Comparative Issues of Cathode Materials for Li-Ion Batteries



Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Framework | Compound | Specific capacity a (mAh g−1) | Average potential (Vvs. Li0/Li+) |
|---|---|---|---|
| Layered | LiCoO2 | 272 (140) | 4.2 |
| LiNi1/3Mn1/3Co1/3O2 | 272 (200) | 4.0 | |
| Spinel | LiMn2O4 | 148 (120) | 4.1 |
| LiMn3/2Ni1/2O4 | 148 (120) | 4.7 | |
| Olivine | LiFePO4 | 170 (160) | 3.45 |
| LiFe1/2Mn1/2PO4 | 170 (160) | 3.4/4.1 |
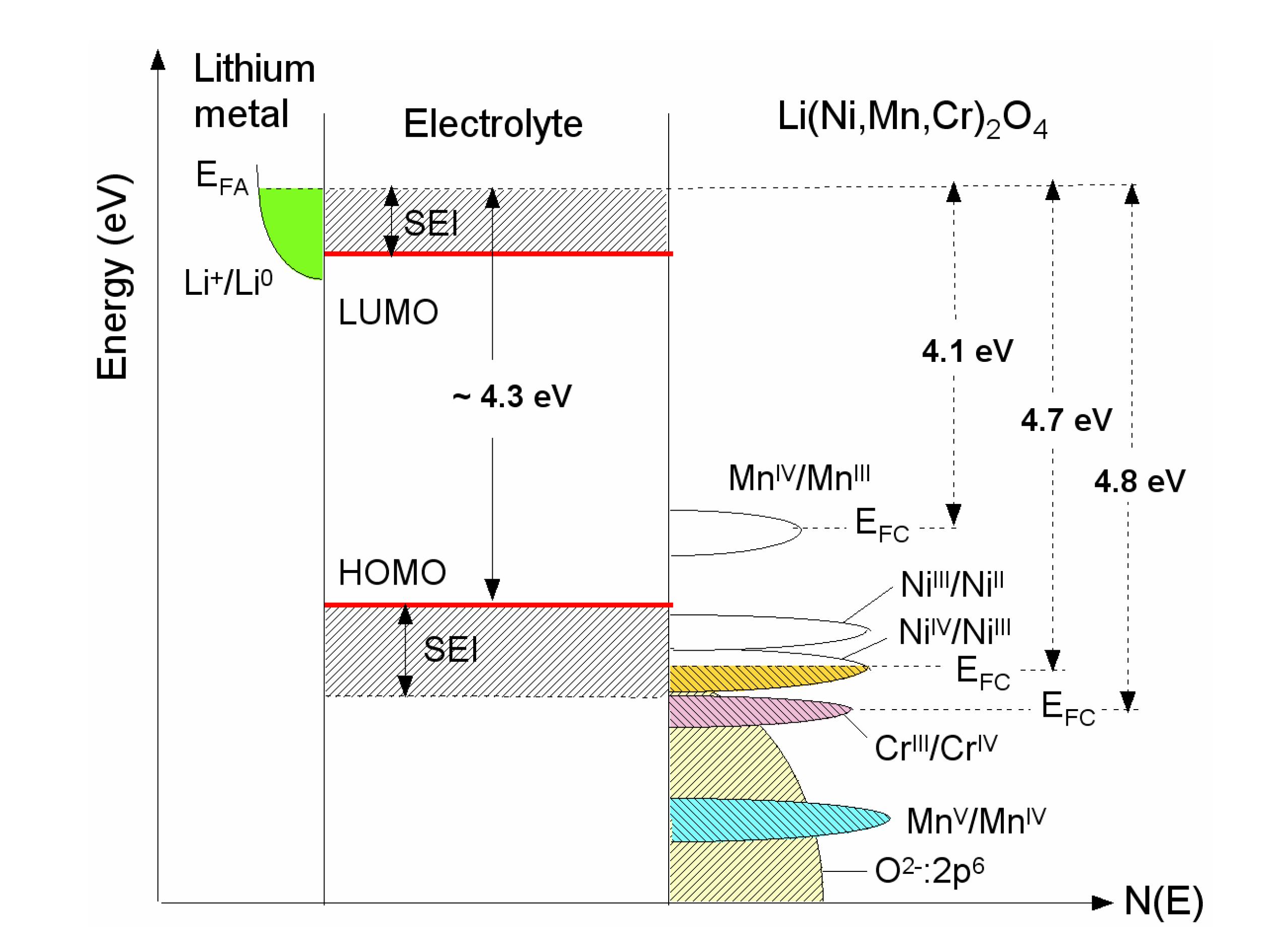
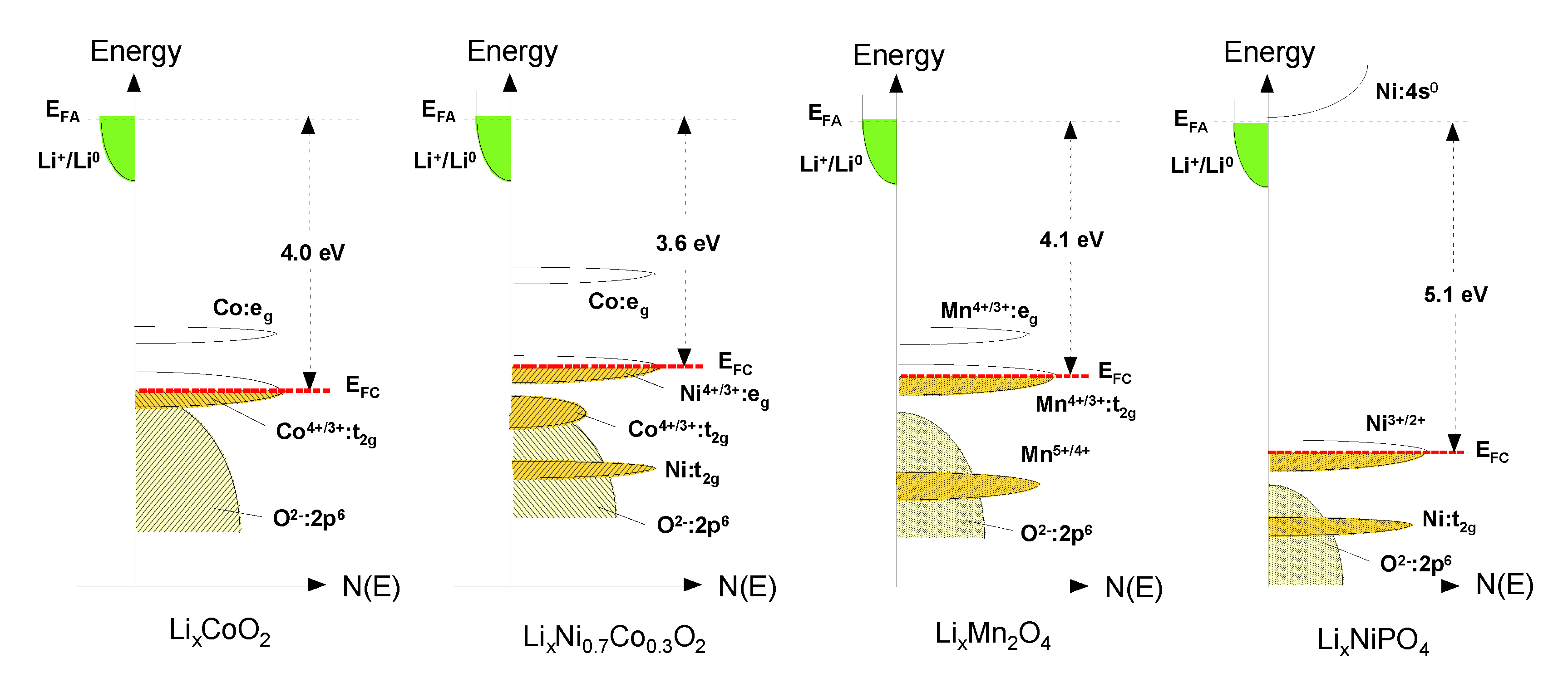
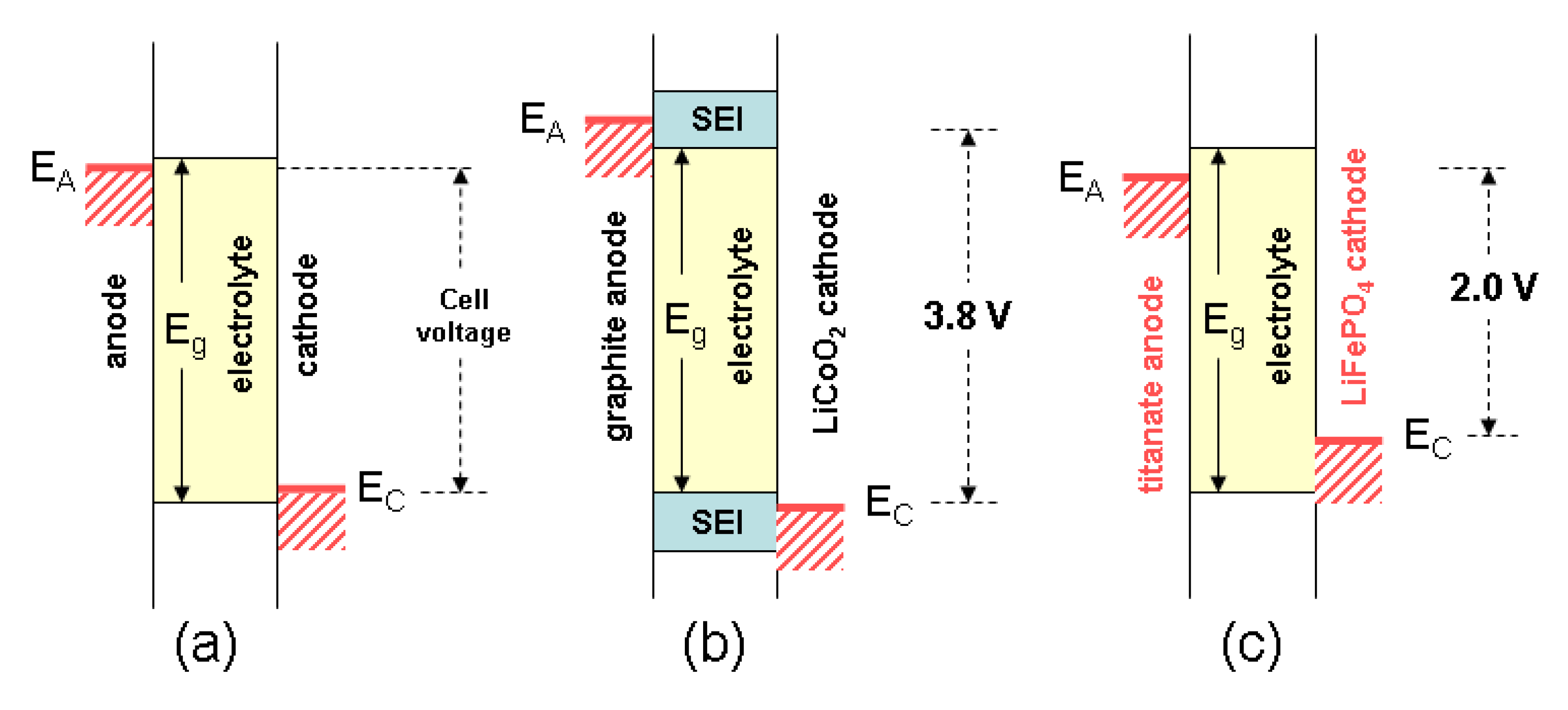
2. The Cell Potential (Goodenough Diagram)


3. Crystal Structure and Electronic Properties

3.1. Layered Compounds
3.2. LiMn2O4 (LMO)
3.3. LiNi0.5Mn1.5O4 (LNM)
3.4. Olivine LiFePO4 (LFP)

4. Electrochemical Properties and Phase Diagram
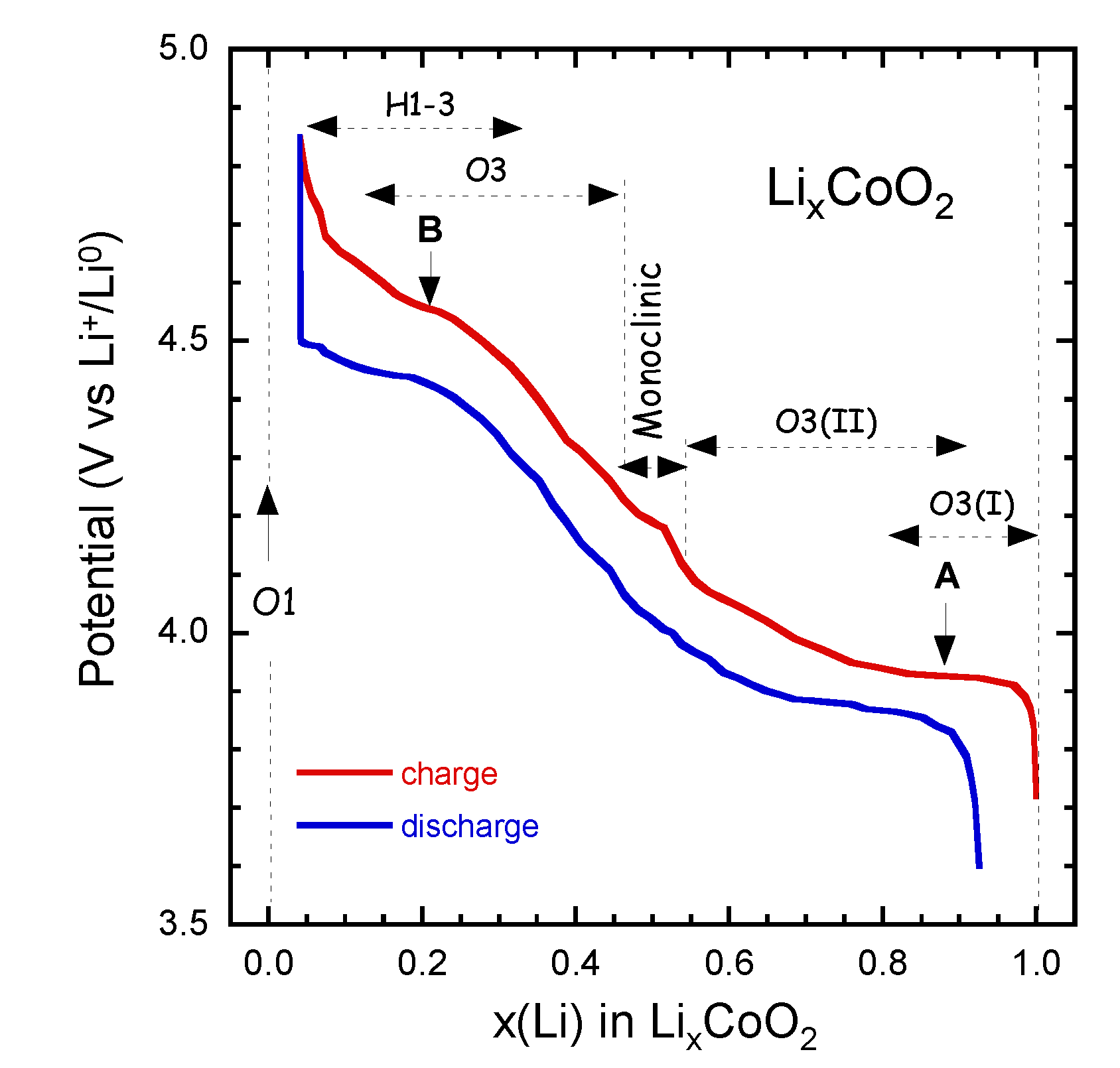
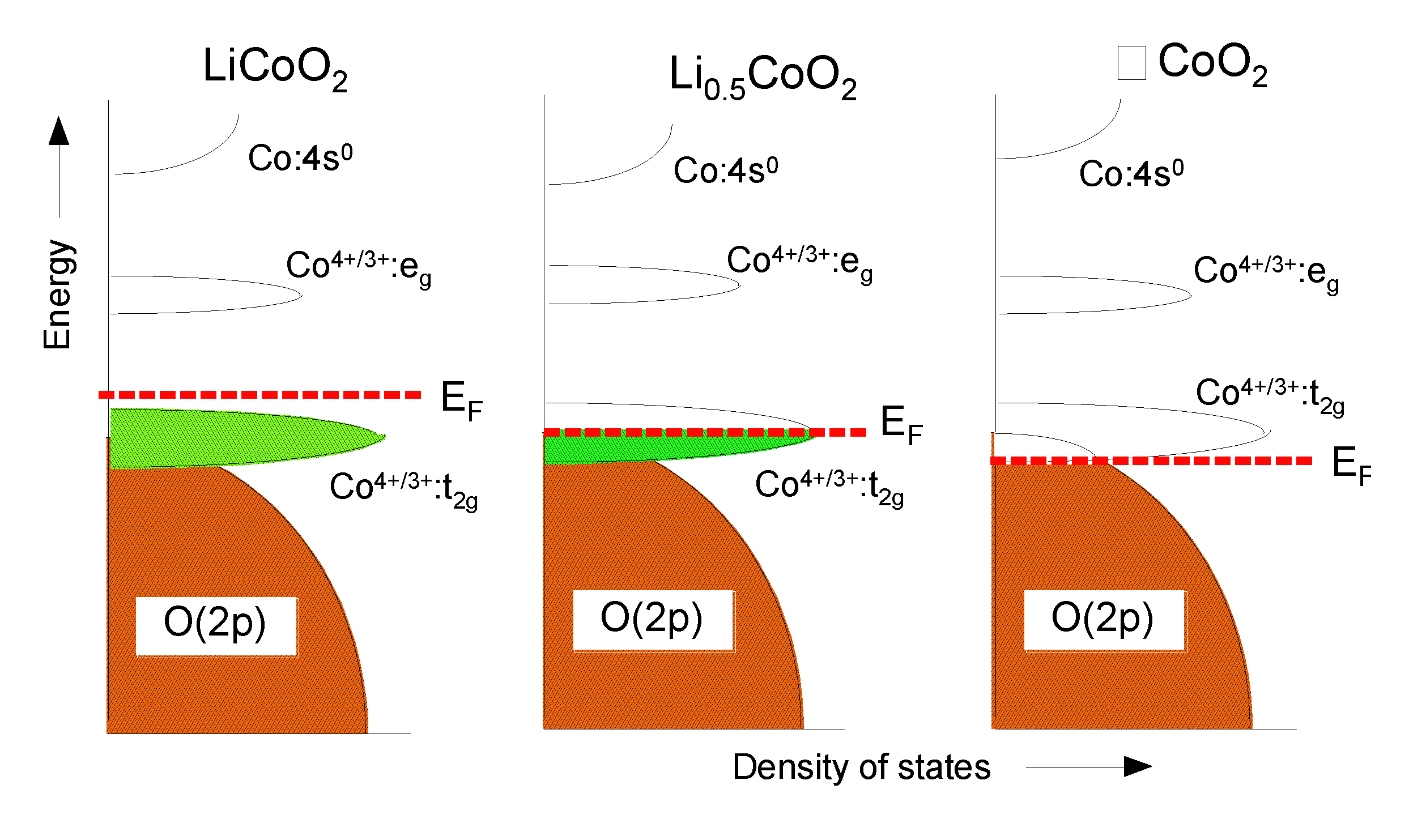
4.1. Lithium Cobaltate (LCO)

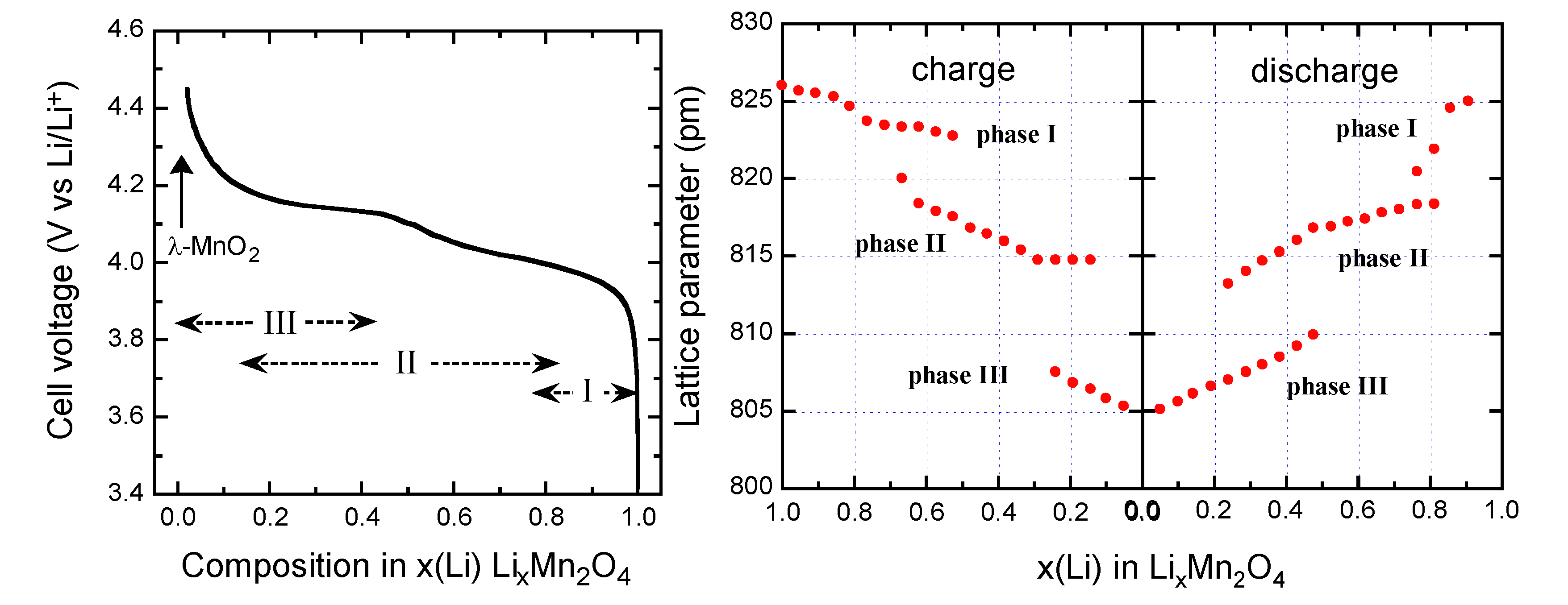
4.2. Lithium Manganese Spinel (LMO)

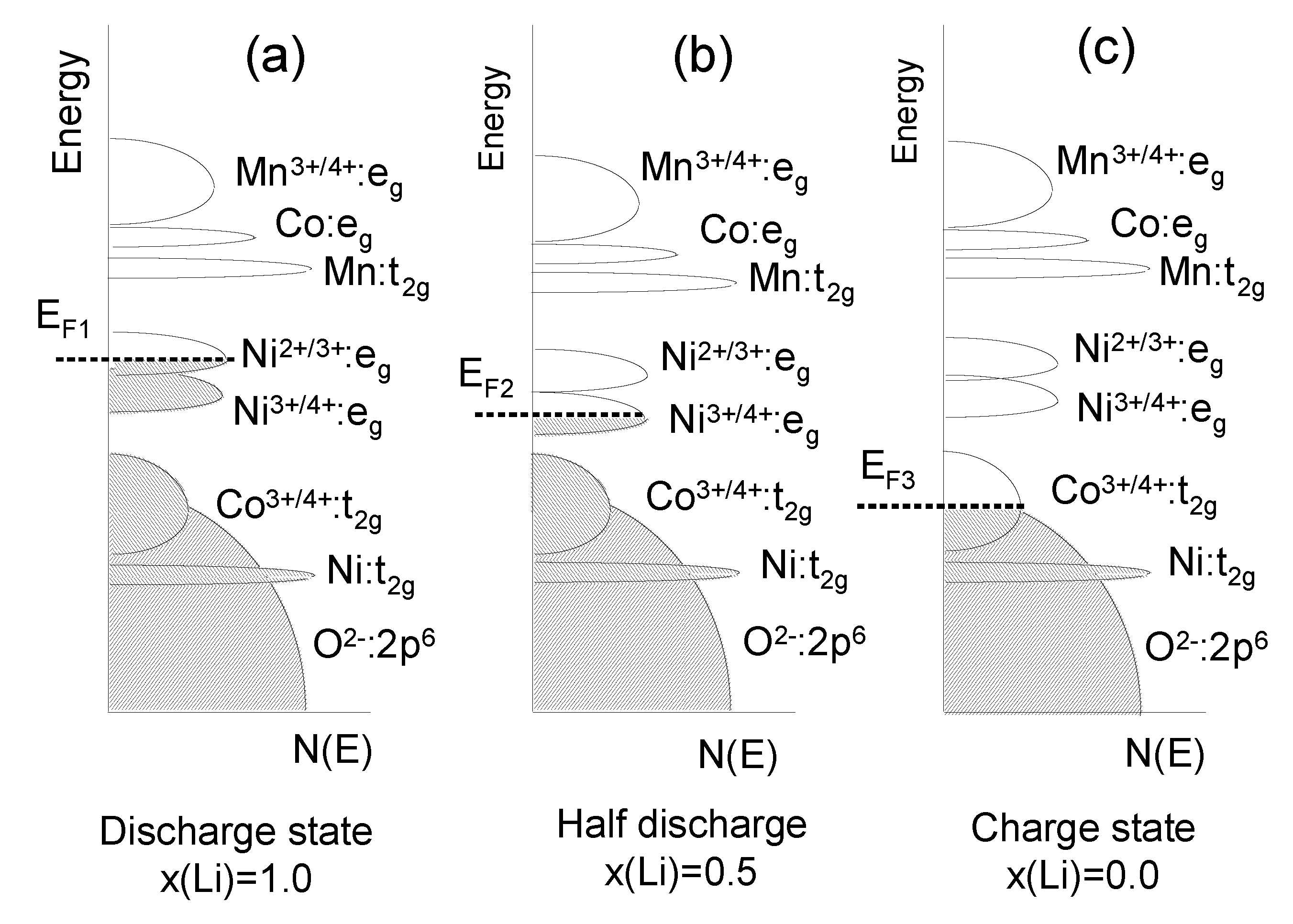
4.3. Lithium Mn-Ni-Co Oxides
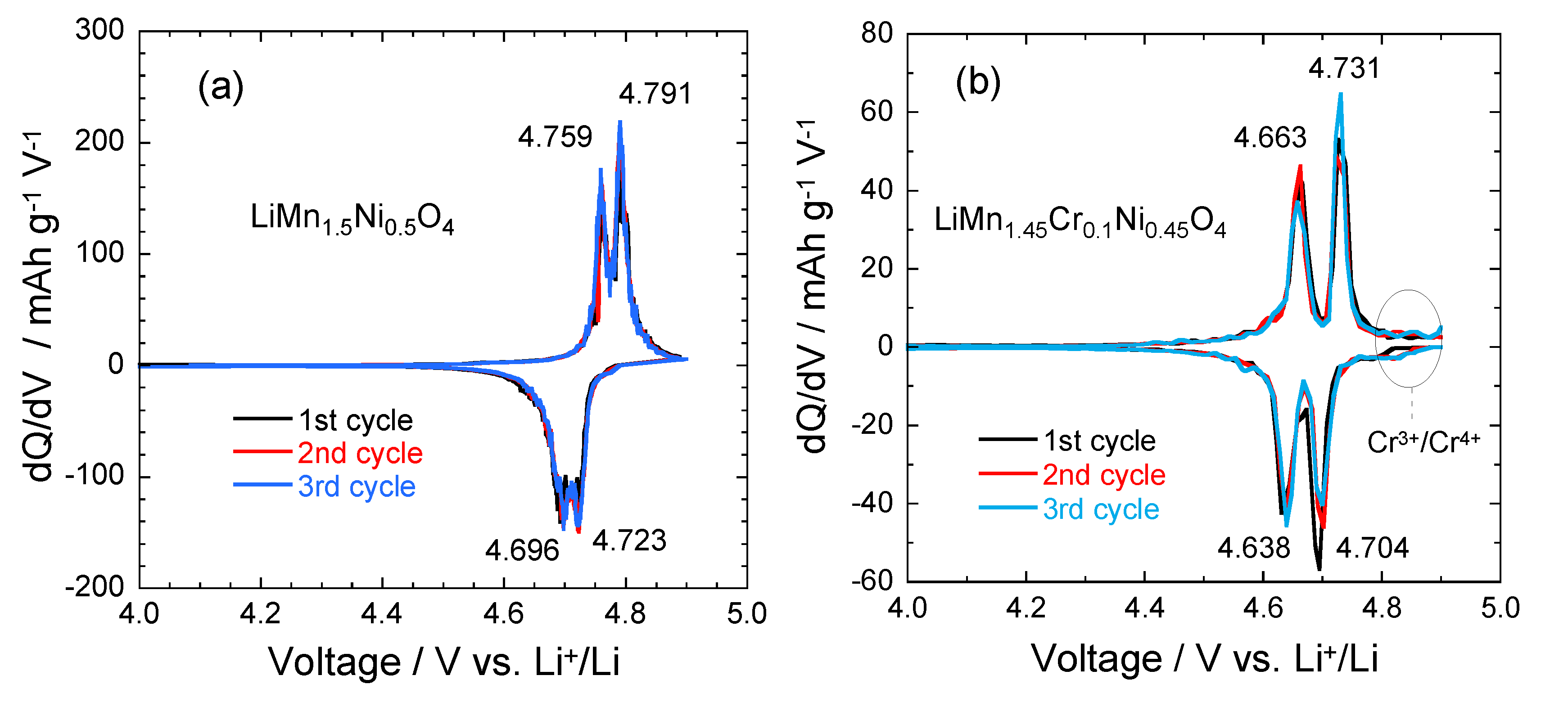
4.4. LMN and Doped-LMN Electrodes

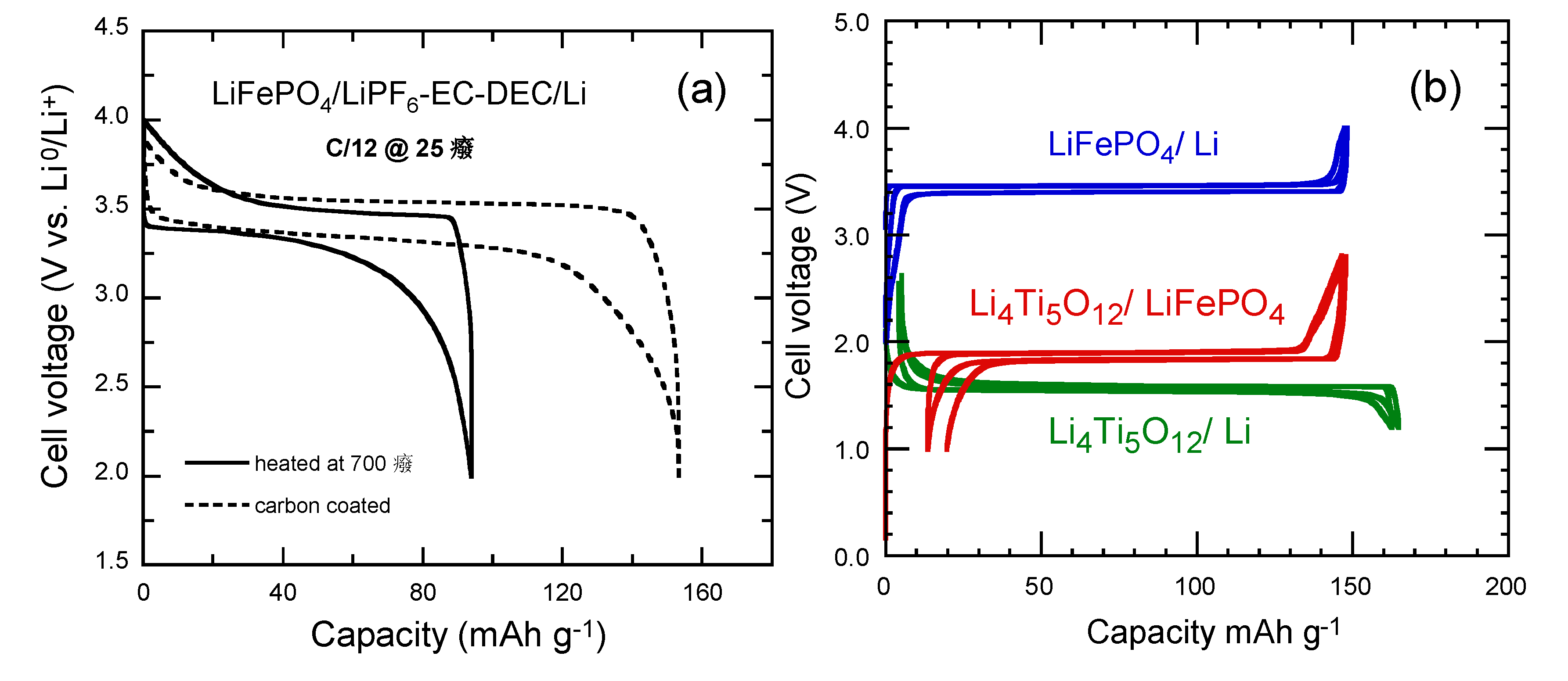
4.5. Lithium Iron Phosphate (LiFePO4)
 10−14 cm2 s−1) of LFP may result in losses in capacity during high-rate discharge. However, the reduction of the LFP particles to the nanosize provides short Li+-ion diffusion paths within the positive electrode. In addition, the synthesis of carbon-coated LFP remarkably enhances the electrical conduction between particles ensuring high rate capability and preventing particles agglomeration [58,59,60].
10−14 cm2 s−1) of LFP may result in losses in capacity during high-rate discharge. However, the reduction of the LFP particles to the nanosize provides short Li+-ion diffusion paths within the positive electrode. In addition, the synthesis of carbon-coated LFP remarkably enhances the electrical conduction between particles ensuring high rate capability and preventing particles agglomeration [58,59,60]. 

5. Safety Issues
5.1. Loss of Oxygen in LixCoO2
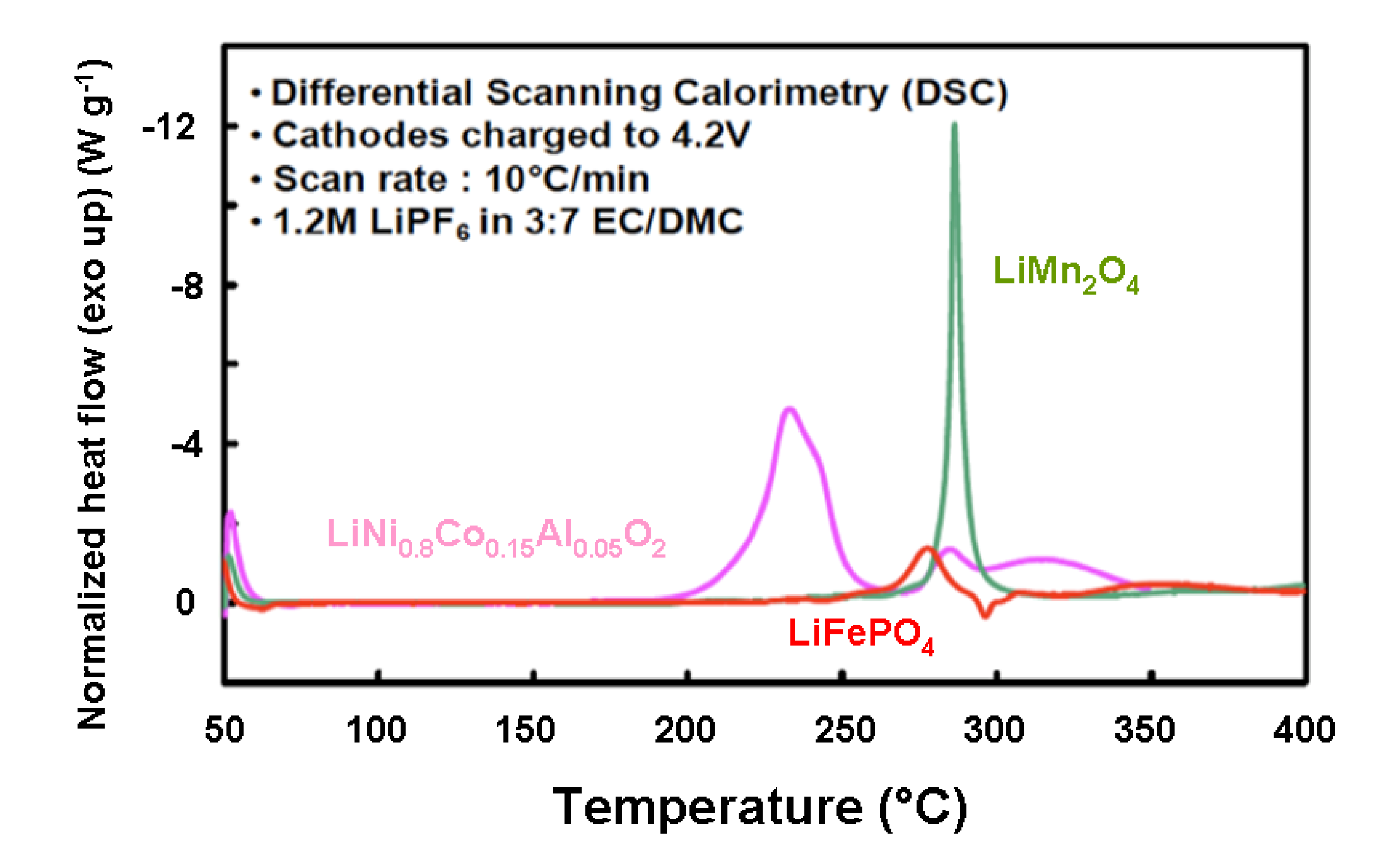
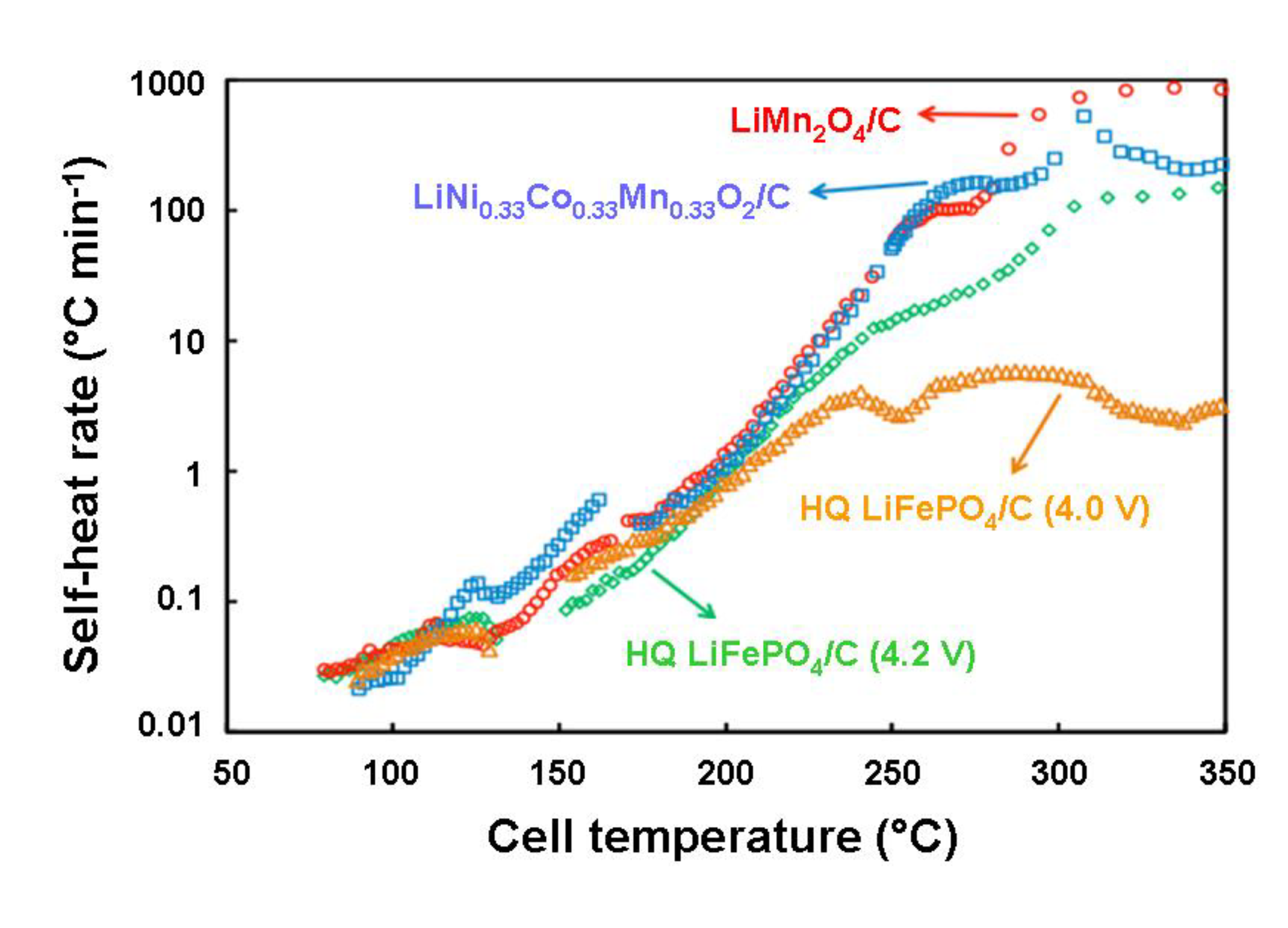
5.2. Comparative Safety Issues


| Cathode material | Onset T (°C) | Overall ∆H (J g−1) |
|---|---|---|
| LiNi0.8Co0.15Al0.05O2 | 170 | −941 |
| LiMn2O4 | 264 | −439 |
| LiFePO4 | 245 | −250 |

