
First-Principles View on Photoelectrochemistry: Water-Splitting as Case Study


Competence Centre for Catalysis and the Department of Physics, Chalmers University of Technology,412 96 Gothenburg, Sweden
*
Author to whom correspondence should be addressed.
Inorganics 2017, 5(2), 37; https://doi.org/10.3390/inorganics5020037
Submission received: 30 March 2017
/
Revised: 19 May 2017
/
Accepted: 24 May 2017
/
Published: 1 June 2017
(This article belongs to the Special Issue Photochemical Water Splitting)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Photoelectrochemistry is truly an interdisciplinary field; a natural nexus between chemistry and physics. In short, photoelectrochemistry can be divided into three sub-processes, namely (i) the creation of electron-hole pairs by light absorption; (ii) separation/transport on the charge carriers and finally (iii) the water splitting reaction. The challenge is to understand all three processes on a microscopic scale and, perhaps even more importantly, how to combine the processes in an optimal way. This review will highlight some first-principles insights to the above sub-processes, in particular as they occur using metal oxides. Based on these insights, challenges and future directions of first-principles methods in the field of photoelectrochemistry will be discussed.
1. Introduction
“Water will be the coal of the future” states Cyrus Harding, one of the protagonists in Jules Verne’s novel “The Mysterious Island” [1]. Interestingly this is the conclusion after the protagonists have discussed the fact that the known coal reserves will eventually be depleted. The analogy to today’s crude oil is obvious. Based on the simple fact that it takes millions of years to produce fossil fuel, whereas the extraction, in comparison, is very fast, the depletion of crude oil is certain [2,3,4], although the exact time remains debatable.
The scenario of depleting crude oil reserves is in stark contrast to the increase in global energy consumption. In 2015, the global power consumption was estimated to be a total of 17 TW [5], and projections for 2050 ranges between 28 and 31 TW [6,7]. This increased demand of energy is know as the TW challenge [8,9,10], and alternative energy solutions must be able to contribute to the TW level in order to make an appreciable impact at a global scale [11,12].
In light of possible peak oil production, increased global energy consumption and humanity’s environmental footprint, we need to find a renewable source of energy that enables us to move away from fossil fuels. An obvious candidate is the Sun. For instance, more energy from sunlight strikes Earth in one hour than what society currently consumes in a year [13]; thus the Sun easily rises to the TW challenge. Solar energy can be harvested in many different ways [14,15,16,17]. For instance, photosynthesis, which play a crucial role for life on Earth, is able to transform sunlight to chemical energy in the form of biomass. If the biomass is produced from plants with low water and fertilizer requirements, and without competition with ordinary food production, then biomass can contribute to the TW challenge by e.g., use of conventional thermal power plants. Other conventional methods like wind and wave power, which is an indirect form of solar energy, can generate electricity, which is also true for photovoltaic cells. Today, silicon based solar cells has an energy conversion efficiencies of approximately 15% to 25% [18], but there exist several of similar techniques. For instance, aqueous dye-sensitized solar cells (DSSCs) works as an artificial photosynthetic system converting solar energy into electricity, and have recently emerged as an alternative to silicon-based solar cells owing to the low cost, synthetic flexibility, ease of fabrication, and short energy payback time. Moreover, the conversion efficiencies have recently exceeded 14% [19]. The most recent studies on such systems can be found in Ref. [18,20,21,22]. Yet another method to utilize the energy of sunlight is to use the energy to break chemical bonds, thereby storing energy in a chemical form. This is the basic challenge in water-splitting, i.e, with the help of sunlight split water into hydrogen and oxygen [23,24,25]. Cyrus Harding explains to his fellow castaways “water will one day be employed as fuel, that hydrogen and oxygen which constitute it, used singly or together, will furnish an inexhaustible source of heat and light, of an intensity of which coal is not capable” [1]. In case of photocatalysis , a photocatalyst is used to capture photons with higher energies than the band gap of the material, i.e., excite electrons from valence band to conduction band [26,27,28,29,30]. These photogenerated carriers can participate the reduction and oxidation reactions of water to hydrogen and oxygen. In order to achieve this, the band positions of the photocatalyst must lie on both sides of the reduction and oxidation potentials of water. In the case of photoelectrochemistry [23,24,25] the electrochemical setup provide the possibility of applying an external potential that can further assist in the overall reaction, i.e., provide additional driving force to the photogenerated carriers to split water.
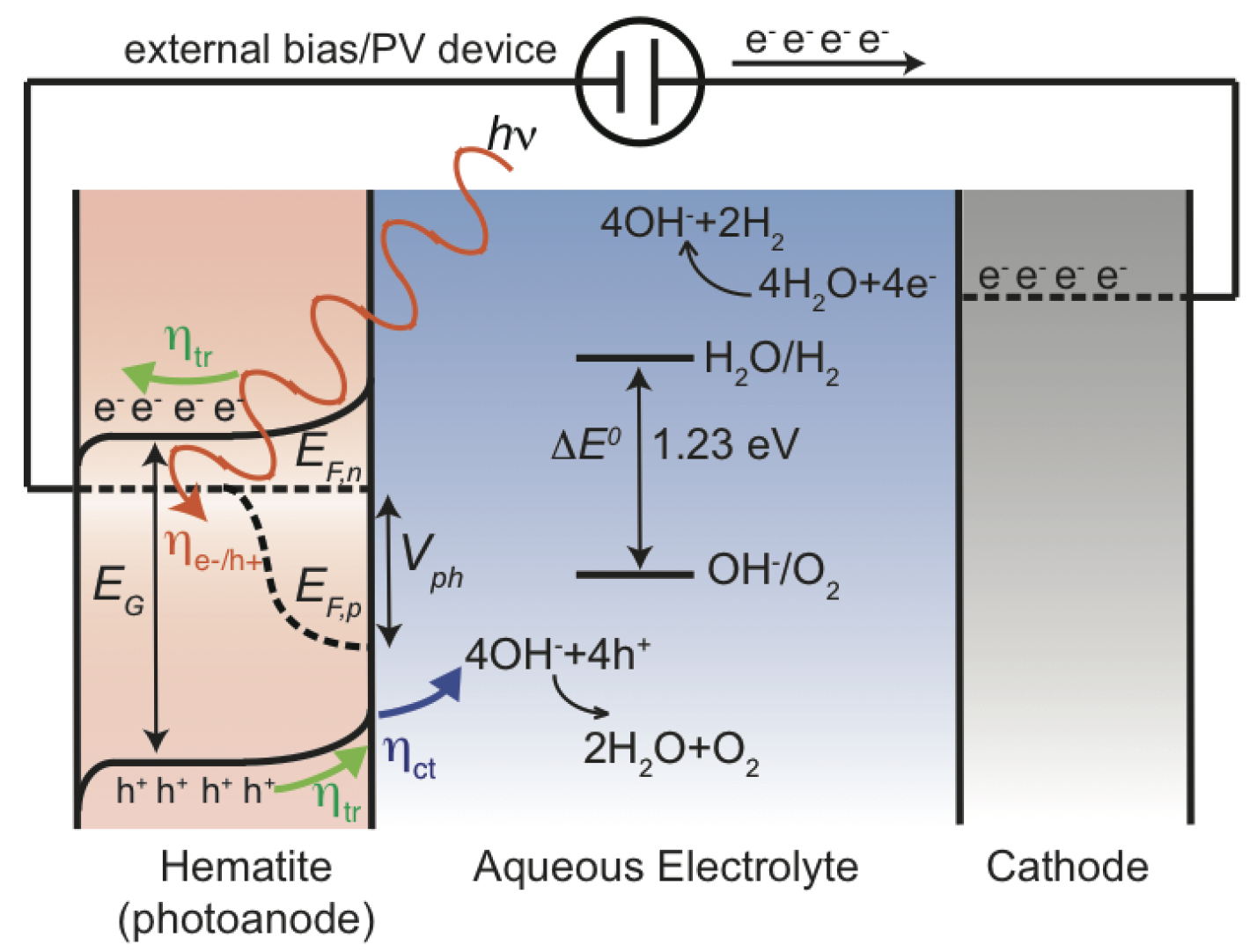
In 1972, Honda-Fujishima published a ground-breaking study in which hydrogen was produced from water using TiO as the photoanode material [31], proving that it is indeed possibly to do photoelectrochemical water splitting. A schematic energy diagram of the basic principles of water splitting by a photoelectrochemical cell using a photoanode (photoactive semiconductor material) for the oxygen evolution reaction (OER) and a cathode for the hydrogen evolution reaction (HER) is shown in Figure 1. When light interacts with the photoanode, the electrons in the valence band have the possibility to cross the forbidden band-gap and, thus, be excited into the conduction band while the holes remain in the valence band. The holes will move towards the interface of the photoanode and electrolyte where they will participate in the water splitting, while the electrons will move to the cathode side and participate in the HER. The free energy change in the water splitting reaction (2HO → 2H + O) amounts to 4.92 eV, which in a four-electron process implies that each electron-hole needs to bring 1.23 eV to the reaction. Hence, the photoanode material must absorb light to make its electrode potential higher than 1.23 V, which puts a lower bound on the band-gap. There exist several of excellent textbooks [32,33,34] and reviews [23,35,36,37,38,39] describing the above process in detail.
