
η1:η2-P-Pyrazolylphosphaalkene Complexes of Ruthenium(0)

Department of Chemistry, University of Sussex, Brighton BN1 9QJ, UK
*
Author to whom correspondence should be addressed.
Inorganics 2016, 4(4), 30; https://doi.org/10.3390/inorganics4040030
Submission received: 9 September 2016
/
Revised: 23 September 2016
/
Accepted: 27 September 2016
/
Published: 30 September 2016
(This article belongs to the Special Issue Organophosphorus Chemistry 2016)
Abstract
:An extended range of novel ruthenium phosphaalkene complexes of the type [Ru{η1-N:η2-P,C-P(pz′)=CH(SiMe2R)}(CO)(PPh3)2] (R = Tol, C6H4CF3-p; pz′ = pzMe2, pzCF3, pzMe,CF3; R = Me, C6H4CF3-p; pz′ = pzPh) have been prepared from the respective ruthenaphosphaalkenyls [Ru{P=CH(SiMe2R)}Cl(CO)(PPh3)2] upon treatment with Lipz′. Where R = C6H4CF3-p and pz′ = pzMe2 the complex is characterized by single crystal X-ray diffraction, only the second example of such species being structurally characterized. This indicates enhanced pyramidalisation of the alkenic carbon center when compared with precedent data (R = Me, pz′ = pz) implying an enhanced Ru→π*PC contribution, which can be correlated with the greater donor power of pzMe2. This is similarly reflected in spectroscopic data that reveal significant influence of the pyrazolyl substituents upon the phosphaalkene, stronger donors imparting significantly enhanced shielding to phosphorus; in contrast, a much lesser influence if noted for the silyl substituents.
1. Introduction
After almost half a century of study, the chemistry of low-coordinate phosphorus continues to fascinate both organic and inorganic chemists alike [1,2,3,4,5,6,7,8]. Dominated by the isolobal and isoelectronic relationship between phosphorus and the “CH” fragment, the chemistry of phosphacarbons is often familiar from their carbo-centric and nitrogenous counterparts, yet they simultaneously embody appreciable dichotomy in terms of their underlying electronic and chemical nature. Nowhere is this more apparent than in the chemistry of phosphaalkenes (RP=CR′R″) and phosphaalkynes (RC≡P), which rank among the most heavily studied classes of phosphacarbon. Reactivity is in each case dominated by the high-energy π-systems, though in the case of phosphaalkenes this is often competitive with the lone-pair, which lies close—albeit marginally lower—in energy. In contrast, the phosphaalkyne lone-pair is appreciably stabilized, but can be engaged chemically under appropriately directing conditions.
The study of these compounds is, however, often complicated by an intrinsic lack of stability, which restricts the range of available substrates and necessitates some synthetic ingenuity. Such difficulties are often addressed by the imposition of steric bulk, as a means of imparting kinetic stability, which has proven particularly effective in precluding homo-oligomerisation of phosphaalkenes. The same approach has typically been cited in the development of kinetically stabilized phosphaalkynes (e.g., tBuC≡P, AdC≡P), however, in these cases the bulk is often sufficiently remote from the reactive π-system as to preclude it being the sole stabilizing influence. Moreover, even bulky phosphaalkynes, e.g., R3SiC≡P (R = Ph [9], Me [10,11]) often exhibit only limited stability, in some instances comparable to that of unencumbered systems (e.g., MeC≡P [12]). Significantly, the formally related phosphaethynyloate ion “O–C≡P−” is isolable as a sodium salt [13,14], which exhibits appreciable stability despite the lack of any steric “protection”; this fact is attributed to electronic influences, with the preferential adoption of an O=C≡P− type structure. Taken alongside computational studies of the isolated cyaphide ion (“C≡P−”), which indicate an intrinsically unstable hypovalent “[C=P]−” structure [15], this would imply that electronics serve an equally important role in imparting stability to low-coordinate phosphacarbons. Indeed, in the context of phosphaalkenes—which remain the longest and most heavily studied of the phosphacarbons—the incorporation of π-donor substituents (e.g., NR2, OR) and/or conjugate π-systems has long been employed as an alternative means of stabilization [16].
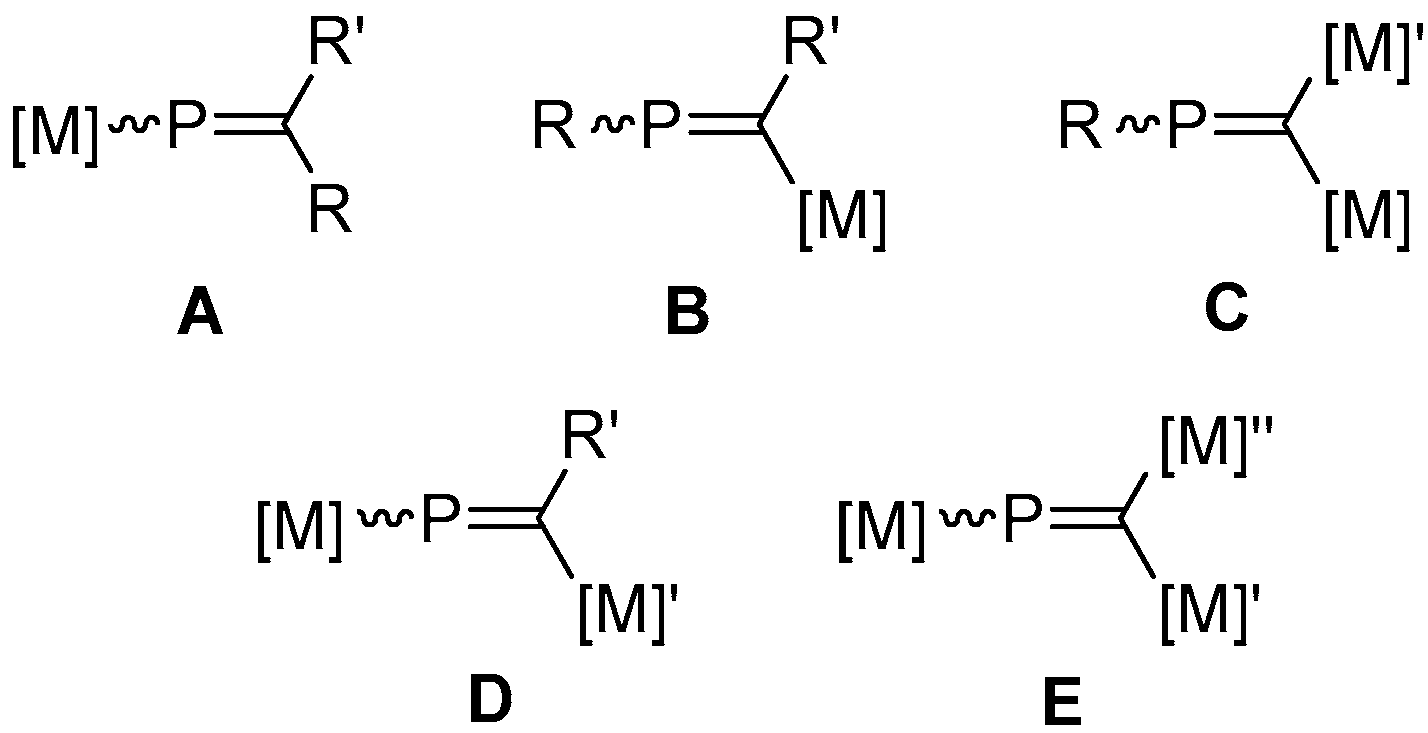
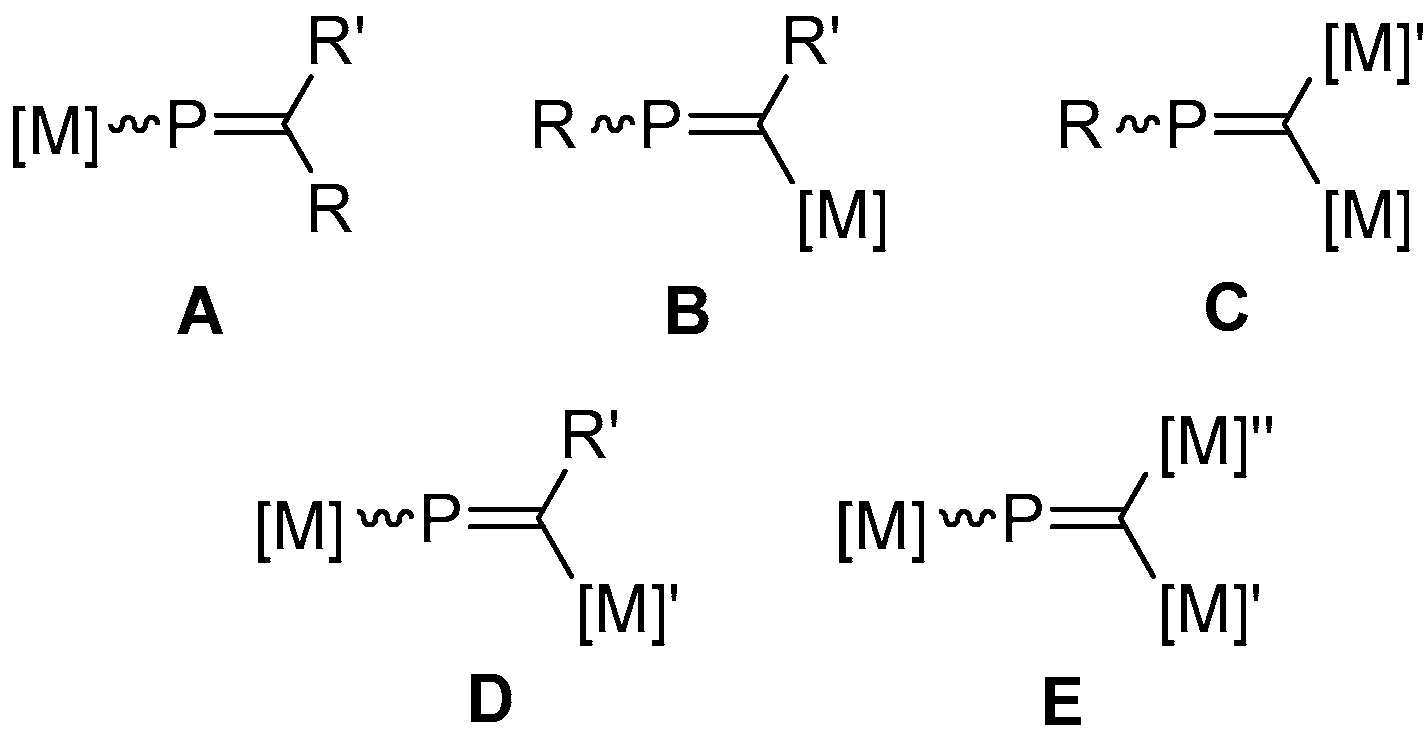
In seeking to further stabilize, and chemically exploit phosphaalkenes, the incorporation of transition metal fragments has proven to be a particularly valuable tool. Prominently, the coordination of phosphaalkenes [17] in an η1-fashion allows for selective reactivity of the π-system to be developed, by sequestering the lone-pair and precluding its competitive reaction [18]. Somewhat less extensively studied, η2-coordination—common among carbo-centric analogues—has also been employed with phosphaalkenes [19,20], discrimination between η1 and η2 being achieved through judicious selection of the metal fragment, though bridging-η1:η2 complexes have also been described [21]. Moreover, both transition metal and, to a lesser extent, main-group fragments can be incorporated as discrete substituents on the phosphaalkenic core to afford a range of metallaphosphaalkenyl complexes (Figure 1) [22,23]. First described in 1985 [24], such systems remain relatively rare, though—with the exception of type E—all possible motifs have been realized, with P-metalla- (type A) and C-metalla- (type B) systems the most heavily studied.