6. Concluding Remarks
| Application | Positive electrode | Negative electrode | Remarks |
|---|---|---|---|
| High energy | LiCoO2 (L) LiNi yCozM1−y−zO2 (L) LiMn2O4 (S) | Graphite, Si, SnO x, CoOx, FeOx, CuOx, NiOx, etc. | M = Mn, Al, Cr named “LMO” |
| High power | LiMn2− yAlyO4+δ (S) LiNiyMnyCo2−yO2 (L) LiFePO4 (O) | hard carbon, graphite Li4Ti5O12 (S) | named “NMC” “LFP//LTO” cell |
| Long cycle life | LiMn2− yAlyO4+δ (S) LiFePO4 (O) LiFe1−yMnyPO4 (O) | Graphite Li4Ti5O12 (S) Li4Ti5O12 (S) | named “LFP” |
 , where L is the particle size and the diffusion coefficient of Li+ ion in the host lattice. As an example, for 2-µm LiFePO4 particles τ = 83 h, while decreasing the particle to 40 nm reduces τ to 13 s. The volume changes caused by Li+ insertion/extraction are better accommodated by nanosized particles due to faster strain relaxation. However, a few disadvantages are: (i) the need of surface coating for either minimizing the reaction at the electrode-electrolyte interface for LCO and LMO or enhanced the electrical contact between particles for LFP; (ii) the increase of the specific surface area of nanoparticles could enhance the rate reaction at the interfaces, especially for LCO material and (iii) the lower density of the electrode material, which reduces the volumetric capacity.
, where L is the particle size and the diffusion coefficient of Li+ ion in the host lattice. As an example, for 2-µm LiFePO4 particles τ = 83 h, while decreasing the particle to 40 nm reduces τ to 13 s. The volume changes caused by Li+ insertion/extraction are better accommodated by nanosized particles due to faster strain relaxation. However, a few disadvantages are: (i) the need of surface coating for either minimizing the reaction at the electrode-electrolyte interface for LCO and LMO or enhanced the electrical contact between particles for LFP; (ii) the increase of the specific surface area of nanoparticles could enhance the rate reaction at the interfaces, especially for LCO material and (iii) the lower density of the electrode material, which reduces the volumetric capacity.Conflicts of Interest
References
- Nagaura, T.; Tozawa, K. Lithium ion rechargeable battery. Prog. Batter. Sol. Cells 1990, 9, 209–211. [Google Scholar]
- Mizushima, K.; Jones, P.C.; Wiseman, P.J.; Goodenough, J.B. LixCoO2 (0 < x < 1): A new cathode material for batteries of high energy density. Mater. Res. Bull. 1980, 15, 783–789. [Google Scholar] [CrossRef]