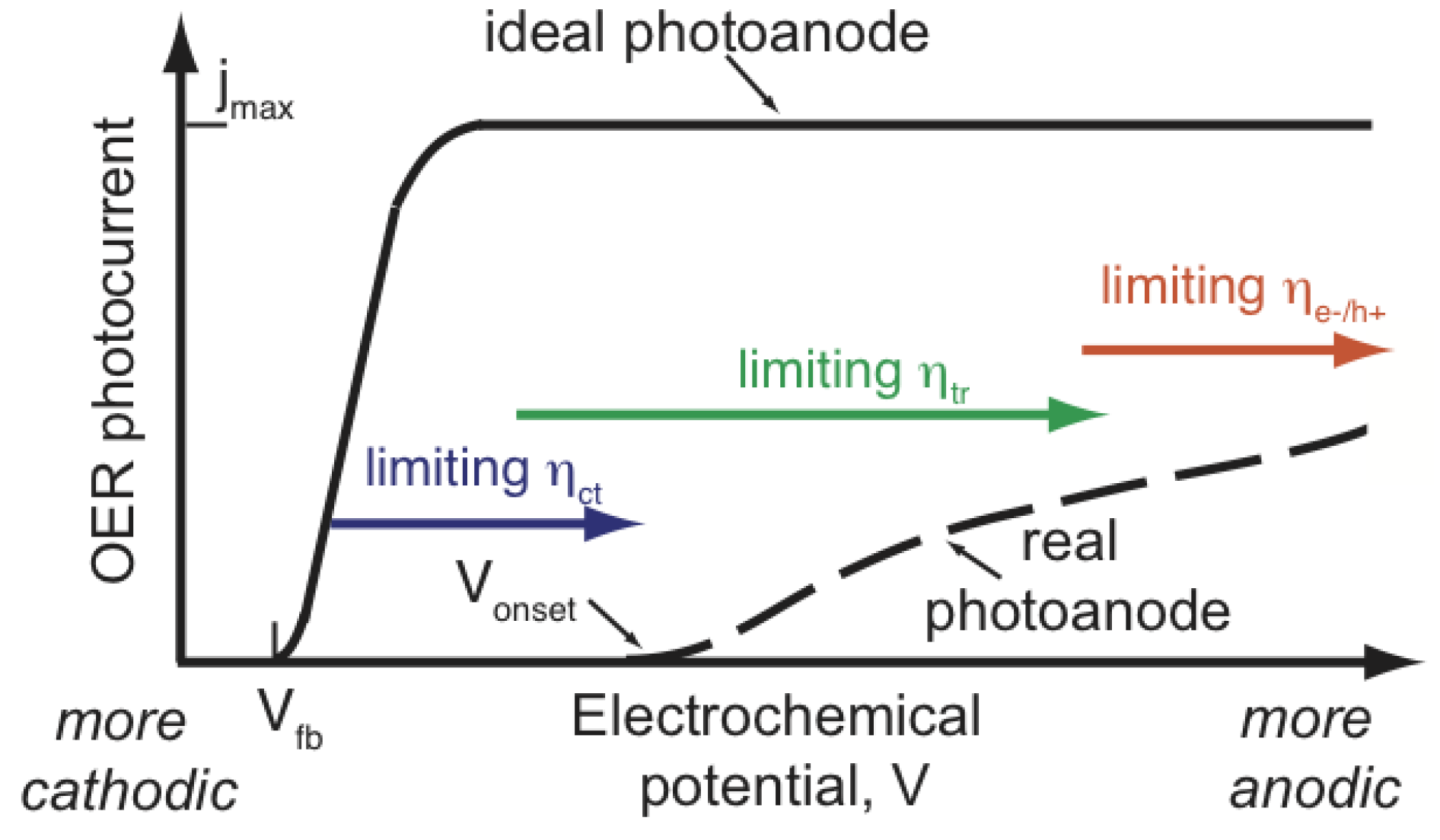
Therefore, what are the criteria that an ideal photoanode material needs to fulfill? Simply put, there are five criteria: (1) by necessity, the band-gap must be larger than 1.23 eV, however, with respect to energy losses, a more practical band-gap is around 2.0 eV, which still allows the photoanode to be working under visible light as half of the incoming solar energy comes from visible light and shorter wavelengths; (2) band edge positions should straddle the redox potentials of water; (3) electron-hole mobility and lifetime that allow the electron-hole pair to reach the active sites, e.g., materials with few defects and small particle size inhibit electron-hole recombination; (4) the rate of oxygen evolution reaction must be larger than any competing recombination reaction where the activity and the number of active sites become crucial; (5) the material needs to be stable in an aqueous environment and under illuminating conditions i.e., to avoid anion from the catalyst itself being oxidized by photogenerated holes instead of water. However, in practice, the performance gap exists between a real photoanode and an ideal photoanode shown in Figure 2. The main performance limiting factors for increasing anodic potential are indicated by arrows. Although, there exist promising candidates, such as tungsten oxide and hematite, the challenge to find one such material remains to be solved [41]. For instance, owing to the relatively large band-gap (3.2 eV) of TiO [42], only a fraction of the solar spectrum is utilized, which ultimately puts some hard limitation of the use of TiO to resolve the TW challenge [35].
Density functional theory (DFT) is based on two theorems [43] that in principle are a recast of the Schrödinger equation into a functional form and instead of dealing with the many-body wave-function focus on the density. Several excellent reviews [44,45] and textbooks [46,47,48] discuss the fundamentals of DFT, and a brief discussion on the methodologies of DFT are provided in the appendix part of this paper. Being a first-principles method, DFT is able to predict many properties of any atomic system, and there exist several of successful examples in the literature. In the last decade, the predictive power of first-principles has been used in high-throughput computational materials studies [49,50]. With the historical increase in available computer power, it is clear that first-principles will become the first starting point when a new material property is desired or even when an unknown material is needed.
This review focus on how first-principles methods can address the processes involved in photoelectrochemistry, namely photoexcitation of electron-hole pairs, separation and transport of the holes to the semiconductor/electrolyte interface where the oxygen evolution reaction takes place. The hope is that by providing further understanding of the underlying mechanisms, this will assist in the development of new or improved photoanode materials. This review is far from complete, and some aspects will not get the attention they deserve, for which the authors can only apologize. In more specific terms, only a limited number of first-principles studies of how dopants affect light absorption, carrier transport, and catalytic reactions are included, although the study of dopants are the bread-and-butter of first-principles. Further, the focus is on standard DFT, and only briefly are results from time-dependent DFT and molecular dynamics accounted for. In addition, experimental literature is not discussed in-depth. These limitations are not because these aspects are not important. On the contrary, all of them deserve their own extensive review. However, to make this review tractable these limitations are included nonetheless.
2. Processes Involved in Photoelectrochemistry
There are many microscopic processes involved in photoelectrochemistry, such as light-matter interaction, transport phenomena, charge-transfer reactions, solid-liquid interaction, and many more. In an attempt to provide an overview of the processes involved and how to tackle them with first-principles methods, we will limit ourselves to three separate major processes. These will be (i) creation of electron-hole pairs, (ii) charge carrier transport and (iii) electrochemical surface reactions.
2.1. Creation of Electron-Hole Pairs
As light penetrates the photo-active semiconductor, the electronic structure of the material will interact with the propagating electromagnetic wave [51,52]. The interaction might result in an electronic excitation where an electron in an occupied band is transferred to an unoccupied band. In the case of a semiconductor, the occupied (valance) band and unoccupied (conduction) band is separated by a band-gap. The band-gap efficiently hinders the dissipation of energy, as the lack of accessible electronic states quenches many of the common dissipation channels (carrier–carrier interaction and the phonon coupling) [51]. Generally, one can classify band-gaps into two classes: direct and indirect band-gaps. The difference lies in the prerequisite for the photon adsorption. In the case of a direct band-gap, the electron can transfer to the conduction band without any change in its momentum, whereas in the case of an indirect band-gap, the transfer also requires the creation/annihilation of one (or many) lattice phonon(s), which makes the probability for this process much less as compared to the probability in a direct band-gap.
