
Product Selectivity in Homogeneous Artificial Photosynthesis Using [(bpy)Rh(Cp*)X]n+-Based Catalysts

Institute of Inorganic Chemistry I, University of Ulm, 89081 Ulm, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2017, 5(2), 35; https://doi.org/10.3390/inorganics5020035
Submission received: 2 March 2017
/
Revised: 15 May 2017
/
Accepted: 19 May 2017
/
Published: 25 May 2017
(This article belongs to the Special Issue Photochemical Water Splitting)
Abstract
:Due to the limited amount of fossil energy carriers, the storage of solar energy in chemical bonds using artificial photosynthesis has been under intensive investigation within the last decades. As the understanding of the underlying working principle of these complex systems continuously grows, more focus will be placed on a catalyst design for highly selective product formation. Recent reports have shown that multifunctional photocatalysts can operate with high chemoselectivity, forming different catalysis products under appropriate reaction conditions. Within this context [(bpy)Rh(Cp*)X]n+-based catalysts are highly relevant examples for a detailed understanding of product selectivity in artificial photosynthesis since the identification of a number of possible reaction intermediates has already been achieved.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Based on mankind’s tremendous impact on planet earth, the scientific question of whether a new division of geological time should be defined and consequently be termed “anthropocene” has emerged [1]. Due to the mostly detrimental influences of human activity on our environment, as well as for economic reasons, catalysis has played a major role in the development of industrial chemistry [2]. The continuous enhancement of catalysts is also driven by the intellectual insatiable desire of keeping up with nature’s highly selective operating enzymes, whose activity has been optimized to perfection during millions of years of evolution. Within this context, highly stereoselective catalysis in organic synthesis has been one of the most outstanding achievements during the last decades of catalyst development [3].
On the other side, nature is additionally not only able to perform ground state chemistry, but also uses light energy as a primary resource for several energy demanding chemical conversions. This is accompanied by the challenging handling of excited states and highly reactive intermediates. Due to a precise arrangement of functional proteins within the thylakoid membrane of green plants, the light driven water splitting of natural photosynthesis generates the indispensable ubiquitous oxidant O2, as well as reducing equivalents in the form of NADPH along with the energy carrier ATP, and can therefore be ascribed from a chemist’s point of view as one of nature’s biggest masterpieces [4,5,6]. Although light nowadays has its application as a key reagent or energy source in selective chemical transformations [7], the knowledge about product selectivity with respect to artificial photosynthesis is still in its infancy.
In this feature article, recent work regarding the product distribution of homogeneous artificial photosynthetic systems capable of generating different reaction products at the catalytic center will be discussed, with a clear focus on RhCp*-based catalysts. In order to pave the way towards developing an understanding of the reaction outcome of the different systems, overviews of the underlying reaction mechanisms at the catalytic centers will be provided.
2. General Considerations on Product Selectivity within Artificial Photosynthetic Systems and Selected Aspects of the Homogeneous Photocatalytic CO2 Reduction
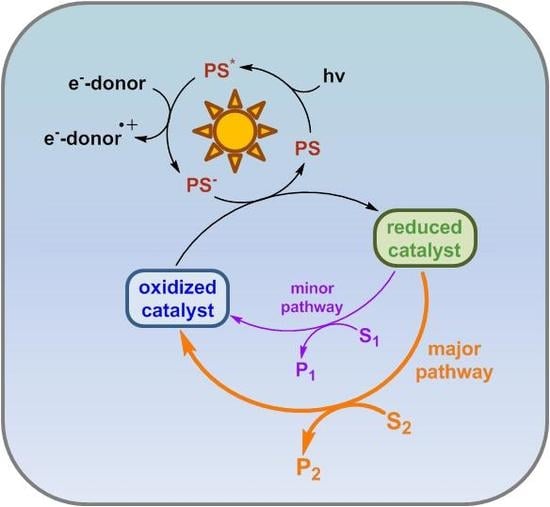
Due to its highly sophisticated architecture and the presence of two catalytic centers, natural photosynthesis is able to drive the overall water splitting reaction. By virtue of the complex interplay of light absorption, electron transfers, and catalysis, typical artificial photosynthetic systems focus on one of the redox half reactions. Besides the photosensitizer (PS), which absorbs the light, and the catalytic center (CC), that drives the substrate conversion after electron transfer reactions with the excited photosensitizer (PS*), a sacrificial agent has to be added in order to close the catalytic cycle. As depicted in Figure 1, an artificial photosynthetic system can be multifunctional, if the activated catalytic center (CCa) is able to convert different substrates into different redox products. Therefore, the catalytic cycles have to exhibit a common branching point. However, the branching point of the catalytic cycles does not necessarily need to be the catalytically active species itself; a simple catalytically inactive reaction intermediate could be envisaged instead.
From Figure 1, the general idea of how the product distribution can be principally switched between different reaction outcomes becomes evident. If the amount of product B needs to be maximized, catalytic pathway 2 should be avoided as effectively as possible. The easiest approach to achieve this would be simply not adding substrate C to the system. In cases where this is not possible (usually due to the presence of protons as a substrate for hydrogen evolution catalysis), k1 needs to be much higher than k2, which could be accomplished by choosing appropriate reaction conditions, such as the right pH value, light intensity, water content, etc., or by designing a more sophisticated ligand environment around CC, which would allow substrate A to react much faster with CCa than substrate C, based on steric or electronic factors.
One prominent example for the possible generation of different redox products is the photocatalytic reduction of carbon dioxide, whose general mechanism will be briefly discussed in the following section for the purpose of illustrating the general catalysis scheme of Figure 1.
Due to the unavoidable depletion of fossil fuels, the conversion of CO2 into valuable chemicals would establish a sustainable supply of man-made energy carriers. Hence, a lot of work has been done in order to design highly active photocatalysts using solar energy for driving CO2 reduction reactions.
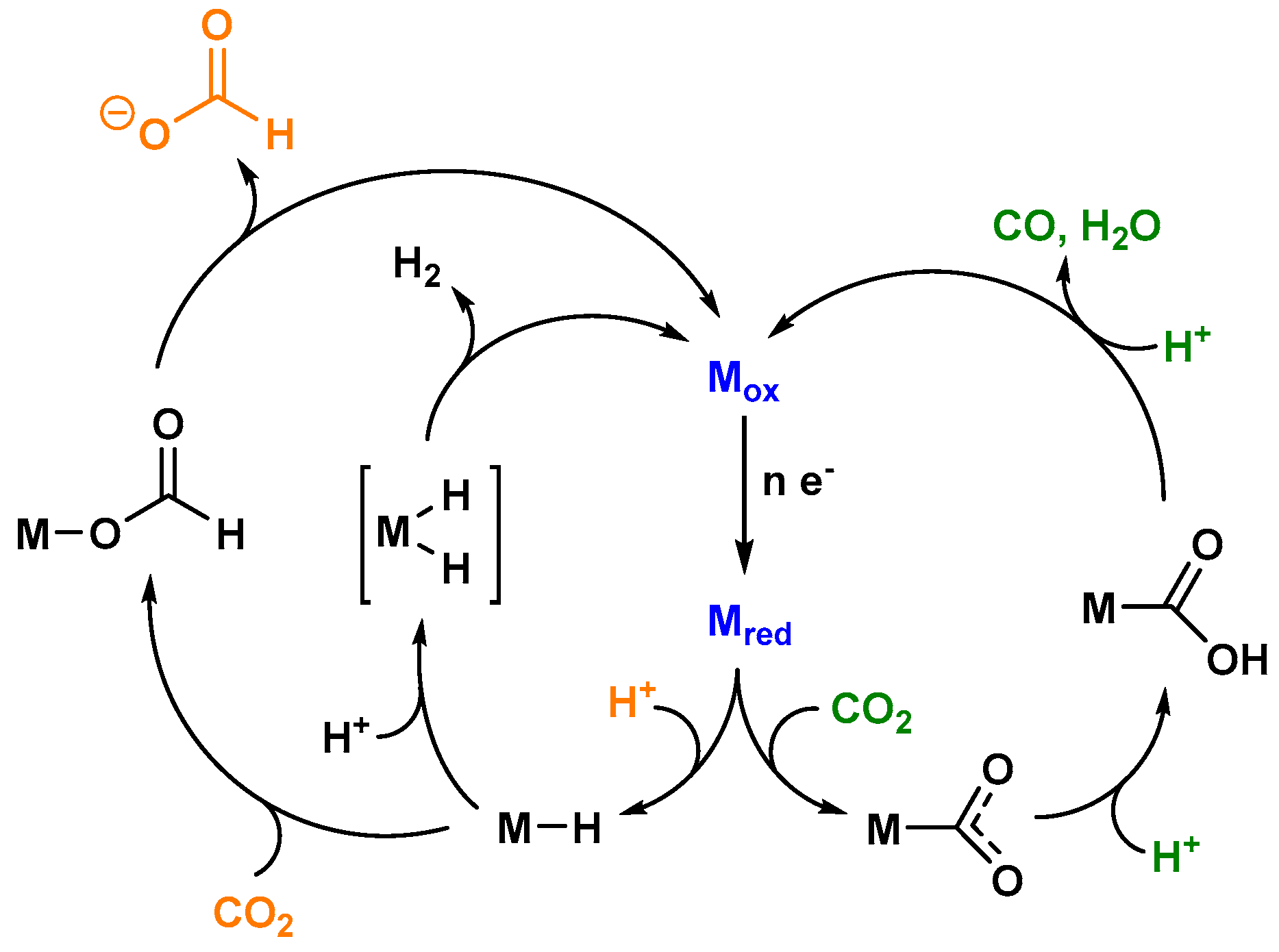
A broad variety of coordination compounds (bearing Mn, Fe, Co, Ni, Ru, Re, or Ir as the central metal) have been found to function as catalysts for the photocatalytic conversion of CO2, mainly generating the two electron reduced carbon-based products CO and HCOOH, as well as H2 as the side product from the competing proton reduction [8]. As described above, the detection of multiple redox products indicates the presence of at least one reactive species, appearing in two or more catalytic cycles. Figure 2 shows a simplified reaction scheme for the key intermediates of the photocatalytic CO2 reduction processes [9].
A coordination compound in an oxidized state (Mox) represents the starting point of the overall cycle. A light driven electron accumulation onto Mox yields the reduced intermediate Mred, which either takes up a proton to generate a metal hydride species or directly binds carbon dioxide, forming a metal carboxylate species. The latter liberates via an initial protonation step of the reduced carbon species CO, as well as water. HCOO− is produced by the insertion of carbon dioxide into the metal hydride bond, forming a labile formato complex. In addition, the closing of the catalytic cycle can also be achieved by the reaction of M–H with a second proton, thereby generating molecular hydrogen.
The chemoselectivity of any process will mainly be determined by differences in the reaction kinetics at the branching points [9]. Crucial factors herein are predominantly the energy content of the relevant species generated from the common branching point intermediate with the appropriate substrates and the available concentration of the latter. Coherently low energy barriers and high substrate concentrations for a specific dark reaction following the light driven generation of a certain branching point species will increase the amount of a certain reaction product in an artificial photocatalytic system.
With a much more sophisticated ligand environment at the catalytic centers in comparison to the man-made ones, nature drives the transformation of CO2 into valuable chemicals using earth abundant metals. A key moiety of several enzymes dealing with the incorporation of CO2 into organic substrates is the metal carbamato species, which can be found in rubisco and biotin-dependent enzymes [10]. Its formation can be interpreted similarly to the insertion of carbon dioxide into the M–H bond shown in Figure 2, indicating that the latter represents a very important step for the generation of useful carbon containing chemicals.
It should be noted that the above discussed general mechanism is only a highly superficial view on the actual complex sequence of elementary steps underlying the overall photocatalytic CO2 reduction processes; interested readers are referred to several selected review articles [8,9,11,12,13,14]. Nevertheless, the simplified reaction scheme depicted in Figure 2 was chosen in order to clarify the role of catalytic intermediates exhibiting multiple reaction pathways.
Although principally at least three different reduction products can be obtained by performing a photocatalytic carbon dioxide reduction, several systems have been found to exhibit a high chemoselectivity for either CO or formate production. Transition metal complexes based on the [(bpy)Re(CO)3X]n+ structural motive mostly form carbon monoxide as the main product in water-free solvent systems, with chemoselectivities reaching almost 100% [15,16,17,18,19,20]. Interestingly a supramolecular heterodinuclear Ru–Re photocatalyst showed an opposed product selectivity towards formate generation in an aqueous solution [21]. This solvent dependent behavior could be attributed to the different reaction kinetics for the formation of the intermediates M–H and M–CO2 from the common branching point species Mred, as depicted in Figure 2. As the proton concentration in an aqueous solution is higher than in an organic solution, the formation of M–H will be accelerated and consequently, the yield of HCOO− increases.
Impressive selectivities and TONs (turnover numbers) have also recently been obtained with a cobalt cryptate as the catalyst [22]. In contrast to the mentioned Re complexes, the Ru-based catalyst of the general composition [(NN)Ru(CO)2Cl2] or [(NN)2RuX2] (X = CO, Cl, DMF; NN = N,N-chelating diimine ligand) shows a selectivity towards formate production in the majority of cases, although sometimes CO can also be obtained as the major product [8,23,24,25].
Since access to the much more valuable higher reduced carbon species, such as methanol using highly efficient and durable photocatalytic processes, would represent a major turning point in the global energy crisis, further developments in the field of visible light driven CO2 reduction and an increase in the knowledge of the underlying reaction mechanisms are highly likely to occur.
3. Application of [(bpy)Rh(Cp*)X]n+-Like Coordination Compounds as Multifunctional Redox Catalysts
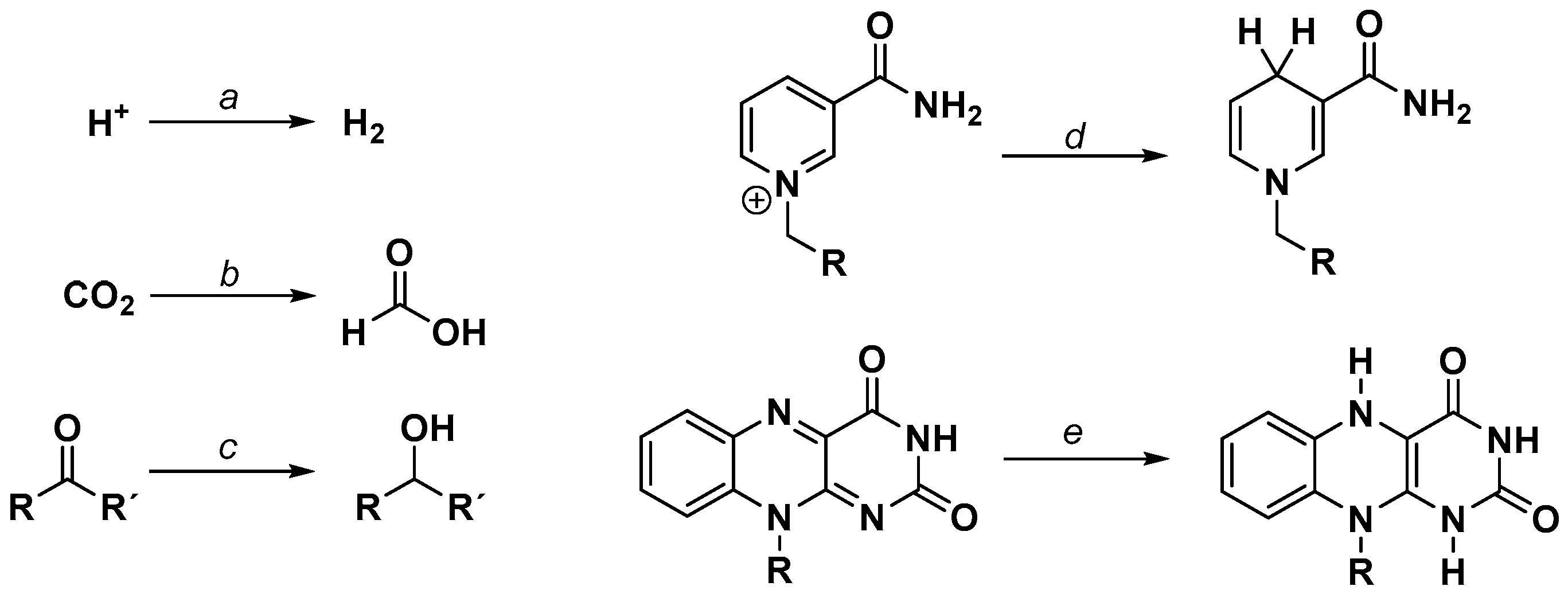
As measured by the amount of possible selective redox transformations, [(NN)Rh(Cp*)X]n+-like catalysts hold a leading position within artificial photosynthetic systems (Cp* = pentamethylcyclopentadienyl, X = Cl, OH, H2O). As depicted in Figure 3, a broad number of substrates can be reduced using [(NN)Rh(Cp*)X]n+-like structures as catalysts by means of chemical, electrochemical, or photochemical methods. In addition, imines [26,27,28] and α,β-unsaturated carbonyl compounds [29] can also be reduced with this type of catalyst, as well as nitrogen containing heterocycles such as quinolines or quinoxalines [30,31]. To illustrate the high potential of this organometallic rhodium-based catalyst, a fully homogeneous system for the total photocatalytic water splitting using [Co4(H2O)2(PW9O34)2]10− as the water oxidation catalyst and [(bpy)Rh(Cp*)(H2O)]2+ as the NAD+ reducing catalyst using [Ru(bpy)3]2+ as a photosensitizer has been reported [32]. However, only turnover numbers (TONs) below 10 were found for the Rh catalyst and the Ru chromophore.
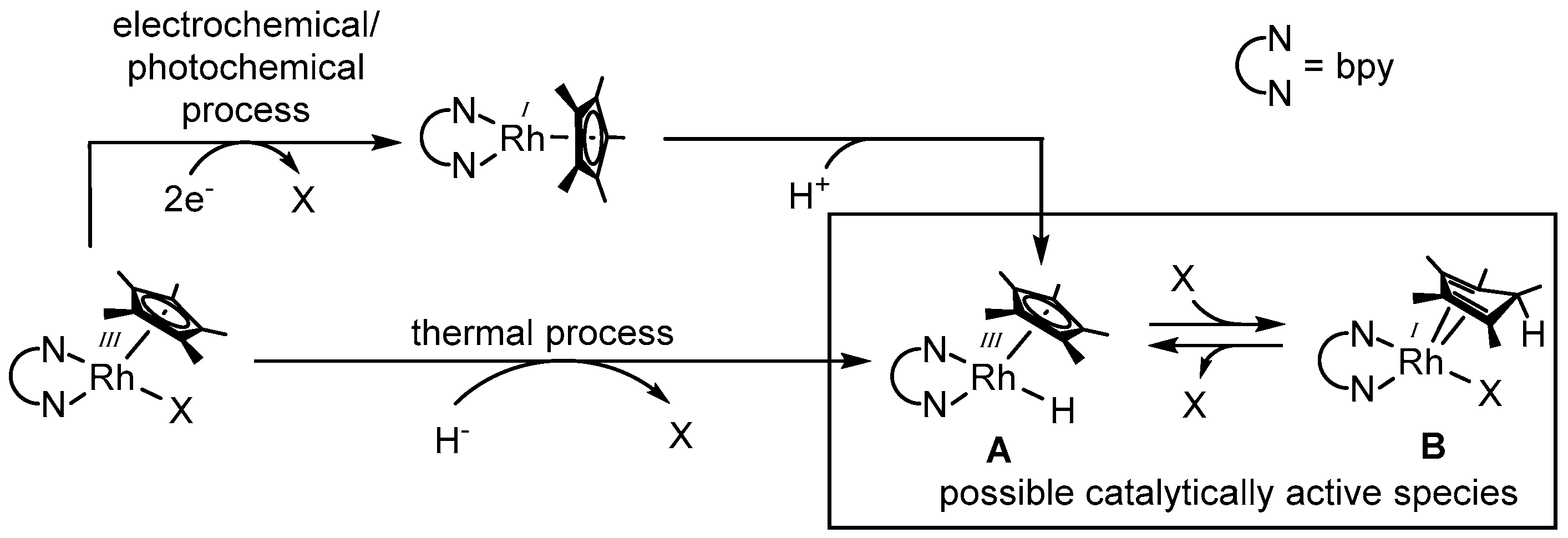
As shown in Figure 4, the generation of a catalytically active species can be achieved on the one hand by the thermal decomposition of a suitable hydride donor in the coordination sphere of the catalyst using formate most frequently [56], but also alcohols [44] and phosphite [57], or on the other hand, by a sequential two electron reduction via an electrochemical or photochemical pathway and a concomitant oxidative addition of a proton.