Recently, as part of an extended program investigating transition metal compounds featuring low-coordinate phosphacarbons with potential for conjugation [25,26,27,28], we have prepared and studied a range of ruthenaphosphaalkenyl complexes of the type [Ru{P=CH(SiMe2R)}Cl(CO)(PPh3)2] (R = Me 1a, Ph 1b, Tol 1c) [29,30]. These are prepared by hydroruthenation of phosphaalkynes R3SiC≡P, following from methodology developed initially by Hill and Jones for tBuC≡P and related systems [31,32,33], and superficially related to Nixon’s independent reduction of [(Ph3P)2Pt(η2-P≡CtBu)] with Schwartz’s reagent [34]. Notably, we have described the first structural data for ruthenaphosphaalkenyls, which demonstrate the phosphaalkenyl moieties to behave as classical 1-electron donors, within a square-based pyramidal metal coordination sphere. Significantly, this precludes, in the ground state at least, any augmenting P→Ru donation from the lone-pair (i.e., phosphavinylidene character), which is consistent with precedent reactivity studies that demonstrate a nucleophilic phosphorus center [35,36,37,38,39]; indeed, in common with these precedent reports, we have found 1a–b to engage in reaction with electrophilic fragments at phosphorus [30,40].
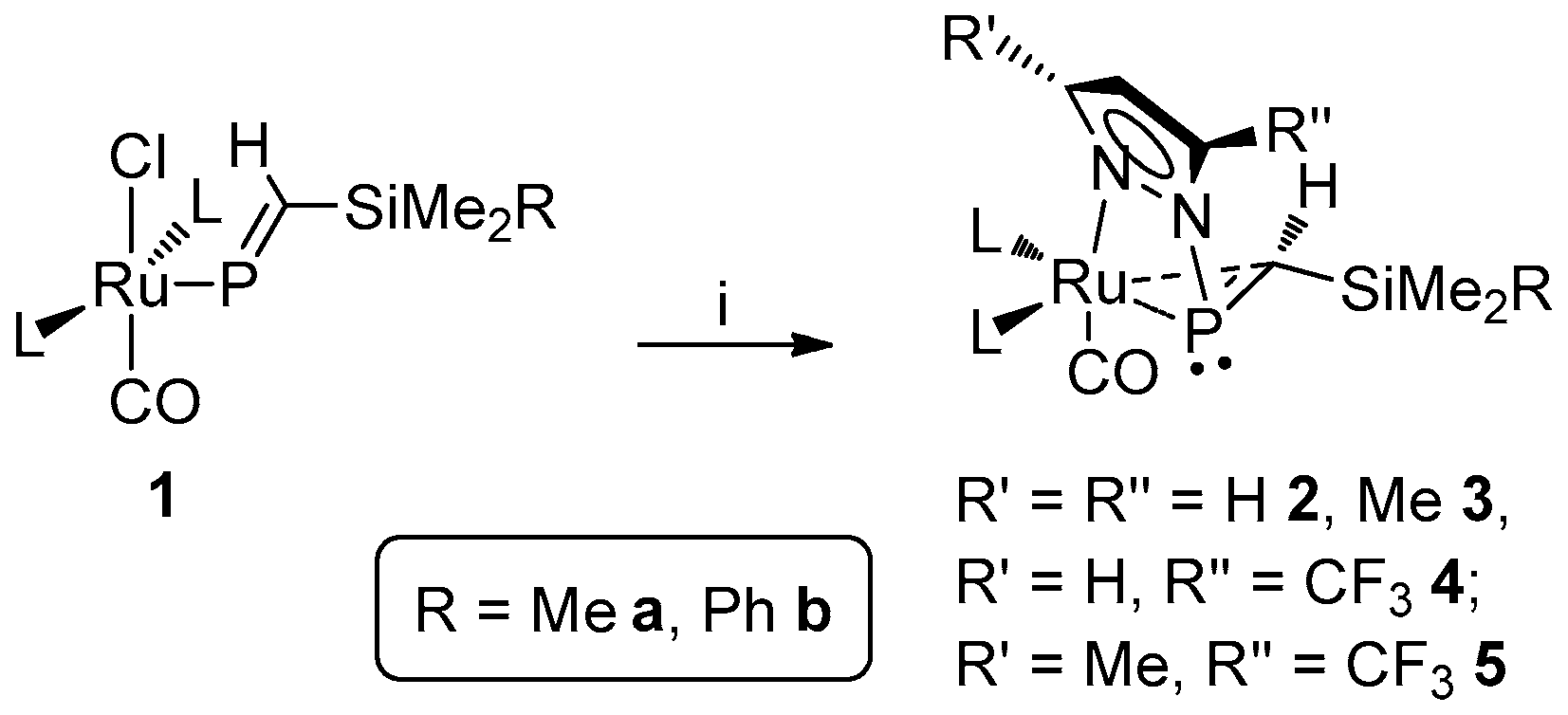
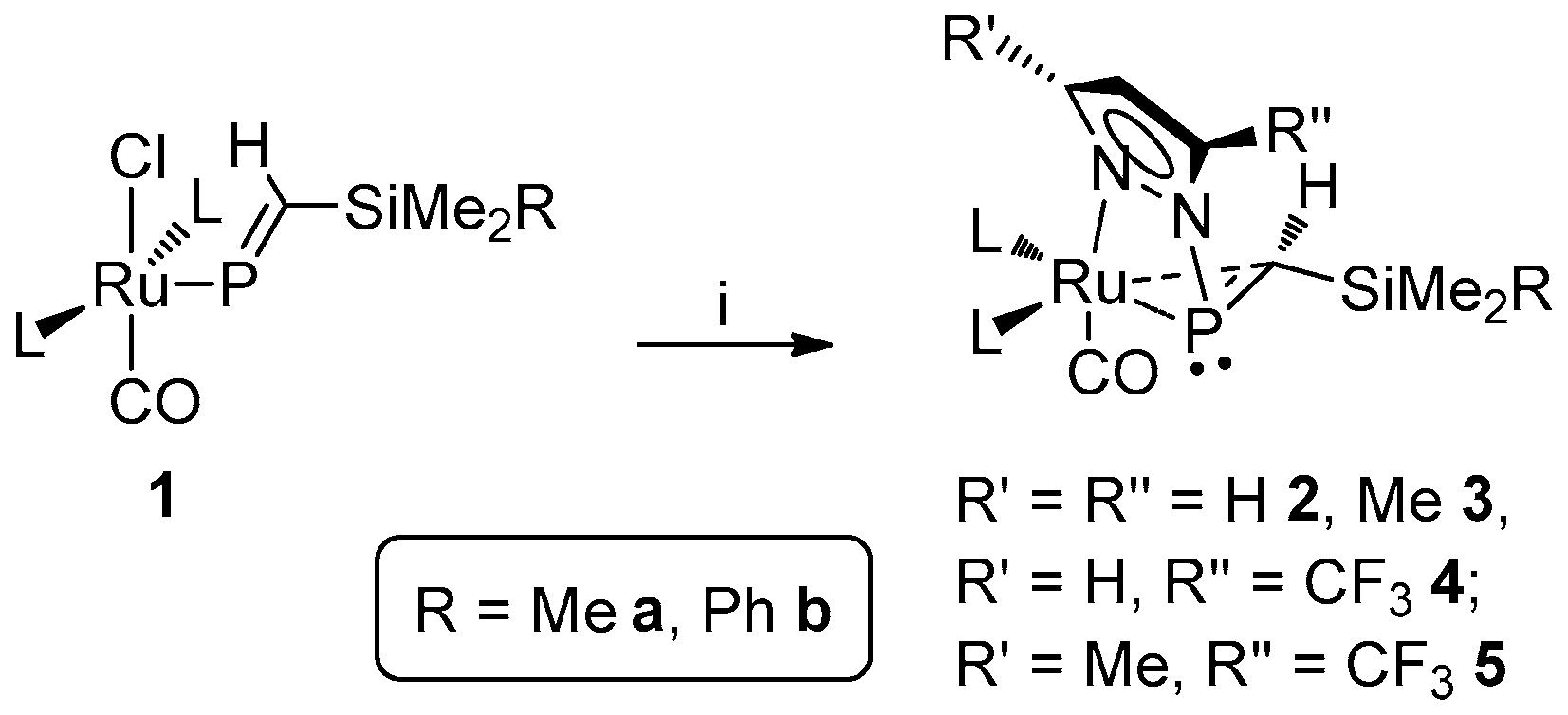
Notwithstanding, we have additionally observed an unusual reaction to proceed when 1a or 1b is exposed the lithium pyrazolates Li[pz′] (pz′ = pz, pzMe2, pzCF3, pzMe,CF3), giving rise to the unprecedented phosphaalkene complexes [Ru{η1-N:η2-P,C-P(pz′)=CH(SiMe2R)}(CO)(PPh3)2] (Scheme 1) [29,30], which feature an apparent 3-member (Ru–P–C) metallacyclic core, bridged by a pyrazolyl moiety. Also replicated using Hill’s [Ru{P=CH(tBu)}Cl(CO)(PPh3)2], this chemistry would perhaps imply ambiphilicty for the ruthenaphosphaalkenyl motif, if not directly of the alkenyl phosphorus center. On the basis of available data, we reasoned the resulting complexes to be best formulated as involving an η2-phosphaalkene moiety, in line with the Dewar–Chatt–Duncanson model, with a dominant contribution from dπ→π*(PC) retro-donation to metal-ligand binding. The precise extent of the latter might reasonably be influenced by the nature of the phosphaalkene substituents, or indeed those of the pyrazolyl moiety, resulting variations in the atom-specific parameters (e.g., NMR shielding effects) offering a potential means of quantification. In seeking to assess the influence of such factors, and thus potentially to elaborate means of control over reactivity, we report herein the synthesis and characterization of an extended range of these novel pyrazolylphosphaalkene complex.