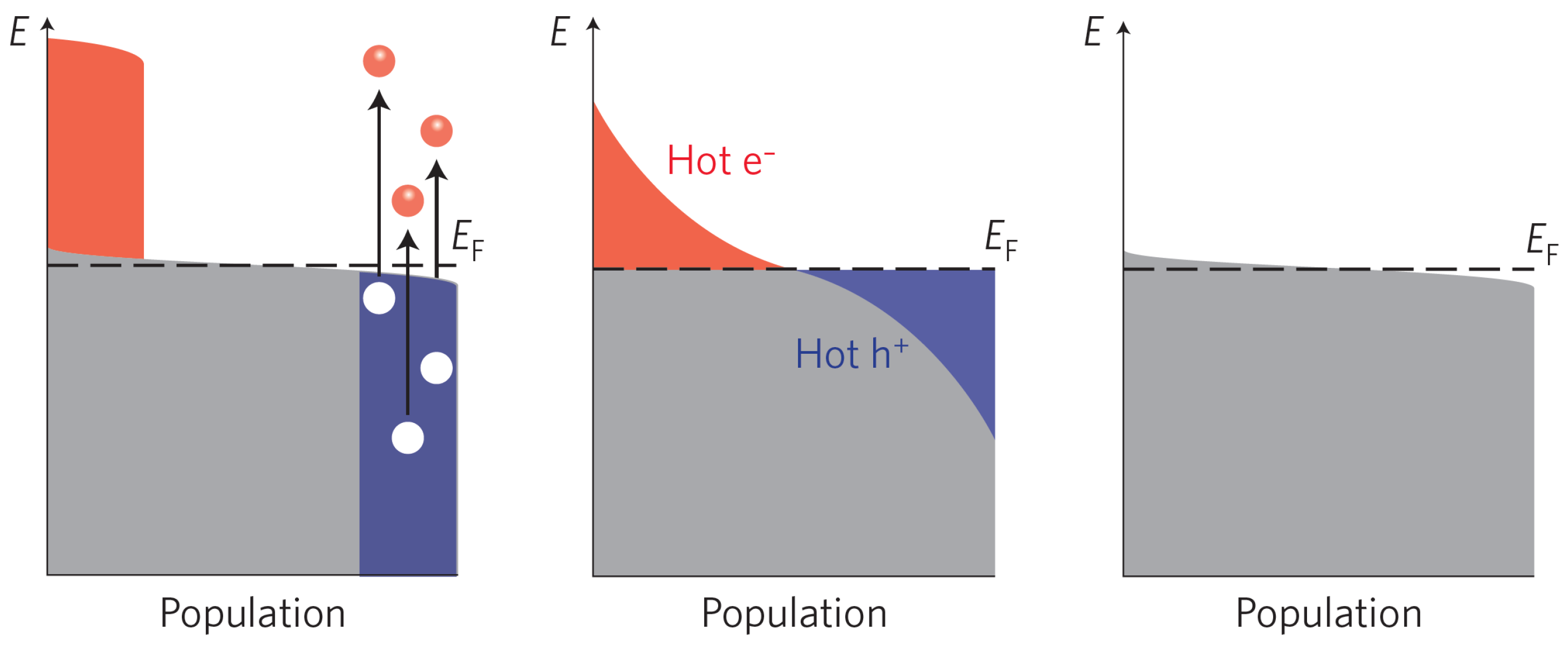
It is, of course, possible to transfer electrons deeper down in the valance band and/or to higher states in the conduction band if the photon energy is larger than the band-gap. These electron-hole pairs are in a non-equilibrium state, but thanks to carrier–carrier interaction and phonon coupling, the additional energy is quickly dissipated. The cooling process normally goes through the following steps [53,54,55]; first, the electron and the hole reach equilibrium by their respective carrier–carrier collisions, resulting in two different temperatures defined by the distribution of energy of the respective carrier distribution. This temperature is always higher than the phonon temperature, thereby giving rise to the terms hot electrons and hot holes. This relaxation process is very rapid (1–10 fs). Second, the hot carriers are equilibrating with the phonons, through carrier–phonon interactions thereby transferring excess energy to the heating of the photoanode material. The relaxation process occurs on the time scale of 1–100 ps. The last step involves the electron-hole recombination, either radiativley (luminescence) or nonradiatively (heat). A schematic of these processes is shown in Figure 3. Assuming that the above dissipation channels are efficient, the theoretical power conversion of a photo-active semiconductor depends only on the band-gap and the incident light spectrum [42]. Although there exists ways to modify the solar spectra, for instance combining low-energy photons to create a higher energy photon (photon fussion) [56], the most obvious parameter to optimize is the band-gap.
DFT is a proven method, able to explain, predict and guide experimental results; however, in the case of band-gap design, there is a well-known problem. The most frequently-used approximations for the exchange-correlation functional simply fails at calculating the band-gap of even the simplest materials. For instance, Si is calculated to have a band-gap of 0.52 eV, whereas the experimental value is 1.17 eV [57,58]. There are many extensions to circumvent this problem, such as, DFT + U, hybrid functionals and random-phase approximation [59]. However, there is also work being done on the simple semi-local functionals; see for instance [60,61], which describe PBEsol and GLLB-SC. The latter has recently been compared to more advanced methods, in this case GW [62], with very good agreement [63]. Using the GLLB-SC has allowed the use of high-throughput screening [63] and machine-learning techniques [64] to find materials with suitable band-gaps for water-splitting.
The fundamental and optical band-gap of semiconductors can be modified not only by changing material properties (structure, composition, etc.), but also through doping. The description of dopants in semiconductors using DFT requires accurate description of the band structure and the charge states of transition levels [65]. The defect energy levels can be described with the obtained single-particle Kohn-Sham eigenvalues, while determining the transition between different charge states can be evaluated using the approach described by Lany and Zunger [66] or Van de Valle and Neugebauer [67]. However, the well-known band gap problem and the insufficient cancellation of the self-interaction energy are the two main drawbacks of DFT [68]. DFT + U or hybrid functionals represents a general and practical solution to the problems, although limited by offhand parameters, such as, the U value in DFT + U or the fractional of exact Hatree-Fock exchange in hybrid functionals [69,70,71,72]. Technically, when modeling doped systems, corrections of finite-size effects are required in supercell calculations, especially in the case of charged defects [73] or when Moss-Burstein-type band-filling effects are important [74,75]. Many approaches and schemes have been applied to address this, readers are referred to Ref. [66] and references therein for details. Dopant can either stay at interstitial site or substitute one of the host atom in bulk materials depending on the magnitude of the formation energy of different configurations, which can be calculated using DFT. In cubic system, the lattice parameters are often linearly dependent on the dopant concentration following the empirical Vegard law [76,77,78]. For other systems, equilibrium volume can be obtained by fitting the DFT calculated energies to the Birch–Murnaghan equation of states [79,80]. There exist numerous examples from the literature where first-principles have been used to guide which doped atoms to use to improve the band-gap of a given material. For instance, Asahi et al. [81,82] showed that nitrogen doped into substitutional sites of TiO resulted in a band-gap narrowing, as the p states of N contribute to the narrowing by mixing with O 2p states. Cristiana et al. [83] have studied the defect states in reduced and n-type doped TiO based on DFT + U and hybrid functionals (B3LYP). Both localized/delocalized solutions close in energy were found in the calculations, indicating that measurable temperature effects in determining the nature of the defect states can be used in experiment. Theoretical studies of carbon doped TiO (both rutile and anatase) have been conducted by Kamisaka et al. [84] and Cristiana et al. [85] as carbon can work as both the anion doping and cation doping. Both groups found that carbon impurities induce several localized occupied states contributing to the observed red shift of the absorption edge. Moreover, Cristiana et al. [85] also found that carbon doping might favor the formation of O vacancy in TiO. Co-doping with appropriate elements could significantly enhance the photocatalytic properties in TiO owing to the existence of synergistic effects between doping elements, which can increase the visible light absorption and improve the separation efficiency of the electron-hole pairs [86,87,88]. For more detailed discussion about mono-doping and co-doping effects on the photocatalytic properties of TiO, we refer the reader to the review articles of Ref. [89,90,91]. Meng et al. [92] used Cu dopants to move the conduction band minimum (CBM) of -FeO above the hydrogen redox level, while Ti dopants create very shallow donor levels and chang the band-gap slightly. The above examples show that first-principles can be used to provide explanations of what is happening at the atomic level. However, first-principles should be able to provide guidelines for the search for new materials with improved band-gaps. The next example indicates what we can expect in the near future.
2.2. Charge Carrier Transport
Once the electron-hole pair is created, it needs to be separated; however, this is not a straight-forward process. The electron-hole pair feels a screened Coulomb attraction, and if it is strong enough, the electron-hole pair can be referred to as an exciton [93]. The attraction will affect the spatial distribution and transport properties of both the electron and the hole. However, as the wavefunction of the electron-hole pair becomes uncorrelated, the electron-hole pair can be viewed as free carriers. The charge carrier with the lowest effective mass (remember the second derivative of the dispersion relation) will exhibit a larger root-mean-square motion as compared to the heavier charge carrier. This implies that the lighter carrier will effectively create a charge separation, known as the photo-Dember effect [94]. This is the main effect that determines carrier transport under zero-field conditions.