- Thackeray, M.M.; David, W.I.F.; Bruce, P.G.; Goodenough, J.B. Lithium insertion into manganese spinels. Mater. Res. Bull. 1983, 18, 461–472. [Google Scholar] [CrossRef]
- Thackeray, M.M.; Johnson, P.J.; de Picciotto, L.A.; Bruce, P.G.; Goodenough, J.B. Lithium extraction from LiMn2O4. Mater. Res. Bull. 1984, 19, 179–187. [Google Scholar] [CrossRef]
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Peres, J.P.; Weill, F.; Delmas, C. Lithium-vacancy ordering in the monoclinic LixNiO2 (0.50 < x < 0.75) solid solution. Solid State Ion. 1999, 116, 19–27. [Google Scholar] [CrossRef]
- Ohzuku, T.; Makimura, Y. Layered lithium insertion material of LiCo1/3Ni1/3Mn1/3O2 for lithium-ion batteries. Chem. Lett. 2001, 30, 642–643. [Google Scholar] [CrossRef]
- Tarascon, J.M.; McKinnon, W.R.; Coowar, F.; Bowmer, T.N.; Amatucci, G.; Guyomard, D. Synthesis conditions and oxygen stoichiometry effects on Li insertion into the spinel LiMn2O4. J. Electrochem. Soc. 1994, 141, 1421–1431. [Google Scholar] [CrossRef]
- Zaghib, K.; Mauger, A.; Groult, H.; Goodenough, J.B.; Julien, C.M. Advanced electrodes for high power Li-ion batteries. Materials 2013, 6, 1028–1049. [Google Scholar] [CrossRef]
- Goodenough, J.B. Design considerations. Solid State Ion. 1994, 69, 184–198. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Kim, Y. Challenges for rechargeable Li batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Liu, D.; Han, J.; Dontigny, M.; Charest, P.; Guerfi, A.; Zaghib, K.; Goodenough, J.B. Redox behaviors of Ni and Cr with different counter cations in spinel cathodes for Li-ion batteries. J. Electrochem. Soc. 2010, 157, A770–A775. [Google Scholar] [CrossRef]
- Goodenough, J.B. Oxides cathodes. In Advances in Lithium-Ion Batteries; Kluwer Academic/Plenum: New York, NY, USA, 2002; pp. 135–154. [Google Scholar]
- Manthiram, A. Chemical and Structural Stabilities of Layered Oxide Cathodes. In New Trends in Intercalation Compounds for Energy Storage, NATO Science Series; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2002; pp. 177–192. [Google Scholar]
- Thackeray, M.M. Structural considerations of layered and spinel lithiated oxides for lithium io batteries. J. Electrochem. Soc. 1995, 142, 2558–2563. [Google Scholar] [CrossRef]