The molecular structure of the actual active rhodium catalyst has been under debate during the last years. Since the late 1980s, the catalytically active species has been described as a RhIII–H (structure A in Figure 4) [33,45]. The experimental proof of the existence of this species has been performed by Steckhan et al. [51], as well as Fukuzumi et al. [60], using a 6,6′-dimethylated bipyridine as the N,N-chelating ligand. However, as for other sterically less demanding N,N-chelating ligands, the RhIII–H moiety has so far not been observed. The independent discovery of an alternative possible catalytically active species (see structure B in Figure 4) by Pitman et al. [61], as well as Quintana et al. [62], established the potential presence of multiple reactive species involved in the reductive transformation of several substrates.
Due to the endo orientation of the hydrogen atom, the conversion of structure A into structure B can be regarded as the reductive elimination of a proton from the rhodium center onto a carbon atom of the Cp* ligand. As species A was detected at low temperatures and the conversion into species B occurs upon warming, the thermodynamically favorable formation of the latter most likely proceeds via the initial generation of A [61,62].
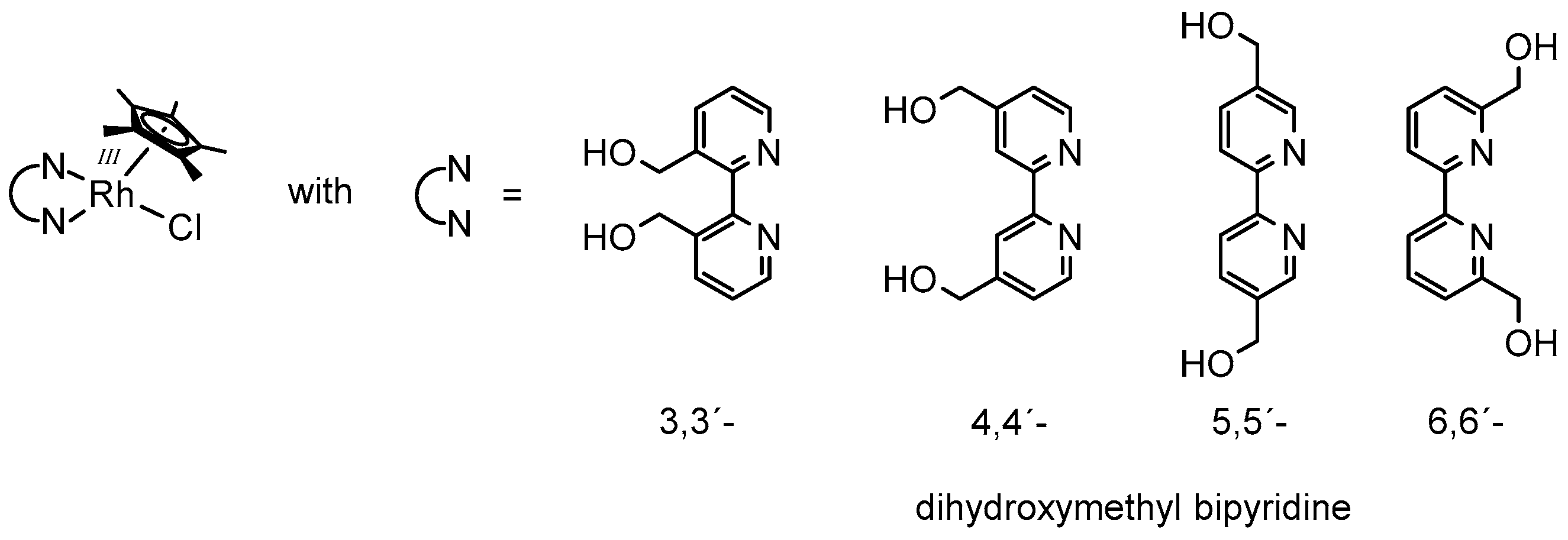
In a very recent report, the influence of the nature of the N,N-chelating ligand on the structure of the catalytically active species was discussed [63]. Similarly to the bulky 6,6′-dimethylated bipyridine, the 6,6′-dihydroxymethyl bipyridine described in this study also shows a 1H-NMR signal attributable as RhIII–H.
However, when using 3,3′-, 4,4′-, or 5,5′-dihydroxymethyl bipyridine as the N,N-chelating ligand (see Figure 5), the formation of the reduced Cp* ligand (Cp*H) was observed instead. These findings were rationalized by the enlarged Cp*-Rh distance in the case of the 6,6′-dihydroxymethyl bipyridine due to steric interactions with the Cp* ligand, which might finally prohibit hydrogen transfer onto the five-membered ring.
As depicted in Figure 4, access to the catalytically active species is not only possible via a thermal process by formally abstracting “H−” from a suitable donor, but also by means of photo- and electrochemistry. Thereby, the generation of structure A is accomplished via the successive transfer of two electrons onto the catalyst, followed by the loss of ligand X and the oxidative addition of a proton from the solvent.
Detailed investigations demonstrated that the special ligand environment of the [(NN)RhIII(Cp*)X]n+-like catalyst grants it a favorable electrochemistry for applications in homogeneous photo- and electrocatalysis. Access to the reactive [(NN)RhI(Cp*)] intermediate is accomplished via a two electron reduction at a rather low cathodic voltage (ranging from −700 to −900 mV against Ag/AgCl depending on the N,N-chelating ligand) [51]. After the addition of one electron, the primarily obtained coordinatively unstable RhII species disproportionates extremely rapidly, leading to the desired RhI compound (for the majority of the applied complexes [64]), as this process is merely connected with the easy loss of a weakly bound monodentate ligand [33,37,65]. In addition, a crystal structure of the reduced d8 intermediate [(bpy)RhI(Cp*)] revealed that the bpy and the Cp* plane are lying perpendicular to each other, as depicted in Figure 4, different to the piano-stool like structure of the d6 species [(bpy)RhIII(Cp*)X]n+ [66].
As the thermal, as well as the photo- or electrochemical, route leads to intermediate A, substrate conversions that were to date only reported using the thermal approach should also be realizable using electrochemical or photochemical methods under appropriate reaction conditions. This would further expand the already wide range of applications of [(bpy)Rh(Cp*)X]n+-like catalysts in homogeneous photocatalytic systems.
3.1. Reduction of Carbonyl Compounds to Primary or Secondary Alcohols
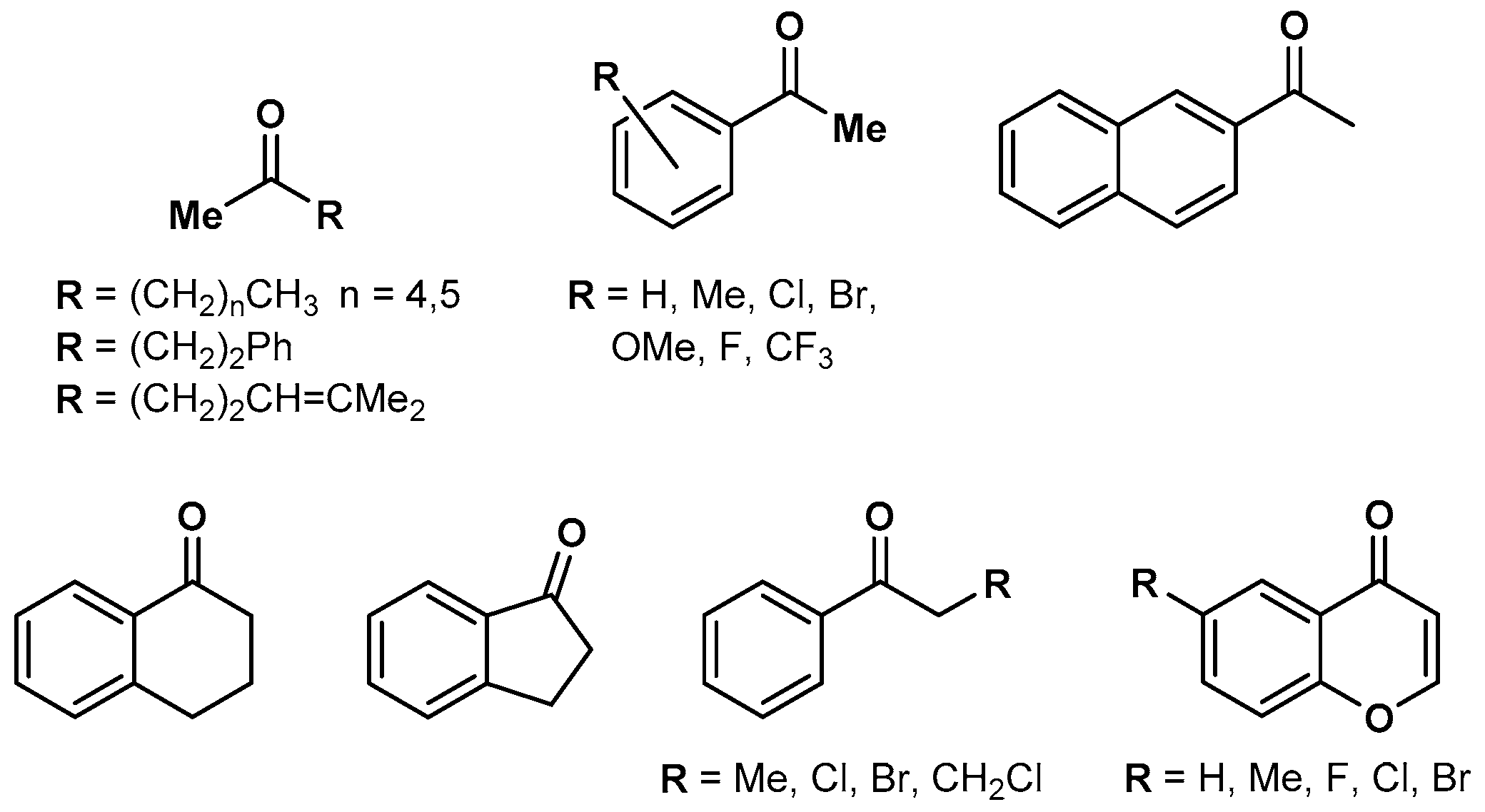
A typical substrate for transfer hydrogenations are carbonyl compounds. Within several thermal processes, the use of chiral N,N-chelating ligands coordinated to the Rh(Cp*) moiety allowed ketone reductions with high enantioselectivity, indicating that the ligand-induced asymmetry of the whole catalyst is sufficiently preserved at the rhodium center to sterically discriminate between the two different enantiotopic faces of the ketone [42,43,44]. In addition, chromenones, a group of cyclic α,β-unsaturated ketones, were reduced to the fully saturated chiral alcohol with high ee-values, using HCOONa as a hydride donor [29]. A number of these stereoselectively reducible ketones are depicted in Figure 6.
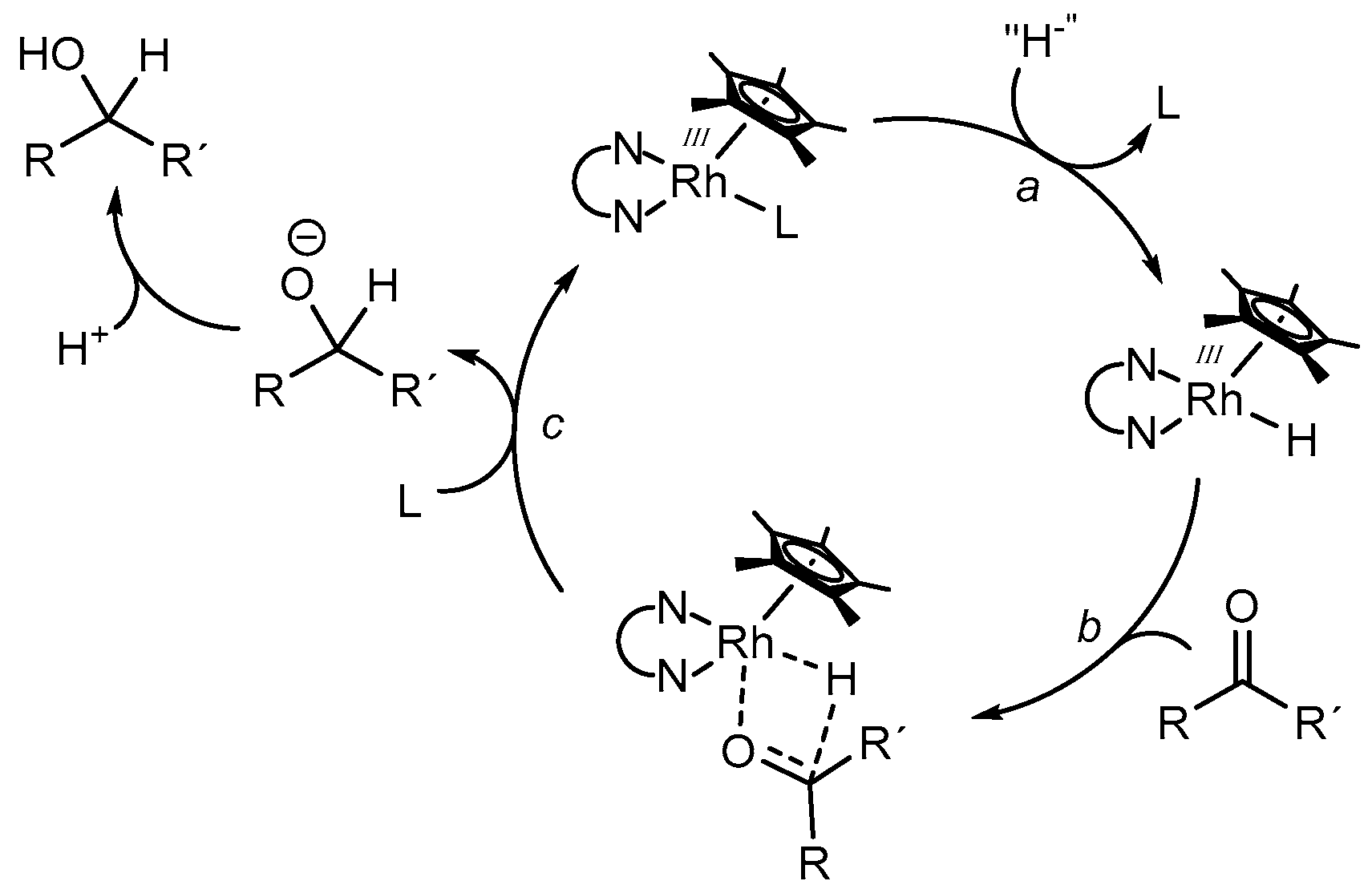
In order to evaluate a rational reaction scheme for the rhodium catalyzed reduction of carbonyl compounds to the appropriate alcohols, Leiva et al. performed concentration dependent experiments using a variety of aldehydes and ketones, as well as sodium formate as a hydride donor and [(bpy)Rh(Cp*)(H2O)](OTf)2 as a catalyst [41]. Although the formation of the catalytically active [(bpy)Rh(Cp*)H]+ species was proposed to be the rate limiting step, the structure of the substrates had a significant effect on the observable TOFs (turnover frequencies, i.e., turnover number per time). Generally, aldehydes were converted faster than ketones due to the higher electrophilicity of the carbon atom. In addition, higher reaction rates were observed for substrates with an increased electron density on the oxygen atom, indicating that the binding of the substrate into the coordination sphere of the catalyst is important and may take place in concert with the hydride transfer reaction. From these observations, a simplified reaction mechanism was proposed, as depicted in Figure 7. After the replacement of a ligand L for a hydrido ligand (reaction a) via the decomposition of formate, coordination of the carbonyl compound occurs (reaction b). Following a hydride transfer via a four-membered transition state, the replacement of the alkoxide ligand (or alcohol after protonation) for L regenerates the initial Rh species (reaction c).
Besides the above delineated thermal transfer hydrogenation reactions in which the catalytically active species is present in a significant concentration due to the excess of hydride donors, a selective reduction of aldehydes in the presence of ketones via a homogeneous photocatalytic process was described by Ghosh et al. [26]. Using proflavine as a photosensitizer, [(bpy)Rh(Cp*)Cl]Cl as a catalyst, and TEOA as a sacrificial electron donor, the catalytically active species was generated very slowly, which allowed the maintenance of a continuously low Rh–H amount and an efficient kinetic discrimination of aldehydes and ketones by steric factors. Contrary to the mechanistic investigations of the thermal process described above, the authors suggested that within the photocatalytic process, the hydride transfer from the rhodium center onto the carbonyl compound represents the rate determining step.
3.2. Use of [(bpy)Rh(Cp*)X]n+ Catalysts for Hydrogen Evolution and CO2 Reduction
Reduced carbon species, as well as molecular hydrogen, are prime targets for the storage of light energy. Due to the rather low volumetric energy density of hydrogen, methods for its incorporation into materials, as well as its temporary introduction into carrier molecules, are under intensive investigation [67,68]. Within this context, formic acid is one possibility of transiently storing H2 [69]. In order to use molecular hydrogen as a green fuel, efficient catalytic liberation from the storage materials is an important prerequisite for this technology. It has been shown that [(bpy)Rh(Cp*)(H2O)]2+ is able to selectively decompose formic acid into H2 and CO2 via a rhodium formato and a rhodium hydride intermediate without the production of CO [60]. Interestingly, a fast H/D exchange of the Rh-H intermediate was observed, illustrating the bivalent protic and hydridic nature of the metal bound hydrogen. The pKa value for the acid base equilibrium of [(NN)Rh(Cp*)H]+/[(NN)Rh(Cp*)]0 + H+ has been determined to lie between seven and eight, depending of the structure of the N,N-chelating ligand [33,58,70].
[(bpy)Rh(Cp*)X]n+ and its derivatives are also efficient catalysts for the electrochemical generation of dihydrogen. In accordance with recently published calculations [62], hydrogen evolution is only observed at low pH values or by the use of strong acids [33,34,35,36,38]. The liberation of molecular hydrogen from the Rh-H intermediate has been ascribed as the rate determining step of the overall catalysis, whereas the rate constants for the disproportionation reaction yielding the [(bpy)Rh(Cp*)]0 intermediate and its protonation to form [(bpy)Rh(Cp*)H]+, are multiple orders of magnitude higher [33,65]. Concerning the activity of these Rh-based catalysts, two successive papers reported a faradayic efficiency of 100% for hydrogen formation at −0.55 V vs. SCE by using a polymeric Rh(Cp*) catalyst in an aqueous solution of pH 1 [34,35]. This correlated to a TON of 353 after 14 h.
Therefore, the photocatalytic generation of H2 using an intermolecular system containing [Ru(bpy)3]2+ as a photosensitizer, [(bpy)Rh(Cp*)(H2O)]2+ as a catalyst, and sodium ascorbate as an electron donor showed a strong dependence of the catalytic activity on the proton concentration, with an optimum pH value of 3.6. A negligible formation of hydrogen was observed at pH values higher than five and lower than two, which indicates on the one hand, that a sufficient thermodynamic driving force for the proton hydride reaction needs to be generated, which can be achieved by a high proton concentration, and that on the other hand ascorbate rather than ascorbic acid acts as the electron donor [37]. Additionally, molecular hydrogen was also evolved by thermally produced [(bpy)Rh(Cp*)H]+ in the presence of photoactivated platinum nanoparticles [71]. Herein the rhodium hydrido complexes served as electron donors for the catalytically active particles.
Despite intensive work in the field of catalytic hydrogen evolution using [(bpy)Rh(Cp*)X]n+-based catalysts, a detailed understanding of the important proton hydride reaction liberating H2 and the closing of the catalytic cycle remains unclear. Besides the possibility of a simple acid base reaction outside of the rhodium coordination sphere, the binding of a second proton to the intermediates A or B of Figure 4 has been hypothesized, resulting in a Rh assisted intramolecular liberation of hydrogen [62].
In addition to the evolution of H2, the reduction of carbon dioxide has also been achieved by electro- and photochemical methods using [(NN)Rh(Cp*)X]n+ catalysts. Besides the direct CO2 reduction in the coordination sphere of the catalyst, which will be described here, an indirect pathway by the assistance of an enzyme, powered by the [(NN)Rh(Cp*)X]n+ driven regeneration of nicotinamide cofactors will be discussed in the next section.
The electrochemical reduction of CO2 has been performed using [(bpy)RhIII(Cp*)Cl]+ as a catalyst in aqueous acetonitrile solution [39]. In addition to the generation of formic acid, dihydrogen in a significant and carbon monoxide in a negligible amount were formed. The authors claim that under their experimental conditions, the actual catalytically active species can be described as the neutral, threefold reduced complex [(bpy)RhII(Cp*)H], indicating the necessity of an increased hydricity of the metal bound hydrogen for CO2 reduction.
Surprisingly, there is only one report about the direct photocatalytic reduction of carbon dioxide using [(NN)Rh(Cp*)X]n+ as a catalyst [40]. CO2 has been reduced by [(bpy)Rh(Cp*)Cl]Cl in a mixture of MeCN:TEOA = 5:1, using [Ru(bpy)3]2+ as a photosensitizer. After 10 h of irradiation, formic acid has been detected as the sole reduced carbon species (TON = 125), besides the significant evolution of hydrogen (TON = 55).