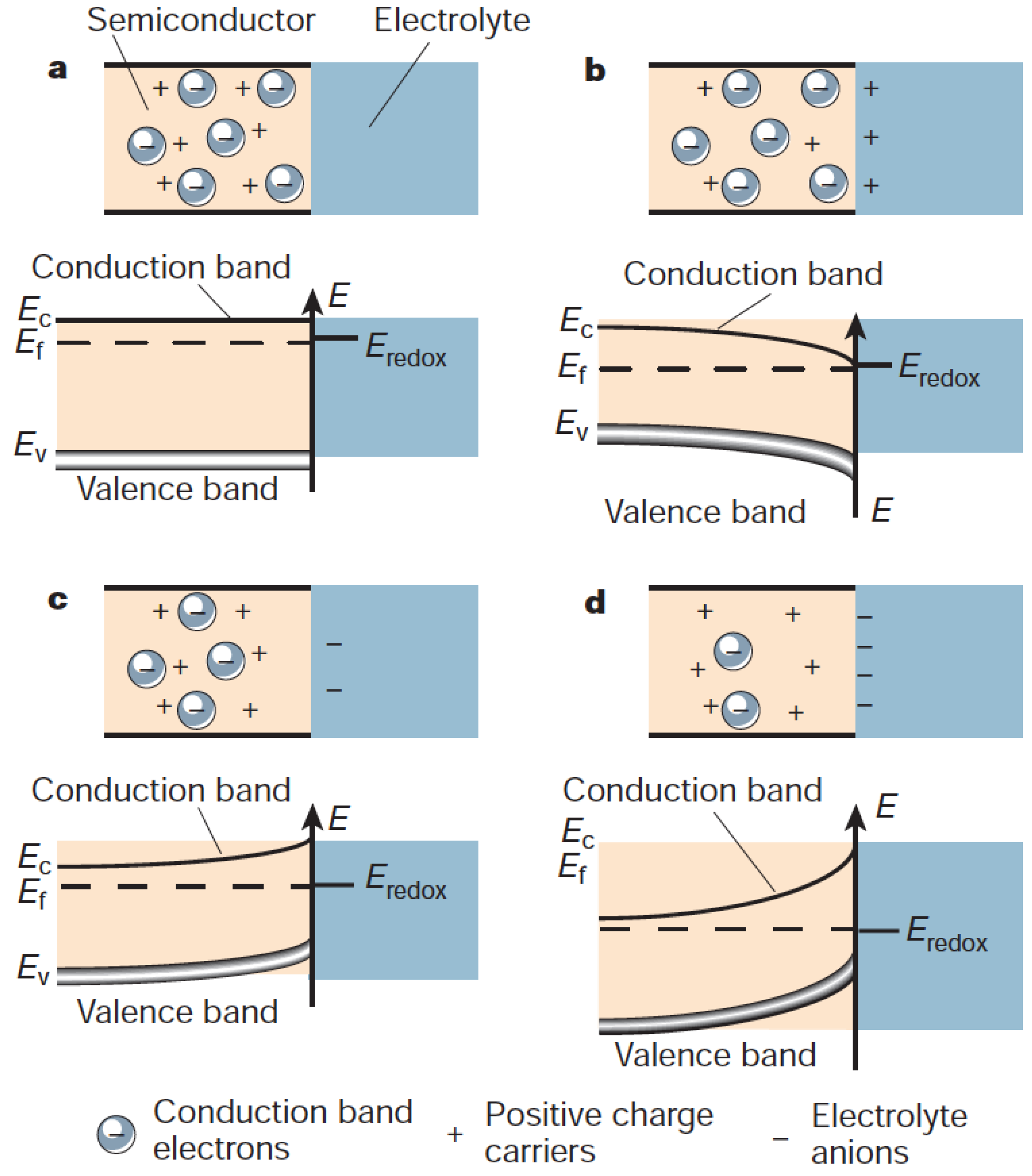
However, this separation is rarely sufficient enough to be the main mechanism in photoelectrochemistry; instead, it is the presence of an interface that allows for an efficient separation of the electron-hole pairs. This is well-known from the semiconductor physics where interfaces, in the form of p-n junctions, are used to separate electron-hole pairs. Over the interface, a space charge layer is normally formed, owing to the difference in electrochemical potentials [95]. As the system equilibrates, i.e., electrons flow from higher potential to lower until the compensating field is sufficient to stop the flow, resulting in a parabolic bending of the semiconductor bands [95].
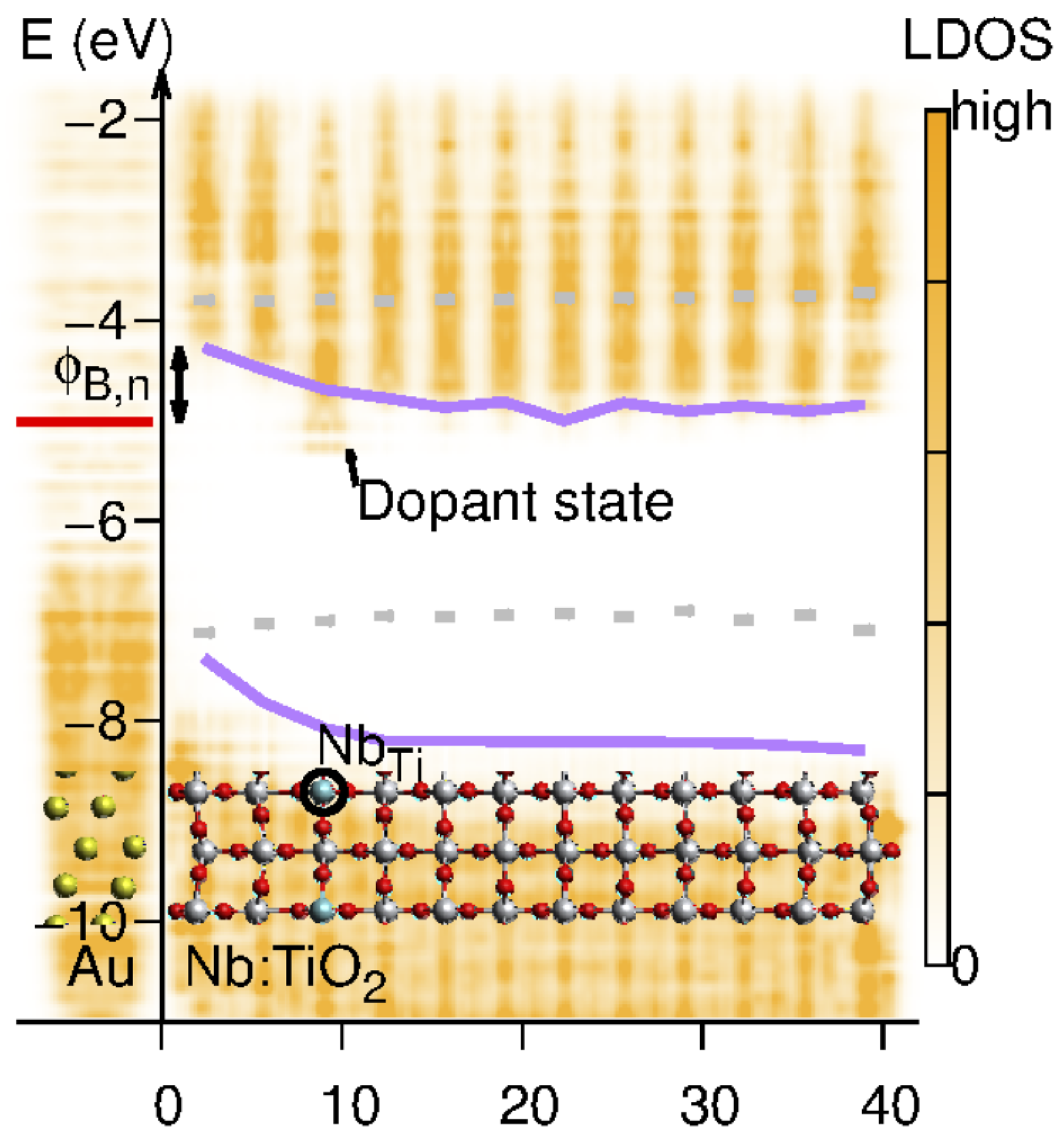
In a photoelectrochemical system, the interface is instead between the semiconductor and the liquid phase, see Figure 4. However, owing to the mobile charge carriers in the liquid, a Schottky junction is formed, which has a redistribution of the charge on the electrolyte side, effectively forming the Helmholtz layer. The field in the semiconductor can extend several of hundred Angstroms, depending on the doping level of the semiconductor. Conventional models all assume a uniform dopant distribution, and it remains unclear how the composition and atomic structure of the semiconductor affect the electronic structure, i.e., barrier height and band bending, on the atomic level. It should be noted, however, that recent advances [96,97,98,99] have made it possible to characterize the Schottky band bending at the nanometer scale and have revealed important deviations from predictions made on the conventional Schottky model [100]. Instead, the results, which depend on materials’ properties, dopant compositions and concentrations, can be qualitatively interpreted in the inhomogeneous Schottky barrier height model [101,102,103]. Interestingly theory studies [104] show that the band bending is inhomogeneous and highly localized to the defect region, see Figure 5. This indicates a much more complicated band bending picture in real systems.
This field will assist in the separation of the photogenerated electron-hole pairs and drive the minority carrier to the surface, while picking up the excess energy from the field. This implies that the carriers can arrive at the surface with an excess energy corresponding the degree of bend banding, i.e., hot carriers. Note that in the space-charge layer, the carrier-carrier interaction is much less, and in principle only the phonon channel is open for the dissipation of energy. In contrast, the majority carrier will experience both the carrier-carrier and phonon-scattering processes during its propagation through the semiconductor material.
As the electron or hole is transported through the lattice, the lattice responds to this charge by delocalizing it over many atoms (a large polaron) or by localizing it over just a few atoms (a small polaron). As the extra charge carrier will fill or empty states with the bonding or antibonding characteristics of the lattice atoms, the charge influences bond lengths and angles in the material. If the charge is delocalized, the influence is small because it is spread over many atoms, while a highly localized charge dramatically distorts the lattice. One way to view the process is to consider the carriers dragging a cloud of phonons along as it propagates through the lattice. This gives rise to an understanding of the phenomena of self-trapping. A small polaron will shift the surrounding lattice to its new position with in a few lattice vibrations. This increases the stability of the state, creating a deeper potential well that must be overcome to transfer the polaron to a neighboring lattice site, i.e., there is a large reorganization energy associated with small polarons [105,106].
The excited charge carriers (electrons/holes) transport mainly through the hopping of small polarons in many metal oxides [105,107]. The description of the mobility of the small polarons are within the framework of the Marcus theory [108], of which the associated parameters can be calculated with quantum-mechanical calculation methods such as DFT, including DFT + U, hybrid functionals and -self consistent field (SCF) [109,110] to describe the localization of the trapped carriers [111,112,113,114,115,116,117].
