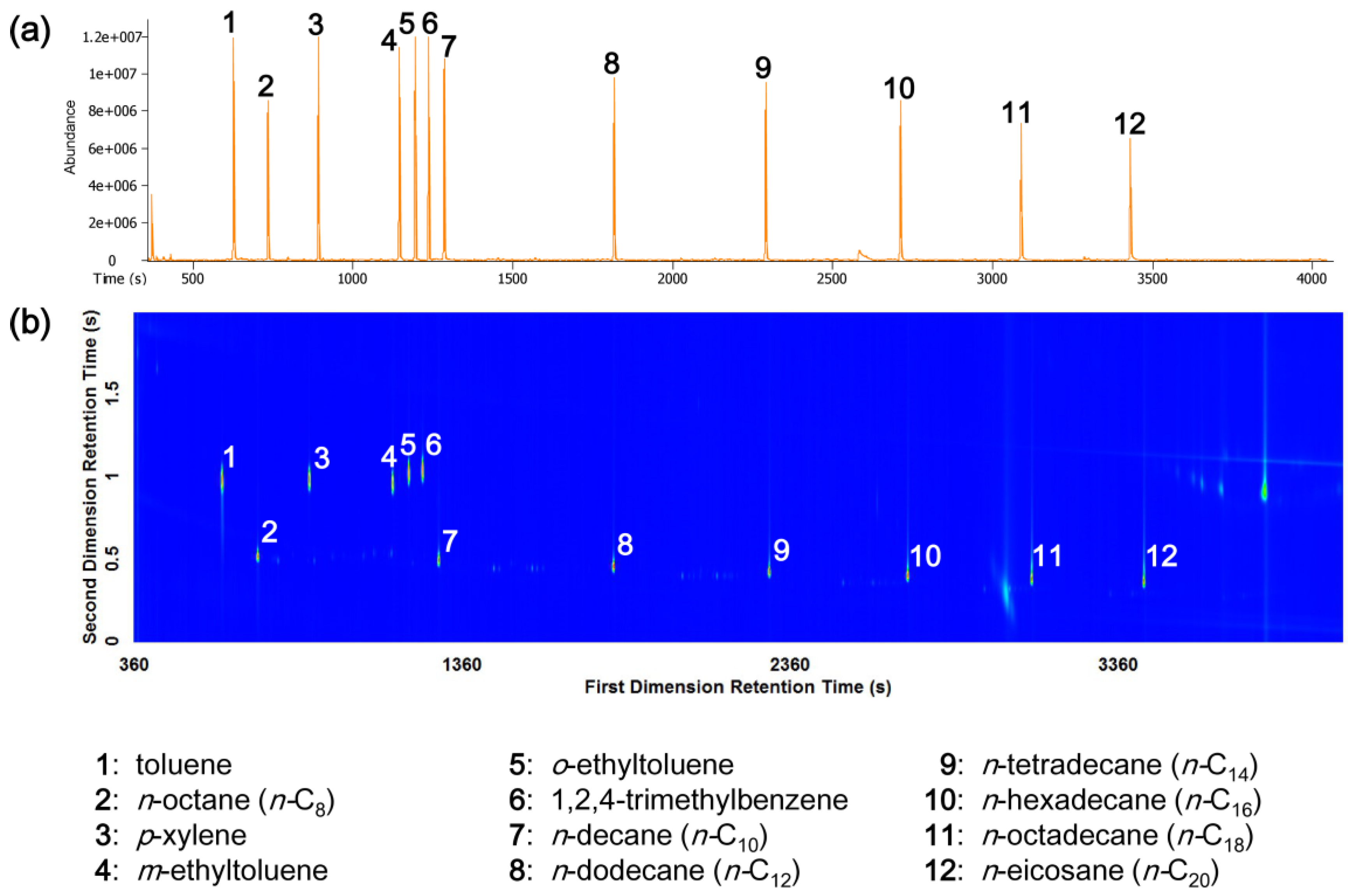
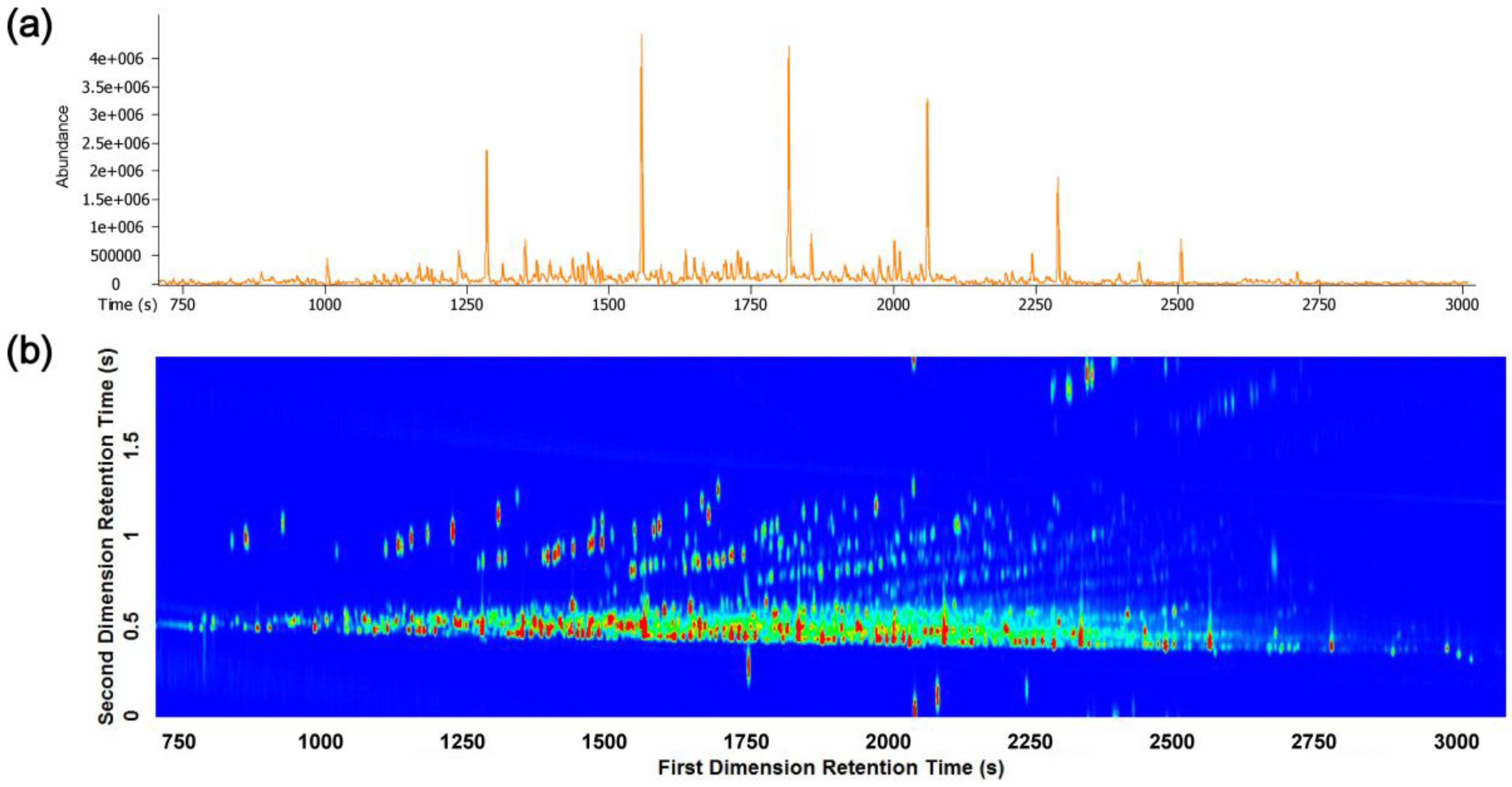
3.1. GC-TOFMS and GC×GC-TOFMS Analysis
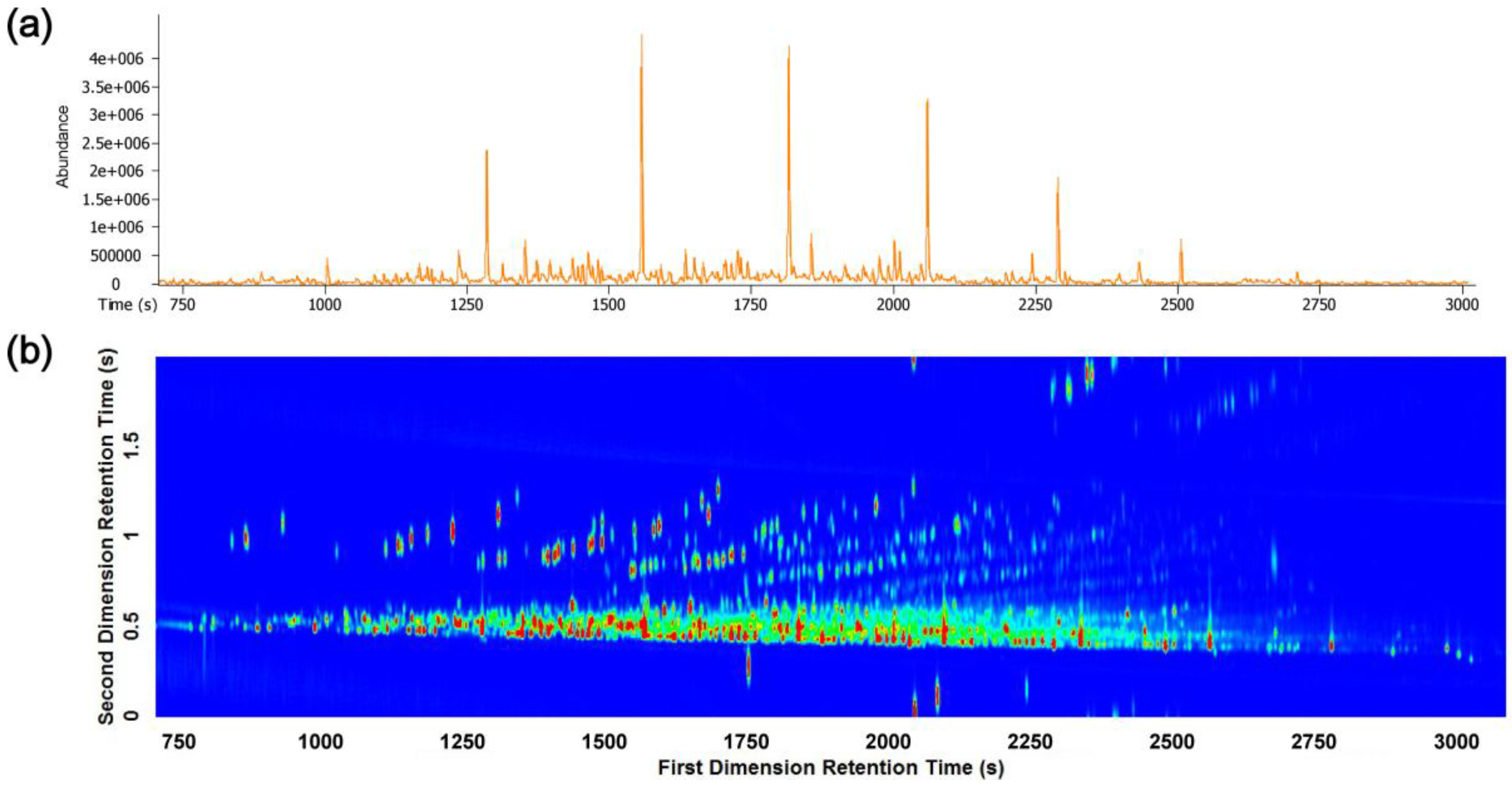
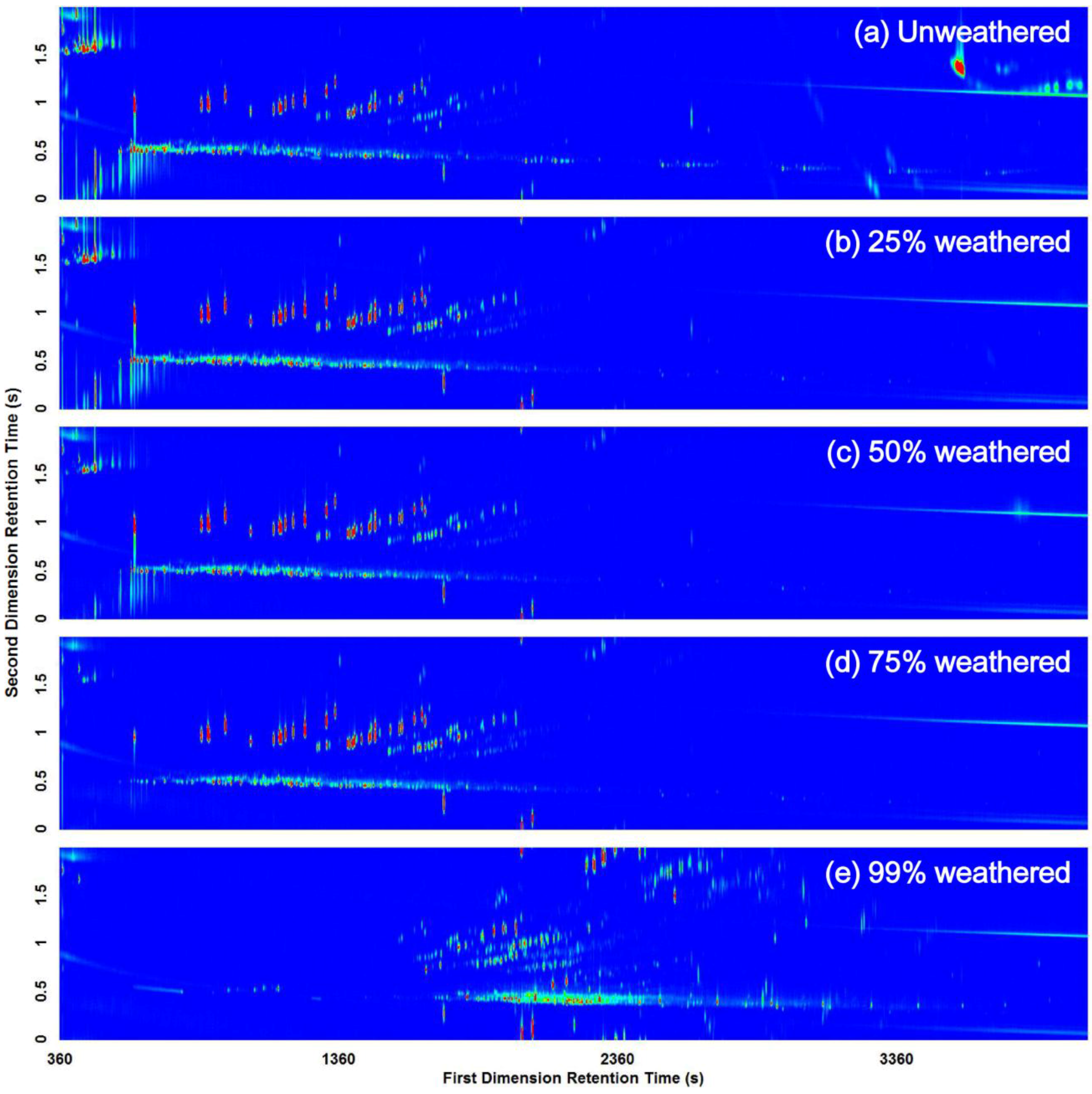
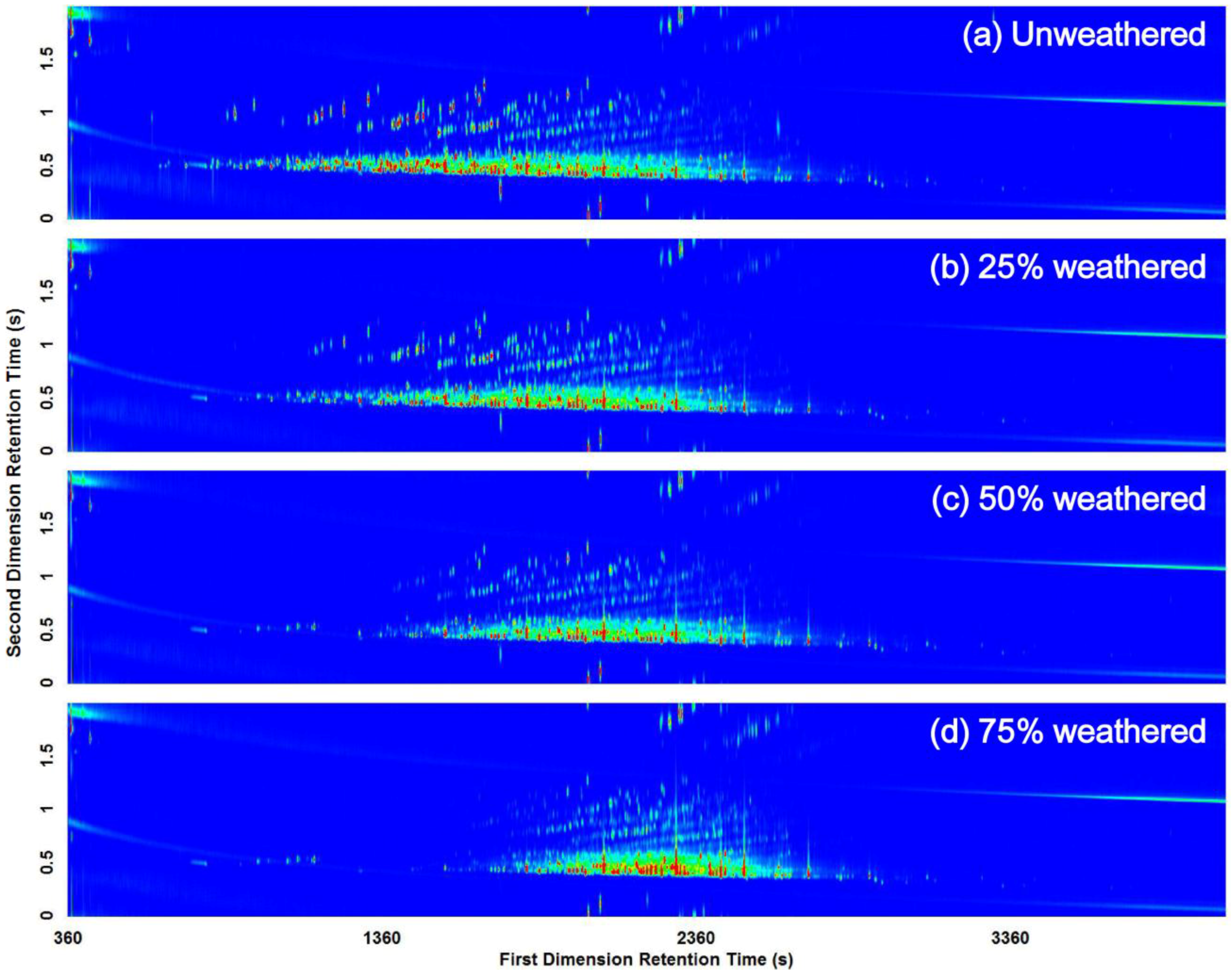
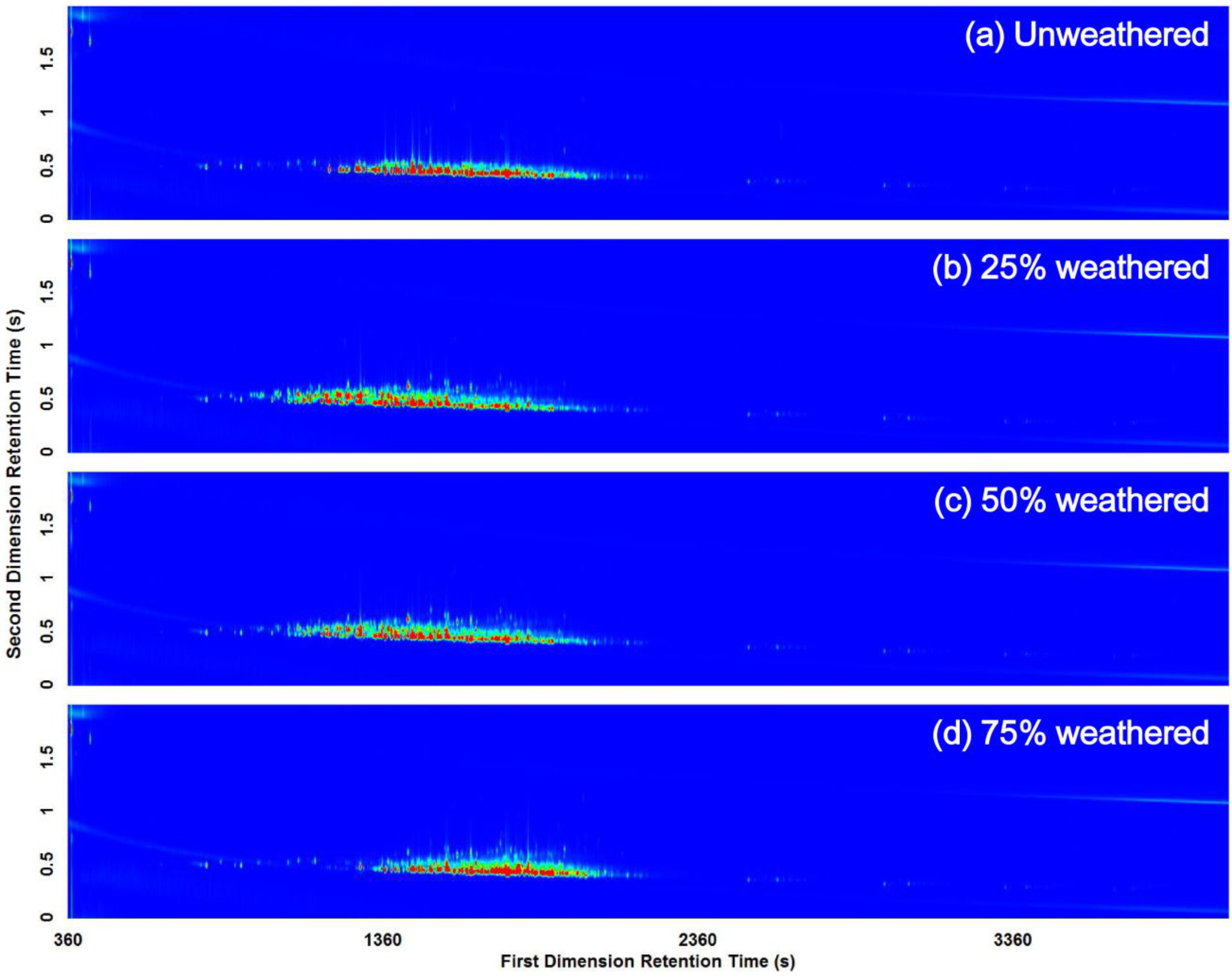
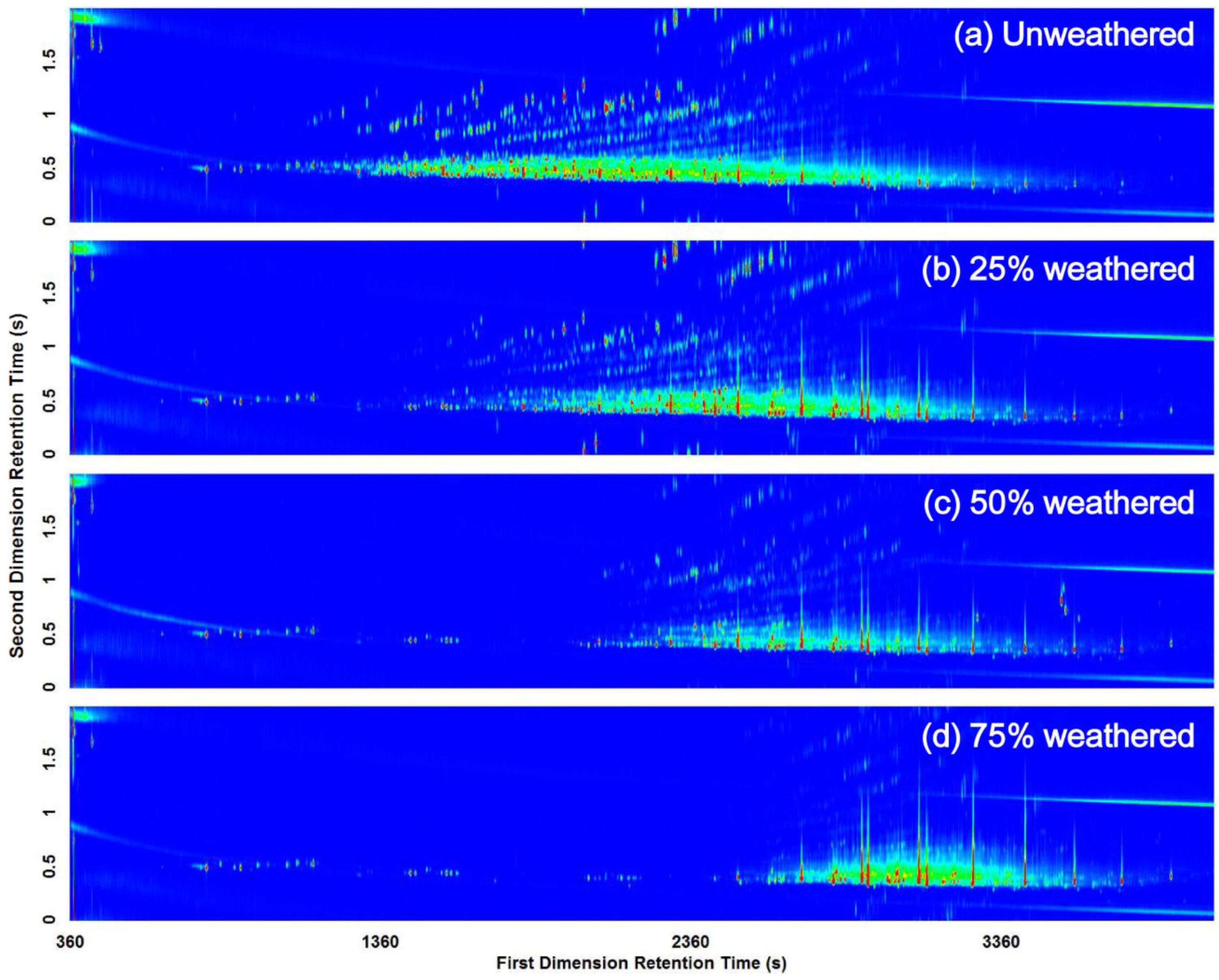