In addition to the above mentioned electrocatalytic and photocatalytic methods of converting CO2 into valuable chemicals, the direct hydrogenation of CO2 using H2, thereby generating formic acid, is also feasible using [(bpy)Rh(Cp*)X]n+-based catalysts. In contrast to the Rh complex with the unsubstituted bpy ligand, a marked increase in catalytic activity by the introduction of pH sensitive OH functionalities at the para positions was observed [72]. Deprotonation of the hydroxyl groups under basic conditions generated the oxyanionic species, resulting in an increased electron density at the metal center, which is responsible for the enhancement of catalytic activity (from TON = 216 to 1800). Interestingly, for the iridium analog, the increase of catalytic performance was drastically higher, increasing from a TON of 105 to 190,000 under optimized conditions.
3.3. NAD(P)H Formation Using the [(bpy)Rh(Cp*)X]n+ Motive
A very interesting property of [(NN)Rh(Cp*)X]n+ catalysts is their highly regioselective production of 1,4-dihyro nicotinamide from N-substituted nicotinamide cations. As the coenzymes NAD+ and NADP+ also exhibit this moiety, [(NN)Rh(Cp*)X]n+ complexes allow the construction of semiartificial biocatalytic systems powering enzymatic reduction reactions by transition metal catalyst driven NAD(P)H regeneration. For a detailed overview of the different biochemical applications of these [(bpy)Rh(Cp*)X]n+-based cofactor regeneration systems, the interested reader is referred to several review articles [73,74,75].
It should be noted that other rhodium complexes like [Rh(bpy)3]3+ and [Rh(terpy)2]3+, as well as several cobalt complexes, can act as regioselective catalysts for 1,4-dihyro nicotinamide formation too [76,77,78,79,80,81]. However, as for most of these catalysts only very small TONs were observed, [(NN)Rh(Cp*)X]n+ has achieved outstanding significance in selective NAD(P)+ reduction catalysis due to its much higher reactivity.
The thermal production of NAD(P)H has most often been performed using formate as a hydride donor [45,46,47,49,82]. Intensive mechanistic studies performed by Lo et al. using different nicotinamide derivatives, altered in the 1- as well as the 3-position of the pyridine ring, point to a catalytic mechanism, as depicted in Figure 8 [83,84]. As the initial step, formate has to replace the rhodium bound monodentate ligand L. It has been shown that the presence of strong σ donor ligands such as NH3 decreases the rate of thermal NAD+ reduction as they compete with formate for the available Rh binding site [46,51,56,59]. A similar but pH dependent behavior was suggested for L = H2O; upon increasing the pH value, the NADH generation slowed down [56]. Since the pKa value of the metal bound water was determined as 8.2 [70], higher pH values of the solution led to the formation of the hydroxo ligand, which is a stronger σ donor than water, and replacement by formate thus becomes more difficult.
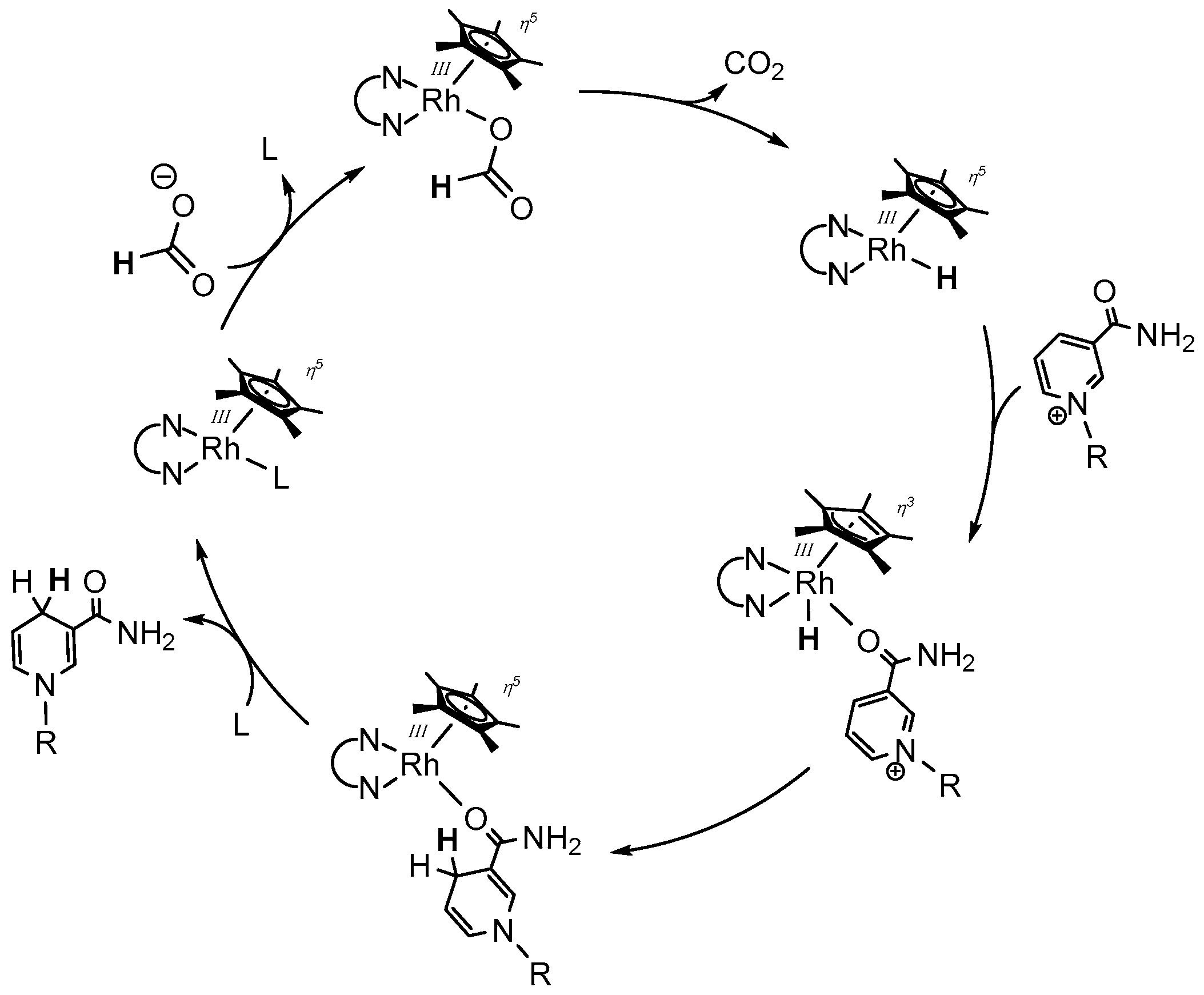
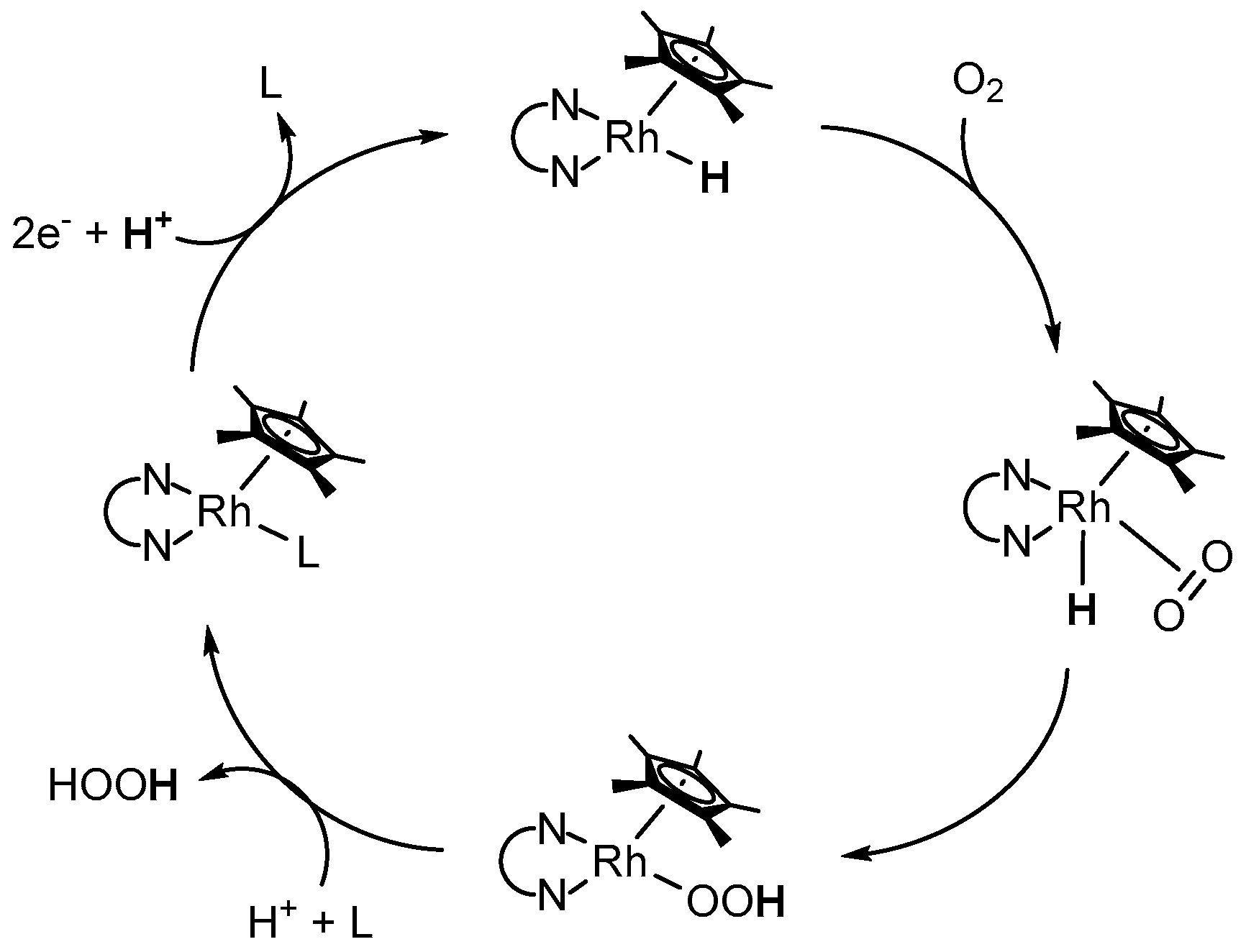
However, after the coordination of formate, decomposition affords CO2 and the catalytically active RhIII–H species; the latter has been reported to produce hydrogen peroxide in the presence of molecular oxygen with the concomitant reformation the initial RhIII–L species [53,56]. The authors proposed a reaction scheme, in which O2 inserts into the Rh–H bond of the electrogenerated [(NN)Rh(Cp*)H]+ species, as depicted in Figure 9 [53]. After protonation and ligand exchange, the initial Rh catalyst gets reformed and H2O2 is liberated.
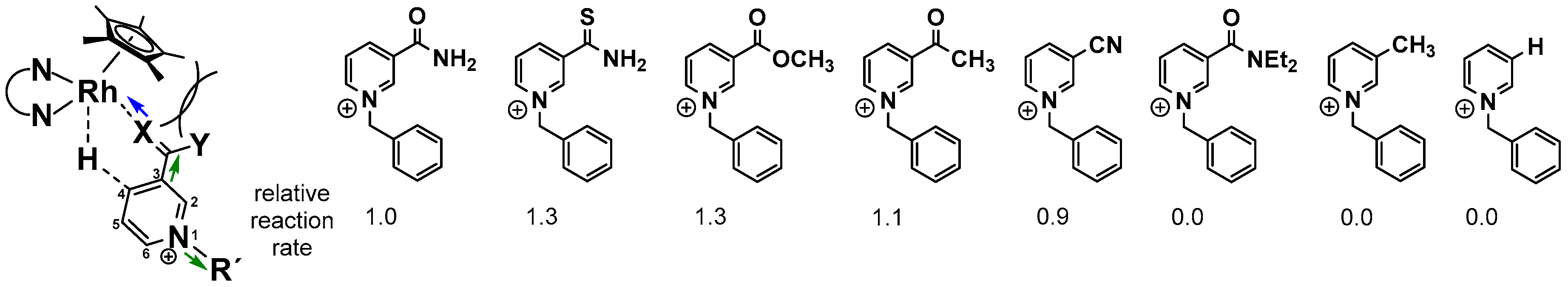
After the formation of the catalytically active RhIII–H species, coordination of the cationic nicotinamide has been suggested to proceed in combination with a binding mode change of the Cp* ligand from η5 to η3, since the ring slippage would retain the formal 18 valence electron count on the central metal (see Figure 8). The arrangement of the ligands around the rhodium atom in this intermediate stage is responsible for the highly regioselective production of the 1,4-reduced nicotinamide, due to the formation of an energetically favourable six membered transition state (see Figure 10). The regioselectivity has been reported to be at least as high as 95% for the 1,4 product. Only traces of the 1,6 isomer are produced, which are formed putatively by a subsequent [(NN)Rh(Cp*)X]n+ catalyzed isomerization of the 1,4-dihydro form. Closing of the catalytic cycle proceeds finally by the displacement of the reduced nicotinamide by another ligand.
As depicted in Figure 10, several nicotinamide-related structural parameters affect the rate of nicotinamide reduction [83,84]. Of highest importance is the necessity of a σ donor moiety in the 3-position of the pyridine ring, as with an unsubstituted or 3-methylated derivative, no product formation was observed. Furthermore, an increased σ donor ability accelerates the overall catalysis by making the metal bound hydrogen more hydridic. Substitution patterns which make the carbon atom at C-4 more electrophilic also increase the reaction rates (green arrows in Figure 10).
Similar results have been obtained by the means of electrochemical and photochemical methods [85]. Finally, larger R substituents and the carbamoyl nitrogen inhibit product formation due to steric interactions with the Cp* ligand, as marked in Figure 10.
Depending on the reduction potential of the RhIII/RhI couple, the catalytic activity for NADH production varies in the thermal and the electrochemical approach in the opposite direction. Whereas a high reduction potential is beneficial in terms of the electrocatalytic activity, the same is detrimental when operating thermally by using formate as a hydride donor. As the electron density on the metal increases due to a more basic and electron rich N,N-chelating ligand, its reduction becomes more difficult, but the subsequent addition of a proton is accelerated [51,54]. The latter property can also be viewed as a higher amount of protonated catalytically active [(NN)RhIII(Cp*)H]+ at a given pH value, since its deprotonation yields the transiently inactive neutral [(NN)Rh(Cp*)] species. By operating thermally using formate as a hydride donor, the lower catalytic activity with an increased reduction potential could be interpreted by the different mechanism of generating the catalytically active species. The necessary coordination of the formate ligand, as well as the subsequent hydride abstraction, will be energetically more demanding if the electron density on the rhodium atom will be increased as a consequence of having a more electron rich N,N-chelating ligand bound to the metal [63].
Inactivation of the rhodium catalyst has been reported for the electrocatalytic reduction of NAD+ in the presence of amino or thiol groups [86]. Consequently, the mutual inactivation of [(bpy)Rh(Cp*)(H2O)]2+ and an enzyme bearing cysteine and lysine residues on the accessible protein surface has been observed [87]. A detailed electrochemical investigation on the influence of amino acids on the electrocatalytic activity of [(bpy)Rh(Cp*)(H2O)]2+ for NADH formation revealed that the strongest inhibitory effect arose from cysteine, histidine, and tryptophane [88]. Although all these detrimental interactions have been observed, a detailed understanding of the underlying molecular processes is still missing. In the case of thermal NADH regeneration using formate as a hydride donor, the lack of catalytic activity can be tentatively explained by a thermodynamically unfavourable ligand exchange from a thiolato ligand to formate [87]. Based on electrochemical studies, it has been revealed that the binding of a chloride ion to the two electron reduced neutral species [(NN)RhI(Cp*)] can occur, forming the anionic [(NN)RhI(Cp*)Cl]− species [35]. We hypothesize a similar binding of sulfur or nitrogen containing groups to the rhodium center, which could finally prevent the necessary oxidative addition of a proton as a key step for the generation of the catalytically active species.
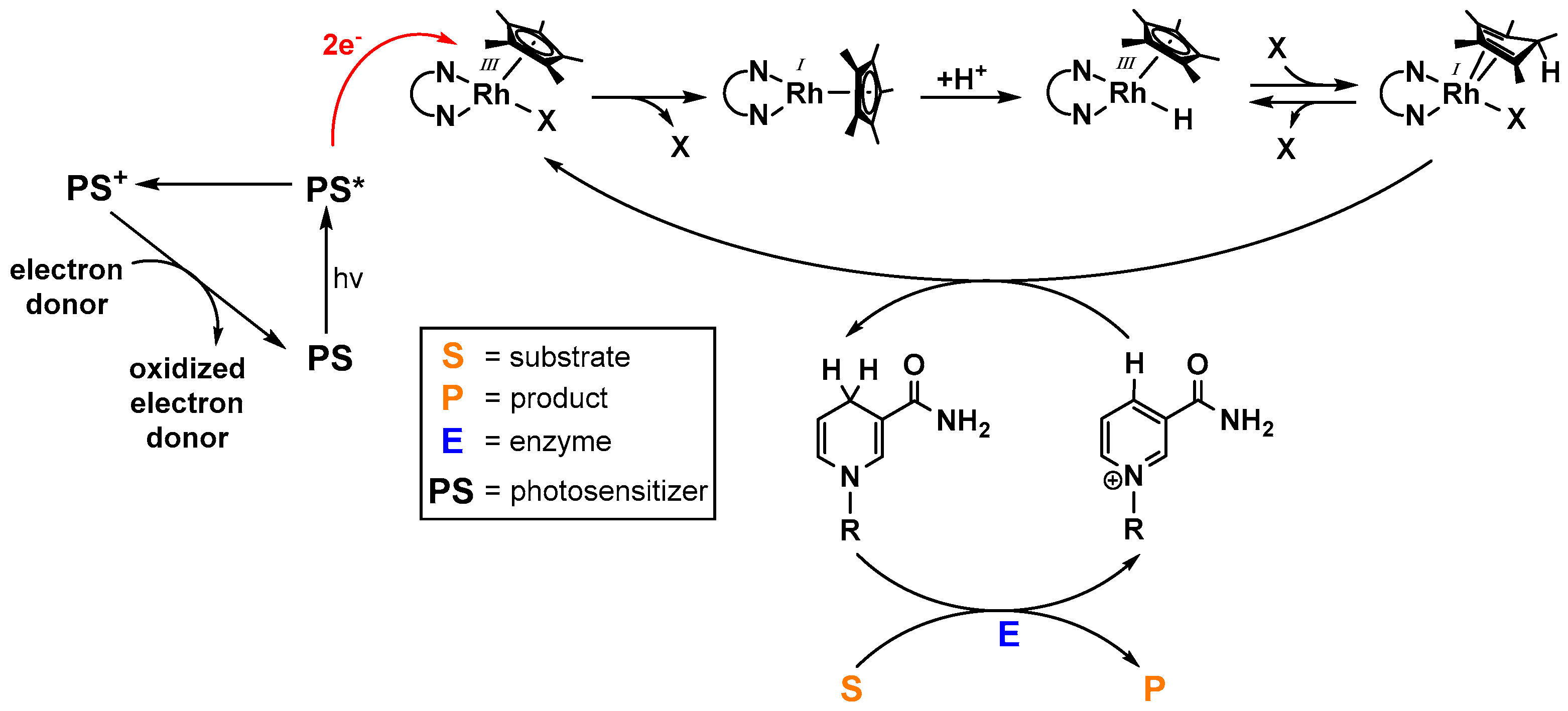
As the photochemical generation of NAD(P)H imitates photosystem I of the natural photosynthesis and consequently allows the light driven powering of enzymatic transformations, intensive efforts have been undertaken in order to optimize the catalytic activity of [(NN)Rh(Cp*)X]n+-based systems (an overview of the working principle is depicted in Figure 11) [55]. A broad variety of photosensitizers have been applied in these systems, ranging from small organic dyes [85,89,90,91] to functionalized graphenes [92,93,94,95], photoredoxactive polymers [96,97], red light absorbing tin porphyrines from Knör and coworkers [98], ruthenium polypyridine complexes [32,97,99], and different solid photoactive materials [100,101,102,103,104,105,106,107,108,109,110].
Identically to the photocatalytic generation of hydrogen, the catalytically active rhodium complex is most likely formed via the fast disproportionation of the intermediate RhII species [33,37]. Since the underlying mechanism for the generation of the protonated active catalyst is similar with respect to the electrochemical and the photochemical approach, the same reactivity trends for different N,N-chelating ligands are assumed to be true. This means, in other words, that the more electron rich the catalyst is, the faster the proton uptake becomes, which consequently increases the catalytic activity. However, it should be noted that a higher reduction potential of the RhIII/RhI couple could lead to decreased electron transfer rates from the excited photosensitizer onto the catalyst, thereby overcompensating the positive effect described above. Finally, if the catalytically active species is formed, the light independent transfer of the hydride onto the cationic nicotinamide substrate proceeds either via the mechanism described by Lo. et al. [83,84] or via the reduced Cp* ligand as hypothesized by Pitman et al. [61].