2. Results and Discussion
2.1. Synthesis and Characterization of η2-Pyrazolylphosphaalkene Complexes
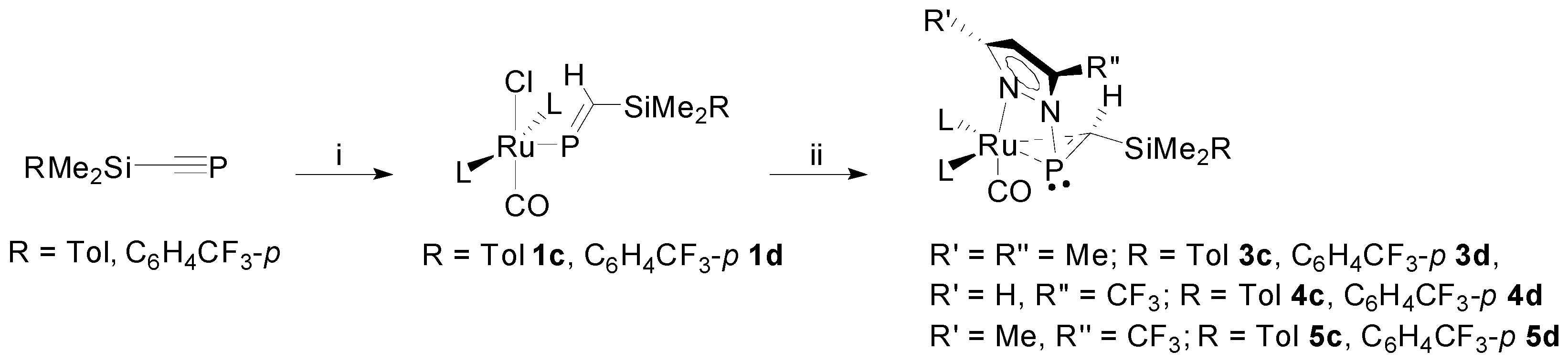
As we have previously described [29,40], the ruthenaphosphaalkenyls 1c–d were prepared via hydroruthenation of the respective phosphalkynes RMe2SiC≡P with [RuHCl(CO)(PPh3)3] (Scheme 2). Each system is readily identified on the basis of characteristic spectroscopic signatures, viz. (i) the heavily deshielded 31P{1H}-NMR resonance for the phosphaalkenyl moiety; (ii) consistently integrating resonances for two equivalent PPh3 ligands; (iii) HMBC correlations (1H–29Si; 1H–31P) confirming the presence of the silyl group and alkenic proton; (iv) infrared band for the retained carbonyl (νCO 1930–1950 cm−1), characteristic of a ruthenium(II) carbonyl complex.
The reactions of 1c–d with Li[pzMe2] each afford a single complex (3c–d respectively), which is spectroscopically comparable to those obtained similarly from 1a and 1b [29,30]. Thus, the characteristic resonances for the phosphaalkenyl and PPh3 ligands are lost, being replaced by three new, mutually coupling, resonances (1:1:1 ratio) in the region 50–30 ppm, which is commonly associated with saturated (λ3σ3) phosphorus centers. In each case, the lower frequency resonance is identified as that associated with the phosphacarbon, on the basis of a 1H–31P-HMBC correlation to the, now appreciably shielded, “CH(SiMe2R)” moiety (identified from 1H–29Si-HMBC spectra and a significant 1JPC coupling (~70 Hz)). The latter is observed at δH ~1.7 and δC ~42, which, though appreciably lower frequency than for a classical alkenyl “CH”, remains somewhat deshielded for a fully saturated “alkyl” moiety. Indeed, as was previously noted for 2a–b and 3a–b, the magnitude of the 1JCH coupling (~130–140 Hz) is intermediate between those of C2H4 (156 Hz) and CH4 (125 Hz) [41], while 1JPC are essentially unperturbed from those of the parent phosphaalkenyls, consistent with the case of a π-bound phosphaalkene. This scenario is further supported by a significant reduction in νCO for the retained carbonyl (1900–1910 cm−1), indicative of increased density at metal and, on the basis of calculated force constants, consistent with reduction of the metal to Ru(0) [42,43,44,45,46].
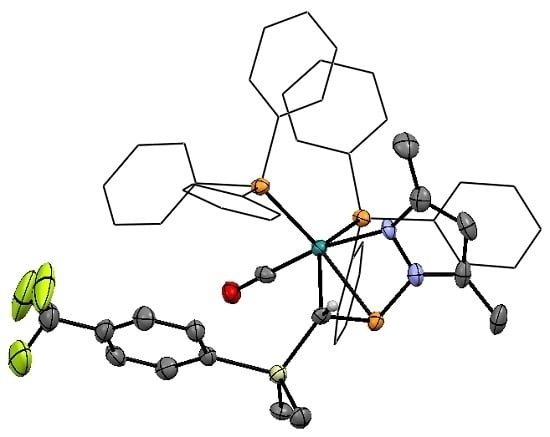
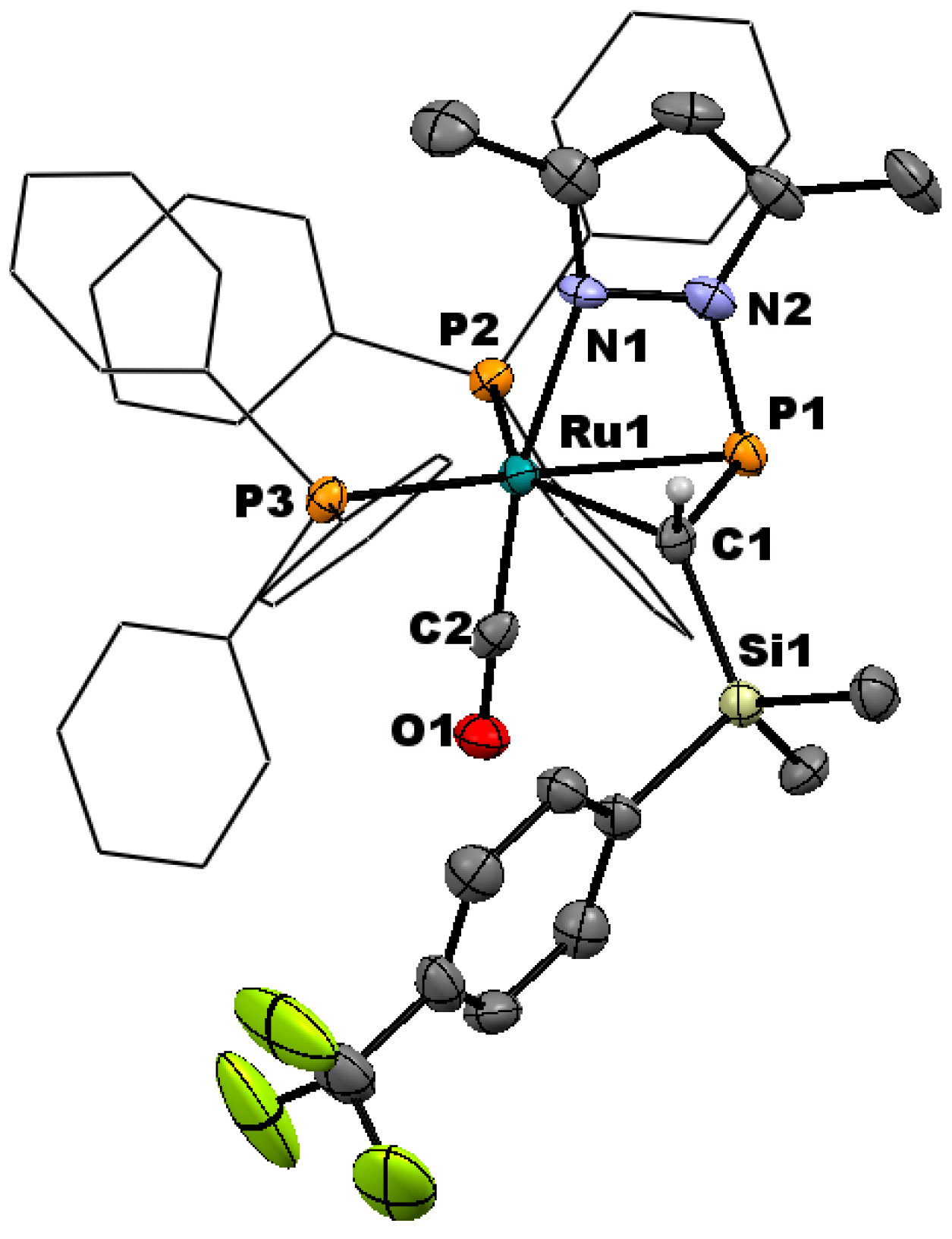
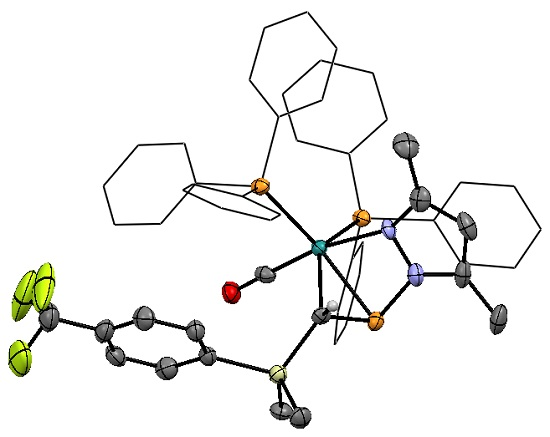
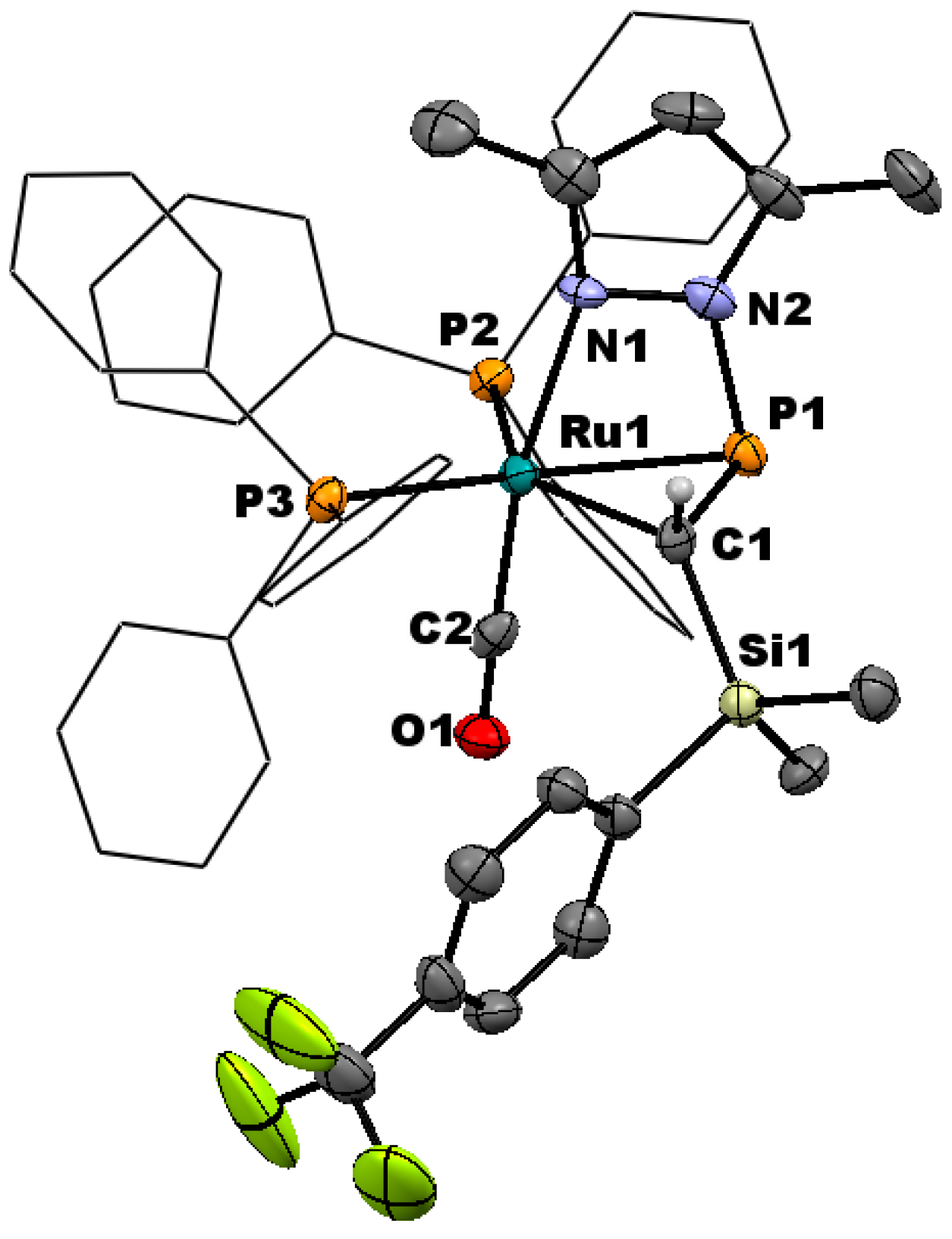
Though augmented by computational data, the direct structural characterization of η2-pyrazolylphosphaalkene complexes has been limited previously to the single crystal X-ray structure of pyrazole derivative 2a and a low-quality structure of 3a that served to confirm connectivity [30]. However upon prolonged standing at ambient temperature, a concentrated CDCl3 solution of 3d yielded X-ray quality single crystals of the chloroform solvate (Figure 2).