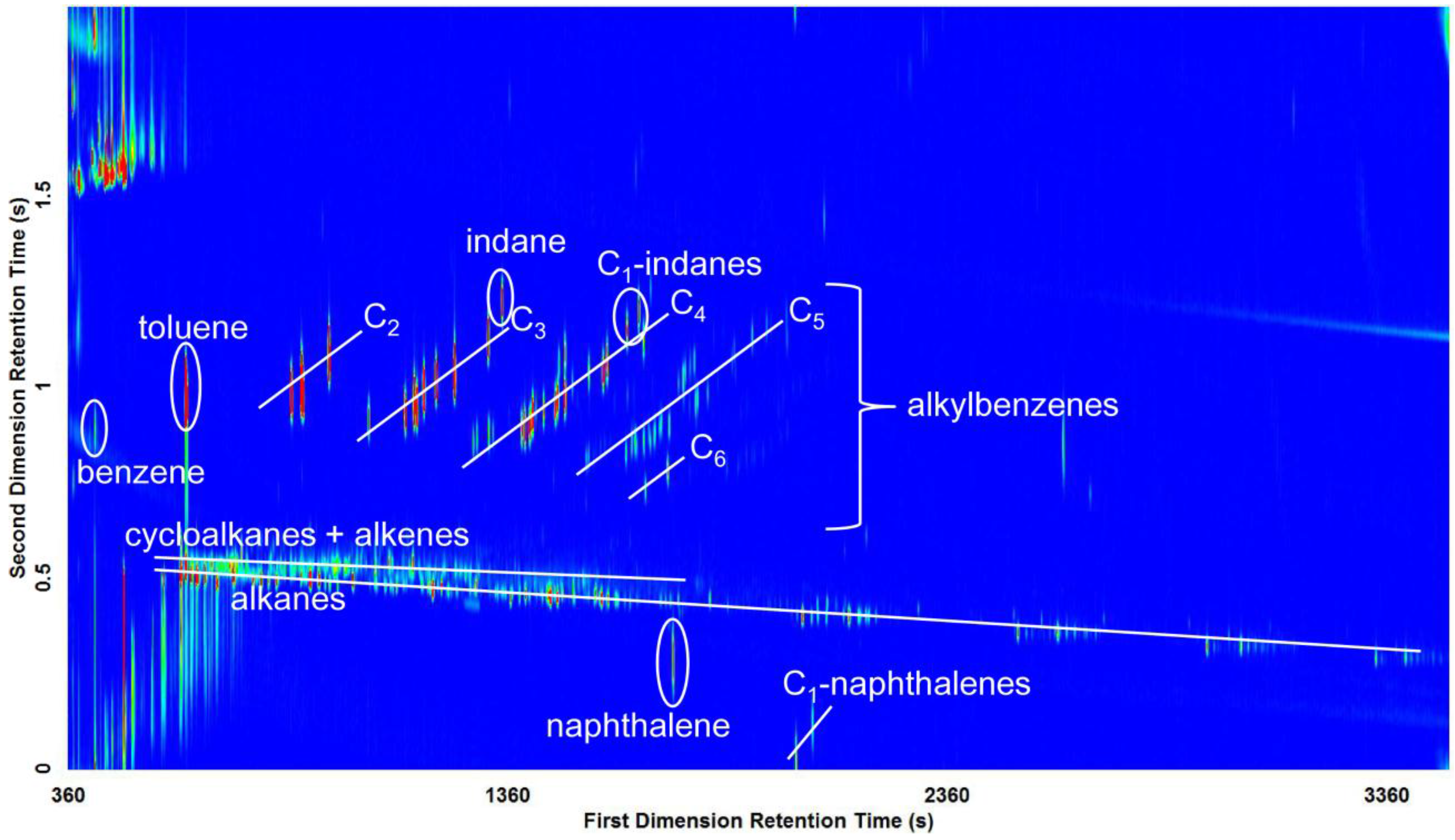
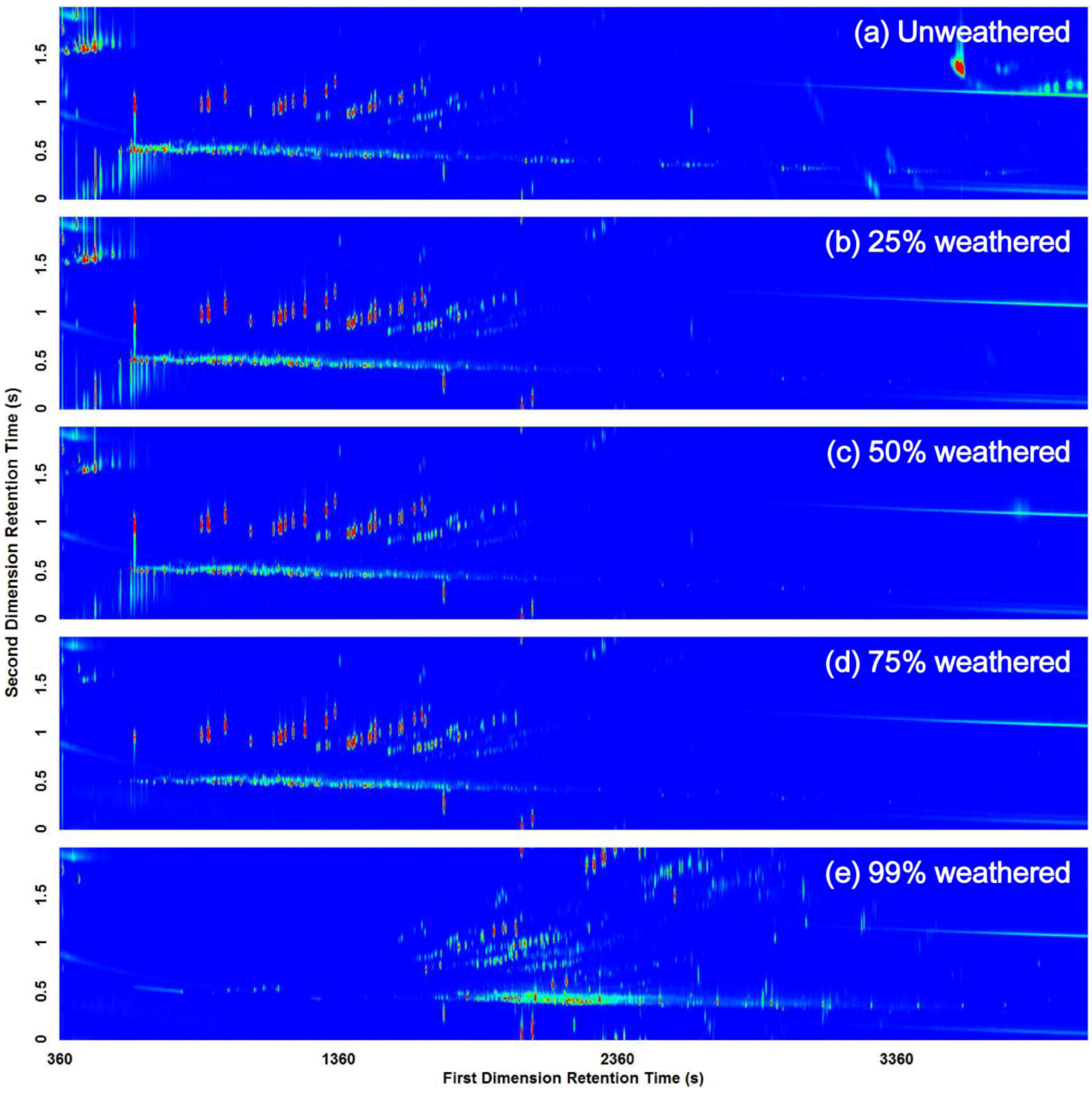
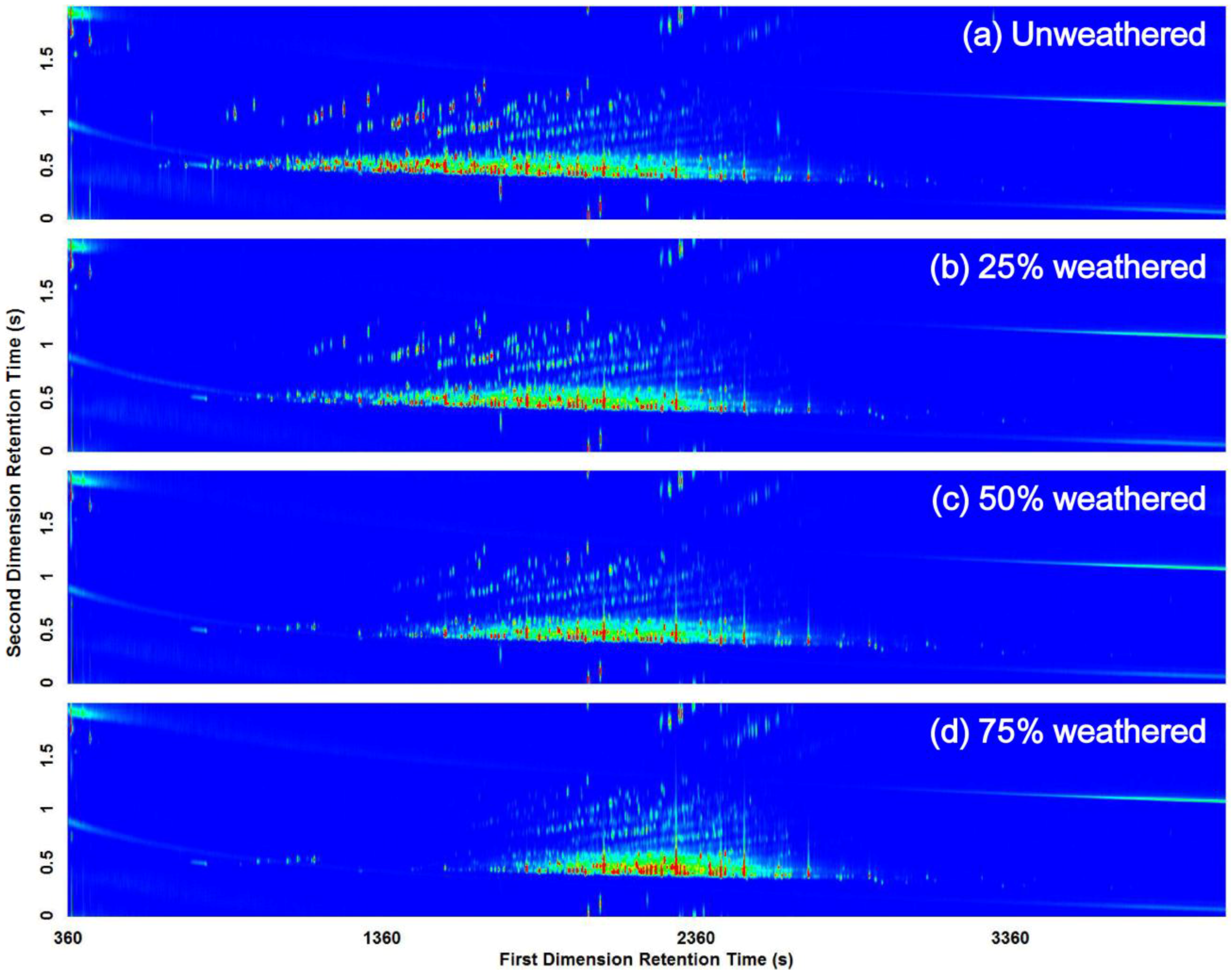
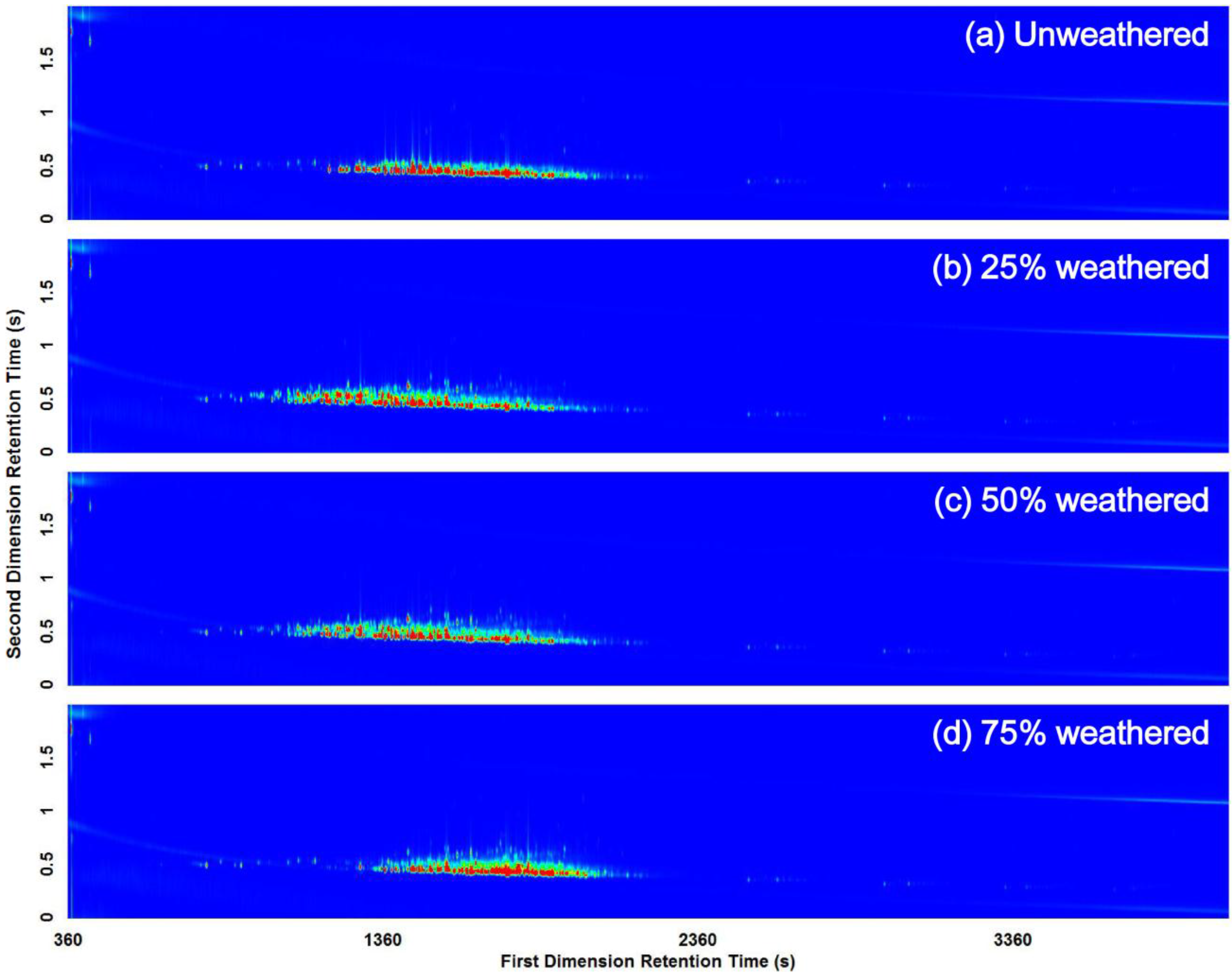
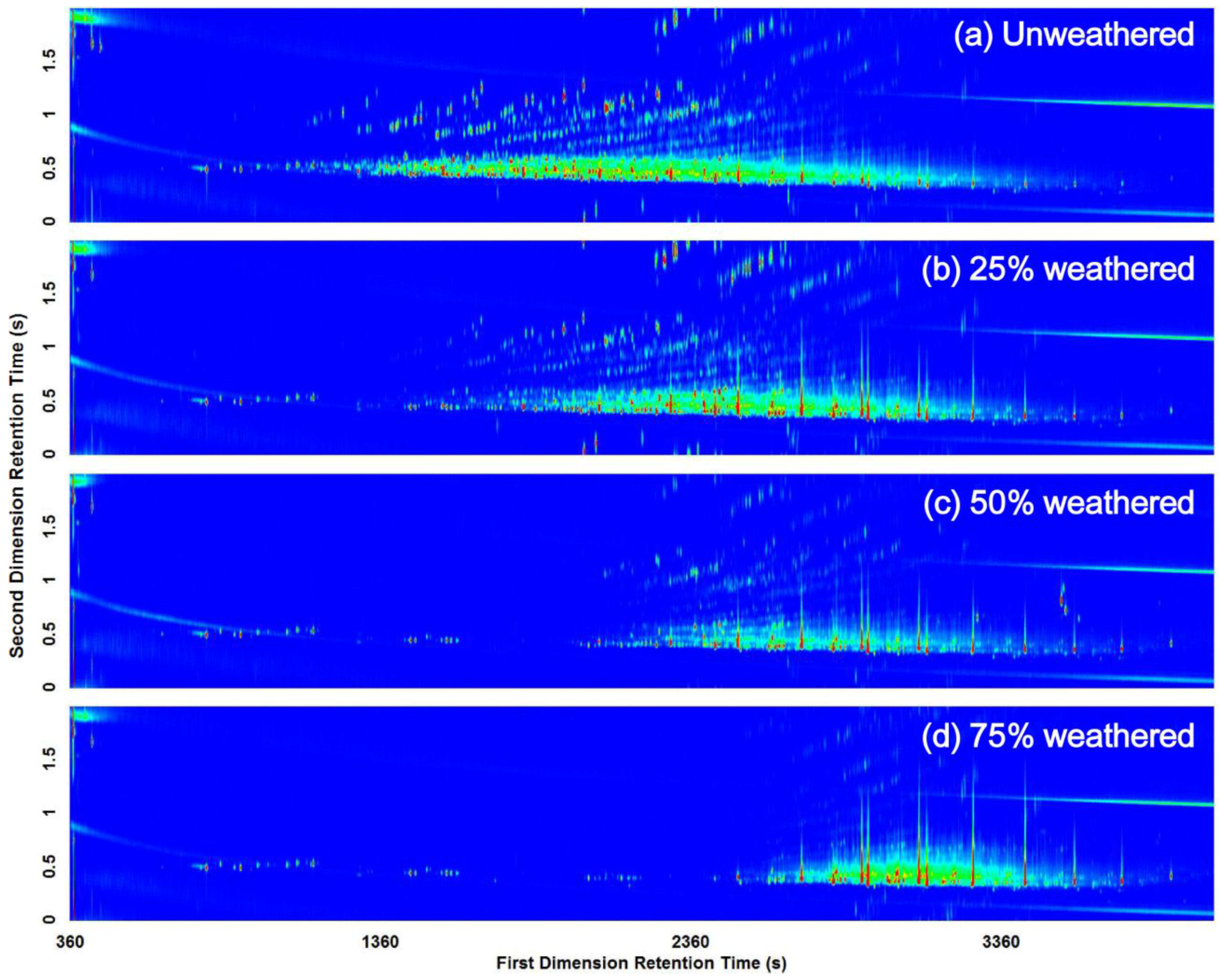
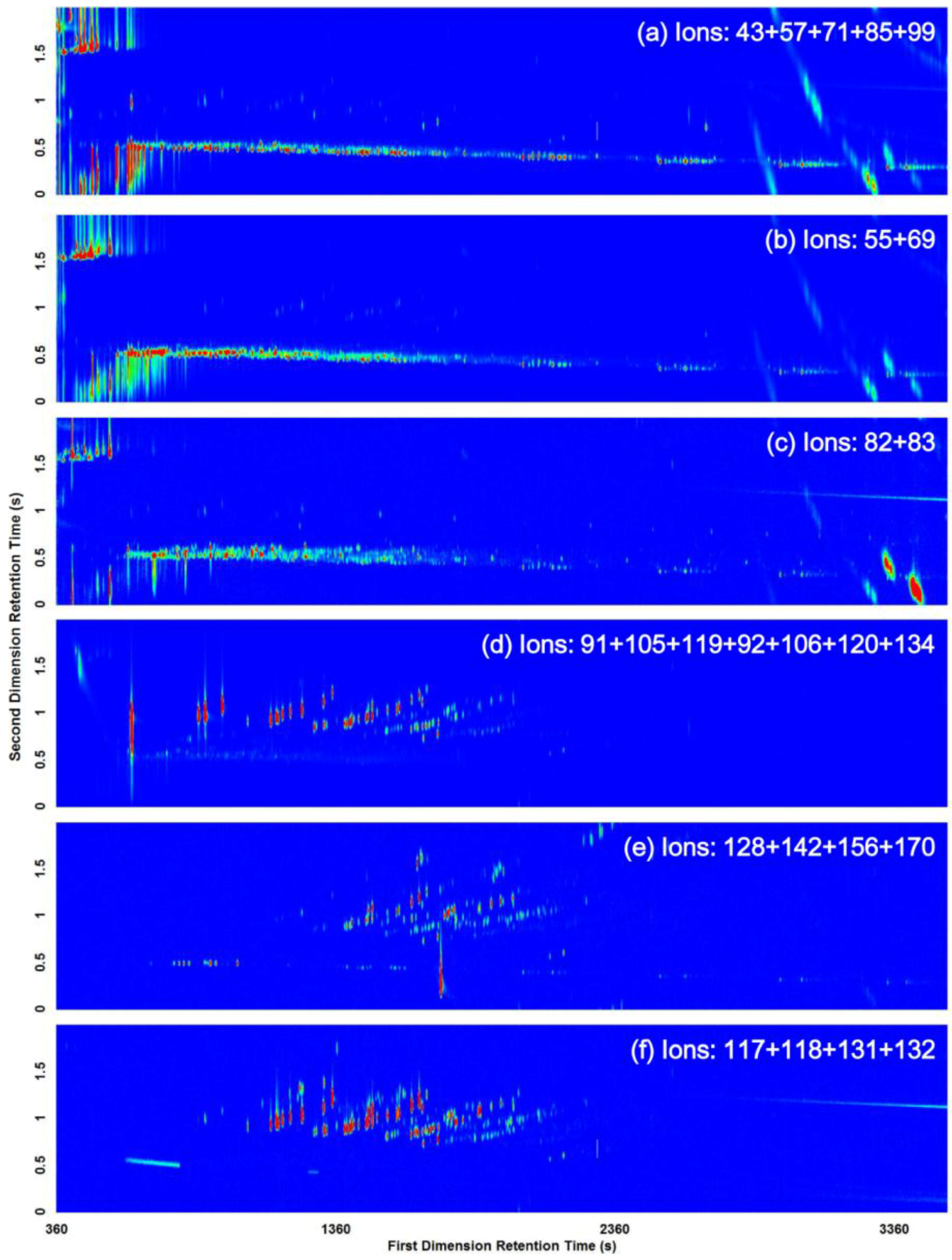
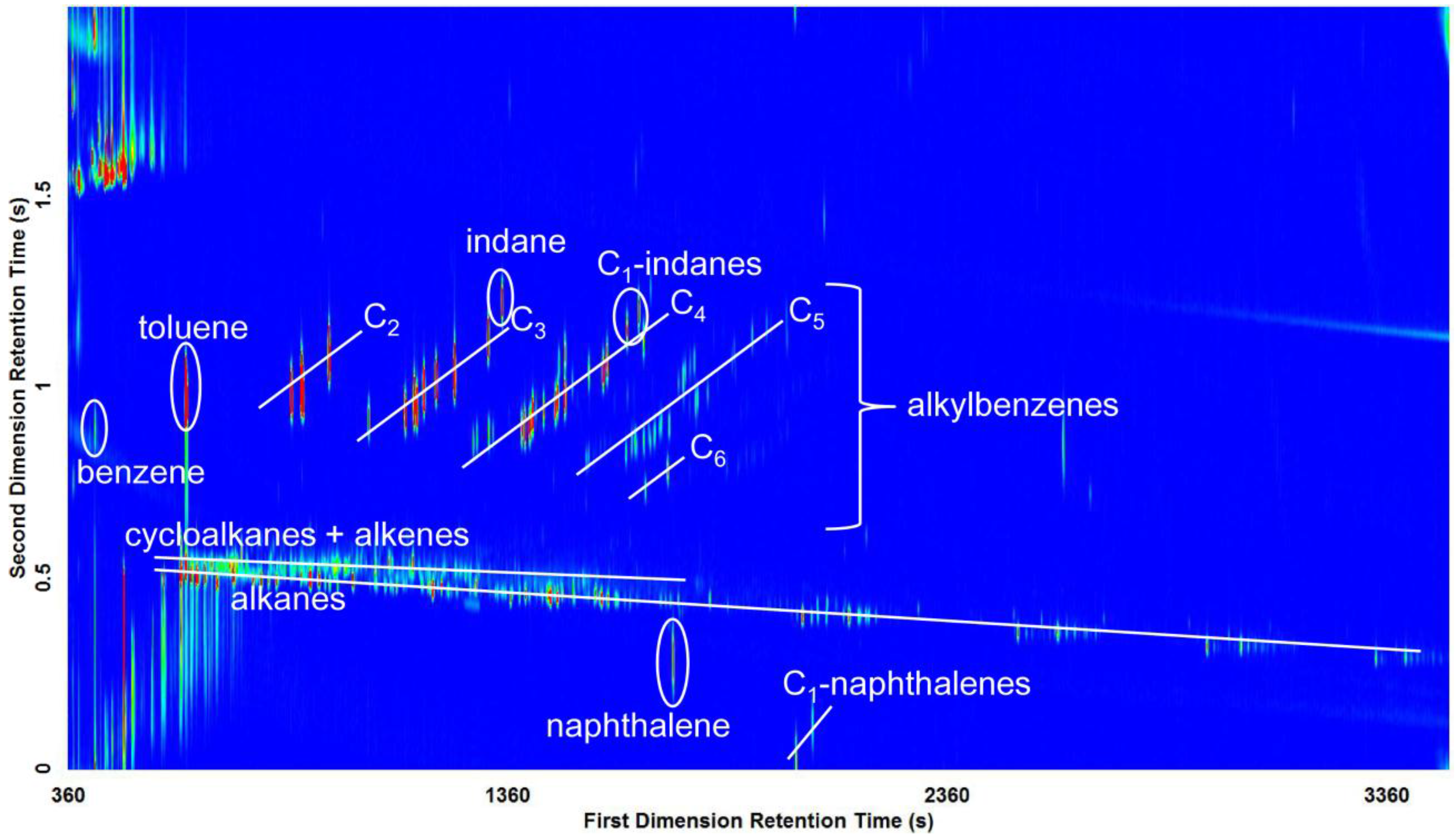
3.2. Visual Pattern Recognition