- Julien, C. Local cationic environment in lithium nickel–cobalt oxides used as cathode materials for lithium batteries. Solid State Ion. 2000, 136–137, 887–896. [Google Scholar] [CrossRef]
- Arroyo y de Dompablo, M.E.; Marianetti, C.; van der Ven, A.; Ceder, G. Jahn-Teller mediated ordering in layered LixCoO2 compounds. Phys. Rev. B 2001, 63, 144104. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Meng, X.; Tang, Y.; Banis, M.N.; Yang, J.; Hu, Y.; Li, R.; Cai, M.; Sun, X. Significant impact on cathode performance of lithium-ion batteries by precisely controlled metal oxide nanocoatings via atomic layer deposition. J. Power Sources 2013, 247, 57–69. [Google Scholar]
- Li, X.; Liu, J.; Banis, M.N.; Lushington, A.; Li, R.; Caib, M.; Sun, X. Atomic layer deposition of solid-state electrolyte coated cathode materials with superior high voltage cycling behavior for lithium ion battery application. Energy Environ. Sci. 2014, 7, 768–778. [Google Scholar] [CrossRef]
- Ohzuku, T.; Ueda, A.; Nagayama, M.; Iwakashi, Y.; Komori, H. Comparative study of LiCoO2, LiNi1/2Co1/2O2 and LiNiO2 for 4 volt secondary lithium cells. Electrochim. Acta 1993, 38, 1159–1167. [Google Scholar] [CrossRef]
- Delmas, C.; Saadoune, I. Electrochemical and physical properties of LixNi1−yCoyO2 phases. Solid State Ion. 1992, 53–56, 370–375. [Google Scholar] [CrossRef]
- Rougier, A.; Saadoune, I.; Gravereau, P.; Willmann, P.; Delmas, C. Effect of cobalt substitution on cationic distribution in Li electrode materials. Solid State Ion. 1996, 90, 83–90. [Google Scholar] [CrossRef]
- Julien, C.; El-Farh, L.; Rangan, S.; Massot, S. Synthesis of LiNi1−yCoyO2 cathode materials prepared by a citric acid-assisted sol-gel method for lithium batteries. J. Sol-Gel Sci. Technol. 1999, 15, 63–72. [Google Scholar] [CrossRef]
- Li, W.; Curie, J. Morphology effects on the electrochemical performance of LiNi1−xCoxO2. J. Electrochem. Soc. 1997, 144, 2773–2779. [Google Scholar] [CrossRef]
- Ohzuku, T.; Kitagawa, M.; Hirai, T. Electrochemistry of manganese dioxide in lithium nonaqueous cell: III. X-ray diffractional study on the reduction of spinel-related manganese dioxide. J. Electrochem. Soc. 1990, 137, 769–775. [Google Scholar] [CrossRef]
- Yonemura, M.; Kamiyama, T.; Kawamoto, Y.; Kanno, R. Phase transitions and low-temperature structure of lithium manganese oxide spinel. Mater. Trans. 2004, 45, 2048–2055. [Google Scholar] [CrossRef]