When working with oxidized nicotinamide cofactors as substrates for electrochemical or photochemical reduction processes, special care must be taken in order to prevent the undesirable formation of enzymatically inactive isomers [94], as depicted in Figure 12, since they limit the practical applicability of visible light driven cofactor reductions due to lower enzymatic substrate (S) to product (P) conversions as a consequence of an inefficient NAD(P)H regeneration.
It has been shown that under photocatalytic conditions and in the absence of the rhodium catalyst, the formation of complex product mixtures is possible [110]. Steckhan and coworkers identified the reduced nicotinamide cofactor product during photoredox catalysis with [Ru(bpy)3]2+ as the chromophore in the absence of a rhodium catalyst as the dimeric (NAD)2 species [73]. They propose its formation by radical coupling of the neutral, one electron reduced NAD molecules [111] (see Figure 12), which are formed after electron transfer from the photoreduced chromophore.
As these dimeric species and the bioactive NADH all show a characteristic UV absorbance band at 340 nm, unambiguous identification of the biologically consumable product is not possible using only UV/Vis spectroscopy [112].
Since the quantity of bioinactive reduced NAD species is commonly small, it can be rationalized that the electron transfer from a highly energy rich excited chromophore to the catalyst is faster than to the oxidized nicotinamide, probably due to the easier reduction of the RhIII center. Although the light driven redox reactions of Figure 12 are depicted to proceed via the excited chromophore, they could also take place via the reduced photosensitizer due to a reductive quenching of the excited state by a suitable electron donor.
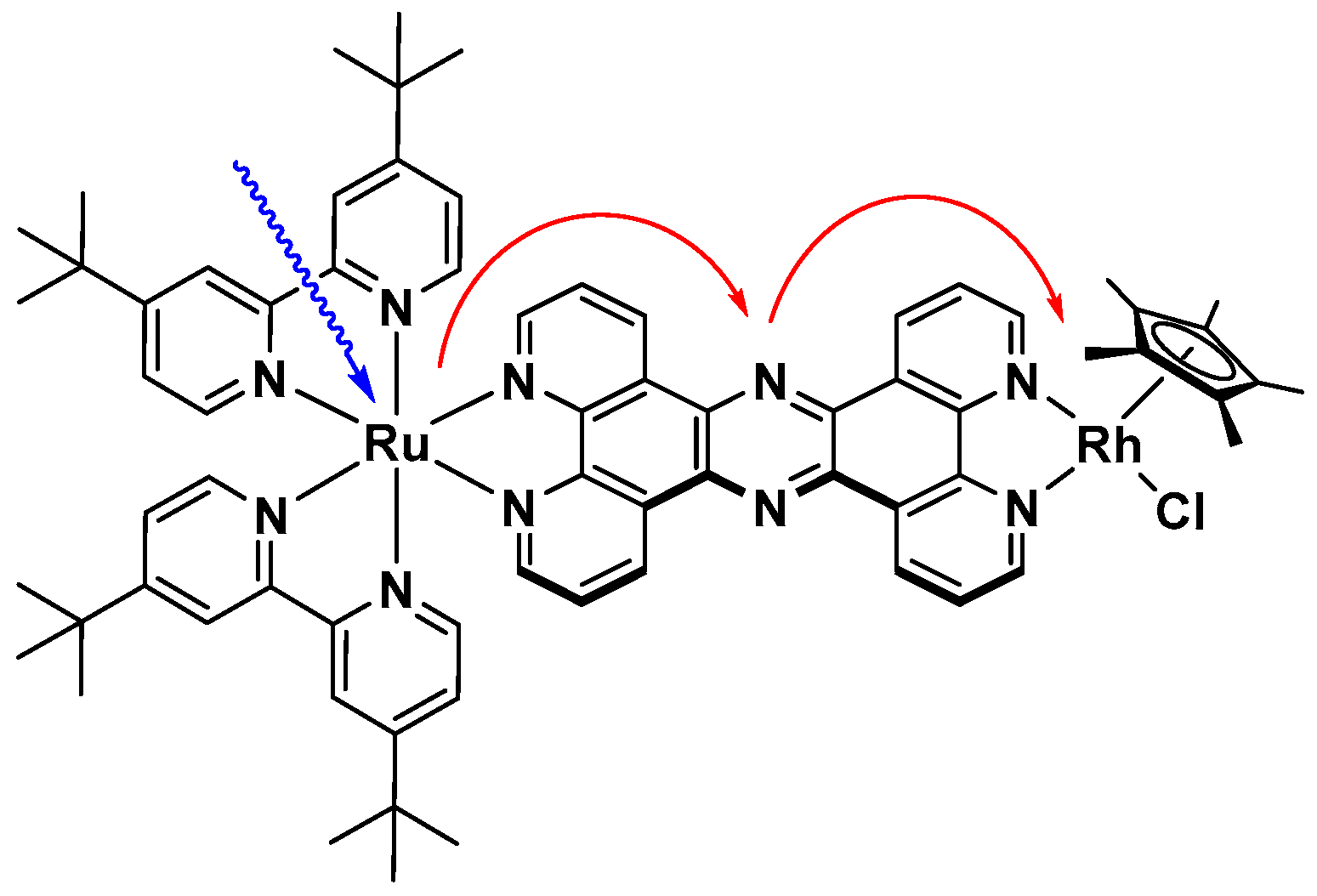
However, the immanent problem of the direct one electron reduction of the nicotinamide cofactors using intermolecular homogenous photocatalytic systems might be solved by oligonuclear coordination compounds. These could exhibit, due to their molecular preorganisation of the chromophore and catalyst, much faster collision independent electron transfers onto the latter, which would consequently decrease the amount of bioinactive NAD(P) dimers, even at low catalyst concentrations. An example of these catalysts is depicted in Figure 13, where a photoredox active ruthenium polypyridine chromophore is linked to a [(NN)Rh(Cp*)Cl] catalyst via a tetrapyridophenazine bridging ligand [99]. The molecular catalyst exhibits suitable redox potentials for intramolecular electron transfers [113].
Figure 12 also shows that reduced nicotinamide cofactors are exposed to oxidative decomposition during a photocatalytic process since they are stronger reductants than the commonly used tertiary amines such as triethylamine or triethanolamine [114]. Ishitani and coworkers successfully used artificial NADH derivatives (BNAH = N-benzyl nicotinamide) as sacrificial electron donors for the photocatalytic reduction of CO2. They observed that under the utilized conditions, BNAH acts as a simple one electron donor. Therefore, they hypothesized that the dimeric species (BNA)2, despite its higher reduction potential in comparison to the monomeric BNAH, represents the endpoint of the photooxidation pathway. This was ascribed to fast back electron transfers as a consequence of the rather slow decomposition kinetics of (BNA)2 [23,24,115].
In contrast to the findings of the Ishitani group, where BNAH and (BNA)2 act as competing excited state quenchers during the photocatalysis, several other reports indicate that the dimeric species can also serve as an effective electron donor. Irradiation of C60 and C70 fullerenes led to the generation of the reduced carbon allotropes with quantum yields exceeding unity, showing the ability of (BNA)2 to act as a two electron reductant with the concomitant formation of the oxidized BNA+ [116,117,118]. Even the generation of a two electron reduced tetranuclear Ru complex under visible light irradiation has been reported using the dimer as a sacrificial electron donor [119].
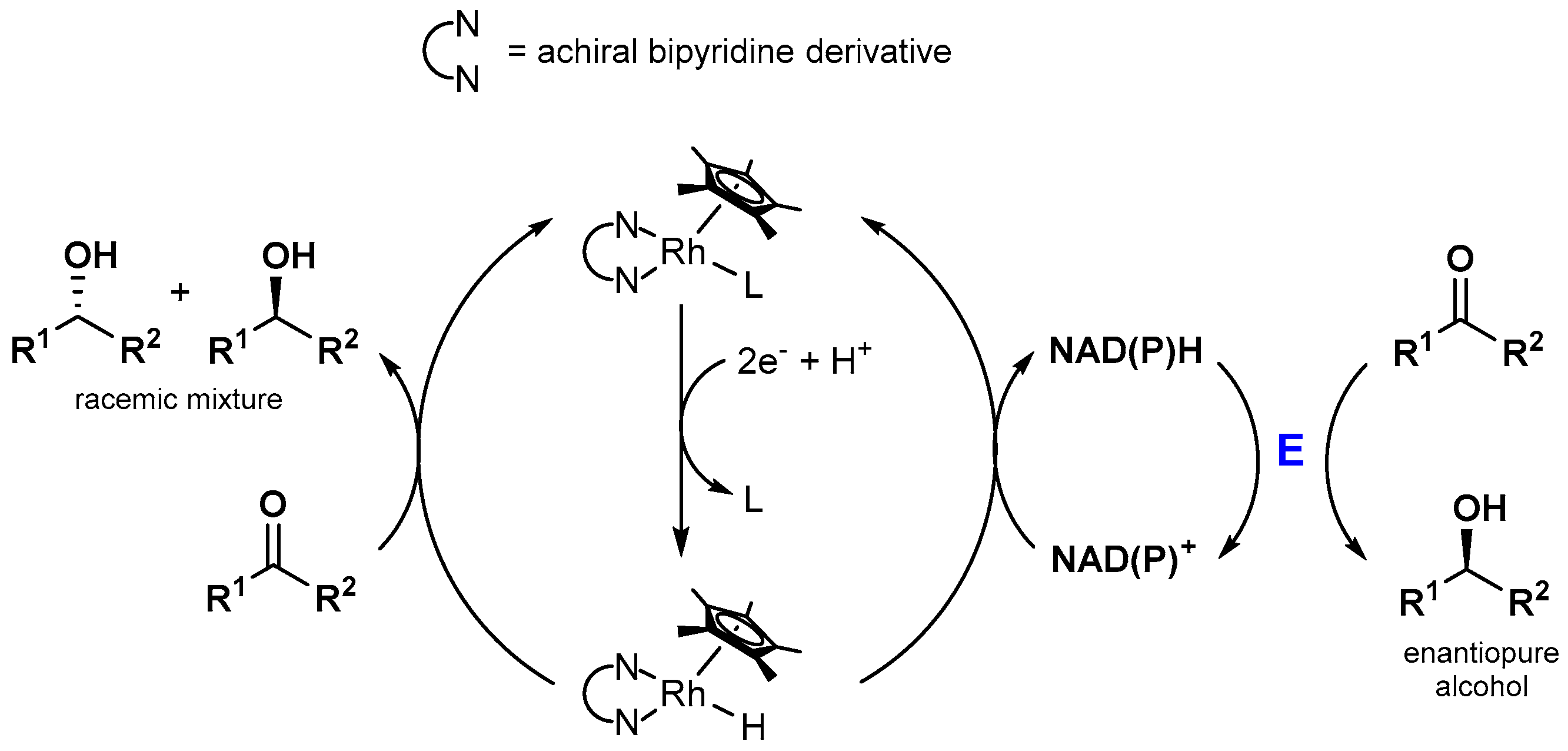
Since enzyme catalyzed reactions are highly interesting due to aspects such as stereoselectivity or high substrate conversion rates at low temperatures, the photocatalytic production of NAD(P)H is often coupled to a biocatalytic dark reaction, as schematically drawn in Figure 11. Interestingly, simple control experiments almost always reveal that the highly effective substrate conversion is only possible in the presence of the enzyme. This indicates that the catalytically active [(NN)Rh(Cp*)H]+ species exhibits high chemoselectivity for NAD(P)H generation over ketone [93,94] or CO2 [95,97,104,106] reduction under the applied conditions, although the latter substrates are added in high excess. In terms of enzymatic stereoselective alcohol formation, this chemoselectivity is highly beneficial, as shown in Figure 14. The achiral rhodium catalyst is not able to produce significant amounts of the racemic product mixture and therefore does not decrease the enantiomeric excess of the overall system. However, the lack of product formation in the absence of an enzyme in the case of the CO2 reduction shows that direct carbon-based solar fuel production using [(NN)Rh(Cp*)X]n+ catalysts could be accompanied by significant obstacles.
3.4. Hypothesis of an Overall Reaction Scheme for the Competing Generation of Molecular Hydrogen and Reduced Nicotinamide Cofactors: Application of [(bpy)Rh(Cp*)X]n+ as a Catalytic Center for Selective Homogeneous Photocatalysis
As described above, [(NN)Rh(Cp*)X]n+ catalysts exhibit different reaction rates for the reduction of various substrates, which finally leads to a high chemoselectivity under appropriate conditions. This behavior apparently has to be correlated with the underlying intertwined catalytic cycles and the energies of important intermediates and transition states, which allows the rhodium catalyst to efficiently discriminate between various reaction partners by virtue of thermodynamic reasons.
A lot of effort has been focused on elucidating the catalytic mechanisms of [(NN)Rh(Cp*)X]n+ complexes in the presence of several substrates. The following discussion intends to create a coherent picture of the intricate mechanisms of hydrogen and NADH production from the available literature. Special attention will be given to slightly basic conditions, since the TONs for NAD+ reduction have been observed to be higher by a factor of 100 in contrast to the TONs for hydrogen production using a heterobimetallic photocatalyst [99]. Based on other literature reports, this finding could be anticipated, since for the photocatalytic formation of hydrogen, an optimal pH value of 3.6 has been found [37]. Although under electrochemical conditions the lowering of the pH value was reported to increase the rate of hydrogen generation [34], for the formation of NADH, a maximum of catalytic activity was observed at neutral pH conditions [56].
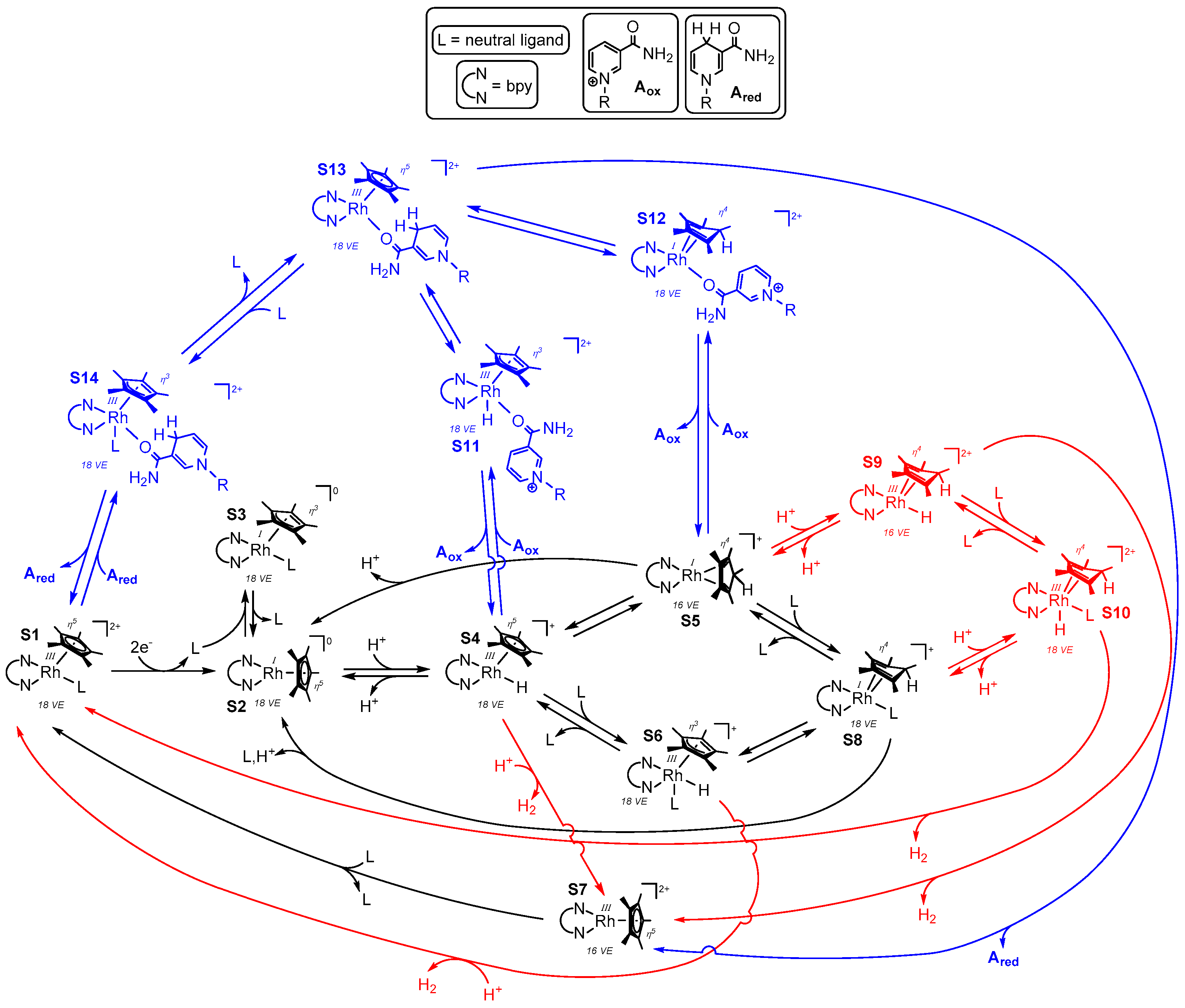
As depicted in Figure 15, the different proposed mechanistic cycles for H2 formation and selective NAD+ reduction are unified in one scheme, together with some additional possible intermediates, which could also represent transition states on the road to the actual involved species. The coloration of Figure 15 is aimed to discriminate reaction steps and intermediates that refer to both catalytic cycles (black), the NAD(P)H formation (blue), and to the evolution of hydrogen (red).
Starting from species S1, S2 is formed after the reduction by two electrons (or by the reaction with an appropriate electron donor, as depicted in Figure 4). Reversible coordination of a ligand L might lead to the formation of species S3, as previously assumed by Chardon-Noblat et al. [35]. By changing the binding mode of the Cp* ligand from η5 to η3, the 18 valence electron count would be preserved on the central metal in S3. After an oxidative addition of a proton at the electron rich rhodium center [62], S2 converts into S4. Depending on the N,N-chelating ligand, the pKa value of the metal bound (S4) or possibly Cp* bound hydrogen in S5 and S8 varies between seven and eight [58,70]. In accordance with these findings, pH dependent electrochemical investigations showed that by increasing the pH value from seven to 10, the reversibility of the RhIII/RhI couple increases, which could be ascribed to the lower formation of species S4 [51].
Direct hydrogen evolution from species S4 could be achieved by an acid base reaction out of the coordination sphere yielding species S7. As the latter is coordinatively unsaturated and exhibits a valence electron count of 16, S7 might be relatively high in energy, and either a powerful driving force such as a very low pH value or an alternative pathway of hydrogen evolution via S6 occurs, where the preformation of S1, as well as the rearomatization of the Cp* ligand, could facilitate the simple acid base reaction.
Although these reactions might occur, the transformation of S4 to S5 has been reported to be thermodynamically favourable [62] and crystal structures of the 18 valence electron species S8 have been obtained [61,62]. Therefore, hydrogen evolution might proceed via a metal assisted intramolecular H2 elimination from either S9 or via the intermediate S10, which could facilitate the release of the product as described above. However, as S4 and S5 should stay in equilibrium with each other, it might be possible that hydrogen evolution only occurs via the repopulation of S4 and an intermolecular acid base reaction.
Based on the work of Lo et al., it is clear that for the generation of the 1,4-reduced dihydronicotinamides, binding of the carbamoyl moiety via the oxygen atom to the rhodium catalyst is essential [83,84]. As for the evolution of hydrogen, the generation of NAD(P)H could proceed via S4 or S5, yielding either the ring slipped intermediate S11 as proposed by Lo et al. or species S12 as proposed by Pitman et al. [61]. The subsequent selective hydride transfer onto the bound nicotinamide could generate intermediate S13 in both cases, which again could lose the reaction product directly via S7 or ligand assisted via S14, where the latter could represent a transition state between S13 and S1.
As S5 has a lower energy content than S4, the high chemoselectivity of NAD(P)H formation over H2 generation at neutral (i.e., cofactor and enzyme compatible) pH values might be caused by the electronic situation of the intermediate S5. Its HOMO is a RhI centered dz2 orbital and the high electron density on the metal is stabilized by π backbonding into the π-accepting Cp*H ligand. The preference for S5 over S4 therefore seems to arise from an energetically well balanced electron density distribution, although such an interplay could have also be assumed for the interaction of the electron poor RhIII center in S4 and the electron rich σ-donating Cp* ligand.