In common with the precedent structure of 2a, compound 3d exhibits distorted trigonal-bipyramidal geometry about ruthenium, with PPh3 ligands and the η2-phosphaalkene lying in the equatorial plane. The carbonyl adopts an axial position, along with the metal-pyrazolyl bond, the latter being marginally distorted from linearity (∠CCO–Ru–N 170.2(2)°) due to the strain of bridging the Ru–phosphacarbon linkage. Internal angles for the phosphacarbon moiety indicate partial pyramidalization of the carbon center, while the unusually tight angles about phosphorus can be attributed to the constraint of the bridging pyrazolyl group. Taken together with the P–C linkage 1.782(5) Å, which is intermediate between a single and double bond [47,48], these data are wholly consistent with our previous conclusion of η2-binding, rather than a discrete metallacyclic species. The C≡O linkage is marginally elongated relative to the structurally characterized ruthenium(II) phosphaalkenyls [30], consistent with a more electron rich metal center, and is comparable to those of other established Ru(0) carbonyls, including that previously reported for 2a. Indeed, within the bounds of uncertainty, the molecular geometry of 3d is fully comparable to that of 2a, with the exception of a marginally contracted P–N linkage (1.788(4) Å, cf. 1.809(5) Å 2a), which might reasonably be attributed to the increased donor strength of pzMe2 relative to pz, as is reflected in the spectroscopic features (vide infra). Moreover, marginally greater pyramidalization about carbon is apparent in 3d (∠Si–C–P 113.4(2)° cf. 116.7(3)° 2a), which might similarly reflect the enhanced donor strength of pzMe2 leading to an increase in retro-donation from the metal.
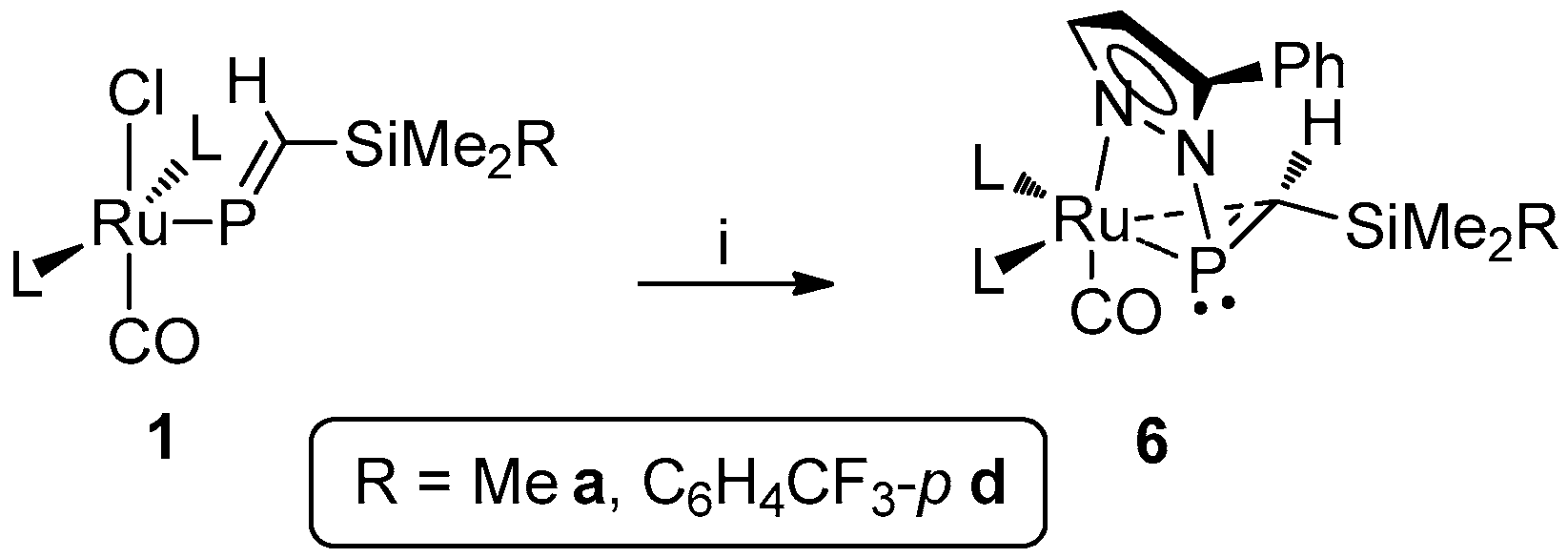
In analogous fashion to the synthesis of 3, treatment of phosphaalkenyls with Li[pz′] (pz′ = pzCF3, pzMe,CF3, pzPh) led to formation of the respective η2-phosphaalkene complexes 4c–d, 5c–d (Scheme 2), 6a and 6d (Scheme 3); the SiMe3 (a) and SiMe2Ph (b) derivatives of 4 and 5 have been previously described [29]. In contrast to 2 and 3, the asymmetrically substituted pyrazolates offer potential for positional isomerism; however, only a single isomer is observed in each case. Though non-trivial in lieu of crystallographic data, assignment of the specific isomer obtained can be achieved with recourse to spectroscopic data. Thus, for each of 4 and 5 a notable coupling (JFP ~20 Hz) is apparent between the CF3 substituent and the phosphacarbon only; this would imply proximity of the two moieties, strongly suggesting that the bulkier CF3 substituent (cf. H or Me) is more favorably positioned away from the sterically encumbered metal center. This is supported by significant deshielding of the phosphacarbon center, consistent with proximity to an electron withdrawing substituent. Indeed, we find the resonant frequency of the phosphacarbon center to be very sensitive to the pyrazolyl substituents (vide infra), most significantly so for that at the proximal site.
Assignment of the isomeric preference in 6a and 6d is a more complex undertaking, given the lack of any direct spectroscopic handle, though the same steric arguments can reasonably be applied. A more electron-withdrawing character is apparent, relative to pyrazole itself (δP 64.4 6a, cf. 58.7 2a), which conforms to expectation and is again consistent with the precedent systems, though the modest level of deshielding is inadequate to unequivocally confirm substituent proximity to the phosphacarbon. Nonetheless, we believe this to be the most likely scenario.
2.2. Spectroscopic Features and Trends
The key NMR spectroscopic data for all compounds 2, 3, 4 and 5, along with 6a and 6d are summarized in Table 1. As one would anticipate, the pzMe2 derivatives impart significant shielding to the phosphacarbon “P” center, which resonates around 25 ppm to lower frequency than for the pz systems; conversely, the presence of an electron-withdrawing CF3 substituent leads to appreciable deshielding of this site (ΔδP ~+20), though this is moderated in the pzMe,CF3 derivatives by competitive induction from the methyl. All complexes featuring a methyl substituent proximal to the metal also exhibit additional shielding of one PPh3 ligand (ΔδP −3) compared to their non-alkylated analogues; significantly, the extent of this effect is quantitatively comparable whether the second substituent is methyl or trifluoromethyl, which would imply a localized effect, and also offers further support for the isomeric assignment. The presence of a methyl proximal to the metal also leads to an appreciable decrease in the magnitude of the alkenic C–H coupling constant. This implies reduced s-character in this linkage, consistent with an increased contribution from dπ→π*(CP) retrodonation (the result of increased electron density at the metal) and thus greater pyramidalization; indeed, this concurs with structural data for 3d (vide supra).
Influence of the pyrazole upon the alkenic carbon center is also apparent, but exhibits a less defined trend; in contrast, the nature of the silyl substituent clearly holds significance, the SiMe3 derivatives being notably more deshielded than their SiMe2Ar analogues, distinctions between the latter being ill defined. This is consistent with 13C-NMR studies of vinyl- [49] and aryl-silanes [50], for which enhanced deshielding of the α-carbon follows from increasing methyl substitution at silicon, presumably reflecting the relative “inductive effect”, given the greater electronegativity of carbon relative to silicon. While carbocentric systems show the reverse shielding trend for the β-center, in the case of 2–6 enhanced deshielding of phosphorus is again noted for the SiMe3 derivatives. Though the relatively lower electronegativity of phosphorus (cf. carbon) might reasonably be invoked in accounting for this disparity, indirect effects via the synergic metal-ligand binding mode (such complexes have not been considered for the carbocentric silanes) cannot be discounted. Indeed, it is noted that analogues of 2 and 3 featuring a tert-butyl [29], rather than silyl, substituent exert a significant shielding effect upon the phosphorus center (δP 38.8 (pz); 14.2 (pzMe2)), yet deshield the α-carbon nucleus (δC 81.6 (pz), 79.8 (pzMe2)) even more effectively than does SiMe3, which would appear counter-intuitive on the basis of purely electronegativity arguments. The relative contributions of ligand→metal σ donation, and dπ→π*(CP) retrodonation for each center must thus be considered contributory, though inadequate data are currently available to quantify this.