- Zhong, Q.M.; Bonakdarpour, A.; Zhang, M.J.; Gao, Y.; Dahn, J.R. Synthesis and electrochemistry of LiNixMn2−xO4. J. Electrochem. Soc. 1997, 144, 205–213. [Google Scholar]
- Liu, D.; Hamel-Paquet, J.; Trottier, J.; Barray, F.; Gariépy, V.; Hovington, P.; Guerfi, A.; Mauger, A.; Julien, C.M.; Goodenough, J.B.; et al. Synthesis of pure phase disordered LiMn1.45Cr0.1Ni0.45O4 by a post-annealing method. J. Power Sources 2012, 217, 400–406. [Google Scholar] [CrossRef]
- Geller, S.; Durand, J.L. Refinement of the structure of LiMnPO4. Acta Crystallogr. 1960, 13, 325–331. [Google Scholar] [CrossRef]
- Santorro, R.P.; Newnham, R.E. Antiferromagnetism in LiFePO4. Acta Crystallogr. 1967, 22, 344–347. [Google Scholar] [CrossRef]
- Moring, J.; Kostiner, E. The crystal structure of NaMnPO4. J. Solid State Chem. 1986, 61, 379–383. [Google Scholar] [CrossRef]
- Zaghib, K.; Mauger, A.; Goodenough, J.B.; Gendron, F.; Julien, C.M. Design and properties of LiFePO4 positive electrode materials for Li-ion batteries. In Advanced Materials and Methods for Lithium-ion Batteries; Zhang, S.S., Ed.; Transworld Research Network: Trivandrum, India, 2007; pp. 115–149. [Google Scholar]
- Laubach, S.; Laubach, S.; Schmidt, P.C.; Ensling, D.; Schmid, S.; Jaegermann, W.; Thisen, A.; Nikolowski, K.; Ehrenberg, H. Changes in the crystal and electronic structure of LiCoO2 and LiNiO2 upon Li intercalation and de-intercalation. Phys. Chem. Chem. Phys. 2009, 11, 3278–3289. [Google Scholar] [CrossRef]
- Reimers, J.N.; Dahn, J.R. Electrochemical and in situ X-ray diffraction studies of lithium intercalation in LixCoO2. J. Electrochem. Soc. 1992, 139, 2091–2097. [Google Scholar] [CrossRef]
- Van der Ven, A.; Aydinol, M.K.; Ceder, G.; Kresse, G.; Hafner, J. First-principles investigation of phase stability in LixCoO2. Phys. Rev. B 1998, 58, 2975–2987. [Google Scholar] [CrossRef]
- Chen, Z.; Dahn, J.R. Methods to obtain excellent capacity retention in LiCoO2 cycled to 4.5 V. Electrochim. Acta 2004, 49, 1079–1090. [Google Scholar] [CrossRef]
- Amatucci, G.G.; Schmutz, C.N.; Blyr, A.; Sigala, C.; Gozdz, A.S.; Larcher, D.; Tarascon, J.M. Materials effects on the elevated and room temperature performance of C/LiMn2O4 Li-ion batteries. J. Power Sources 1997, 69, 11–25. [Google Scholar] [CrossRef]
- Thackeray, M.M. Manganese oxides for lithium batteries. Prog. Solid State Chem. 1997, 25, 1–71. [Google Scholar] [CrossRef]