However, the crucial difference between hydrogen evolution catalysis and nicotinamide reduction catalysis might be the disruption of the well balanced electron density distribution in S5. Binding of the nicotinamide to S5 yields S12, where an electron rich RhI metal is still bound to the π-accepting Cp*H ligand, indicating that the distribution of the electron density is still efficiently ensured. On the contrary, the interaction of a proton with the dz2 HOMO of S5 would lead to the formation of S9, where a formally electron poor RhIII center is bound to the π-accepting Cp*H ligand. Due to the possibly much less effective interplay of mutual stabilizing effects in S9 compared to S12, the energy barrier for the formation of the latter would be lower, which could explain the different observed reaction rates for hydrogen production and NAD(P)H formation at neutral pH values [99].
Finally it should be noted that the hypothesized reasons for the observed chemoselectivity of [(NN)Rh(Cp*)X]n+ catalysts are based on literature reports and would need further theoretical calculations as support. Moreover, the low substrate (i.e., H+ or H3O+) concentration for the hydrogen evolution catalysis at pH 7 in comparison to commonly used NAD(P)+ concentrations, which are generally in the μM to mM range, might additionally play a significant role in the observed reaction rate differences.
4. Conclusions
Our aim of this feature article was to summarize the broad and interesting chemistry of [(NN)Rh(Cp*)X]n+ catalysts, as well as to raise the readers’ awareness for general concepts and examples of chemoselective catalysis within homogeneous artificial systems. By comparing the results of different catalytic approaches for the reductive transformation of similar substrates and by providing a mechanistic overview over the mostly independently discussed but intertwined catalytic cycles of hydrogen evolution catalysis and NAD(P)H formation, we hope to contribute to a fruitful development of these multifunctional rhodium-based catalysts in order to create pathways for the effective solar driven generation of sustainable energy carriers.
Due to the high selectivity of the catalytically active Rh species towards NAD(P)H formation at neutral pH conditions, the practical application of these photocatalytic systems could be possible by their integration into biotechnological processes. Within this context, the availability of “cheap” electrons for the regeneration of the oxidized photosensitizer is crucial. Herein the immobilization of fully active molecular systems at suitable electrode surfaces could be one of the methods employed to achieve this critical prerequisite.
Acknowledgments
Alexander K. Mengele acknowledges generous financial support by a Chemiefonds-Stipendium of Fonds der Chemischen Industrie.
Author Contributions
The conceptual design and writing of the manuscript was done by Alexander K. Mengele. Suggestions regarding the scope of the article as well as critical proof reading was performed by Sven Rau.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Monastersky, R. The human age. Nature 2015, 519, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Armor, J.N. A history of industrial catalysis. Catal. Today 2011, 163, 3–9. [Google Scholar] [CrossRef]
- Yang, H.; Yang, X.; Tang, W. Transition-metal catalyzed asymmetric carbon–carbon cross-coupling with chiral ligands. Tetrahedron 2016, 72, 6143–6174. [Google Scholar] [CrossRef]
- Renger, G. The light reactions of photosynthesis. Curr. Sci. 2010, 98, 1305–1319. [Google Scholar]
- Liu, Z.; Yan, H.; Wang, K.; Kuang, T.; Zhang, J.; Gui, L.; An, X.; Chang, W. Crystal structure of spinach major light-harvesting complex at 2.72 Å resolution. Nature 2004, 428, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, N. Photochemical Reactions as Key Steps in Organic Synthesis. Chem. Rev. 2008, 108, 1052–1103. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Takeda, H.; Ishitani, O. Photocatalytic reduction of CO2 using metal complexes. J. Photochem. Photobiol. C 2015, 25, 106–137. [Google Scholar] [CrossRef]
- Sutin, N.; Creutz, C.; Fujita, E. Photo-Induced Generation of Dihydrogen and Reduction of Carbon Dioxide Using Transition Metal Complexes. Comments Inorg. Chem. 1997, 19, 67–92. [Google Scholar] [CrossRef]
- Walther, D.; Ruben, M.; Rau, S. Carbon dioxide and metal centres: From reactions inspired by nature to reactions in compressed carbon dioxide as solvent. Coord. Chem. Rev. 1999, 182, 67–100. [Google Scholar] [CrossRef]
- Fujita, E. Photochemical carbon dioxide reduction with metal complexes. Coord. Chem. Rev. 1999, 185–186, 373–384. [Google Scholar] [CrossRef]
- Morris, A.J.; Meyer, G.J.; Fujita, E. Molecular Approaches to the Photocatalytic Reduction of Carbon Dioxide for Solar Fuels. Acc. Chem. Res. 2009, 43, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Ishitani, O. Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies. Coord. Chem. Rev. 2010, 254, 346–354. [Google Scholar] [CrossRef]
- Doherty, M.D.; Grills, D.C.; Muckerman, J.T.; Polyansky, D.E.; Fujita, E. Toward more efficient photochemical CO2 reduction: Use of scCO2 or photogenerated hydrides. Coord. Chem. Rev. 2010, 254, 2472–2482. [Google Scholar] [CrossRef]
- Tamaki, Y.; Koike, K.; Morimoto, T.; Ishitani, O. Substantial improvement in the efficiency and durability of a photocatalyst for carbon dioxide reduction using a benzoimidazole derivative as an electron donor. J. Catal. 2013, 304, 22–28. [Google Scholar] [CrossRef]
- Tamaki, Y.; Koike, K.; Morimoto, T.; Yamazaki, Y.; Ishitani, O. Red-Light-Driven Photocatalytic Reduction of CO2 using Os(II)–Re(I) Supramolecular Complexes. Inorg. Chem. 2013, 52, 11902–11909. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Imori, D.; Morimoto, T.; Koike, K.; Ishitani, O. High catalytic abilities of binuclear rhenium(I) complexes in the photochemical reduction of CO2 with a ruthenium(II) photosensitiser. Dalton Trans. 2016, 45, 14668–14677. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, Y.; Ishitani, O. Iridium(III) 1-Phenylisoquinoline Complexes as a Photosensitizer for Photocatalytic CO2 Reduction: A Mixed System with a Re(I) Catalyst and a Supramolecular Photocatalyst. Inorg. Chem. 2016, 55, 5702–5709. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Tamaki, Y.; Ueno, K.; Kato, E.; Nishikawa, T.; Ohkubo, K.; Yamazaki, Y.; Morimoto, T.; Ishitani, O. Photocatalytic Reduction of Low Concentration of CO2. J. Am. Chem. Soc. 2016, 138, 13818–13821. [Google Scholar] [CrossRef] [PubMed]
- Rohacova, J.; Ishitani, O. Rhenium(I) trinuclear rings as highly efficient redox photosensitizers for photocatalytic CO2 reduction. Chem. Sci. 2016, 7, 6728–6739. [Google Scholar] [CrossRef] [PubMed]
- Nakada, A.; Koike, K.; Nakashima, T.; Morimoto, T.; Ishitani, O. Photocatalytic CO2 Reduction to Formic Acid Using a Ru(II)–Re(I) Supramolecular Complex in an Aqueous Solution. Inorg. Chem. 2015, 54, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, T.; Huang, H.-H.; Wang, J.-W.; Zhong, D.-C.; Lu, T.-B. A Dinuclear Cobalt Cryptate as a Homogeneous Photocatalyst for Highly Selective and Efficient Visible-Light Driven CO2 Reduction to CO in CH3CN/H2O Solution. Angew. Chem. Int. Ed. 2017, 56, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Morimoto, T.; Koike, K.; Ishitani, O. Photocatalytic CO2 reduction with high turnover frequency and selectivity of formic acid formation using Ru(II) multinuclear complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 15673–15678. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Koike, K.; Ishitani, O. Highly efficient, selective, and durable photocatalytic system for CO2 reduction to formic acid. Chem. Sci. 2015, 6, 7213–7221. [Google Scholar] [CrossRef]
- Kuriki, R.; Matsunaga, H.; Nakashima, T.; Wada, K.; Yamakata, A.; Ishitani, O.; Maeda, K. Nature-Inspired, Highly Durable CO2 Reduction System Consisting of a Binuclear Ruthenium(II) Complex and an Organic Semiconductor Using Visible Light. J. Am. Chem. Soc. 2016, 138, 5159–5170. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.; Slanina, T.; König, B. Visible light photocatalytic reduction of aldehydes by Rh(III)–H: A detailed mechanistic study. Chem. Sci. 2015, 6, 2027–2034. [Google Scholar] [CrossRef]
- Blacker, A.J.; Clot, E.; Duckett, S.B.; Eisenstein, O.; Grace, J.; Nova, A.; Perutz, R.N.; Taylor, D.J.; Whitwood, A.C. Synthesis and structure of ‘‘16-electron’’ rhodium(III) catalysts for transfer hydrogenation of a cyclic imine: Mechanistic implications. Chem. Commun. 2009, 44, 6801–6803. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Han, J.; Lee, E.S.; Choi, E.B.; Lee, H.-K. Enantioselective Synthesis of Cyclic Sulfamidates by Using Chiral Rhodium-Catalyzed Asymmetric Transfer Hydrogenation. Org. Lett. 2010, 12, 4184–4187. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Lin, Z.; Wang, Q.; Wang, X.; Cun, L.; Yuan, W.; Zhu, J.; Deng, J. Rh(II)-Cp*-TsDPEN catalyzed aqueous asymmetric transfer hydrogenation of chromenones into saturated alcohol: C=C and C=O reduction in one step. Tetrahedron Lett. 2012, 53, 3828–3830. [Google Scholar] [CrossRef]
- Wang, C.; Li, C.; Wu, X.; Pettman, A.; Xiao, J. pH-Regulated Asymmetric Transfer Hydrogenation of Quinolines in Water. Angew. Chem. Int. Ed. 2009, 48, 6524–6528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qiu, R.; Xue, X.; Pan, Y.; Xu, C.; Li, H.; Xu, L. Versatile (Pentamethylcyclopentadienyl) rhodium-2,2’-Bipyridine (Cp*Rh-bpy) Catalyst for Transfer Hydrogenation of N-Heterocycles in Water. Adv. Synth. Catal. 2015, 357, 3529–3537. [Google Scholar] [CrossRef]
- Ryu, J.; Nam, D.H.; Lee, S.H.; Park, C.B. Biocatalytic Photosynthesis with Water as an Electron Donor. Chem. Eur. J. 2014, 20, 12020–12025. [Google Scholar] [CrossRef] [PubMed]
- Kölle, U.; Grätzel, M. Organometallic Rhodium(III) Complexes as Catalysts for the Photoreduction of Protons to Hydrogen on Colloidal TiO2. Angew. Chem. Int. Ed. Engl. 1987, 26, 567–570. [Google Scholar] [CrossRef]
- Cosnier, S.; Deronzier, A.; Vlachopoulos, N. Carbon/Poly{pyrrole-[(C5Me5)RhIII(bpy)Cl]+} Modified Electrodes; a Molecularly-based Material for Hydrogen Evolution (bpy = 2,2′-bipyridine). J. Chem. Soc. Chem. Commun. 1989, 1259–1261. [Google Scholar] [CrossRef]
- Chardon-Noblat, S.; Cosnier, S.; Deronzier, A.; Vlachopoulos, N. Electrochemical properties of [(C5Me5)RhIII(L)Cl]+ complexes (L = 2,2′-bipyridine or 1,10-phenanthroline derivatives) in solution and in related polypyrrolic films. Application to electrocatalytic hydrogen generation. J. Electroanal. Chem. 1993, 352, 213–228. [Google Scholar] [CrossRef]
- Caix, C.; Chardon-Noblat, S.; Deronzier, A.; Moutet, J.-C.; Tingry, S. (Pentamethylcyclopentadienyl) (polypyridyl) rhodium and iridium complexes as electrocatalysts for the reduction of protons to dihydrogen and the hydrogenation of organics. J. Organomet. Chem. 1997, 540, 105–111. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Kobayashi, T.; Suenobu, T. Photocatalytic Production of Hydrogen by Disproportionation of One-Electron-Reduced Rhodium and Iridium–Ruthenium Complexes in Water. Angew. Chem. Int. Ed. 2011, 50, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, J.D.; Gupto, A.; Warren, J.J.; Brunschwig, B.S.; Gray, H.B. Noncovalent Immobilization of Electrocatalysts on Carbon Electrodes for Fuel Production. J. Am. Chem. Soc. 2013, 135, 18288–18291. [Google Scholar] [CrossRef] [PubMed]
- Caix, C.; Chardon-Noblat, S.; Deronzier, A. Electrocatalytic reduction of CO2 into formate with [(η5-Me5C5)M(L)CI]+ complexes (L = 2,2′-bipyridine ligands; M = Rh(III) and Ir(III)). J. Electroanal. Chem. 1997, 434, 163–170. [Google Scholar] [CrossRef]
- Chambers, M.B.; Wang, X.; Elgrishi, N.; Hendon, H.C.; Walsh, A.; Bonnefoy, J.; Canivet, J.; Quadrelli, E.A.; Farrusseng, D.; Mellot-Draznieks, C.; et al. Photocatalytic Carbon Dioxide Reduction with Rhodium-based Catalysts in Solution and Heterogenized within Metal–Organic Frameworks. ChemSusChem 2015, 8, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Leiva, C.; Lo, H.C.; Fish, R.H. Aqueous organometallic chemistry. 3. Catalytic hydride transfer reactions with ketones and aldehydes using [Cp*Rh(bpy)(H2O)](OTf)2 as the precatalyst and sodium formate as the hydride source: Kinetic and activation parameters, and the significance of steric and electronic effects. J. Organomet. Chem. 2010, 695, 145–150. [Google Scholar]
- Montalvo-González, R.; Chávez, D.; Aguirre, G.; Parra-Hake, M.; Samanathan, R. Asymmetric Transfer Hydrogenation of Ketones in Aqueous Solution Catalyzed by Rhodium(III) Complexes with C3-Symmetric Fluorene-Ligands Containing Chiral (1R,2R)-Cyclohexane-1,2-diamine. J. Braz. Chem. Soc. 2010, 21, 431–435. [Google Scholar] [CrossRef]
- Himeda, Y.; Onozawa-Komatsuzaki, N.; Sugihara, H.; Arakawa, H.; Kasuga, K. Transfer hydrogenation of a variety of ketones catalyzed by rhodium complexes in aqueous solution and their application to asymmetric reduction using chiral Schiff base ligands. J. Mol. Catal. A Chem. 2003, 195, 95–100. [Google Scholar] [CrossRef]
- Thorpe, T.; Blacker, J.; Brown, S.M.; Bubert, C.; Crosby, J.; Fitzjohn, S.; Muxworthy, J.P.; Williams, J.M.J. Efficient rhodium and iridium-catalysed asymmetric transfer hydrogenation using water-soluble aminosulfonamide ligands. Tetrahedron Lett. 2001, 42, 4041–4043. [Google Scholar] [CrossRef]
- Ruppert, R.; Herrmann, S.; Steckhan, E. Very Efficient Reduction of NAD(P)+ with Formate catalysed by Cationic Rhodium Complexes. J. Chem. Soc. Chem. Commun. 1988, 1150–1151. [Google Scholar] [CrossRef]
- Steckhan, E.; Herrmann, S.; Ruppert, R.; Thömmes, J.; Wandrey, C. Continuous Generation of NADH from NAD+ and Formate Using a Homogeneous Catalyst with Enhanced Molecular Weight in a Membrane Reactor. Angew. Chem. Int. Ed. Engl. 1990, 29, 388–390. [Google Scholar] [CrossRef]
- Westerhausen, D.; Herrmann, S.; Hummel, W.; Steckhan, E. Formate-Driven, Non-Enzymatic NAD(P)H Regeneration for the Alcohol Dehydrogenase Catalyzed Stereoselective Reduction of 4-Phenyl-2-butanone. Angew. Chem. Int. Ed. Engl. 1992, 31, 1529–1531. [Google Scholar] [CrossRef]
- Hollmann, F.; Kleeb, A.; Otto, K.; Schmid, A. Coupled chemoenzymatic transfer hydrogenation catalysis for enantioselective reduction and oxidation reactions. Tetrahedron Asymmetry 2005, 16, 3512–3519. [Google Scholar] [CrossRef]
- Canivet, J.; Süss-Fink, G.; Stepnicka, P. Water-Soluble Phenanthroline Complexes of Rhodium, Iridium and Ruthenium for the Regeneration of NADH in the Enzymatic Reduction of Ketones. Eur. J. Inorg. Chem. 2007, 4736–4742. [Google Scholar] [CrossRef]
- Ruppert, R.; Herrmann, S.; Steckhan, E. Efficient Indirect Electrochemical in-situ Regeneration of NADH: Electrochemically Driven Enzymatic Reduction of Pyruvate Catalyzed by D-LDH. Tetrahedron Lett. 1987, 28, 6583–6586. [Google Scholar] [CrossRef]
- Steckhan, E.; Herrmann, S.; Ruppert, R.; Dietz, E.; Frede, M.; Spika, E. Analytical Study of a Series of Substituted (2,2′-Bipyridyl) (pentamethylcyclopentadienyl)rhodium and -iridium Complexes with Regard to Their Effectiveness as Redox Catalysts for the Indirect Electrochemical and Chemical Reduction of NAD(P)+. Organometallics 1991, 10, 1568–1577. [Google Scholar] [CrossRef]
- Cosnier, S.; Gunther, H. A polypyrrole [RhIII(C5Me5)(bpy)Cl]+ modified electrode for the reduction of NAD+ cofactor. J. Electroanal. Chem. 1991, 315, 307–312. [Google Scholar] [CrossRef]
- Hollmann, F.; Schmid, A.; Steckhan, E. The First Synthetic Application of a Monooxygenase Employing Indirect Electrochemical NADH Regeneration. Angew. Chem. Int. Ed. 2001, 40, 169–171. [Google Scholar] [CrossRef]
- Hildebrand, F.; Kohlmann, C.; Franz, A.; Lütz, S. Synthesis, Characterization and Application of New Rhodium Complexes for Indirect Electrochemical Cofactor Regeneration. Adv. Synth. Catal. 2008, 350, 909–918. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, J.H.; Park, C.B. Coupling Photocatalysis and Redox Biocatalysis toward Biocatalyzed Artificial Photosynthesis. Chem. Eur. J. 2013, 19, 4392–4406. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, F.; Witholt, B.; Schmid, A. [Cp*Rh(bpy)(H2O)]2+: A versatile tool for efficient and non-enzymatic regeneration of nicotinamide and flavin coenzymes. J. Mol. Catal. B Enzym. 2003, 19–20, 167–176. [Google Scholar] [CrossRef]