3. Materials and Methods
3.1. General Methods
All manipulations were performed under anaerobic conditions using standard Schlenk line and glovebox (MBraun, Germany) techniques, working under an atmosphere of dry argon or dinitrogen respectively. Solvents were distilled from appropriate drying agents and stored over either molecular sieves (4 Å, for DCM and THF) or potassium mirrors. General reagents were obtained from Sigma-Aldrich (Gillingham, UK) or Fisher Scientific (Loughborough, UK) and purified by appropriate methods before use, precious metal salts were obtained from STREM (Cambridge, UK). [RuHCl(CO)(PPh3)3] [51], RMe2SiCH2PCl2 (R = Me, Ph [52], Tol [29], C6H4CF3 [39]), RMe2SiC=P [29,30,40] and [Ru{P=CH(SiMe2R)}Cl(CO)(PPh3)2] (R = Me [29,30], Ph [29], Tol, C6H4CF3-p [40]) were prepared as previously described. Unless otherwise stated, NMR spectra were recorded at 303 K on a Varian VNMRS 400 (1H 399.50 MHz, 13C 100.46 MHz, 19F 375.87, 31P 161.71 MHz, 29Si 79.37 MHz) spectrometer (Varian, Yarnton, UK). All spectra are referenced to external Me4Si, 85% H3PO4 or CFCl3 as appropriate. Carbon-13 spectra were assigned by recourse to the 2D (HSQC, HMBC) spectra; phosphaalkenic proton and silicon shifts were determined indirectly by 1H–31P and 1H–29Si correlation (HMBC). Elemental analyses were obtained by Mr. S. Boyer of the London Metropolitan University Elemental Analysis Service.
3.2. X-Ray Crystallography
Single crystal X-ray diffraction data were recorded on an Agilent Xcalibur Eos Gemini Ultra diffractometer (Agilent Technologies, Yarnton, UK) with CCD plate detector using Cu-Kα (λ = 1.54184) radiation. Structure solution and refinement were performed using SHELXS [53] and SHELXL [53] respectively, running under Olex2 [54].
3.3. Syntheses and Characterisation
[Ru{η1-N:η2-P,C-P(pzMe2)=CH(SiMe2Tol)}(CO)(PPh3)2] (3c): In a representative procedure, following literature precedent for compounds 3a and 3b [29,30], HpzMe2 (12.7 mg, 0.13 mmol) as solution in THF was treated, at ambient temperature, with a single equivalence of nBuLi (0.06 cm3, 2.5 M in hexanes) and the mixture stirred for 5 min. The resulting solution was added directly to a solution of [Ru{P=CH(SiMe2Tol)}Cl(CO)(PPh3)2] (1c, 67 mg, 0.08 mmol) at ambient temperature and the mixture stirred for 1h. Volatiles were removed under reduced pressure, then the residue extracted with CH2Cl2 and the resulting solution filtered; the solvent was removed under reduced pressure and the resulting solid dried in vacuo, before being redissolved in CDCl3 for spectroscopic analysis. 1H NMR (CDCl3): δH 0.06 (s, 3H, SiMe2), 0.15 (s, 3H, SiMe2), 0.42 (s, 3H, Pz*–Me), 1.75 (m, 1H, CHSi (1JC–H = 135.31 Hz), 1.95 (s, 3H, Pz*–Me), 2.34 (s, 3H, CH3), 5.04 (s, 1H, Pz*–H4), 7.08–7.42 (m, 35H, Ar–H). 13C{1H} NMR (CDCl3): δC −0.9 (d, 3JC–P = 7.6 Hz, SiCH3), 0.16 (d, 3JC–P = 8.5 Hz, SiCH3), 9.6 (d, 3JC–P = 5.3 Hz, Pz–CH3), 11.9 (s, Pz–CH3), 21.6 (s, CH3), 41.8 (ddd, JC–P = 4.5, 31.3, 78.6 Hz, CHSi (1JC–H = 136.3 Hz)), 105.0 (d, JC–P = 2.7 Hz, Pz*–C4), 127.6 (d, JC–P = 8.87 Ha, Ar–C), 127.8 (d, JC–P = 8.87 Hz, ArC), 128.2 (s, ArC), 128.6 (m, ArC), 128.9 (d, JC–P = 6.7 Hz, ArC), 130.4 (d, JC–P = 13.0 Hz, ArC), 133.8 m, ArC), 133.6–134.4 (m, ArC), 137.4 (s, ArC), 138.2 (d, JC–P = 30.7 Hz, ArC), 138.4 (d, JC–P = 31.8 Hz, ArC), 145.2 (s, Pz*–C5), 152.4 (s, Pz*–C3), 209.5 (br, CO). 31P{1H} NMR (CDCl3): δP 46.7 (d, JP–P = 16.9 Hz), 39.1 (dd, JP–P = 50.1, 16.7 Hz), 32.6 (d, JP–P = 50.4 Hz, P=C). 29Si{1H} NMR (CDCl3): δSi −5.3.
[Ru{η1-N:η2-P,C-P(pzMe2)=CH(SiMe2C6H4CF3-p)}(CO)(PPh3)2] (3d): In comparable fashion to 3c, from the respective phosphaalkenyl (1d). 1H NMR (CDCl3): δH −0.02 (s, 3H, SiCH3), 0.18 (s, 3H, SiCH3), 0.42 (s, 3H, Pz*–Me), 1.66 (m, 1H, CHSi (1JC–H = 135.3 Hz), 1.95 (s, 3H, Pz*–Me), 5.07 (s, 1H, Pz*–H4), 7.09–7.34 (m, 35H, Ar–H), 7.45–7.56 (m, 4H, C6H4CF3). 19F NMR (CDCl3): δF −63.06 (s). 13C{1H} NMR (CDCl3): δC −1.3 (d, 3JC–P = 8 Hz, SiCH3), 0.3 (d, 3JC–P = 8 Hz, SiCH3), 9.6 (d, 3JC–P = 5.3 Hz, Pz–CH3), 11.9 (s, Pz–CH3), 39.8 (br. (1JC–H = 135.3 Hz) CHSi), 105.2 (d, JC–P = 2.9 Hz, Pz*C4), 123.8 (q, 2JC–F = 3.9 Hz, CCF3), 130.9 (q, 1JC–F = 240.0 Hz, CF3), 127.6–129.1, 134.1–135.5, 137.2–138.1 (3 × m, PPh3, C6H4), 145.3 (d, 2JC–P = 1.5 Hz, Pz*–C5), 152.7 (s, Pz*–C3), 209.7 (br, CO). 31P{1H} NMR (CDCl3): δP 46.6 (d, 2JP–P = 16.2 Hz, PPh3), 38.6 (dd, 2JP–P = 54.4, 16.2 Hz, PPh3), 32.1 (d, 2JP–P = 51.4 Hz, P=C). 29Si{1H} NMR (CDCl3): δSi −4.5. νCO = 1913 cm−1. Anal. Found: C, 62.90; H, 5.01; N, 2.90; Calcd for C52H48F3N2OP3RuSi: C, 62.71; H, 4.86; N, 2.81. Crystal data for 3d: C53H48F3N2OP3RuSi·CHCl3, Mw = 1115.42, triclinic, P-1 (No. 2), a = 10.5494(5), b = 11.5375(6), c = 21.871(1) Å, α = 76.877(4), β = 82.084(4), γ = 85.941(4), V = 2565.46(2) Å3, Z = 2, Dc = 1.444 Mg m−3, μ(Cu-Kα) = 5.439 mm−1, T = 173(2) K, 9464 independent reflections, full-matrix F2 refinement R1 = 0.0567, wR2 = 0.1894 on 8126 independent absorption corrected reflections [I > 2σ(I); 2θmax = 143.4°], 607 parameters. The empirical absorption correction was conducted using spherical harmonics, as implemented in the SCALE3 ABSPACK scaling algorithm (CryAlisPro Version 1.171.38.41). CCDC 1502285.
[Ru{η1-N:η2-P,C-P(pzCF3)=CH(SiMe2Tol)}(CO)(PPh3)2] (4c): In comparable fashion to 3, but commencing from HpzCF3. 1H NMR (CDCl3) δH −0.1 (s, 3H, SiCH3), 0.1 (s, 3H, SiCH3), 1.90 (m, 1H, CHSi), 2.35 (br, 3H, CH3), 5.30 (s, 1H, Pz–H3), 5.57 (1 H, s, Pz–H4), 7.04–7.48 (m, 35H, C6H5). 19F NMR (CDCl3) δF −60.4 (d (4JF–P = 17.6 Hz)).13C{1H} NMR (CDCl3) δC −0.9 (d, 3JC–P = 10 Hz, SiCH3), −0.25 (d, 3JC–P = 5 Hz, SiCH3), 21.9 (s, CH3), 45.4 (ddd, JC–P = 80.5, 31.0, 4.8 Hz, (1JC–H = 134 Hz), SiCH), 105.2 (s, Pz–C4), 119.1 (q, 1JC–F 269 Hz, CF3), 128.2–128.6, 128.7–129.2, 133.7–134.4, 137.6–138.0 (4 × m, CH), 140.9 (s, Pz–C3), 210.5 (t, 2JC–P = 13 Hz, C≡O). 31P{1H} NMR (CDCl3) δP 75.0 (dq, 2JP–P = 44.5 Hz, 4JP–F = 17.50 Hz, P=CH), 47.9 (d, 2JP–P = 17.5 Hz, PPh3), 41.3 (dd, 2JP–P = 44.5, 17.5 Hz, PPh3). 29Si{1H} NMR (CDCl3) δ −5.6. Anal. Found: C, 62.10; H, 4.85; N, 2.91; Calcd for C51H46F3N2OP3RuSi: C, 62.39; H, 4.72; N, 2.85.