- Xia, Y.; Zhou, Y.; Yoshio, M. Capacity fading on cycling of 4 V Li/LiMn2O4 cells. J. Electrochem. Soc. 1997, 144, 2593–2600. [Google Scholar] [CrossRef]
- Dai, Y.; Cai, L.; White, R.E. Capacity fade model for spinel LiMn2O4 electrode. J. Electrochem. Soc. 2013, 160, A182–A190. [Google Scholar] [CrossRef]
- Jang, D.H.; Oh, S.M. Electrolyte effects on spinel dissolution and cathodic capacity losses in 4-V Li/LixMn2O4 rechargeable cells. J. Electrochem. Soc. 1997, 144, 3342–3348. [Google Scholar] [CrossRef]
- Gnanaraj, J.S.; Pol, V.G.; Gedanken, A.; Aurbach, D. Improving the high-temperature performance of LiMn2O4 spinel electrodes by coating the active mass with MgO via a sonochemical method. Electrochem. Commun. 2003, 5, 940–945. [Google Scholar] [CrossRef]
- Walz, K.A.; Johnson, C.S.; Genthe, J.; Stoiber, L.C.; Zeltner, W.A.; Anderson, M.A.; Thackeray, M.M. Elevated temperature cycling stability and electrochemical impedance of LiMn2O4 cathodes with nanoporous ZrO2 and TiO2 coatings. J. Power Sources 2010, 195, 4943–4951. [Google Scholar] [CrossRef]
- Kannan, A.M.; Manthiram, A. Surface chemically modified LiMn2O4 cathodes for lithium-ion batteries. Electrochem. Solid-State Lett. 2002, 5, A167–A169. [Google Scholar] [CrossRef]
- Amatucci, G.G.; Blyr, A.; Sigala, C.; Alfonse, P.; Tarascon, J.M. Surface treatments of Li1+xMn2−xO4 spinels for improved elevated temperature performance. Solid State Ion. 1997, 104, 13–25. [Google Scholar] [CrossRef]
- Lee, D.J.; Lee, K.S.; Myung, S.T.; Yashiro, H.; Sun, Y.K. Improvement of electrochemical properties of Li1.1Al0.05Mn1.85O4 achieved by an AlF3 coating. J. Power Sources 2011, 196, 1353–1357. [Google Scholar] [CrossRef]
- Gummow, R.J.; de Kock, A.; Thackeray, M.M. Improved capacity retention in rechargeable 4V lithium/lithium manganese oxide (spinel) cells. Solid State Ion. 1994, 69, 59–67. [Google Scholar] [CrossRef]
- Park, O.K.; Cho, Y.; Yoo, H.C.; Song, H.K.; Cho, J. Who will drive electric vehicles, olivine or spinel? Energy Environ. Sci. 2011, 4, 1621–1633. [Google Scholar] [CrossRef]
- Thackeray, M.M.; Shao-Horn, Y.; Kahaian, A.J.; Kepler, K.D.; Skinner, E.; Vaughey, J.T.; Hackney, S.A. Structural fatigue in spinel electrodes in high voltage (4 V) Li/LixMn2O4 cells. Electrochem. Solid-State Lett. 1998, 1, 7–9. [Google Scholar]