- Grau, M.M.; Poizat, M.; Arends, I.W.C.E.; Hollmann, F. Phosphite-driven, [Cp*Rh(bpy)(H2O)]2+-catalyzed reduction of nicotinamide and flavin cofactors: Characterization and application to promote chemoenzymatic reduction reactions. Appl. Organomet. Chem. 2010, 24, 380–385. [Google Scholar] [CrossRef]
- Van Esch, J.H.; Hoffmann, M.A.M.; Nolte, R.J.M. Reduction of Nicotinamides, Flavins, and Manganese Porphyrins by Formate, Catalyzed by Membrane-Bound Rhodium Complexes. J. Org. Chem. 1995, 60, 1599–1610. [Google Scholar] [CrossRef]
- Hollmann, F.; Lin, P.-C.; Witholt, B.; Schmid, A. Stereospecific Biocatalytic Epoxidation: The First Example of Direct Regeneration of a FAD-Dependent Monooxygenase for Catalysis. J. Am. Chem. Soc. 2003, 125, 8209–8217. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, S.; Kobayashi, T.; Suenobu, T. Efficient Catalytic Decomposition of Formic Acid for the Selective Generation of H2 and H/D Exchange with a Water-Soluble Rhodium Complex in Aqueous Solution. ChemSusChem 2008, 1, 827–834. [Google Scholar] [PubMed]
- Pitman, C.L.; Finster, O.N.L.; Miller, A.J.M. Cyclopentadiene-mediated hydride transfer from rhodium complexes. Chem. Commun. 2016, 52, 9105–9108. [Google Scholar] [CrossRef] [PubMed]
- Aguirre Quintana, L.M.; Johnson, S.I.; Corona, S.L.; Villatoro, W.; Goddard, W.A., III; Takase, M.K.; VanderVelde, D.G.; Winkler, J.R.; Gray, H.B.; Blakemore, J.D. Proton–hydride tautomerism in hydrogen evolution catalysis. Proc. Natl. Acad. Sci. USA 2016, 113, 6409–6414. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, V.; Sivanesan, D.; Yoon, S. Correlation between the Structure and Catalytic Activity of [Cp*Rh(Substituted Bipyridine)] Complexes for NADH Regeneration. Inorg. Chem. 2017, 56, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Fiedler, J.; Reinhardt, R.; Kaim, W. Metal vs. Ligand Reduction in Complexes of Dipyrido[3,2-a:2′,3′-c]phenazine and Related Ligands with [(C5Me5)ClM]+ (M = Rh or Ir): Evidence for Potential Rather Than Orbital Control in the Reductive Cleavage of the Metal-Chloride Bond. Inorg. Chem. 2004, 43, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Kölle, U.; Kang, B.-S.; Infelta, P.; Comte, P.; Grätzel, M. Elektrochemische und pulsradiolytische Reduktion von (Pentamethylcyclopentadienyl)(polypyridyl)rhodium-Komplexen. Chem. Ber. 1989, 122, 1869–1880. [Google Scholar] [CrossRef]
- Nakai, H.; Jeong, K.; Matsumoto, T.; Ogo, S. Catalytic C–F Bond Hydrogenolysis of Fluoroaromatics by [(η5-C5Me5)RhI(2,2′-bipyridine)]. Organometallics 2014, 33, 4349–4352. [Google Scholar] [CrossRef]
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.-L.; Xu, Q. Liquid organic and inorganic chemical hydrides for high-capacity hydrogen storage. Energy Environ. Sci. 2015, 8, 478–512. [Google Scholar] [CrossRef]
- Joó, F. Breakthroughs in Hydrogen Storage—Formic Acid as a Sustainable Storage Material for Hydrogen. ChemSusChem 2008, 1, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Kölle, U.; Fränzl, H. Oxidation of Alcohols by [Cp*Rh(ppy)(OH)]+. Monatsh. Chem. 2000, 131, 1321–1326. [Google Scholar] [CrossRef]
- Kim, S.; Lee, G.Y.; Baeg, J.-O.; Kim, Y.; Kim, S.-J.; Kim, J. Visible-Light-Driven Photoproduction of Hydrogen Using Rhodium Catalysts and Platinum Nanoparticles with Formate. J. Phys. Chem. C 2014, 118, 25844–25852. [Google Scholar] [CrossRef]
- Himeda, Y. Conversion of CO2 into Formate by Homogeneously Catalyzed Hydrogenation in Water: Tuning Catalytic Activity and Water Solubility through the Acid–Base Equilibrium of the Ligand. Eur. J. Inorg. Chem. 2007, 22, 3927–3941. [Google Scholar] [CrossRef]
- Hollmann, F.; Arends, I.W.C.E.; Buehler, K. Biocatalytic Redox Reactions for Organic Synthesis: Nonconventional Regeneration Methods. ChemCatChem 2010, 2, 762–782. [Google Scholar] [CrossRef]
- Kohlmann, C.; Märkle, W.; Lütz, S. Electroenzymatic synthesis. J. Mol. Catal. B Enzym. 2008, 51, 57–72. [Google Scholar] [CrossRef]
- Quinto, T.; Köhler, V.; Ward, T.R. Recent Trends in Biomimetic NADH Regeneration. Top. Catal. 2014, 57, 321–331. [Google Scholar] [CrossRef]
- Wienkamp, R.; Steckhan, E. Indirect Electrochemical Regeneration of NADH by a Bipyridinerhodium(I) Complex as Electron-Transfer Agent. Angew. Chem. Int. Ed. Engl. 1982, 21, 782–783. [Google Scholar] [CrossRef]
- Wienkamp, R.; Steckhan, E. Selective Generation of NADH by Visible Light. Angew. Chem. Int. Ed. Engl. 1983, 22, 497. [Google Scholar] [CrossRef]
- Umeda, K.; Nakamura, A.; Toda, F. Photochemical Reduction of NAD+ to 1,4-NADH without an Enzyme. J. Chem. Soc. Chem. Commun. 1990, 885–886. [Google Scholar] [CrossRef]
- Umeda, K.; Nakamura, A.; Toda, F. Catalytic Mechanism and Activity of Bis(2,2′:6′,2′′-terpyridine) rhodium(III) for the Reduction of NAD+ into NADH in a Photosensitized Reaction System. Bull. Chem. Soc. Jpn. 1993, 66, 2260–2267. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.-P. Electrochemical regeneration of nicotinamide cofactor using a polypyrrole rhodium bis-terpyridine modified electrode. J. Mol. Catal. 1997, 79, 133–140. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, S.; Lee, J.; Baeg, J.-O.; Kim, J. Photochemical Production of NADH Using Cobaloxime Catalysts and Visible-Light Energy. Inorg. Chem. 2012, 51, 8057–8063. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.C.; Fish, R.H. Biomimetic NAD+ Models for Tandem Cofactor Regeneration, Horse Liver Alcohol Dehydrogenase Recognition of 1,4-NADH Derivatives, and Chiral Synthesis. Angew. Chem. Int. Ed. 2002, 41, 478–481. [Google Scholar] [CrossRef]
- Lo, H.C.; Buriez, O.; Kerr, J.B.; Fish, R.H. Regioselective Reduction of NAD+ Models with [Cp*Rh(bpy)H]+: Structure—Activity Relationships and Mechanistic Aspects in the Formation of the 1,4-NADH Derivatives. Angew. Chem. Int. Ed. 1999, 38, 1429–1432. [Google Scholar] [CrossRef]
- Lo, H.C.; Leiva, C.; Buriez, O.; Kerr, J.B.; Olmstead, M.M.; Fish, R.H. Bioorganometallic Chemistry. 13. Regioselective Reduction of NAD+ Models, 1-Benzylnicotinamde Triflate and β-Nicotinamide Ribose-5′-methyl Phosphate, with in Situ Generated [Cp*Rh(Bpy)H]+: Structure-Activity Relationships, Kinetics, and Mechanistic Aspects in the Formation of the 1,4-NADH Derivatives. Inorg. Chem. 2001, 40, 6705–6716. [Google Scholar] [PubMed]
- Lee, S.H.; Lee, H.J.; Won, K.; Park, C.B. Artificial Electron Carriers for Photoenzymatic Synthesis under Visible Light. Chem. Eur. J. 2012, 18, 5490–5495. [Google Scholar] [CrossRef] [PubMed]
- Walcarius, A.; Nasraoui, R.; Wang, Z.; Qu, F.; Urbanova, V.; Etienne, M.; Göllü, M.; Demir, A.S.; Gajdzik, J.; Hempelmann, R. Factors affecting the electrochemical regeneration of NADH by (2,2′-bipyridyl) (pentamethylcyclopentadienyl)-rhodium complexes: Impact on their immobilization onto electrode surfaces. Bioelectrochemistry 2011, 82, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.; Hollmann, F.; Ho, T.V.; Schnyder, A.; Fish, R.H.; Schmid, A. Bioorganometallic chemistry: Biocatalytic oxidation reactions with biomimetic NAD+/NADH co-factors and [Cp*Rh(bpy)H]+ for selective organic synthesis. J. Organomet. Chem. 2004, 689, 4783–4790. [Google Scholar] [CrossRef]
- Hildebrand, F.; Lütz, S. Stable Electroenzymatic Processes by Catalyst Separation. Chem. Eur. J. 2009, 15, 4998–5001. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Nam, D.H.; Park, C.B. Screening Xanthene Dyes for Visible Light-Driven Nicotinamide Adenine Dinucleotide Regeneration and Photoenzymatic Synthesis. Adv. Synth. Catal. 2009, 351, 2589–2594. [Google Scholar] [CrossRef]
- Lee, S.H.; Nam, D.H.; Kim, J.H.; Baeg, J.-O.; Park, C.B. Eosin Y-Sensitized Artificial Photosynthesis by Highly Efficient Visible-Light-Driven Regeneration of Nicotinamide Cofactor. ChemBioChem 2009, 10, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.H.; Park, C.B. Visible Light-Driven NADH Regeneration Sensitized by Proflavine for Biocatalysis. ChemBioChem 2012, 13, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Baeg, J.-O.; Oh, G.H.; Park, N.-J.; Kong, K.-J.; Kim, J.; Hwang, D.W.; Biswas, S.K. A Photocatalyst–Enzyme Coupled Artificial Photosynthesis System for Solar Energy in Production of Formic Acid from CO2. J. Am. Chem. Soc. 2012, 134, 11455–11461. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Baeg, J.-O.; Park, N.-J.; Yadav, R.K. A Photocatalyst/Enzyme Couple That Uses Solar Energy in the Asymmetric Reduction of Acetophenones. Angew. Chem. Int. Ed. 2012, 51, 11624–11628. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Baeg, J.-O.; Park, N.-J.; Yadav, R.K. A solar light-driven, eco-friendly protocol for highly enantioselective synthesis of chiral alcohols via photocatalytic/biocatalytic cascades. Green Chem. 2014, 16, 4389–4400. [Google Scholar] [CrossRef]
- Yadav, R.K.; Baeg, J.-O.; Kumar, A.; Kong, K.-J.; Oh, G.H.; Park, N.-J. Graphene–BODIPY as a photocatalyst in the photocatalytic–biocatalytic coupled system for solar fuel production from CO2. J. Mater. Chem. A 2014, 2, 5068–5076. [Google Scholar] [CrossRef]
- Oppelt, K.T.; Wöß, E.; Stiftinger, M.; Schöfberger, W.; Buchberger, W.; Knör, G. Photocatalytic Reduction of Artificial and Natural Nucleotide Cofactors with a Chlorophyll-Like Tin-Dihydroporphyrin Sensitizer. Inorg. Chem. 2013, 52, 11910–11922. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, S.; Anjong, T.F.; Jang, H.Y.; Kim, J.-Y.; Lee, C.; Park, S.; Lee, H.J.; Yoon, J.; Kim, J. Artificial Photocatalytic System Using Polydiacetylene-(−NHphen)Ru(bpy)2 for Cofactor Regeneration and CO2 Reduction. J. Phys. Chem. C 2016, 120, 28407–28414. [Google Scholar] [CrossRef]
- Oppelt, K.T.; Gasiorowski, J.; Egbe, D.A.M.; Kollender, J.P.; Himmelsbach, M.; Hassel, A.W.; Sariciftci, N.S.; Knör, G. Rhodium-Coordinated Poly(arylene-ethynylene)-alt-Poly(arylenevinylene) Copolymer Acting as Photocatalyst for Visible-Light-Powered NAD+/NADH Reduction. J. Am. Chem. Soc. 2014, 136, 12721–12729. [Google Scholar] [CrossRef] [PubMed]
- Mengele, A.K.; Seibold, G.M.; Eikmanns, B.J.; Rau, S. Coupling Molecular Photocatalysis to Enzymatic Conversion. 2017; Submitted. [Google Scholar]
- Shi, Q.; Yang, D.; Jiang, Z.; Li, J. Visible-light photocatalytic regeneration of NADH using P-doped TiO2 nanoparticles. J. Mol. Catal. B Enzym. 2006, 43, 44–48. [Google Scholar] [CrossRef]
- Liu, J.; Antonietti, M. Bio-inspired NADH regeneration by carbon nitride photocatalysis using diatom templates. Energy Environ. Sci. 2013, 6, 1486–1493. [Google Scholar] [CrossRef]
- Ryu, J.; Lee, S.H.; Nam, D.H.; Park, C.B. Rational Design and Engineering of Quantum-Dot-Sensitized TiO2 Nanotube Arrays for Artificial Photosynthesis. Adv. Mater. 2011, 23, 1883–1888. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, J.; Zhou, H.; Antonietti, M. Uniform Graphitic Carbon Nitride Nanorod for Efficient Photocatalytic Hydrogen Evolution and Sustained Photoenzymatic Catalysis. ACS Appl. Mater. Interfaces 2014, 6, 8434–8440. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Yadav, R.K.; Kumar, A.; Park, N.-J.; Baeg, J.-O. Functionalized Graphene Quantum Dots as Efficient Visible-Light Photocatalysts for Selective Solar Fuel Production from CO2. ChemCatChem 2016, 8, 3389–3393. [Google Scholar] [CrossRef]
- Huang, X.; Liu, J.; Yang, Q.; Liu, Y.; Zhu, Y.; Li, T.; Tsang, Y.H.; Zhang, X. Microfluidic chip-based one-step fabrication of an artificial photosystem I for photocatalytic cofactor regeneration. RSC Adv. 2016, 6, 101974–101980. [Google Scholar] [CrossRef]
- Yadav, R.K.; Kumar, A.; Park, N.-J.; Kong, K.-J.; Baeg, J.-O. A highly efficient covalent organic framework film photocatalyst for selective solar fuel production from CO2. J. Mater. Chem. A 2016, 4, 9413–9418. [Google Scholar] [CrossRef]
- Liu, J.; Cazelles, R.; Chen, Z.P.; Zhou, H.; Galarneau, A.; Antonietti, M. The bioinspired construction of an ordered carbon nitride array for photocatalytic mediated enzymatic reduction. Phys. Chem. Chem. Phys. 2014, 16, 14699–14705. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Antonietti, M.; Liu, J. Bio-inspired carbon nitride mesoporous spheres for artificial photosynthesis: Photocatalytic cofactor regeneration for sustainable enzymatic synthesis. J. Mater. Chem. A 2014, 2, 7686–7693. [Google Scholar] [CrossRef]
- Lee, S.H.; Ryu, J.; Nam, D.H.; Park, C.B. Photoenzymatic synthesis through sustainable NADH regeneration by SiO2-supported quantum dots. Chem. Commun. 2011, 47, 4643–4645. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Baran, T.; Angelini, A.; Labuz, P.; Macyk, W. An integrated photocatalytic/enzymatic system for the reduction of CO2 to methanol in bioglycerol–water. Beilstein J. Org. Chem. 2014, 10, 2556–2565. [Google Scholar] [CrossRef] [PubMed]
- Land, E.J.; Swallow, A.J. One-Electron Reactions in Biochemical Systems as Studied by Puls Radiolysis. IV. Oxidation of Dihydronicotinamide-adenine dinucleotide. Biochim. Biophys. Acta 1971, 234, 34–42. [Google Scholar] [CrossRef]
- Kiwi, J. Photochemical Generation of Reduced β-Nicotinamide Adenine Dinucleotide (Induced by Light). J. Photochem. 1981, 16, 193–202. [Google Scholar] [CrossRef]
- Mengele, A.K.; Kaufhold, S.; Streb, C.; Rau, S. Generation of a stable supramolecular hydrogen evolving photocatalyst by alteration of the catalytic center. Dalton Trans. 2016, 45, 6612–6618. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, Y.; Odobel, F. Sacrificial electron donor reagents for solar fuel production. Comptes Rendus Chim. 2017, 20, 283–295. [Google Scholar] [CrossRef]
- Tamaki, Y.; Watanabe, K.; Koike, K.; Inoue, H.; Morimoto, T.; Ishitani, O. Development of highly efficient supramolecular CO2 reduction photocatalysts with high turnover frequency and durability. Faraday Discuss. 2012, 155, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, S.; Suenobu, T.; Patz, M.; Hirasaka, T.; Itoh, S.; Fujitsuka, M.; Ito, O. Selective One-Electron and Two-Electron Reduction of C60 with NADH and NAD Dimer Analogues via Photoinduced Electron Transfer. J. Am. Chem. Soc. 1998, 120, 8060–8068. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Suenobu, T.; Hirasaka, T.; Sakurada, N.; Arakawa, R.; Fujitsuka, M.; Ito, O. Enhanced Reactivity of C70 in the Photochemical Reactions with NADH and NAD Dimer Analogues As Compared to C60 via Photoinduced Electron Transfer. J. Phys. Chem. A 1999, 103, 5935–5941. [Google Scholar] [CrossRef]
- Sastre-Santos, Á.; Parejo, C.; Martín-Gomis, L.; Ohkubo, K.; Fernández-Lázaro, F.; Fukuzumi, S. C60 dimers connected through pleiadene bridges: Fullerenes talking to each other. J. Mater. Chem. 2011, 21, 1509–1515. [Google Scholar] [CrossRef]
- Ali, M.M.; Sato, H.; Haga, M.-A.; Tanaka, K.; Yoshimura, A.; Ohno, T. Two-Electron Reduction of [{(bpy)2Ru(dmbbbpy)}3Ru]8+ from (BNA)2 via Photoinduced Electron Transfer [dmbbbpy = 2,2′-Bis(N-methylbenzimidazole-2-yl)-4,4′-bipyridine]. Inorg. Chem. 1998, 37, 6176–6180. [Google Scholar] [CrossRef]
Figure 1.
General diagram for a multifunctional artificial photosynthetic system capable of transforming two different substrates (A and C) to the appropriate redox products (B and D). The system’s main components are the photosensitizer (PS) and the catalytic center (CC, in its activated/inactive form denoted with the subscript a/i). The regeneration of PS by a reaction with a sacrificial agent is omitted for clarity.
Figure 1.
General diagram for a multifunctional artificial photosynthetic system capable of transforming two different substrates (A and C) to the appropriate redox products (B and D). The system’s main components are the photosensitizer (PS) and the catalytic center (CC, in its activated/inactive form denoted with the subscript a/i). The regeneration of PS by a reaction with a sacrificial agent is omitted for clarity.

Figure 2.
Simplified reaction scheme for the competing reduction reactions involved in artificial photocatalytic CO2 reducing systems. The n electrons stem from an excited/reduced photosensitizer.
Figure 2.
Simplified reaction scheme for the competing reduction reactions involved in artificial photocatalytic CO2 reducing systems. The n electrons stem from an excited/reduced photosensitizer.
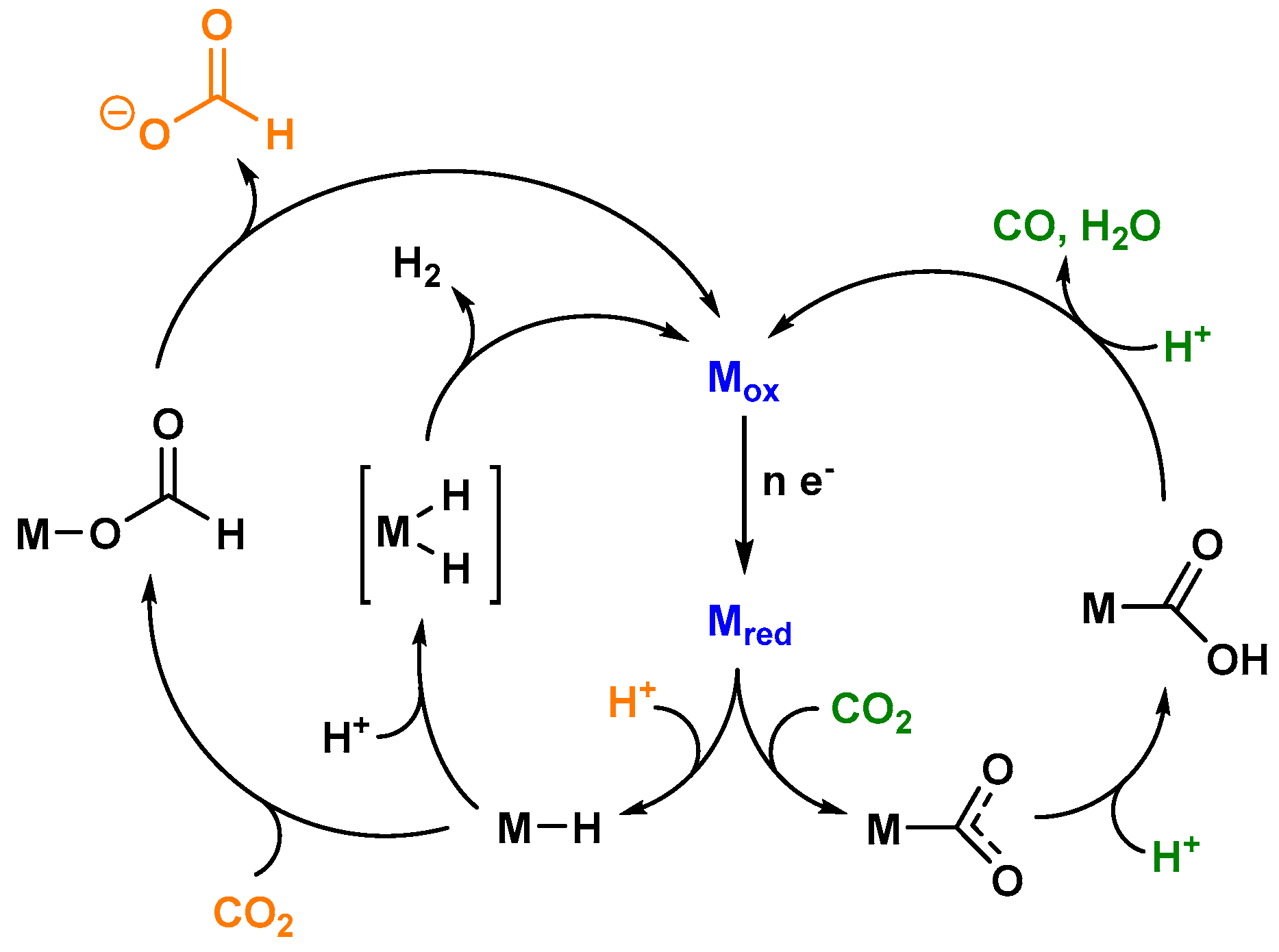
Figure 3.