[Ru{η1-N:η2-P,C-P(pzCF3)=CH(SiMe2C6H4CF3-p)}(CO)(PPh3)2] (4d): As for 4c, commencing from 1d. 1H NMR (CDCl3) δ −0.06 (s, 3H, SiCH3), 0.14 (s, 3H, SiCH3), 1.85 (m, 1H, CHSi), 5.32 (s, 1H, Pz–H3), 5.60 (s, 1H, Pz–H4), 7.05–7.39 (m, 30H, C6H5), 7.5 (d, 2H, JH–F = 8.0 Hz, C6H4), 7.63 (d, 2H, JH–F = 7.6 Hz, C6H4). 19F NMR (CDCl3) δ −60.1 (d (4JF–P = 19.4 Hz)), −62.7 (s, C6H4CF3). 13C{1H} NMR (CDCl3) δ 1.2 (s, SiCH3), 43.8 (br. (1JC–H = 134 Hz), SiCH), 104.9 (s, Pz–C4), 128.0 (t, JC–P = 4.3 Hz, CH), 128.7 (d, JC–P = 7.3 Hz, CH), 128.9 (s, CH), 129.5 (s, CH), 132.2–132.6, 133.5–134.1 (2 × m, CH), 142.8 (s, Pz–C5), 145.4 (br. Pz–C3). CF3 resonances obscured by aromatics. 31P{1H} NMR (CDCl3) δ 37.8 (dd, 2JP–P = 47.4, 15.8 Hz), 47.1 (d, 2JP–P = 15.8 Hz, PPh3), 62.0 (dq, 2JP–P = 47.4 Hz, 4JP–F = 19.40 Hz, P=CH). 29Si{1H} NMR (CDCl3) δ −4.2. νCO = 1913 cm−1.
[Ru{η1-N:η2-P,C-P(pzMe,CF3)=CH(SiMe2Tol)}(CO)(PPh3)2] (5c): As for 4c, but commencing from HpzMe,CF3. 1H NMR (CDCl3): δH −0.03 (s, 3H, SiCH3), 0.17 (s, 3H, SiCH3), 0.55 (s, 3H, Pz–CH3), 1.97 (br, 1H, CHSi), 2.36 (s, 3H, CH3), 5.52 (br. Pz–H4), 7.12–7.50 (m, 34 H, 2 × PPh3, C6H4). 19F NMR (CDCl3): δF −59.9 (d, JF–P = 19.4 Hz). 13C{1H} NMR (CDCl3): δC −1.1 (d, 3JC–P = 7.8 Hz, SiCH3), 0.14 (d, 3JC–P = 8.8 Hz, SiCH3), 11.85 (s, Pz–CH3), 21.6 (m, CH3), 42.1 (br. ddd, JC–P = 28.2, 78.8 Hz, 1JC–H = 137 Hz, SiCH), 105.7 (br m, Pz–C4), 119.5 (br, 1JC–F = 269 Hz, CF3), 127.6−130.3 (m, PPh3), 133.5–135.1 (m, PPh3, C6H4) 137.6 (dd, J = 1.49, 30.1 Hz, Pz–C5), 152.6 (br, Pz–C3), 209.0 (m, CO). 31P{1H} NMR (CDCl3): δP 61.62 (dq, 2JP–P = 45.7, 4JP–F = 19.3 Hz, P=C), 47.2 (d, 2JP–P = 16.4 Hz, PPh3), 38.4 (ddd, 2JP–P = 45.7, 16.4, JP–F = 1.4 Hz, PPh3). 29Si{1H} NMR (CDCl3): δSi −5.7. νCO = 1918 cm−1. Anal. Found: C, 62.42; H, 5.05; N, 2.89; Calcd for C52H48F3N2OP3RuSi: C, 62.71; H, 4.86; N, 2.81.
[Ru{η1-N:η2-P,C-P(pzMe,CF3)=CH(SiMe2C6H4CF3-p)}(CO)(PPh3)2] (5d): As for 5c, but commencing from 1d. 1H NMR (CDCl3): δH 0.01 (s, 3H, SiCH3), 0.21 (s, 3H, SiCH3), 0.56 (s, 3H, Pz–CH3) 1.85 (br, 1H, CHSi), 5.54 (s, 1H, Pz–H4), 7.13–7.28, 7.37–7.46, 7.47–7.53, 7.58–7.67 (4 × m, 34H, PPh3, C6H4). 19F NMR (CDCl3): δF −59.9 (d, J = 20.3 Hz, Pz–CF3), −62.7 (s, CF3-p). 13C{1H} NMR (CDCl3): δC −1.4 (d, 3JC–P = 7.4 Hz, SiCH3), 0.16 (d, 3JC–P = 8.0 Hz, SiCH3), 11.9 (s, Pz–CH3), 40.7 (ddd, JC–P = 32.9, 79.8, 5.0 Hz, 1JC–H = 137 Hz, SiCH), 105.9 (br m, Pz–C4), 119.4 (q, 1JC–F = 270 Hz, CF3), 124.6 (q, 1JC–F = 272 Hz, CF3), 127.6–128.0 (m, CH), 128.6 (d, J = 7 Hz, CH), 128.5–129.1 (m, CH), 133.6–134.3 (m, CH), 137.6 (dd, J = 1.3, 3.2 Hz, Pz–C5), 152.8 (br, Pz–C3), 209.1 (m, CO). 31P{1H} NMR (CDCl3): δP 61.99 (dq, 2JP–P = 57.0, 4JP–F = 18.8 Hz, PPh3), 47.1 (d, 2JP–P = 15.8 Hz, PPh3), 37.8 (dd, 2JP–P = 47.0, 15.9, P=C). 29Si{1H} NMR (CDCl3): δSi −4.16. νCO 1917 cm−1.
[Ru{η1-N:η2-P,C-P(pzPh)=CH(SiMe3)}(CO)(PPh3)2] (6a): In a comparable fashion, commencing from 1a and HpzPh. 1H NMR (CDCl3): δH 0.15 (s, 9H, Si(CH3)3), 1.74 (m, 1H, CHSi), 5.39 (s, 1H, Pz–H3), 5.56 (br. Pz–H4), 7.05–7.55 (m, 35H, PPh3, C6H5,). 13C{1H} NMR (CDCl3): δC 1.4 (d, 3JC–P = 5.5 Hz, Si(CH3)), 47.5 (ddd, 2JP–P = 4.4, 31.0, 80.2 Hz, 1JC–H = 135.9 Hz, SiCH), 103.3 (d, JC–P = 3.2 Hz, PzC4), 127.4 (d, JC–P = 6.6 Hz, CH), 127.7 (d, JC–P = 8.7Hz, CH), 128.1 (d, JC–P = 8.6 Hz, CH), 128.6 (s, CH), 128.8 (d, JC–P = 19.0 Hz, CH), 129.1 (s, CH), 133.9 (m, CH), 137.3 (d, JC–P = 9.7 Hz, ipso-C), 137.7 (d, JC–P = 32.0 Hz, ipso-C), 138.0 (d, JC–P = 32.0 Hz, ipso-CH), 141.0 (s, Pz–C3), 147.7 (br, Pz–C5), 210.7 (m, CO). 31P{1H} NMR (CDCl3): δP 64.4 (d, JP–P = 46.2 Hz, P=C), 47.4 (d, JP–P = 18.5 Hz, PPh3), 41.8 (dd, JP–P = 46.2, 18.4 Hz, PPh3). 29Si{1H} NMR (CDCl3): δSi −0.5. νCO = 1908 cm−1. Anal. Found: C, 65.71; H, 5.18; N, 3.03; Calcd for C46H47N2OP3RuSi: C, 65.62; H, 5.33; N, 2.99.
[Ru{η1-N:η2-P,C-P(pzPh)=CH(SiMe2C6H4CF3-p)}(CO)(PPh3)2] (6d): As for 6a, but commencing from 1d. 1H NMR (CDCl3): δH −0.03 (s, 3H, SiCH3), 0.27 (s, 3H, SiCH3), 1.86 (m, 1H, CHSi), 5.56 (1H, s, Pz–H4), 5.60 (s, 1H, Pz–H5), 7.11–7.68 (m, 39 H, 2 × PPh3, C6H5, C6H4). 19F NMR (CDCl3): δF 62.62 (s, CF3). 13C{1H} NMR (CDCl3): δC −1.3 (d, 3JC–P = 8 Hz, SiCH3), −0.9 (d, 3JC–P = 5 Hz, SiCH3), 45.6 (ddd, JC–P = 5.1, 31.4, 79.5 Hz, 1JC–H = 133.9 Hz, SiCH), 103.5 (d, JC–P = 3.3 Hz, Pz–C4), 124.5 (q, 1JC–F = 272 Hz, CF3), 127.4–130.2, 133.7–134.5 (2 × m, PPh3, C6H5, C6H4), 141.2 (s, Pz–C3), 147.9 (br. Pz–C5), 210.5 (m, CO). 31P{1H} NMR (CDCl3): δP 60.6 (d, 2JP–P = 46.9 Hz, P=C), 47.7 (d, 2JP–P = 17.4 Hz, PPh3), 41.3 (dd, 2JP–P = 46.9, 17.4 Hz, PPh3). 29Si{1H} NMR (CDCl3): δSi −4.9. νCO = 1912. Anal. Found: C, 64.02; H, 4.69; N, 2.71; Calcd for C53H48F3N2OP3RuSi: C, 64.11; H, 4.77; N, 2.80.
4. Conclusions
We have described the synthesis of an extended range of pyrazolyl-bridged η2-phosphaalkene complexes of ruthenium(0), obtained by the reaction of pyrazolates with ruthenaphosphaalkenyl complexes. Spectroscopic and structural data suggest the nature of the pyrazolyl substituents in the 3/5 positions has a significant influence on the nature of the phosphaalkene fragment, which is a balance of direct shielding/deshielding of the phosphorus center, and an indirect influence resulting from donation to the metal, mediated through the extent of metal-ligand retro-donation. In contrast, the nature of the silyl substituents exerts a much smaller, albeit still noticeable, influence. Presently, the available data pool is inadequate to formulate an unequivocally quantitative description of these effects, which are the subject of on-going investigations.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/4/4/30/s1, crystal data for compound 3d in cif format (CCDC 1502285); supplementary file containing illustrative 13C and 31P NMR spectra for compounds 3c (Figures S1 and S2), 4c (Figures S3 and S4), 5d (Figures S5 and S6), 6a (Figures S7 and S8).
Acknowledgments
We thank the Royal Society and University of Sussex (studentship to Victoria K. Greenacre) for financial support. Ian R. Crossley gratefully acknowledges the award of a Royal Society University Research Fellowship. We thank Dr. L. Higham (guest editor) for the invitation to contribute to this issue.