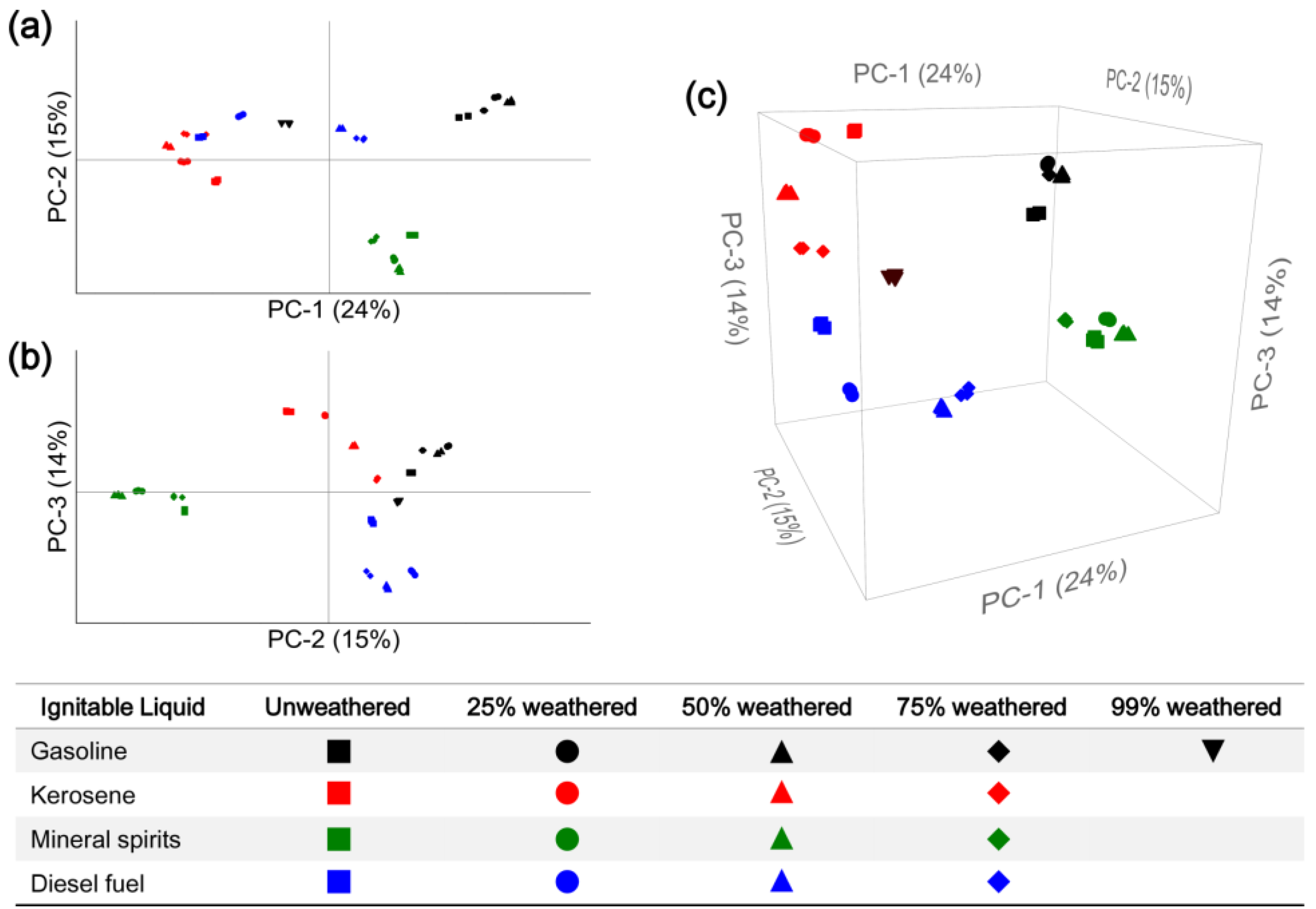
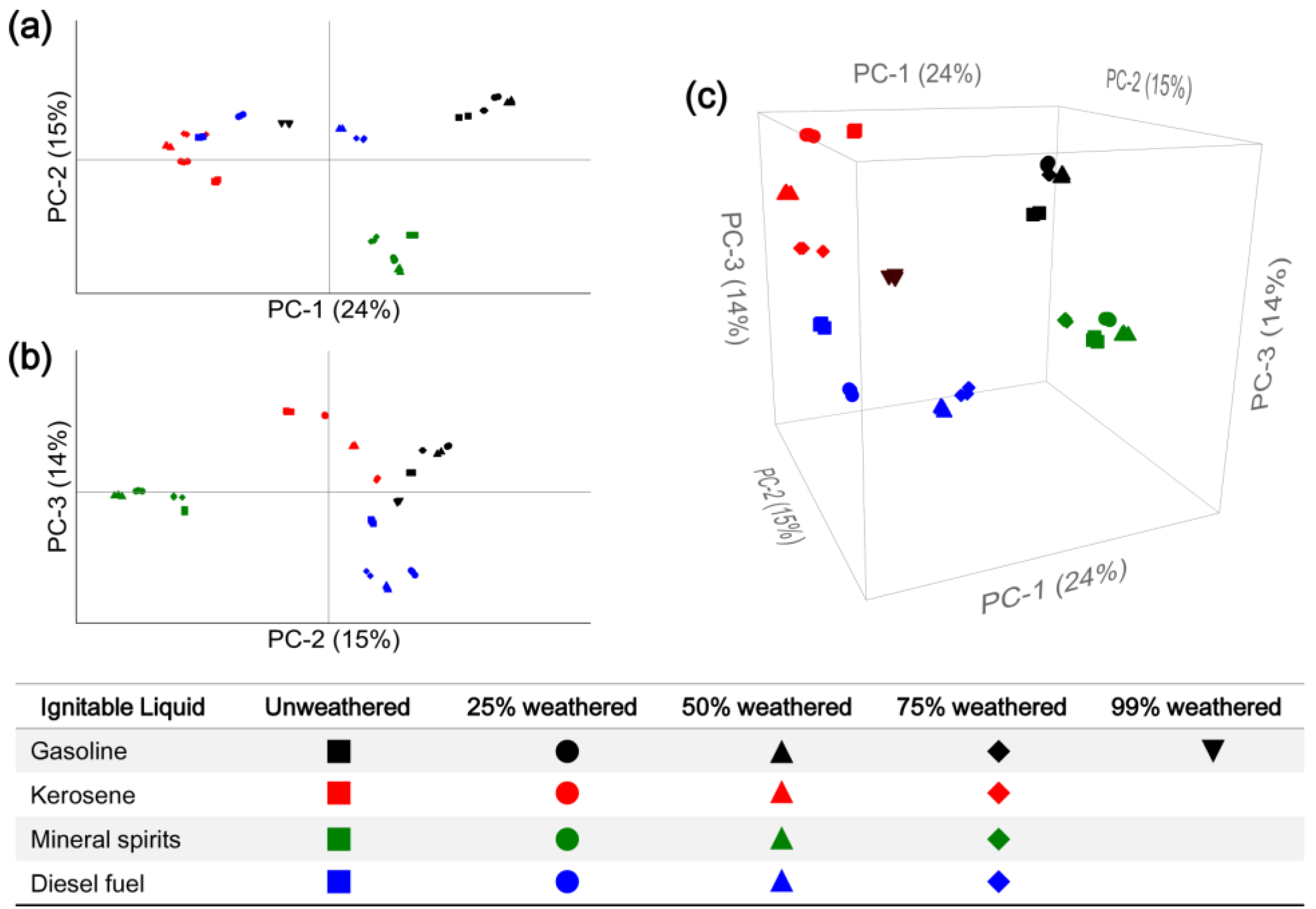
3.3. Computerized Pattern Recognition
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 1D | First dimension |
| 1D | One-dimensional |
| 2D | Second dimension |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| ASTM | American Society of Testing and Materials |
| df | Film thickness |
| EIC | Extracted ion current |
| Fcrit | Critical value |
| GC×GC | Comprehensive two-dimensional gas chromatography |
| GC-qMS | Gas chromatography–quadrupole mass spectrometry |
| GC-TOFMS | Gas chromatography–time-of-flight mass spectrometry |
| i.d. | Inner diameter |
| k | Retention factor |
| NIST | National Institute of Standards and Technology |
| NSW | New South Wales |
| PC-1 | First principal component |
| PC-2 | Second principal component |
| PC-3 | Third principal component |
| PCA | Principal component analysis |
| PFTBA | Perfluorotributylamine |
| SPME | Solid phase microextraction |
| TIC | Total ion current |
| TOFMS | Time-of-flight mass spectrometry |
References
- Drabsch, T. Arson, NSW Parliamentary Library, Briefing Paper No 2/03. Available online: http://www.parliament.nsw.gov.au/prod/parlment/publications.nsf/0/CA9A4BBDED3D32B3CA256ECF00071B74/$File/02-03.pdf (accessed on 21 April 2016).