Selected reduction reactions that can be accomplished by [(bpy)Rh(Cp*)X]n+ under appropriate reaction conditions. Literature: reaction (a) [33,34,35,36,37,38], reaction (b) [39,40], reaction (c) [26,29,41,42,43,44], reaction (d) [45,46,47,48,49,50,51,52,53,54,55], and reaction (e) [48,56,57,58,59].
Figure 3.
Selected reduction reactions that can be accomplished by [(bpy)Rh(Cp*)X]n+ under appropriate reaction conditions. Literature: reaction (a) [33,34,35,36,37,38], reaction (b) [39,40], reaction (c) [26,29,41,42,43,44], reaction (d) [45,46,47,48,49,50,51,52,53,54,55], and reaction (e) [48,56,57,58,59].
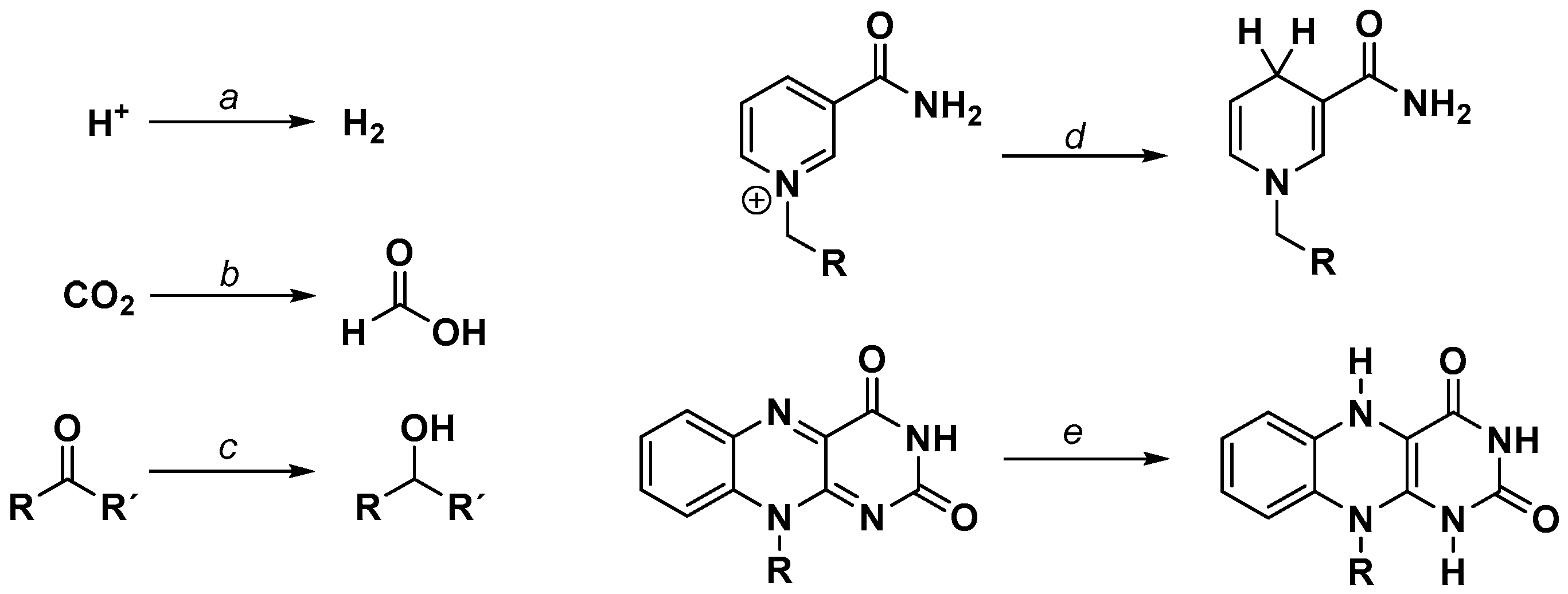
Figure 4.
Simplified access paths to the catalytically active species.
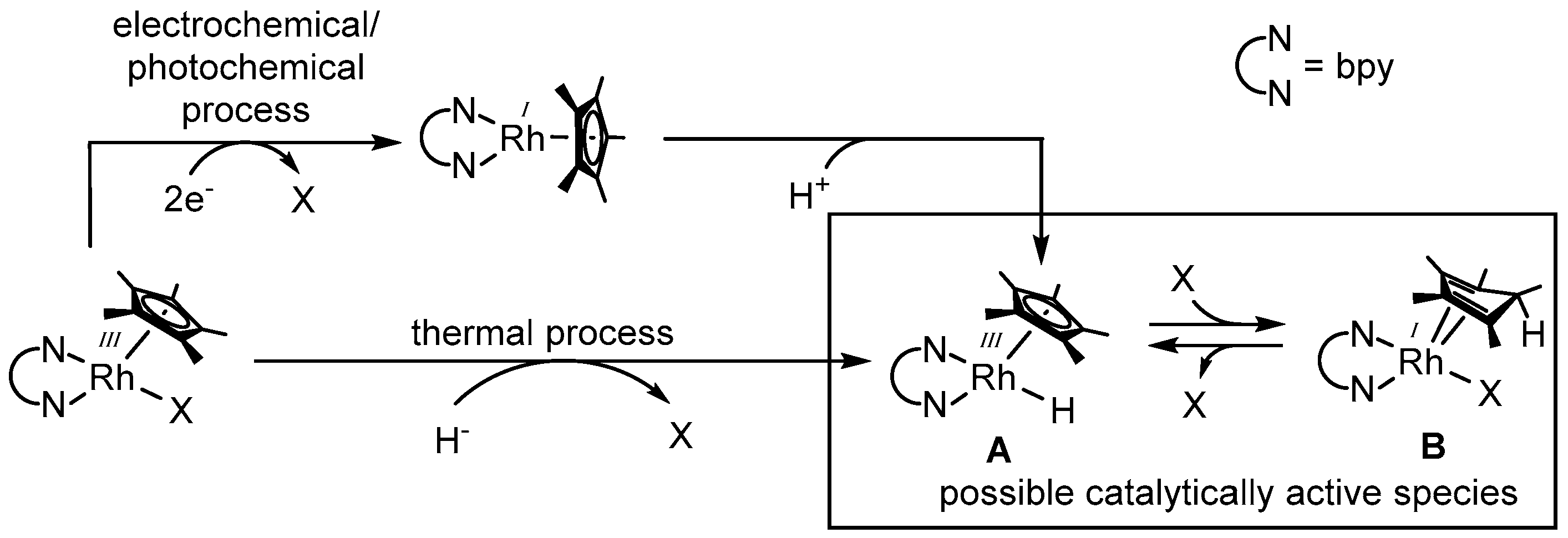
Figure 5.
Molecular structure of different regioisomeric dihydroxymethyl bipyridines bound to the Rh(Cp*)Cl fragment [63].
Figure 5.
Molecular structure of different regioisomeric dihydroxymethyl bipyridines bound to the Rh(Cp*)Cl fragment [63].
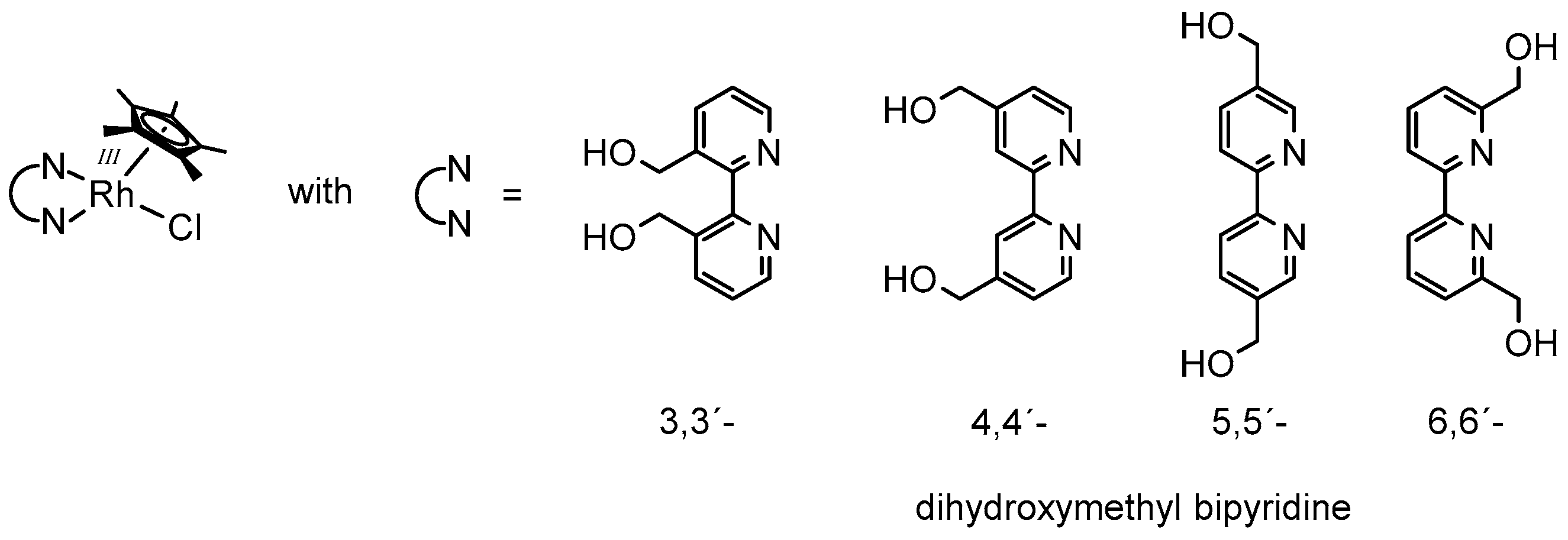
Figure 6.
Range of carbonyl compounds that were reduced stereoselectively to the corresponding alcohols using enantiopure [(NN)Rh(Cp*)X]n+ catalysts and sodium formate as a hydride source [29,42,43,44].

Figure 7.
General reaction scheme for the [(bpy)Rh(Cp*)L]n+ catalyzed reduction of carbonyl compounds. Within step a, the catalytically active species can be generated either by a thermal or by a electro- or photochemical process, as shown in Figure 4.
Figure 7.
General reaction scheme for the [(bpy)Rh(Cp*)L]n+ catalyzed reduction of carbonyl compounds. Within step a, the catalytically active species can be generated either by a thermal or by a electro- or photochemical process, as shown in Figure 4.
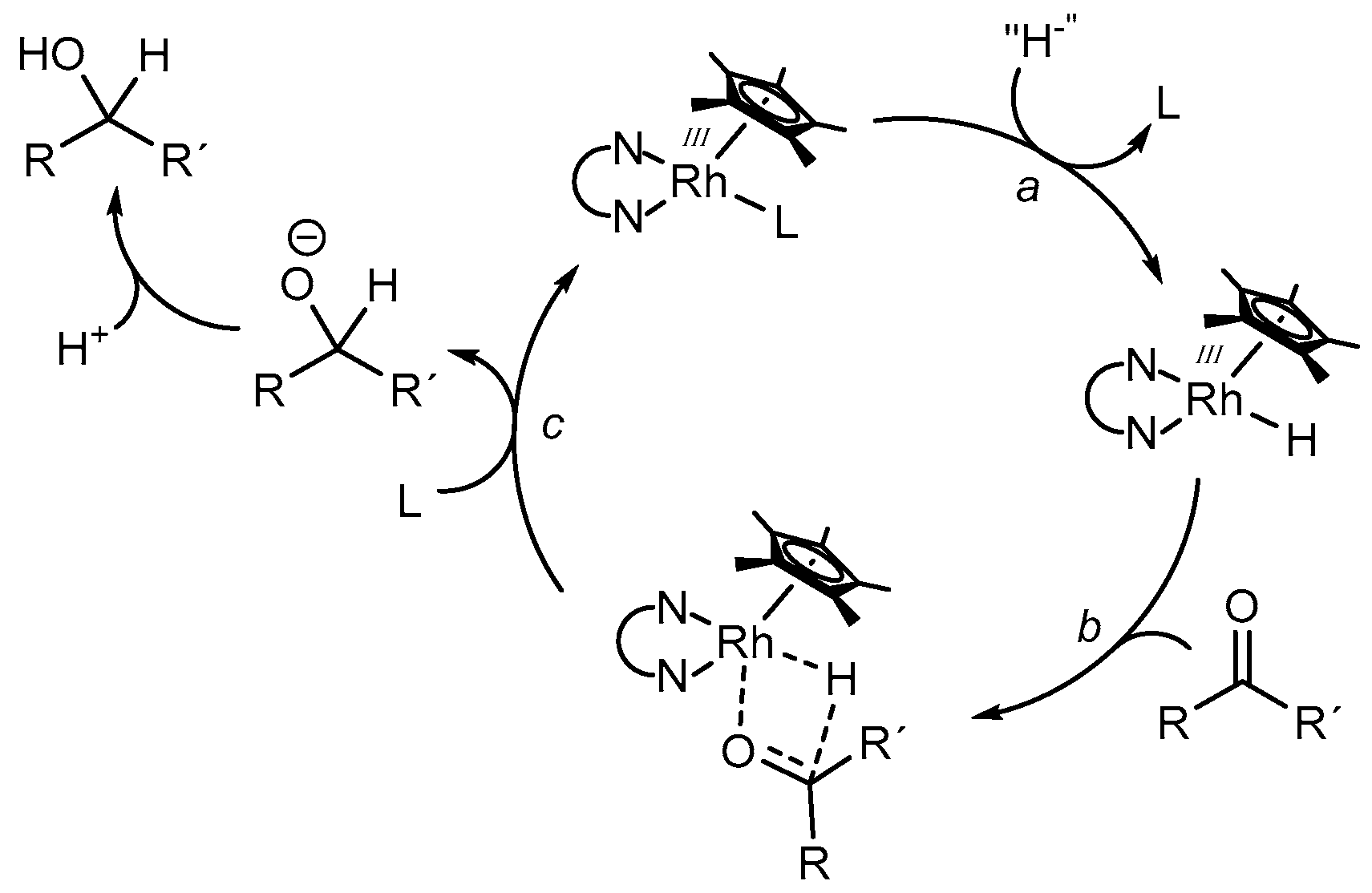
Figure 8.
Mechanism of the formate driven nicotineamide cofactor reduction according to Lo et al. [83,84].

Figure 9.
Proposed reaction mechanism for the generation of hydrogen peroxide upon the electrochemical formation of [(NN)Rh(Cp*)H]+ [53].
Figure 9.
Proposed reaction mechanism for the generation of hydrogen peroxide upon the electrochemical formation of [(NN)Rh(Cp*)H]+ [53].
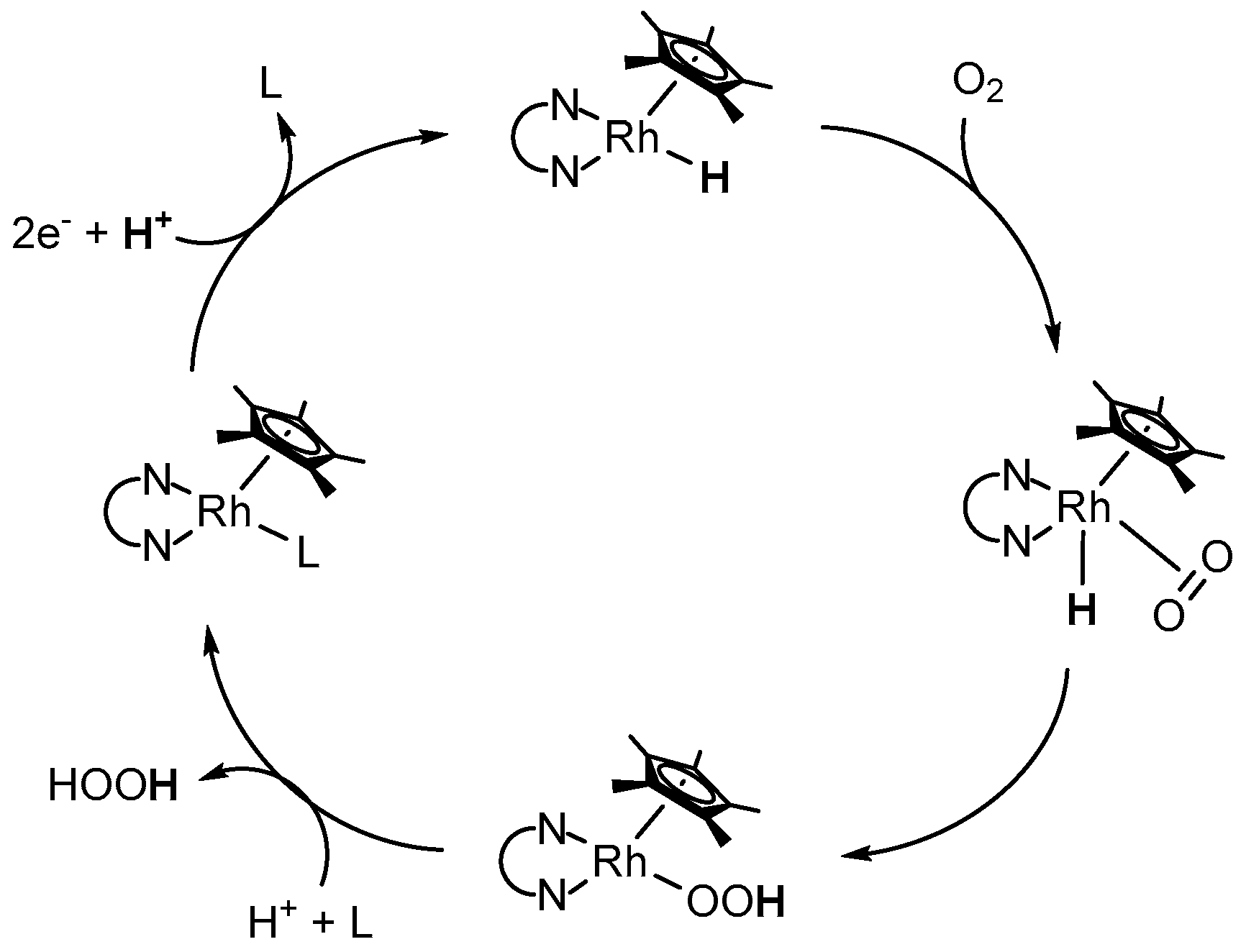
Figure 10.
Schematic structure of the transition state of the regioselective hydride transfer onto the 4-position of the cationic nicotinamide substrate according to Lo et al. and relative reaction rates for selected examples [83,84]. The green arrows indicate the electron accepting properties of the substituents, the blue arrow indicates the σ donor property of the coordinating moiety; steric repulsion is marked as well.
Figure 10.
Schematic structure of the transition state of the regioselective hydride transfer onto the 4-position of the cationic nicotinamide substrate according to Lo et al. and relative reaction rates for selected examples [83,84]. The green arrows indicate the electron accepting properties of the substituents, the blue arrow indicates the σ donor property of the coordinating moiety; steric repulsion is marked as well.
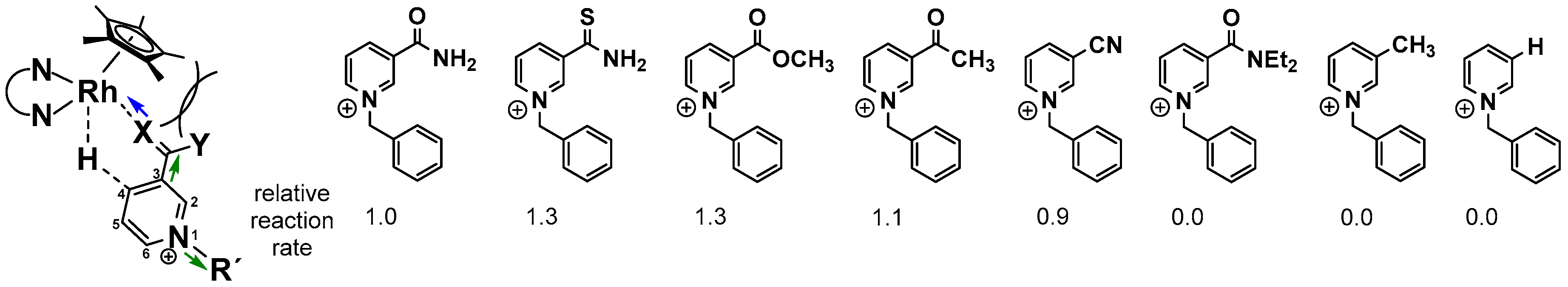
Figure 11.
Schematic reaction scheme for photobiocatalytic systems.