Deskins et al. [118] studied the effect on adding one excess electron to the electronic structure of bare and singly hydroxylated rutile (110) surfaces. The excess electron formed a small polaron with its spin density and associated lattice distortion localized around a single site. Further, surface hydroxyl only perturbs the electronic potential slightly, and both clean and hydroxylated surfaces exhibit similar polaron stability. In the case of holes introduced into the electronic structure, the lattice distortions around hole polarons were found to be larger than around electron polarons [119], and holes were formed by removal of an O(2p) valence electron. Combining DFT and Marcus/Holstein theory of electron/polaron transfer (where reorganization energy and electronic coupling were taken into account) [120,121,122], the hole hopping was shown to be equal in all directions in rutile, whereas in anatase, one direction was found to be adiabatic in character, i.e., thermal processes coupled to phonons. Furthermore, the study [119] showed that holes are thermodynamically more stable in the rutile phase, while electrons are more stable in the anatase phase. In a related study, Valentin et al. [123] showed that in order to describe localized defect states on reduced and hydroxylated TiO(110), it is necessary to go beyond semi-local description of the exchange-correlation functional and use hybrid exchange functionals instead. Even though the electron trapping nature of Ti(OH) groups was verified through lattice distortion [124], no support that these defects also act as hole traps was found. On the other hand, the SCF method [109,110] enable us the addition/substration of the electron density of the specified orbital at each SCF cycle [117]. Within this scheme, Zawadzki et al. [111] has studied the electron/hole self-trapping in different anatase and rutile surface facets. Their results indicate that the position of the hole trapping states in the band-gap varies among rutile and anatase facets. Ji et al. [125] reported that the bridge oxygen is the most stable hole trapping site based on their DFT + U methods. First-principles studies on carrier transport in hematite showed that substitutional doping of Al benefits small polaron migration, thus resulting in an improvement in conductivity compared to the undoped sample [126]. Liao and Carter [127], on the other hand, investigated how the activation energies for hole diffusion were affected by different dopants. The study suggests that hole hopping was via oxygen anions for hematite and that hole carriers are predicted to be attracted to O anions near the dopants. Adelstein et al. [128] studied small polaron mobility in hematite with DFT + U methods. In their work, both the prefactor and activation energies for adiabatic polaron transport were calculated.
The examples above indicate that DFT is able to characterize carrier transport, in particular electron/hole polarons. However, there remains still some development before DFT can be to calculate all details involved in the electron-hole transport process. Especially the difficulty to have an adequate description of the energy difference between a localized and a delocalized state is crucial [129], and the predictive power of DFT is hampered by the need to compare with experiment. In a recent review Shluger et al. [130] give more examples of modeling of electron and hole trapping in metal oxides, and also the discussion concerning the different challenges involved.
The major challenge of the transport of photoexcited carriers in real material is the recombination of electrons and holes [25,36,131]. Crystal structure, crystallinity and particle size strongly affect the separation and migration of the photoexcited carriers. The defects normally work as the trapping sites and recombination centers which is harmful to the transport of electrons and holes. Therefore, highly crystalline and stoichiometric materials have fewer defects with a high photocatalytic activity. On the other hand, the diffusion distance of electrons or holes to surface active sites becomes shorter in nanosized materials which can decrease the recombination probability. Moreover, nanosized materials usually have high surface to volume ratio, which contributes to effective interaction between charge carriers and surface active sites [25].
In addition, the morphology of oxide thin films, defect structures, and the electrolyte composition [40,132,133,134,135,136,137,138] all can affect the transport of the photo-excited carriers. Nanostructured materials such as nanotubes with one dimensional structure can enhance the charge collection efficiency by providing direct transport pathway [132]. Moreover, in the dye-sensitized solar cell, larger surface area can improve the loading of dye molecules which can combine with the increased transport pathways in the nanotudes to improve the carrier collection and electron-hole separation [139,140]. In colloidal semiconductors, such as, TiO and ZnO, semiconducting polymers, and amorphous organic photoconductors, the transport of the carriers can described in terms of a multiple trapping model [141,142,143], which is determined by the chemical composition and microscopic structure of the material [133,136]. In this model, charge transport is treated as the transfer of carriers between localized states or traps, which originates from the defects states associated with oxygen vacancies in noncrystalline TiO [136,137].
2.3. Electrochemical Surface Reactions
Once the hole arrives at the semiconductor-liquid interface, there is a clear analogy with the microscopic processes involved in an electrochemical cell. This is very beneficial as the recent developments in computational electrochemistry have been quite remarkable. The frameworks put-forth by, e.g., the Anderson group [144,145,146,147,148,149], Neurock group [150,151,152,153,154], and the Rossmeisl/Norskov [155,156,157,158,159,160,161,162] group has provided a molecular-level insight into the atomic-scale processes that occur at the vicinity of the anode/cathode surface. As a natural extension, based on the electrochemical framework suggested by Rossmeisl/Norskov group, Valdes et al. [163,164,165] suggested a theoretical framework in which the photo-oxidation of water can be described by first-principles methods. Here, the driving force for the reaction originates from the photoinduced hole at the edge of the valence/conduction band. Hence, in the scale obtained by aligning the energy levels of oxide semiconductors with the redox level of the standard hydrogen electrode, a deep valence band edge energy level will result in a larger thermodynamic driving force in the case of oxidation reactions. Although the framework treats only the thermodynamics of the reaction mechanism, it provides a methodology for a detailed atomistic understanding of photoelectrochemical water-splitting.
The standard hydrogen electrode (SHE) plays a crucial role in the computational electrochemical framework [166], as the SHE is by definition zero when the chemical potential of the H(aq) + e pair is equal to that of 1/2 H in the gas-phase. This circumvents the problem of calculating the energy of solvated protons and electrons and instead focuses on the gas-phase value of H, which is easily described by first-principles. Entropy contributions from the liquid phase are approximated by reference to the equilibrium pressure in contact with liquid water at 298.15 K and 0.0317 bar where the free energy of gas phase water is equal to the free energy of liquid water. This permits the use of gas-phase water to calculate the binding energies and its transformation into liquid-phase water when adding the entropy correction [157,159,161]. The free energy change of the reaction step involving the formation of O is set to the experimentally-obtained value of 4.92 eV per O molecule. The free energy of the reaction intermediates is calculated via DFT by also including the zero-point energy (ZPE) and vibrational contributions. Normally, the entropy contribution is low as the photoelectrochemical conditions are not so high in temperature. Furthermore, the effect of the electrode potential on the adsorption energies is simple to include by adding a stabilization of +eU when appropriate. In principle, the adsorption energy of reaction intermediates can depend on the electrode potential. However, first-principles studies [167] indicate that this effect is small, e.g., the adsorption energies of *O, *H, and *OH are only changed slightly when an electric field in the range of −0.3 V/Å to 0.3 V/Å is used. Assuming a double layer thickness of 3 Å, the range corresponds to a potential of −0.9 V and 0.9 V with respect to the zero potential. Therefore, the primary effect of the electrode potential is to change the (free) energy of the electrons.