- Lee, Y.J.; Wang, F.; Mukerjee, S.; James McBreen, J.; Grey, C.P. 6Li and 7Li magic-angle spinning nuclear magnetic resonance and in situ X-ray diffraction studies of the charging and discharging of LixMn2O4 at 4 V. J. Electrochem. Soc. 2000, 147, 803–812. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Ohzuku, T. Novel lithium insertion material of LiCo1/3Ni1/3Mn1/3O2 for advanced lithium-ion batteries. J. Power Sources 2003, 119–121, 171–174. [Google Scholar] [CrossRef]
- Shaju, K.M.; Subba Rao, G.V.; Chowdari, B.V.R. Performance of layered Li(Ni1/3Co1/3Mn1/3)O2 as cathode for Li-ion batteries. Electrochim. Acta 2002, 48, 145–151. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Jiang, W.-J.; Mauger, A.; Lu, Q.; Gendron, F.; Julien, C.M. Minimization of the cation mixing in Li1+x(NMC)1−xO2 as cathode material. J. Power Sources 2010, 195, 1292–1301. [Google Scholar] [CrossRef]
- Ein-Eli, Y.; Howard, W.F., Jr. LiCuxIICuyIIIMn[2−(x+y)]III,IVO4: 5 V cathode materials. J. Electrochem. Soc. 1997, 144, L205–L207. [Google Scholar] [CrossRef]
- Julien, C.M.; Mauger, A. Review of 5-V electrodes for Li-ion batteries: Status and trends. Ionics 2013, 19, 951–988. [Google Scholar] [CrossRef]
- Kunduraci, M.; Amatucci, G.G. Effect of oxygen non-stoichiometry and temperature on cation ordering in LiMn2−xNixO4 (0.50 ≥ x ≥ 0.36) spinels. J. Power Sources 2007, 165, 359–367. [Google Scholar] [CrossRef]
- Kim, J.-H.; Yoon, C.S.; Myung, S.-T.; Prakash, J.; Sun, Y.-K. Phase transitions in Li1−δNi0.5Mn1.5O4 during cycling at 5 V. Electrochem. Solid-State Lett. 2004, 7, A216–A220. [Google Scholar] [CrossRef]
- Wang, J.; Sun, X. Understanding and recent development of carbon coating on LiFePO4 cathode materials for lithium-ion batteries. Energy Environ. Sci. 2012, 5, 5163–5185. [Google Scholar] [CrossRef]
- Wang, J.; Yang, J.; Tang, Y.; Li, R.; Liang, G.; Sham, T.K.; Sun, X. Surface aging at olivine LiFePO4: A direct visual observation of iron dissolution and the protection role of nano-carbon coating. J. Mater. Chem. A 2013, 1, 1579–1586. [Google Scholar]
- Wang, J.; Yang, J.; Zhang, Y.; Li, Y.; Tang, Y.; Banis, M.N.; Li, X.; Liang, G.; Li, R.; Sun, X. Interaction of carbon coating on LiFePO4: A local visualization study of the influence of impurity phases. Adv. Funct. Mater. 2013, 23, 806–814. [Google Scholar] [CrossRef]