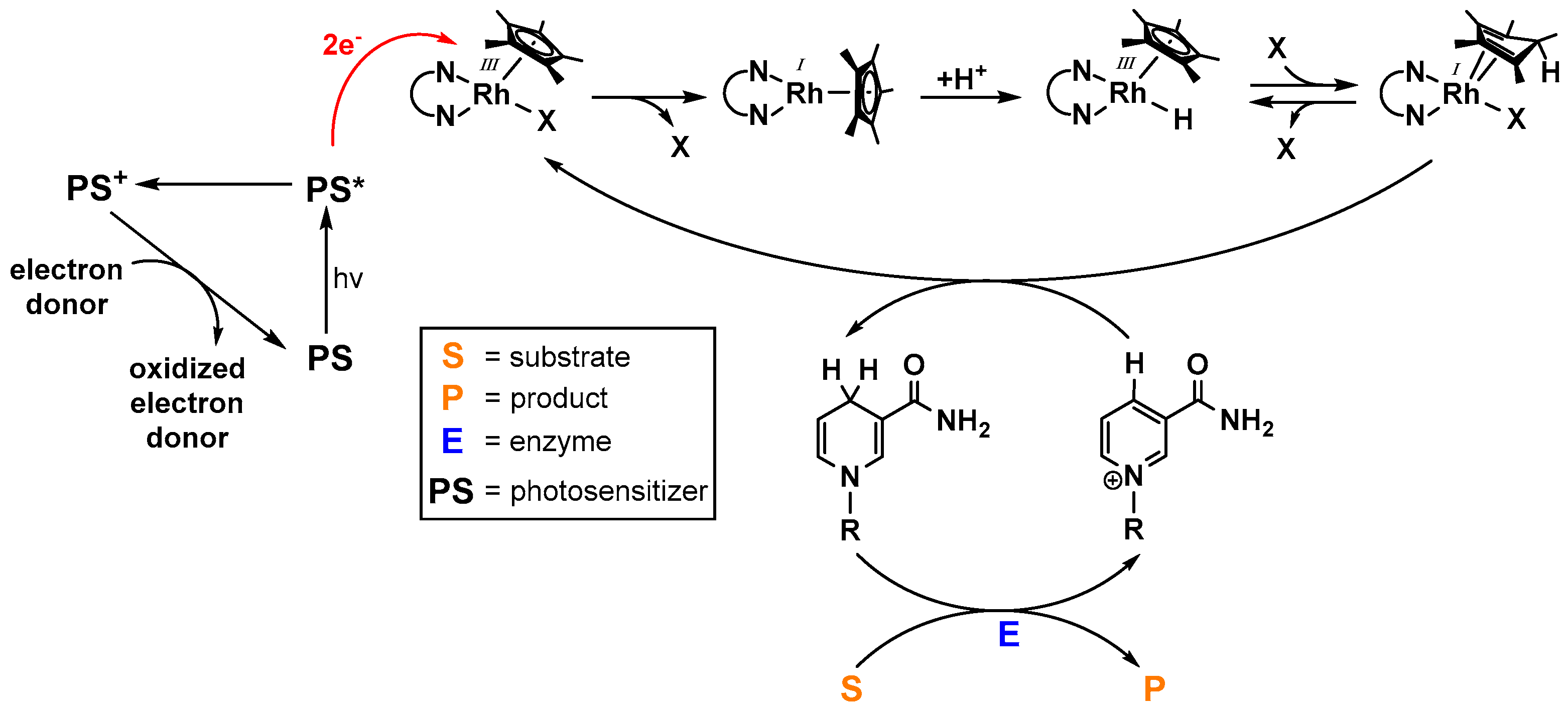
Figure 12.
Schematic overview over the productive and nonproductive light driven photoredox processes within the context of [(NN)Rh(Cp*)X]n+ catalyzed NAD(P)H formation. Rate constants for the electron transfer from the excited chromophore to the Rh catalyst or the oxidized cofactor are expressed as k1 and k2.
Figure 12.
Schematic overview over the productive and nonproductive light driven photoredox processes within the context of [(NN)Rh(Cp*)X]n+ catalyzed NAD(P)H formation. Rate constants for the electron transfer from the excited chromophore to the Rh catalyst or the oxidized cofactor are expressed as k1 and k2.
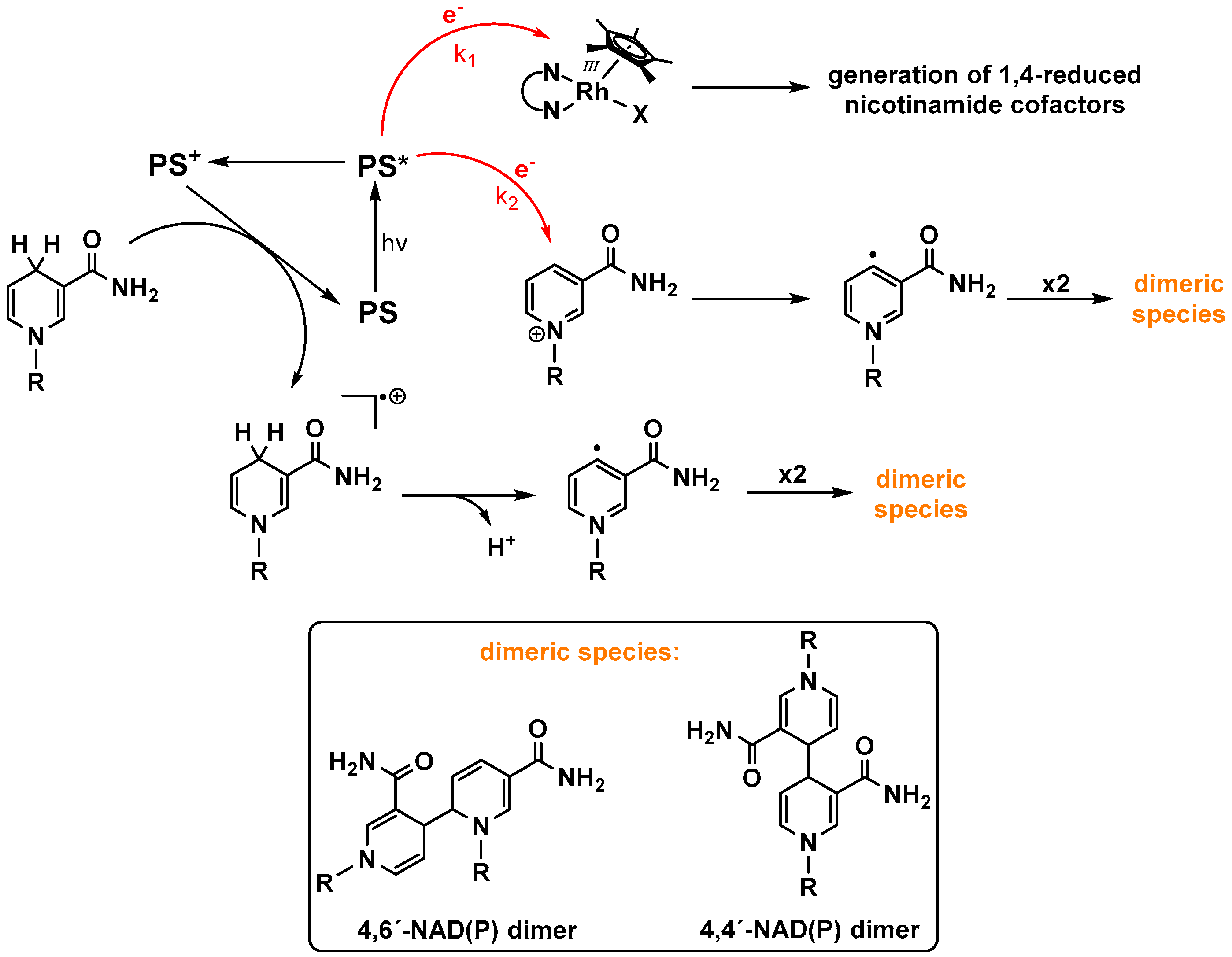
Figure 13.
Molecular structure of a heterodinuclear photocatalyst for the generation of NAD(P)H [99].
Figure 13.
Molecular structure of a heterodinuclear photocatalyst for the generation of NAD(P)H [99].
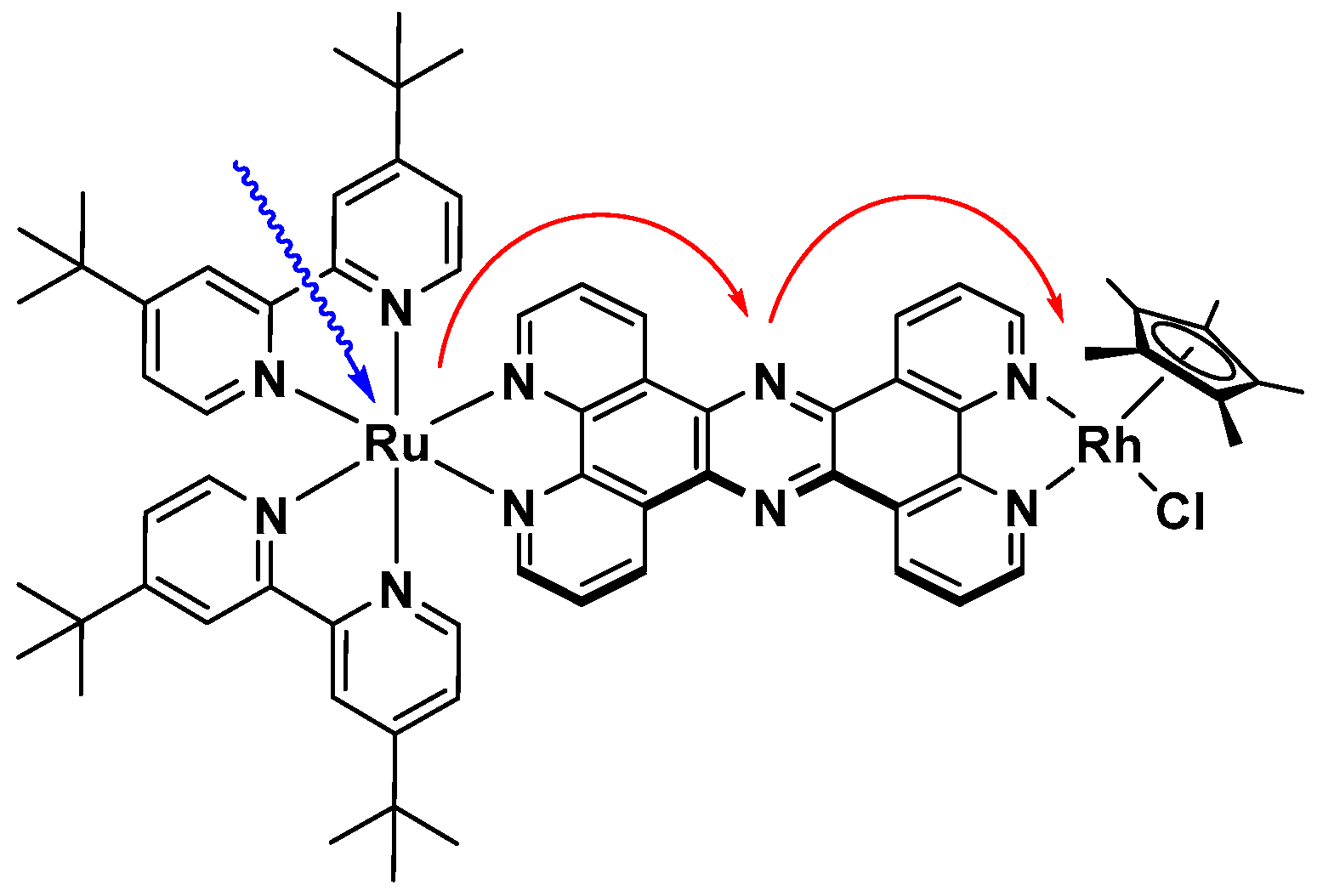
Figure 14.
General reaction scheme for the photobiocatalytic enantioselective alcohol formation using [(NN)Rh(Cp*)L]n+ catalysts for the regeneration of NAD(P)H (E = enzyme).
Figure 14.
General reaction scheme for the photobiocatalytic enantioselective alcohol formation using [(NN)Rh(Cp*)L]n+ catalysts for the regeneration of NAD(P)H (E = enzyme).
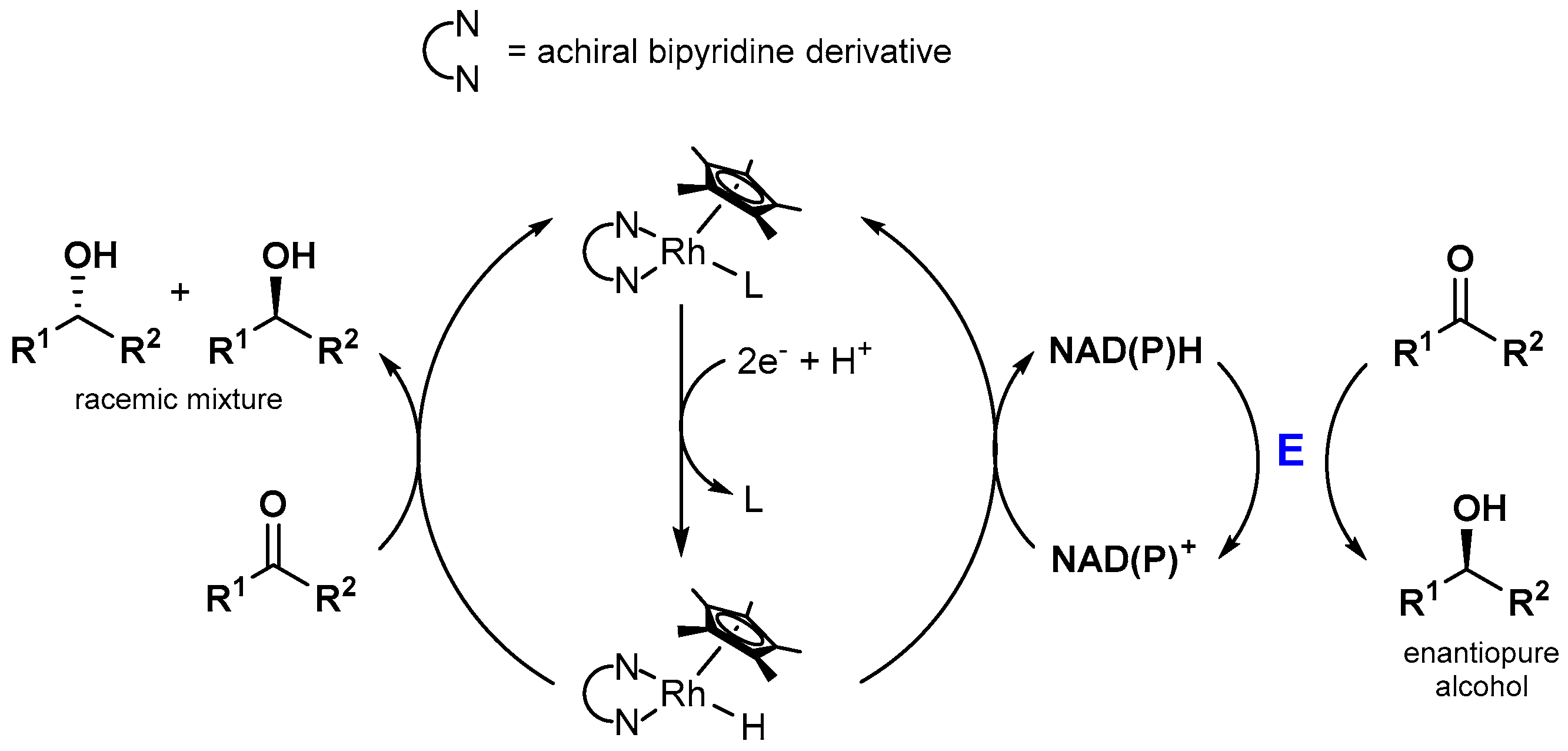
Figure 15.
Hypothesized overall reaction mechanism of competing hydrogen evolution and NAD(P)H formation driven by [(bpy)Rh(Cp*)L]2+. Reaction steps and intermediates that belong to both catalytic cycles are marked in black, the hydrogen and NAD(P)H formation specific parts are red and blue, respectively.
Figure 15.
Hypothesized overall reaction mechanism of competing hydrogen evolution and NAD(P)H formation driven by [(bpy)Rh(Cp*)L]2+. Reaction steps and intermediates that belong to both catalytic cycles are marked in black, the hydrogen and NAD(P)H formation specific parts are red and blue, respectively.
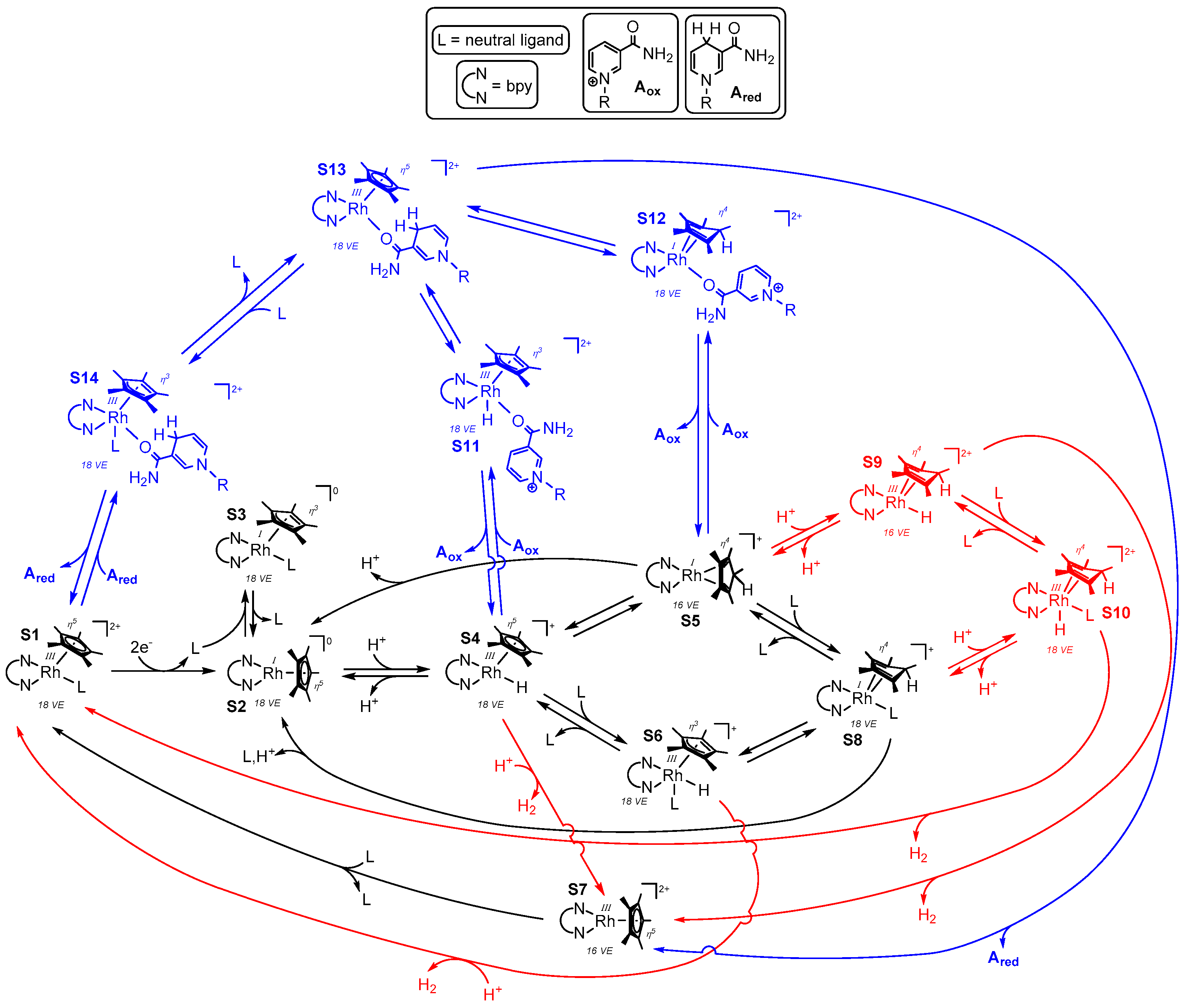
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mengele, A.K.; Rau, S. Product Selectivity in Homogeneous Artificial Photosynthesis Using [(bpy)Rh(Cp*)X]n+-Based Catalysts. Inorganics 2017, 5, 35. https://doi.org/10.3390/inorganics5020035
AMA Style
Mengele AK, Rau S. Product Selectivity in Homogeneous Artificial Photosynthesis Using [(bpy)Rh(Cp*)X]n+-Based Catalysts. Inorganics. 2017; 5(2):35. https://doi.org/10.3390/inorganics5020035
Chicago/Turabian StyleMengele, Alexander K., and Sven Rau. 2017. "Product Selectivity in Homogeneous Artificial Photosynthesis Using [(bpy)Rh(Cp*)X]n+-Based Catalysts" Inorganics 5, no. 2: 35. https://doi.org/10.3390/inorganics5020035
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.