- Hine, G.A. Fire scene investigation: An introduction for chemists. In Analysis and Interpretation of Fire Scene Evidence; Almirall, J.R., Furton, K.G., Eds.; CRC Press LLC: Boca Raton, FL, USA, 2004; pp. 35–70. [Google Scholar]
- ASTM E1386-15. Standard Practice for Separation of Ignitable Liquid Residues from Fire Debris Samples by Solvent Extraction. In Annual Book of ASTM Standards; ASTM International: West Conshohocken, PA, USA, 2015.
- ASTM E1388-12. Standard Practice for Sampling of Headspace Vapors from Fire Debris Samples. In Annual Book of ASTM Standards; ASTM International: West Conshohocken, PA, USA, 2012.
- ASTM E1413-13. Standard Practice for Separation of Ignitable Liquid Residues from Fire Debris Samples by Dynamic Headspace Concentration. In Annual Book of ASTM Standards; ASTM International: West Conshohocken, PA, USA, 2013.
- ASTM E1412-16. Standard Practice for Separation of Ignitable Liquid Residues from Fire Debris Samples by Passive Headspace Concentration With Activated Charcoal. In Annual Book of ASTM Standards; ASTM International: West Conshohocken, PA, USA, 2016.
- ASTM E2154-15a. Standard Practice for Separation and Concentration of Ignitable Liquid Residues from Fire Debris Samples by Passive Headspace Concentration with Solid Phase Microextraction (SPME). In Annual Book of ASTM Standards; ASTM International: West Conshohocken, PA, USA, 2015.
- ASTM E1618-14. Standard Test Method for Ignitable Liquid Residues in Extracts from Fire Debris Samples by Gas Chromatography-Mass Spectrometry. In Annual Book of ASTM Standards; ASTM International: West Conshohocken, PA, USA, 2014.
- Sandercock, P.M.L.; Du Pasquier, E. Chemical fingerprinting of gasoline. 2. Comparison of unevaporated and evaporated automotive gasoline samples. Forensic Sci. Int. 2004, 140, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Zorzetti, B.M.; Shaver, J.M.; Harynuk, J.J. Estimation of the age of a weathered mixture of volatile organic compounds. Anal. Chim. Acta 2011, 694, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.A.; Goodpaster, J.V. The effects of microbial degradation on ignitable liquids. Anal. Bioanal. Chem. 2009, 394, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Almirall, J.R.; Furton, K.G. Characterization of background and pyrolysis products that may interfere with the forensic analysis of fire debris. J. Anal. Appl. Pyrolysis 2004, 71, 51–67. [Google Scholar] [CrossRef]
- Sinkov, N.A.; Sandercock, P.M.L.; Harynuk, J.J. Chemometric classification of casework arson samples based on gasoline content. Forensic Sci. Int. 2014, 235, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Frysinger, G.S.; Gaines, R.B. Forensic analysis of ignitable liquids in fire debris by comprehensive two-dimensional gas chromatography. J. Forensic Sci. 2002, 47, 471–482. [Google Scholar] [PubMed]
- Taylor, C.; Rosenhan, A.; Raines, J.; Rodriguez, J. An arson investigation by using comprehensive two-dimensional gas chromatography-quadrupole mass spectrometry. J. Forensic Res. 2012, 3, 169–179. [Google Scholar]
- Liu, Z.; Phillips, J.B. Comprehensive two-dimensional gas chromatography using an on-column thermal modulator interface. J. Chromatogr. Sci. 1991, 29, 227–231. [Google Scholar] [CrossRef]
- Von Mühlen, C.; Zini, C.A.; Caramão, E.B.; Marriott, P.J. Applications of comprehensive two-dimensional gas chromatography to the characterization of petrochemical and related samples. J. Chromatogr. A 2006, 1105, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Adahchour, M.; Beens, J.; Vreuls, R.; Brinkman, U.A.T. Recent developments in comprehensive two-dimensional gas chromatography (GC×GC) III. Applications for petrochemicals and organohalogens. Trends Anal. Chem. 2006, 25, 726–741. [Google Scholar] [CrossRef]
- Nizio, K.D.; McGinitie, T.M.; Harynuk, J.J. Comprehensive multidimensional separations for the analysis of petroleum. J. Chromatogr. A 2012, 1255, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.G.; Welch, S.M.; Turner, W.E.; Sjödin, A.; Focant, J.F. Cryogenic zone compression for the measurement of dioxins in human serum by isotope dilution at the attogram level using modulated gas chromatography coupled to high resolution magnetic sector mass spectrometry. J. Chromatogr. A 2011, 1218, 3274–3281. [Google Scholar] [CrossRef] [PubMed]
- Beens, J.; Blomberg, J.; Schoenmakers, P.J. Proper tuning of comprehensive two-dimensional gas chromatography (GC×GC) to optimize the separation of complex oil fractions. J. High Resolut. Chromatogr. 2000, 23, 182–188. [Google Scholar] [CrossRef]
- Korytár, P.; Leonards, P.E.G.; de Boer, J.; Brinkman, U.A.T. High-resolution separation of polychlorinated biphenyls by comprehensive two-dimensional gas chromatography. J. Chromatogr. A 2002, 958, 203–218. [Google Scholar] [CrossRef]
- Marriott, P.J.; Massil, T.; Hügel, H. Molecular structure retention relationships in comprehensive two-dimensional gas chromatography. J. Sep. Sci. 2004, 27, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, E.; Dolan, J.A.; Newman, R. Fire Debris Analysis; Academic Press: London, UK, 2008. [Google Scholar]
- Sampat, A.; Lopatka, M.; Sjerps, M.; Vivo-truyols, G.; Schoenmakers, P.; Asten, A. Van The forensic potential of comprehensive two-dimensional gas chromatography. Trends Anal. Chem. 2016, 80, 345–363. [Google Scholar] [CrossRef]
- Martín-Alberca, C.; Ortega-Ojeda, F.E.; García-Ruiz, C. Analytical tools for the analysis of fire debris. A review: 2008–2015. Anal. Chim. Acta 2016, 928, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Dekeirsschieter, J.; Stefanuto, P.-H.; Brasseur, C.; Haubruge, E.; Focant, J.-F. Enhanced characterization of the smell of death by comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry (GC×GC-TOFMS). PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Stefanuto, P.-H.; Perrault, K.; Stadler, S.; Pesesse, R.; Brokl, M.; Forbes, S.; Focant, J.-F. Reading cadaveric decomposition chemistry with a new pair of glasses. ChemPlusChem 2014, 79, 786–789. [Google Scholar] [CrossRef]