- Zaghib, K.; Guerfi, A.; Hovington, P.; Vijh, A.; Trudeau, M.; Mauger, A.; Goodenough, J.B.; Julien, C.M. Review and analysis of nanostructured olivine-based lithium rechargeable batteries: Status and trends. J. Power Sources 2013, 232, 357–369. [Google Scholar] [CrossRef]
- Zaghib, K.; Dontigny, M.; Guerfi, A.; Charest, P.; Rodrigues, I.; Mauger, A.; Julien, C.M. Safe and fast-charging Li-ion battery with long shelf life for power applications. J. Power Sources 2011, 196, 3949–3954. [Google Scholar] [CrossRef]
- Zaghib, K.; Dontigny, M.; Guerfi, A.; Trottier, J.; Hamel-Paquet, J.; Gariepy, V.; Galoutov, K.; Hovington, P.; Mauger, A.; Groult, H.; et al. An improved high-power battery with increased thermal operating range: C-LiFePO4//C-Li4Ti5O12. J. Power Sources 2012, 216, 192–200. [Google Scholar] [CrossRef]
- Julien, C.M.; Zaghib, K.; Mauger, A.; Groult, H. Enhanced electrochemical properties of LiFePO4 aspositive electrode of Li-ion batteries for HEV application. Adv. Chem. Eng. Sci. 2012, 2, 321–329. [Google Scholar] [CrossRef]
- Venkatraman, S.; Shin, Y.; Manthiram, A. Phase relationships and structural and chemical stabilities of charged Li1−xCoO2−δ and Li1−xNi0.85Co0.15O2−δ. Electrochem. Solid State Lett. 2003, 6, A9–A12. [Google Scholar] [CrossRef]
- Zaghib, K.; Dubé, J.; Dallaire, A.; Galoustov, K.; Guerfi, A.; Ramanathan, M.; Benmayza, A.; Prakash, J.; Mauger, A.; Julien, C.M. Enhanced thermal safety and high power performance of carbon-coated LiFePO4 olivine cathode for Li-ion batteries. J. Power Sources 2012, 219, 36–44. [Google Scholar] [CrossRef]
- Bang, H.J.; Joachin, H.; Yang, H.; Amine, K.; Prakash, J. Contribution of the structural changes of LiNi0.8Co0.15Al0.05O2 cathodes on the exothermic reactions in Li-ion cells. J. Electrochem. Soc. 2006, 153, A731–A737. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Julien, C.M.; Mauger, A.; Zaghib, K.; Groult, H. Comparative Issues of Cathode Materials for Li-Ion Batteries. Inorganics 2014, 2, 132-154. https://doi.org/10.3390/inorganics2010132
Julien CM, Mauger A, Zaghib K, Groult H. Comparative Issues of Cathode Materials for Li-Ion Batteries. Inorganics. 2014; 2(1):132-154. https://doi.org/10.3390/inorganics2010132
Chicago/Turabian StyleJulien, Christian M., Alain Mauger, Karim Zaghib, and Henri Groult. 2014. "Comparative Issues of Cathode Materials for Li-Ion Batteries" Inorganics 2, no. 1: 132-154. https://doi.org/10.3390/inorganics2010132