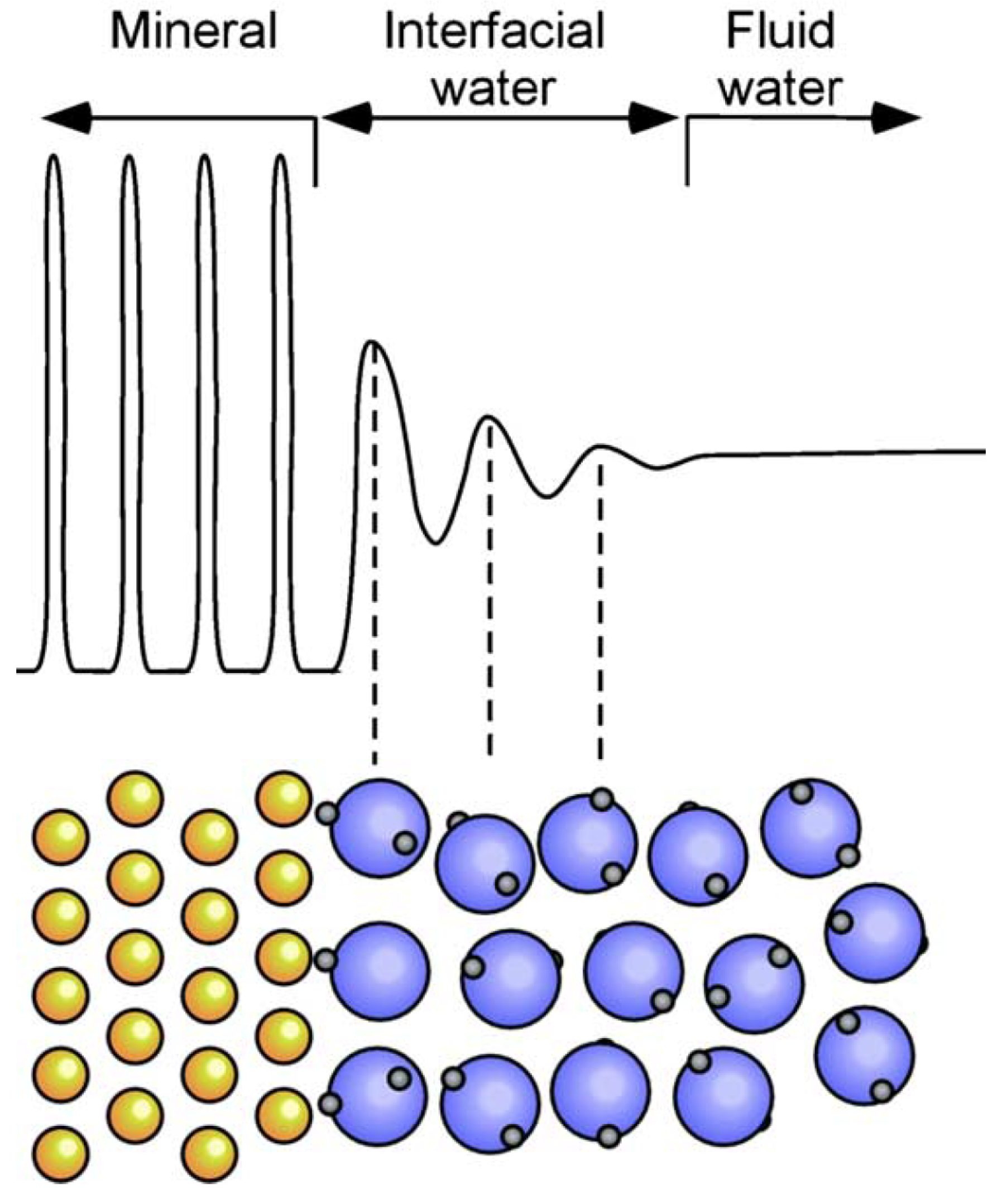
At the photoanode/electrolyte interface, the solvent water molecules may play an important role [168,169,170], see Figure 6 for a schematic overview. Contributions from the liquid phase are normally approximated by including several of water layers in the simulation [140,171,172,173]; however, there also exist more advanced models, e.g., Refs. [174,175]. Ab initio MD (AIMD) has offered key insights into the vibrational motion of higher-frequency modes of water adsorbed to titania [176,177], however, it is difficult to determine lower-frequency translational modes accurately due to the high computational cost of the simulation [178]. Classical Molecular dynamics study of water in contact with TiO surfaces has been performed by Ritwik et al. [179]. It was found that water OH bond lengths, H–O–H bond angles and dipole moments are not affected by the nature of the surface, whereas the orientation of the water molecules in the first and second monolayer is influenced strongly. The water network, with its many hydrogen bonds, will have a large effect on the stability of the reaction intermediates, especially the ones that can contribute with more hydrogen bonds [168]. First-principles results show that OH-containing reaction intermediates, such as, *OH and *OOH, can be stabilized up to 0.6 eV on Pt (111), whereas those of H and O are affected to a lesser extent, ca. 0.1 eV [167].
The main difference between the frameworks of electrochemistry and photoelectrochemistry is the origin of the driving potential [163,164]. In electrochemistry, this can be varied, whereas the photoelectrochemical driving force is the redox potential originating from the photoinduced hole in the valence band. Furthermore, it should be noted that the position of the valence band depends on the pH, however, the same dependence applies to the free energy of each reaction step for water oxidation [169]. Thus, to a first approximation, the thermodynamics of the photoelectrochemical reaction is unchanged by changes in the pH [180].
Pourbaix Surface Diagrams
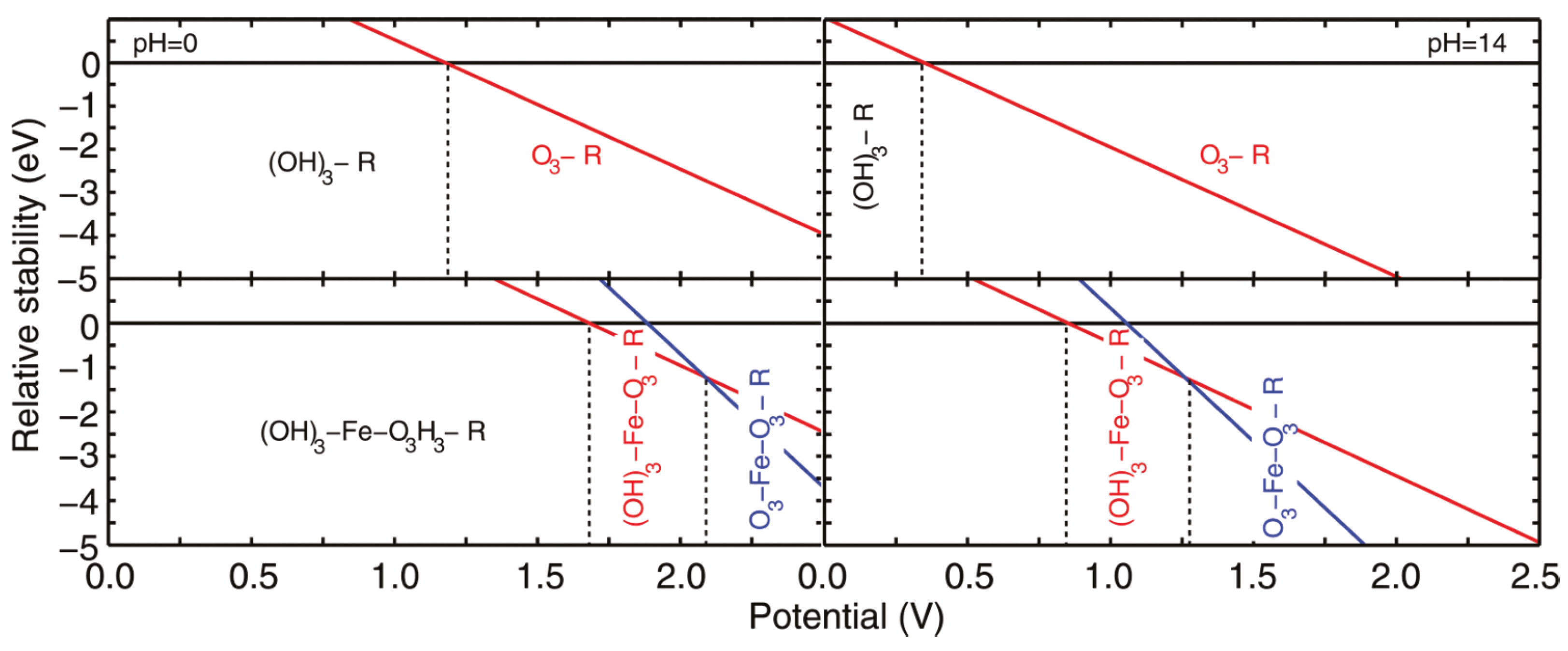
The photoelectrochemical framework allows for the construction of the surface phase diagram, i.e., which reaction intermediates are adsorbed on the surface under specific pH and under dark or light conditions [181,182], e.g., see Figure 7. In electrochemistry, these phase diagrams are called Pourbaix diagrams [183,184,185,186,187] and is today a standard tool when investigating electrochemcial reactions and include even novel techniques, such as machine learning [188]. Although Pourbaix diagrams were originally made for bulk transitions [189], the use of first-principles has shown that it can accurately describe which reaction intermediates are present and which are unstable under electrochemical conditions. The phase diagrams are only applied under stable conditions, which implies that the rate of holes that reach the surface needs to be greater than the back-reaction of water-splitting in order to build up a potential across the photoanode. Furthermore, it should be noted that the potential experienced by a metal nanoparticle at the surface of a photoelectrode during illumination is different from the externally-applied potential, as the quasi-Fermi level of holes shifts to positive potentials, leading to a shift in the Fermi level of the metal particle [169].