- Perrault, K.A.; Nizio, K.D.; Forbes, S.L. A comparison of one-dimensional and comprehensive two-dimensional gas chromatography for decomposition odour profiling using inter-year replicate field trials. Chromatographia 2015, 78, 1057–1070. [Google Scholar] [CrossRef]
- Frysinger, G. GC×GC—A new analytical tool for evironmental forensics. Environ. Forensics 2002, 3, 27–34. [Google Scholar]
- Muscalu, A.M.; Reiner, E.J.; Liss, S.N.; Chen, T.; Ladwig, G.; Morse, D. A routine accredited method for the analysis of polychlorinated biphenyls, organochlorine pesticides, chlorobenzenes and screening of other halogenated organics in soil, sediment and sludge by GC×GC-µECD. Anal. Bioanal. Chem. 2011, 401, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Prebihalo, S.; Brockman, A.; Cochran, J.; Dorman, F.L. Determination of emerging contaminants in wastewater utilizing comprehensive two-dimensional gas-chromatography coupled with time-of-flight mass spectrometry. J. Chromatogr. A 2015, 1419, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Klee, M.S.; Cochran, J.; Merrick, M.; Blumberg, L.M. Evaluation of conditions of comprehensive two-dimensional gas chromatography that yield a near-theoretical maximum in peak capacity gain. J. Chromatogr. A 2015, 1383, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, L.M. Theory of fast capillary gas chromatography—Part 3: Column performance vs. gas flow rate. J. High Resolut. Chromatogr. 1999, 22, 403–413. [Google Scholar] [CrossRef]
- Blumberg, L.M.; Klee, M.S. Optimal heating rate in gas chromatography. J. Microcolumn Sep. 2000, 12, 508–514. [Google Scholar] [CrossRef]
- Sandercock, P.M.; Du Pasquier, E. Chemical fingerprinting of unevaporated automotive gasoline samples. Forensic Sci. Int. 2003, 134, 1–10. [Google Scholar] [CrossRef]
- Giddings, J.C. Two-dimensional separations: concept and promise. Anal. Chem. 1984, 56, 1258A–1270A. [Google Scholar] [CrossRef] [PubMed]
- Górecki, T.; Harynuk, J.; Panić, O. The evolution of comprehensive two-dimensional gas chromatography (GC×GC). J. Sep. Sci. 2004, 27, 359–379. [Google Scholar] [CrossRef] [PubMed]
- Sinkov, N.A.; Johnston, B.M.; Sandercock, P.M.L.; Harynuk, J.J. Automated optimization and construction of chemometric models based on highly variable raw chromatographic data. Anal. Chim. Acta 2011, 697, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.A.; Goodpaster, J.V. Comparing the effects of weathering and microbial degradation on gasoline using principal components analysis. J. Forensic Sci. 2012, 57, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Lopatka, M.; Sigman, M.E.; Sjerps, M.J.; Williams, M.R.; Vivó-Truyols, G. Class-conditional feature modeling for ignitable liquid classification with substantial substrate contribution in fire debris analysis. Forensic Sci. Int. 2015, 252, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Monfreda, M.; Gregori, A. Differentiation of unevaporated gasoline samples according to their brands, by SPME-GC-MS and multivariate statistical analysis. J. Forensic Sci. 2011, 56, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.M.; Hoggard, J.C.; Hope, J.L.; Rainey, P.M.; Hoofnagle, A.N.; Jack, R.M.; Wright, B.W.; Synovec, R.E.; Hospital, C.; Point, S.; et al. Fisher ratio method applied to third-order separation data to identify significant chemical components of metabolite extracts. Anal. Chem. 2006, 78, 5068–5075. [Google Scholar] [CrossRef] [PubMed]
- Mohler, R.E.; Dombek, K.M.; Hoggard, J.C.; Pierce, K.M.; Young, E.T.; Synovec, R.E. Comprehensive analysis of yeast metabolite GC×GC-TOFMS data: Combining discovery-mode and deconvolution chemometric software. Analyst 2007, 132, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Beckstrom, A.C.; Humston, E.M.; Snyder, L.R.; Synovec, R.E.; Juul, S.E. Application of comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometry method to identify potential biomarkers of perinatal asphyxia in a non-human primate model. J. Chromatogr. A 2011, 1218, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Brokl, M.; Bishop, L.; Wright, C.G.; Liu, C.; McAdam, K.; Focant, J.-F. Multivariate analysis of mainstream tobacco smoke particulate phase by headspace solid-phase micro extraction coupled with comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry. J. Chromatogr. A 2014, 1370, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Stefanuto, P.-H.; Perrault, K.A.; Lloyd, R.M.; Stuart, B.; Rai, T.; Forbes, S.L.; Focant, J.-F. Exploring new dimensions in cadaveric decomposition odour analysis. Anal. Methods 2015, 7, 2287–2294. [Google Scholar] [CrossRef]
- Nizio, K.D.; Perrault, K.A.; Troobnikoff, A.N.; Ueland, M.; Shoma, S.; Iredell, J.R.; Middleton, P.G.; Forbes, S.L. In vitro volatile organic compound profiling using GC×GC-TOFMS to differentiate bacteria associated with lung infections: a proof-of-concept study. J. Breath Res. 2016, 10. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
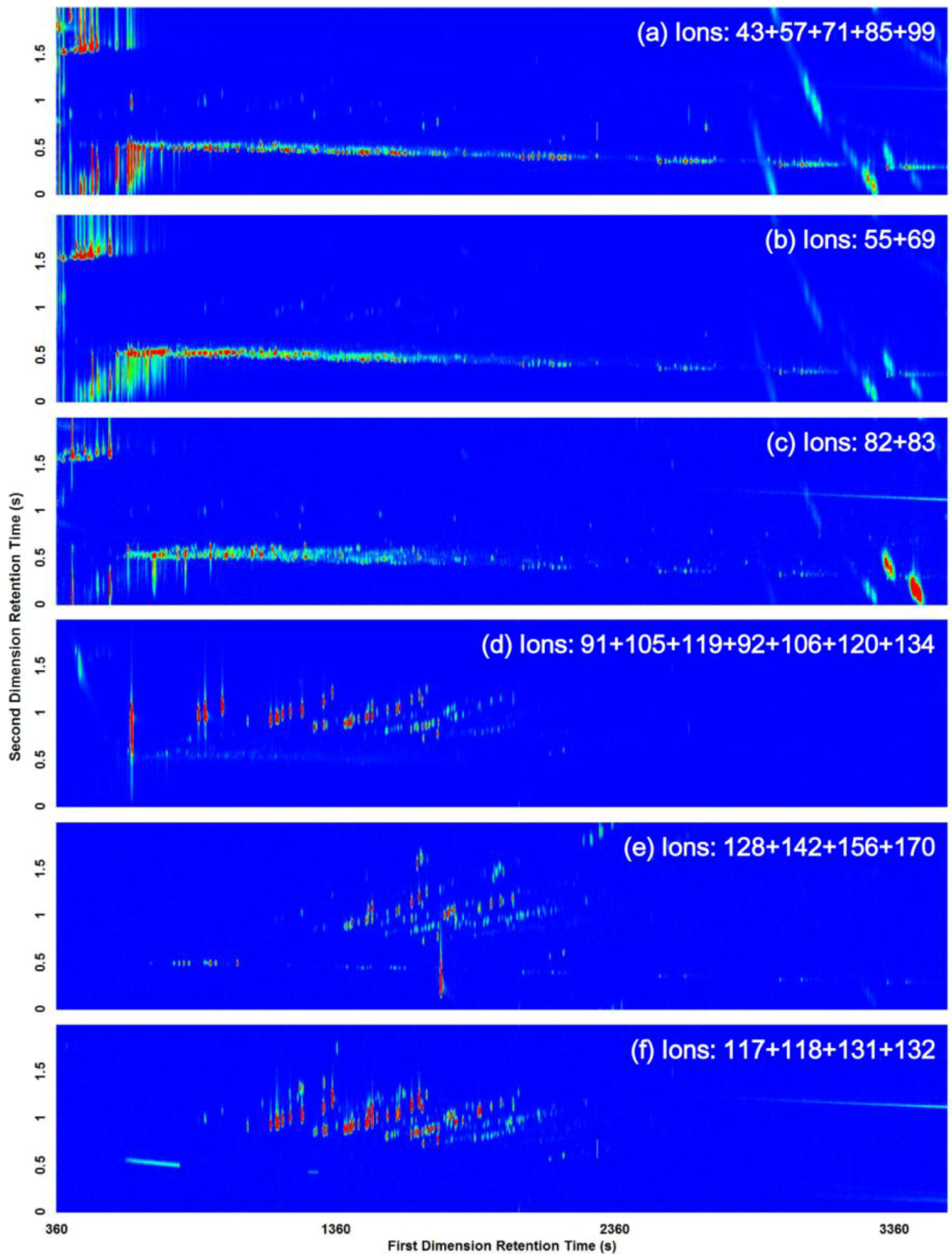
| Compound Class | m/z |
|---|---|
| Alkanes | 43, 57, 71, 85, 99 |
| Cycloalkanes and alkenes | 55, 69 |
| n-Alkylcyclohexanes | 82, 83 |
| Aromatics (i.e., alkylbenzenes) | 91, 105, 119, 92, 106, 120, 134 |
| Alkylnaphthalenes | 128, 142, 156, 170 |
| Indanes | 117, 118, 131, 132 |
| Alkylstyrenes | 104, 117, 118, 132, 146 |
| Alkylanthracenes | 178, 192, 206 |
| Alkylbiphenyls/acenaphthenes | 154, 168, 182, 196 |
| Monoterpenes | 93, 136 |
| Ketones | 43, 58, 72, 86 |
| Alcohols | 31, 45 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nizio, K.D.; Cochran, J.W.; Forbes, S.L. Achieving a Near-Theoretical Maximum in Peak Capacity Gain for the Forensic Analysis of Ignitable Liquids Using GC×GC-TOFMS. Separations 2016, 3, 26. https://doi.org/10.3390/separations3030026
Nizio KD, Cochran JW, Forbes SL. Achieving a Near-Theoretical Maximum in Peak Capacity Gain for the Forensic Analysis of Ignitable Liquids Using GC×GC-TOFMS. Separations. 2016; 3(3):26. https://doi.org/10.3390/separations3030026
Chicago/Turabian StyleNizio, Katie D., Jack W. Cochran, and Shari L. Forbes. 2016. "Achieving a Near-Theoretical Maximum in Peak Capacity Gain for the Forensic Analysis of Ignitable Liquids Using GC×GC-TOFMS" Separations 3, no. 3: 26. https://doi.org/10.3390/separations3030026