Author Contributions
Victoria K. Greenacre identified the specific targets and conducted all experimental work and crystallographic determinations. Ian R. Crossley conceived and led the over-arching research project, contributed to interpretation and wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Waterman, R. Phosphorus chemistry: Discoveries and advances. Dalton Trans. 2016, 45, 1801–1803. [Google Scholar] [CrossRef] [PubMed]
- Mathey, F. Phospha-Organic Chemistry: Panorama and Perspectives. Angew. Chem. Int. Ed. 2003, 42, 1578–1604. [Google Scholar] [CrossRef] [PubMed]
- Dillon, K.B.; Mathey, F.; Nixon, J.F. Phosphorus: The Carbon Copy; Wiley: Chichester, UK, 1998. [Google Scholar]
- Nixon, J.F. Recent developments in the organometallic chemistry of phospha-alkynes, RC-P. Coord. Chem. Rev. 1995, 145, 201–258. [Google Scholar]
- Appel, R. Multiple Bonds and Low Coordination in Phosphorus Chemistry; Regitz, M., Scherer, O.J., Eds.; Thieme: Stutgart, Germany, 1990. [Google Scholar]
- Markovski, L.N.; Romanenko, V.D. Phosphaalkynes and phosphaalkenes. Tetrahedron 1989, 45, 6019–6090. [Google Scholar] [CrossRef]
- Nixon, J.F. Coordination chemistry of compounds containing phosphorus–carbon multiple bonds. Chem. Rev. 1988, 88, 1327–1362. [Google Scholar] [CrossRef]
- Appel, R.; Knoll, F.; Ruppert, I. Phospha-alkenes and Phospha-alkynes, Genesis and Properties of the (p–p) π-Multiple Bond. Angew. Chem. Int. Ed. Engl. 1981, 20, 731–744. [Google Scholar] [CrossRef]
- Cordaro, J.G.; Stein, D.; Rüegger, H.; Grützmacher, H. Making the True “CP” Ligand. Angew. Chem. Int. Ed. 2006, 45, 6159–6162. [Google Scholar] [CrossRef] [PubMed]
- Mansell, S.M.; Green, M.; Kilby, R.J.; Murry, M.; Russell, C.A. Facile preparation of trimethylsilylphosphaalkyne and its conversion to polyphospholide anions. C. R. Chim. 2010, 13, 1073–1081. [Google Scholar] [CrossRef]
- Mansell, S.M.; Green, M.; Russell, C.A. Coordination chemistry of trimethylsilylphosphaalkyne: A phosphaalkyne bearing a reactive substituent. Dalton Trans. 2012, 41, 14360–14368. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Schulten, C.; Stasch, A. The first complexes and cyclodimerisations of methylphosphaalkyne (P ≡CMe). Dalton Trans. 2006, 31, 3733–3735. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Alidori, S.; Puschmann, F.F.; Santiso-Quinones, G.; Benkö, Z.; Li, Z.; Becker, G.; Grützmacher, H.-F.; Grützmacher, H. Sodium Phosphaethynolate as a Building Block for Heterocycles. Angew. Chem. Int. Ed. 2014, 53, 1641–1645. [Google Scholar] [CrossRef] [PubMed]
- Jupp, A.R.; Goichoechea, J.M. The 2-Phosphaethynolate Anion: A Convenient Synthesis and [2+2] Cycloaddition Chemistry. Angew. Chem. Int. Ed. 2013, 52, 10064–10067. [Google Scholar] [CrossRef] [PubMed]
- Mo, O.; Yanez, M.; Guillemin, J.-C.; Riague, E.H.; Gal, J.-F.; Maria, P.-C.; Poliart, C.D. The Gas-Phase Acidity of HCP, CH3CP, HCAs, and CH3CAs: An Unexpected Enhanced Acidity of the Methyl Group. Chem. Eur. J. 2002, 8, 4919–4924. [Google Scholar] [CrossRef]
- Markovski, L.N.; Romanenko, V.D. Phosphaalkynes and phosphaalkenes. Tetrahedron 1989, 45, 6019–6090. [Google Scholar] [CrossRef]
- Le Floch, P. Phosphaalkene, phospholyl and phosphinine ligands: New tools in coordination chemistry and catalysis. Coord. Chem. Rev. 2006, 250, 627–681. [Google Scholar] [CrossRef]
- De Vaumes, R.; Marinetti, A.; Mathey, F. Catalytic hydrogenation of the phosphorus–carbon double bond in phosphaalkene complexes. J. Organomet. Chem. 1991, 413, 411–417. [Google Scholar] [CrossRef]
- Van der Knaap, T.A.; Bickelhaupt, F.; Krasykamp, J.G.; van Koten, G.; Bernards, J.P.C.; Edzes, H.T.; Veeman, W.S.; de Boer, E.; Baerends, E.J. The η1- and η2-coordination in a (phosphaalkene)platinum(0) complex. Organometallics 1984, 3, 1804–1811. [Google Scholar] [CrossRef]
- Kraajkamp, J.G.; van Koten, G.; van der Knaap, T.A.; Bickelhaupt, F.; Stam, C.H. Influence of steric factors on the coordination mode (η1 or η2) of phosphaalkenes to zerovalent Pt(0)L2 centers. X-ray structure of bis(triphenylphosphine)[(2,6-dimethylphenyl)-9-fluorenylidenephosphine]platinum(0)-toluene. Organometallics 1986, 5, 2014–2020. [Google Scholar] [CrossRef]
- Appel, R.; Casser, C.; Knoch, F. Über niederkoordinierte phosphorverbindungen: XXXX. 2,4,6-tri-t-Butylphenylmethylenphosphan,ein vielseitiger ligand in übergangsmetall-komplexen. J. Organomet. Chem. 1985, 293, 213–217. [Google Scholar]
- Weber, L. Recent developments in the chemistry of metallophosphaalkenes. Coord. Chem. Rev. 2005, 249, 741–763. [Google Scholar] [CrossRef]
- Weber, L. Metallophosphaalkenes—from Exotics to Versatile Building Blocks in Preparative Chemistry. Angew. Chem. Int. Ed. Engl. 1996, 35, 271–288. [Google Scholar] [CrossRef]
- Weber, L.; Reizig, K.; Boese, R.; Polk, M. Z-[(η5-C5H5)(CO)2Fe-P=C(OSiMe3)(tBu)], a Phosphaalkenyl-Complex with FeP Single Bond. Angew. Chem. Int. Ed. Engl. 1985, 24, 604–605. [Google Scholar] [CrossRef]
- Saunders, A.J.; Crossley, I.R.; Roe, S.M. Aroylphosphanes: Base-Free Synthesis and Their Coordination Chemistry with Platinum-Group Metals. Eur. J. Inorg. Chem. 2016, 25, 4076–4082. [Google Scholar] [CrossRef]
- Saundersa, A.J.; Crossley, I.R. Synthesis of 3-stannyl and 3-silyl propargyl phosphanes and the formation of a phosphinoallene. Dalton Trans. 2016, 45, 2148–2155. [Google Scholar] [CrossRef] [PubMed]
- Trathen, N.; Leech, M.C.; Crossley, I.R.; Greenacre, V.K.; Roe, S.M. Synthesis and electronic structure of the first cyaphide-alkynyl complexes. Dalton Trans. 2014, 43, 9004–9007. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.J.; Crossley, I.R.; Coles, M.P.; Roe, S.M. Facile self-assembly of the first diphosphametacyclophane. Chem. Commun. 2012, 48, 5766–5768. [Google Scholar] [CrossRef] [PubMed]
- Greenacre, V.K.; Trathen, N.; Crossley, I.R. Ruthenaphosphaalkenyls: Synthesis, Structures, and Their Conversion to η2-Phosphaalkene Complexes. Organometallics 2015, 34, 2533–2542. [Google Scholar] [CrossRef]
- Trathen, N.; Greenacre, V.K.; Crossley, I.R.; Roe, S.M. Ambiphilic Reactivity of a Ruthenaphosphaalkenyl: Synthesis of P-Pyrazolylphosphaalkene Complexes of Ruthenium(0). Organometallics 2013, 32, 2501–2504. [Google Scholar] [CrossRef]
- Bedford, R.B.; Hill, A.F.; Jones, C. Phosphaalkyne Hydrometalation: Synthesis of [RuCl(P=CHtBu)(CO)(PPh3)2]. Angew. Chem. Int. Ed. Engl. 1996, 35, 547–549. [Google Scholar] [CrossRef]
- Bedford, R.B.; Hill, A.F.; Jones, C.; White, A.J.P.; Williams, D.J.; Wilton-Ely, J.D.E.T. Phosphaalkyne Hydrometalation: Synthesis and Reactivity of the Complexes [Ru(PCHCMe3)Cl(CA)(PPh3)2] (A = O, S). Organometallics 1998, 17, 4744–4753. [Google Scholar] [CrossRef]
- Brym, M.; Jones, C. Synthesis, characterisation and reactivity of the first diphosphaalkyne. Dalton Trans. 2003, 3665–3667. [Google Scholar] [CrossRef]
- Benuvenutti, M.H.A.; Cenac, N.; Nixon, J.F. Hydrozirconation of an η2-ligated phosphaalkyne. A new synthetic route to η2-ligated phosphaalkenes. Chem. Commun. 1997, 1327–1328. [Google Scholar] [CrossRef]
- Bedford, R.B.; Hibbs, D.E.; Hill, A.F.; Hursthouse, M.B.; Abdul Malik, K.M.; Jones, C. Complete metal-mediated reduction of the triple bond of a phosphaalkyne: X-ray structure of [Ru(PHFCH2But)Cl(CO)(CNC6H3Me2-2,6)(PPh3)2]BF4·CH2Cl2. Chem. Commun. 1996, 1895–1896. [Google Scholar] [CrossRef]
- Bedford, R.B.; Hill, A.F.; Jones, C.; White, A.J.P.; Williams, D.J.; Wilton-Ely, J.D.E.T. Novel syntheses of heterodinuclear phosphaalkenyl complexes: X-ray structure of [Ru{P(AuPPh3)=CHBut}Cl2(CO)(PPh3)2]. Chem. Commun. 1997, 179–180. [Google Scholar] [CrossRef]
- Hill, A.F.; Jones, C.; White, A.J.P.; Williams, D.J.; Wilton-Ely, J.D.E.T. A metallacyclic λ5-phosphaalkenyl complex of ruthenium(II): X-ray structure of [Ru{κ2-P(=O)CButC(=O)}(CNBut)2(PPh3)2]. Chem. Commun. 1998, 367–368. [Google Scholar] [CrossRef]
- Bedford, R.B.; Hill, A.F.; Jones, C.; White, A.J.P.; Williams, D.J.; Wilton-Ely, J.D.E.T. Co-ordinative activation of phosphaalkynes: Methyl neopentylidene phosphorane complexes of ruthenium(II); crystal structure of [Ru(MeP=CHBut)Cl(I)(CO)(PPh3)2]. J. Chem. Soc. Dalton Trans. 1997, 139–140. [Google Scholar] [CrossRef]
- Hill, A.F.; Jones, C.; White, A.J.P.; Williams, D.J.; Wilton-Ely, J.D.E.T. Mercuriophosphaalkene-P complexes: Crystal structure of [Ru{P(=CHBut)HgC5H4Fe(η-C5H5)}Cl2(CO)(PPh3)2]. J. Chem. Soc. Dalton Trans. 1998, 1419–1420. [Google Scholar] [CrossRef]
- Greenacre, V.K.; Crossley, I.R. Hydrochlorination of Ruthenaphosphaalkenyls: Unexpectedly Facile Access to Alkylchlorohydrophosphane Complexes. Organometallics 2016. Submitted. [Google Scholar]
- Maciel, G.E.; McIver, J.W., Jr.; Ostlund, N.S.; Pople, J.A. Approximate self-consistent molecular orbital theory of nuclear spin coupling. I. Directly bonded carbon–hydrogen coupling constants. J. Am. Chem. Soc. 1970, 92, 1–11. [Google Scholar] [CrossRef]
- Bohanna, C.; Esteruelas, M.A.; Lahoz, F.J.; Onate, E.; Oro, L.A.; Sola, E. Synthesis of Butadiene-Osmium(0) and -Ruthenium(0) Complexes by Reductive Carbon–Carbon Coupling of Two Alkenyl Fragments. Organometallics 1995, 14, 4825–4831. [Google Scholar] [CrossRef]
- Bolton, P.D.; Grellier, M.; Vautravers, N.; Vendier, L.; Sabo-Etienne, S. Access to Ruthenium(0) Carbonyl Complexes via Dehydrogenation of a Tricyclopentylphosphine Ligand and Decarbonylation of Alcohols. Organometallics 2008, 27, 5088–5093. [Google Scholar] [CrossRef]
- Hill, A.F.; Owen, G.R.; White, A.J.P.; Williams, D.J. The Sting of the Scorpion: A Metallaboratrane. Angew. Chem. Int. Ed. 1999, 38, 2759–2761. [Google Scholar] [CrossRef]
- Christian, D.F.; Roper, W.R. Isocyanide complexes of zerovalent ruthenium. Proton addition to a transition-metal base offering alternative base sites. J. Chem. Soc. D Chem. Commun. 1971, 1271–1272. [Google Scholar] [CrossRef]
- Herberhold, M.; Hill, A.F. The coordination chemistry of iminooxosulphuranes VI. Factors affecting coordination geometry in complexes of tosyliminooxosulphurane. J. Organomet. Chem. 1990, 395, 195–206. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. II 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Rakita, P.E.; Worsham, L.S. 13C NMR studies of organosilanes: IV. Vinyl- and allyl-silanes. J. Organomet. Chem. 1977, 139, 135–142. [Google Scholar] [CrossRef]
- Rakita, P.E.; Srebro, J.P.; Worsham, L.S. 13C NMR studies of organosilanes: I. Substituent-chemical-shift parameters for phenylsilanes. J. Organomet. Chem. 1976, 104, 27–37. [Google Scholar] [CrossRef]
- Boniface, S.M.; Clark, G.R.; Collins, T.J.; Roper, W.R. Preparation of octahedral hydrido-aquo-ruthenium(II) complexes, and structural characterisation of hydridoaquodicarbonylbis(triphenylphosphine)-ruthenium(II) tetrafluoroborate. J. Organomet. Chem. 1981, 206, 109–117. [Google Scholar] [CrossRef]
- Averre, C.E.; Coles, M.P.; Crossley, I.R.; Day, I.J. The open-chain triphosphanes RMe2SiCH2P(PR′2)2 (R = Me, Ph; R′ = SiMe3, Cy, Ph). Dalton Trans. 2012, 41, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K. Puschmann, H. OLEX2: A complete structure solution, refinement and analysis. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Figure 1.