2.4. Reaction Mechanism
Although the overall water splitting reaction always evolves oxygen at the anode, the reaction mechanism depends on the pH of the electrolyte. Under acidic conditions, the water oxidation reaction is
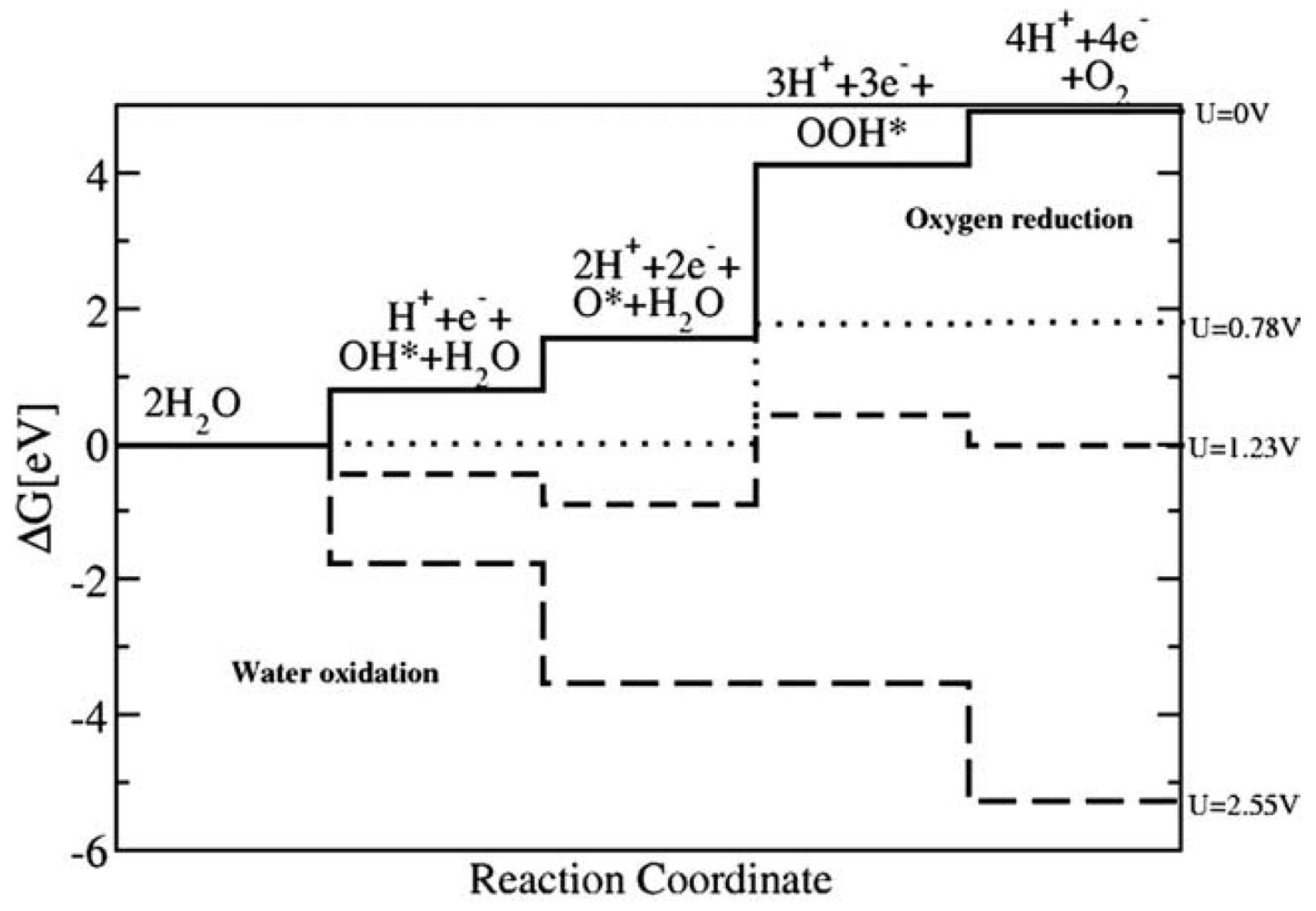
where the following simple reaction mechanism has been suggested in the literature [127,159,161,163,164,181,182,190], see Figure 8 for more details:
Under alkaline conditions, the water oxidation reaction is instead:
and analogously, the reaction mechanism can be written as:
In the theoretical electrochemical framework described above, the same reaction intermediates are present independently of the actual pH of the electrolyte. Direct recombination of oxygen atoms to O was not considered, as this process is expected to be associated with a large barrier on metals and oxides [191].
The described photoelectrochemical framework has been used to study photo-induced water-oxidation on rutile TiO(110) [163], WO (various facets) [164], and FeO(0001) [181,182,190,192,193]. So far, the redox potential from the band edge of the valance band has been sufficient to make the reaction thermodynamically favorable. Only in the case of FeO was the water oxidation prohibited on some surface terminations, however these terminations were not the most stable ones, which implies that during normal operation conditions, these do not play a part [181].
The electrochemical framework that has been described so far does not easily lend itself to the calculation of activation barriers; hence, the kinetics of any reaction are still missing. There exist extensions that deal with this issue. For instance, by varying the number of protons/electrons in the electrolyte, Skulasson et al. [171,194] could determine the activation energy for the hydrogen evolution reaction as a function of electrode potential. Another approach focuses on charge transfer reactions that use a single barrier calculation in an electrochemical environment and use the knowledge of the surface charge at the initial, transition and final states to estimate the barrier [195].
2.5. Overpotential
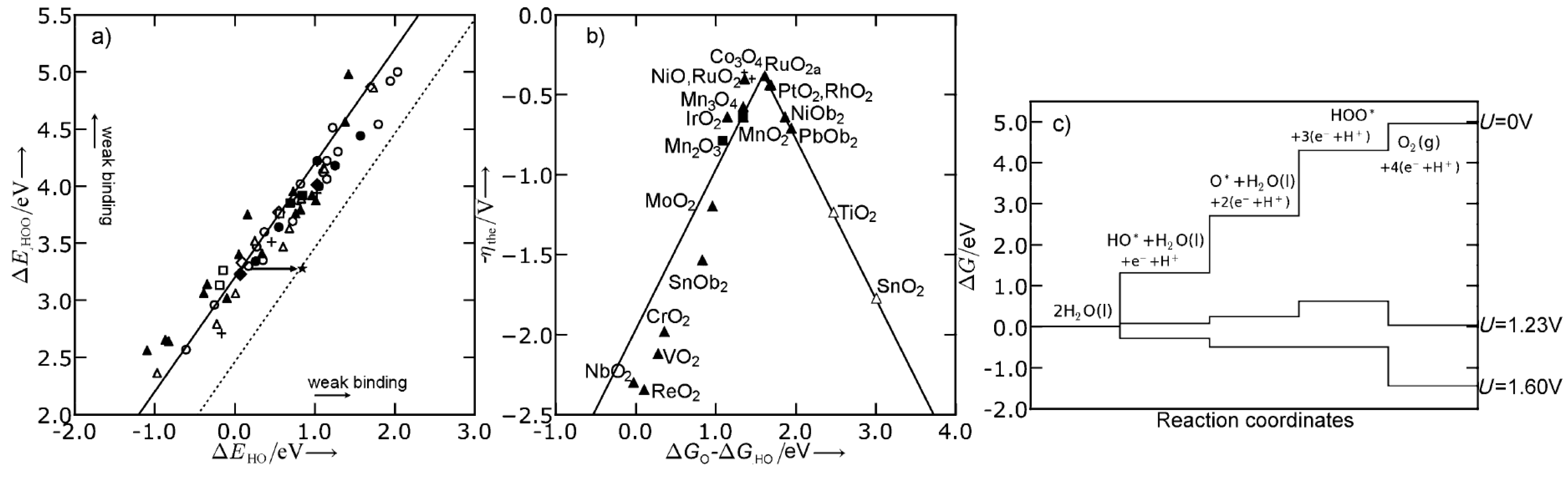
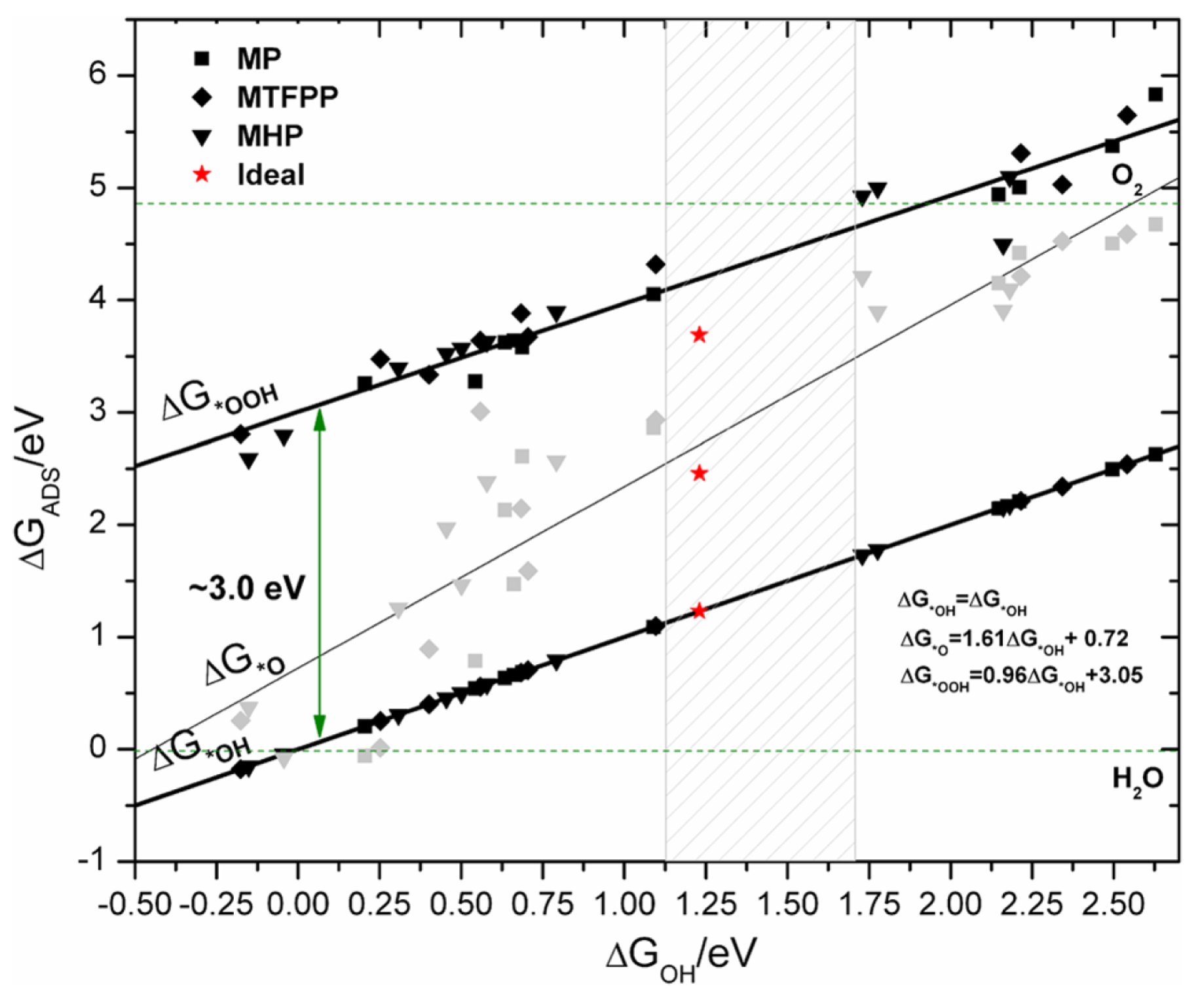
Abild-Pedersen et al. [196] showed the existence of approximately linear relationships between the adsorption energy of hydrogen and non-hydrogen containing species, e.g., OH and O, over many different materials. In combination with Brønsted–Evans–Polanyi relations [197] this has allowed for fast computational screening of heterogeneous catalysts [198]. In electrochemistry, the same linear relations have been used to show that in the oxygen evolution reaction (OER) and the oxygen reduction reaction (ORR), there exists a fundamental overpotential [157,199,200,201,202]. This was done by calculating the difference in Gibbs free energy for each reaction step and, by use of the linear scaling relations, expressing each reaction intermediate as a function of one of these differences or a linear combination [199,200]. In Figure 9, the OER volcano plots over different substrates are shown. Furthermore, the fundamental overpotential, which originates from the fixed distance between the binding of OH and OOH, is also shown. This observation provides an upper limit on how good an OER electrocatalysts can be expected to be.