Metallaphosphaalkenyl motifs.
Scheme 1.
Synthesis of [Ru{η1-N:η2-P,C-P(pz′)=CH(SiMe2R)}(CO)(PPh3)2] (2–5) [29,30] (L = PPh3). Reagents and conditions: (i) Lipz′ (pz′ = pz, pzMe2, pzCF3, pzMe,CF3), THF.
Scheme 2.
Synthesis of [Ru{P=CH(SiMe2R)}Cl(CO)(PPh3)2] (R = Tol 1c, C6H4CF3-p 1d) and their conversion to [Ru{η1-N:η2-P,C-P(pz′)=CH(SiMe2R)}(CO)(PPh3)2] (2–5) (L = PPh3). Reagents and conditions: (i) [RuHCl(CO)(PPh3)3], CH2Cl2, 1 h.; (ii) Lipz′ (pz′ = pzMe2, pzCF3, pzMe,CF3), THF, 1 h.
Scheme 2.
Synthesis of [Ru{P=CH(SiMe2R)}Cl(CO)(PPh3)2] (R = Tol 1c, C6H4CF3-p 1d) and their conversion to [Ru{η1-N:η2-P,C-P(pz′)=CH(SiMe2R)}(CO)(PPh3)2] (2–5) (L = PPh3). Reagents and conditions: (i) [RuHCl(CO)(PPh3)3], CH2Cl2, 1 h.; (ii) Lipz′ (pz′ = pzMe2, pzCF3, pzMe,CF3), THF, 1 h.

Figure 2.
Molecular structure of [Ru{η1-N:η2-P,C-P(pzMe2)=CH(SiMe2C6H4CF3-p)}(CO)(PPh3)2] (3d) in crystals of the chloroform solvate, with thermal ellipsoids at the 50% probability level. Ancillary phenyl rings are reduced, and hydrogen atoms omitted for clarity. Selected bond distance (Å) and angles (°): Ru–P1 2.381(1), Ru–P2 2.359(1), Ru–P3 2.369(1), Ru–C1 2.211(4), Ru–C2 1.831(5), Ru–N1 2.229(3), P1–C1 1.782(5), P1–N2 1.778(4), C1–Si 1.860(4), C2–O 1.155(6); Ru–C1–Si 128.2(2), Ru–C1–P1 72.3(2), Ru–P1–C1 62.2(1), Ru–P1–N2 82.5(1), Ru–C1–H1 112.1(4), P1–C1–Si 113.4(2), P1–C1–H1 112.1(4), C1–P1–N2 96.1(2), P1–Ru–P2 93.6(1), P2–Ru–P3 114.0(1), P3–Ru–C1 107.1(1), P1–Ru–C1 43.5(1), P2–Ru–C2 91.1(1), P3–Ru–C2 89.1(2), C2–Ru–N1 170.2(2).
Figure 2.
Molecular structure of [Ru{η1-N:η2-P,C-P(pzMe2)=CH(SiMe2C6H4CF3-p)}(CO)(PPh3)2] (3d) in crystals of the chloroform solvate, with thermal ellipsoids at the 50% probability level. Ancillary phenyl rings are reduced, and hydrogen atoms omitted for clarity. Selected bond distance (Å) and angles (°): Ru–P1 2.381(1), Ru–P2 2.359(1), Ru–P3 2.369(1), Ru–C1 2.211(4), Ru–C2 1.831(5), Ru–N1 2.229(3), P1–C1 1.782(5), P1–N2 1.778(4), C1–Si 1.860(4), C2–O 1.155(6); Ru–C1–Si 128.2(2), Ru–C1–P1 72.3(2), Ru–P1–C1 62.2(1), Ru–P1–N2 82.5(1), Ru–C1–H1 112.1(4), P1–C1–Si 113.4(2), P1–C1–H1 112.1(4), C1–P1–N2 96.1(2), P1–Ru–P2 93.6(1), P2–Ru–P3 114.0(1), P3–Ru–C1 107.1(1), P1–Ru–C1 43.5(1), P2–Ru–C2 91.1(1), P3–Ru–C2 89.1(2), C2–Ru–N1 170.2(2).
Scheme 3.
Synthesis of [Ru{η1-N:η2-P,C-P(pzPh)=CH(SiMe2R)}(CO)(PPh3)2] (R = Me 6a, C6H4CF3-p 6d) (L = PPh3). Reagents and conditions: (i) LipzPh, THF.
Scheme 3.
Synthesis of [Ru{η1-N:η2-P,C-P(pzPh)=CH(SiMe2R)}(CO)(PPh3)2] (R = Me 6a, C6H4CF3-p 6d) (L = PPh3). Reagents and conditions: (i) LipzPh, THF.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | δP1 | δC2 | δH2 | |||
|---|---|---|---|---|---|---|
| R | P=C | PPh3 | P=C | P=CH (1JCH/Hz)3 | ||
| 24 | 2a | Me | 58.7 | 46.6, 42.0 | 45.1 | 1.59 (137) |
| (pz′ = pz) | 2b | Ph | 57.0 | 47.0, 41.7 | 42.6 | 1.72 (135) |
| 3 | 3a | Me4 | 32.9 | 46.6, 39.2 | 44.9 | 1.62 (123) |
| (pz′ = pzMe2) | 3b | Ph4 | 32.3 | 47.0, 38.9 | 41.8 | 1.77 (128) |
| 3c | C6H4Me-p5 | 32.6 | 46.7, 39.1 | 41.8 | 1.75 (136) | |
| 3d | C6H4CF3-p5 | 32.1 | 46.6, 38.6 | 39.8 | 1.66 (135) | |
| 45 | 4a | Me | 76.6 | 47.7, 41.5 | 47.1 | 1.78 (136) |
| (pz′ = pzCF3) | 4b | Ph | 74.9 | 48.0, 41.3 | 46.7 | 1.91 (136) |
| 4c | C6H4Me-p | 75.0 | 47.9, 41.3 | 45.4 | 1.90 (134) | |
| 4d | C6H4CF3-p | 73.8 | 47.8, 40.9 | 43.8 | 1.82 (134) | |
| 55 | 5a | Me | 64.6 | 46.9, 38.4 | 45.2 | 1.76 (129) |
| (pz′ = pzMe,CF3) | 5b | Ph | 62.7 | 47.2, 38.3 | 41.8 | 1.97 (131) |
| 5c | C6H4Me-p | 61.6 | 47.2, 38.4 | 42.1 | 1.97 (135) | |
| 5d | C6H4CF3-p | 62.0 | 47.1, 37.8 | 40.7 | 1.85 (133) | |
| 65 | 6a | Me | 64.4 | 47.4, 41.8 | 47.5 | 1.74 (137) |
| (pz′ = pzPh) | 6d | C6H4CF3-p | 60.5 | 47.7, 41.3 | 43.7 | 1.78 (136) |
1 Referenced to 85% H3PO4; 2 Referenced to SiMe4; 3 Measured from coupled 1H–13C HSQC spectra; 4 Recorded as solutions in CD2Cl2; 5 Recorded as solutions in CDCl3.
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Greenacre, V.K.; Crossley, I.R. η1:η2-P-Pyrazolylphosphaalkene Complexes of Ruthenium(0). Inorganics 2016, 4, 30. https://doi.org/10.3390/inorganics4040030
AMA Style
Greenacre VK, Crossley IR. η1:η2-P-Pyrazolylphosphaalkene Complexes of Ruthenium(0). Inorganics. 2016; 4(4):30. https://doi.org/10.3390/inorganics4040030
Chicago/Turabian StyleGreenacre, Victoria K., and Ian R. Crossley. 2016. "η1:η2-P-Pyrazolylphosphaalkene Complexes of Ruthenium(0)" Inorganics 4, no. 4: 30. https://doi.org/10.3390/inorganics4040030
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.