The origin of the fundamental overpotential provides directions of how to overcome this limitation. If a catalyst is able to stabilize *OOH with respect to *OH (i.e., make the free energy difference between *OOH and *OH to values closer to 2.46 eV), it will circumvent the fundamental overpotential. One suggested pathway would be to use 3D structures, which are able to differentiate between the intermediates e.g., by confining the OOH group. Hangman porphyrins have been suggested as possible candidates, which indeed have been shown to be good catalysts for OER [203,204,205,206,207]; however, there is no support of this claim from first-principles as of yet [208], see Figure 10.
It is interesting to note that the processes in the biological respiratory system [209] utilize a secondary coordination sphere around the active site, which allows for non-covalent interactions with the reaction intermediates. In fact, the secondary coordination sphere appears to be a necessity in order to overcome energy barriers arising from the triplet ground state of the oxygen [210]. Designing a catalyst material that can mimic the interaction of the secondary coordination sphere remains a challenge, while potentially fruitful if successful.
3. Conclusions
Photoelectrochemistry is a very interesting topic covering various fields in chemistry and physics. Here, we have covered a selection of the many processes involved and discussed how first-principles methods can help in the interpretation and future improvements. The many success stories reveal a promising future, i.e., first-principles calculations will not be used solely to reproduce experimentally-known facts, but will become the standard starting point when a new photoelectrocatalyst for a known reaction is desired, or even when an unknown reaction to obtain a given product is needed.
However, there remain some issues that should be addressed and solved before this promise can come true. The use of more accurate exchange-correlation functionals seems to be able to push first-principles from being explanatory to predicative. This development will most certainly continue, and hopefully, band-gap design will soon be a reality. The problem associated with charge carrier transfer and the connection between DFT calculations of photoelectrocatalysis and dynamical simulations of electron transfer has not yet fully matured, although there exists a number of interesting developments. The use of the standard hydrogen electrode as the reference point has opened up the possibility to use first-principles methods with some easy to electrochemical and photoelectrochemical systems, at least when the information of the thermodynamics is key. The framework around kinetics is still being developed, but even in the light of these remaining issues, the future looks bright for first-principles and its impact on photoelectrochemsitry.
Acknowledgments
The authors want to acknowledge the financial support of the Swedish research council, Formas, and the Chalmers Area of Advance Material Science and Area of Advance Energy are acknowledged.
Conflicts of Interest
The author declares no conflict of interest.
Appendix A. Theoretical Methodologies of Density Functional Theory [211,212]
Appendix A.1. The Hohenberg-Kohn Theorems
The central theorems of DFT are based on the theorems given by Hohenberg and Kohn (HK) [43]:
Theorem A1.
For any system of interacting particles in an external potential , there is a one-to-one correspondence between the potentials and the ground-state particle density . The ground-state expectation value of any observable is a unique functional of the ground-state particle density :
Theorem A2.
For any external potential applied to an interacting particle system, it is possible to define a universal total energy functional of the particle density , written as
where includes all the electron-electron interaction and the kinetic energy of the interacting particle system. The global minimum of this functional is the exact ground-state total energy of the system, , and the particle density that minimizes this functional is the exact ground-state density . The first theorem implies that all ground-state properties of a system can be completely obtained by only the ground-state density. It emphasizes the importance of the ground-state density. The second theorem indicates that the ground-state density can be determined by the minimization of the energy functional, which can further be used to compute other ground-state observables.
Appendix A.2. The Kohn-Sham Equations
The Kohn and Sham ansatz [213] states that the interacting many-body problem can be replaced by a corresponding one-particle non-interacting system, where the total energy functional can be written as
which is the so-called Kohn-Sham functional. The first and second terms in Equation (A3) are the functional of the kinetic energy, and the external potential describing the interaction between nuclei and valence electrons. The third term is the Coulomb potential (Hartee term), and the forth term is the exchange-correlation functional, which includes all many-body effects of exchange and correlation, but also the missing contribution to the kinetic energy of interacting electrons. This is the only term that cannot be evaluated exactly, and should be described by different approximations. The last term is the energy contribution from the interaction of nuclei. Using the variational principle to minimize the Kohn-Sham functional with respect to the density leads to the one-particle Kohn-Sham equations
where are the eigenvalues, are the K-S orbitals and is the K-S potential,
with the exchange-correlation potential defined by
One thing that we should bear in mind is that the Kohn-Sham equations describe non-interacting electrons. The Kohn-Sham orbitals, , have no physical meaning. But the density obtained from the Kohn-Sham equation and the density of the system of interacting electrons should be the same. If the exchange-correlation potential is defined, the ground state density and energy can be obtained by solving the single-particle Kohn-Sham equations. However, the effective potential depends on the electron density, which depends on the Kohn-Sham orbitals, which, in turn, depends on the effective potential, therefore, one needs to solve the Kohn-Sham equations in a self-consistent manner.
Appendix A.3. The Exchange and Correlation Functionals
The main challenge for practical use of the Kohn-Sham equations is to find a good approximation for the exchange-correlation functional. Numerous approximations have been proposed. Most commonly used exchange-correlation functionals will be briefly discussed in the following part.
In the local-density approximation (LDA) approximation, the exchange-correlation functional energy is evaluated from the exchange-correlation energy for the homogeneous electron gas [214]. The as a function of density within each volume can be assumed as the one derived from the uniform electron gas for that density. The total exchange-correlation energy of the system can be written as
where is the exchange-correlation energy density of a homogenous gas with density . In practice, the exchange and correlation energy of the homogeneous electron gas can be calculated separately. The form of the exchange energy is well-known with a simple analytical expression [212] and the correlation energy has been calculated accurately with quantum Monte Carlo method [215].
In principle, LDA can only be valid for systems with slowly varying densities. Calculations performed with this method for atoms, molecules and solids show that Equation (A7) also works surprisingly well for these systems. However, LDA indeed has some accuracy problems. For example, it tends to overestimate cohesive energies and underestimate the lattice constants for metals and insulators [216,217].
One way to improve LDA is to include the gradient of the density instead of only including the local density . This is the so-called generalized gradient approximation (GGA) [218,219,220]. Such a functional can be described as
where is a functional of the density and the gradient of the density and the is the exchange energy of a homogeneous electron gas. The GGA-functionals have many parameterizations for example, such as B88 by Becke [221], PW91 by Perdew and Wang [222] and PBE by Perdew, Burke and Enzerhof [223]. With the inclusion of the gradient of the density , GGA can result in better agreement with experiment than LDA for many properties of molecule and solids such as geometries and ground state energies.
Recently, the functional for “GGAs for solids” (e.g., AM05 [224] and PBEsol [60]) have been proposed. They produce rather accurate lattice constants and surface energies, but give poor atomization energies.
The meta-GGAs are the third generation functionals, which includes the second derivative of the density, , and/or the kinetic energy density, , in the exchange and correlation potential. Functionals of this type are, for example, TPSS [225], revTPSS [226] and AM06-L [227]. The meta-GGAs improve the accuracy of results further and predicting good agreement with experiment for lattice constants, surface energies as well as atomization energies.
A mixing scheme of some exact exchange with the exchange and correlation from DFT is employed in hybrid functionals. Becke proposed an adiabatic connection formula which can be used as a starting point for a hybrid functional [228].
where is the exchange-correlation energy and scales the contribution from exact exchange. This formula is the connection between the non-interacting system and the fully interacting one with density . In the = 0 limit, it restores to the Kohn-Sham non-interacting particles system. B3LYP (Becke, three-parameter, Lee-Yang-Parr) [229,230,231] is most widely used especially in quantum chemistry, where three parameters (determined by fitting to experiment) are employed to treat the mixing of exact exchange and the DFT exchange-correlation. The Heyd-Scuseria-Ernzershof (HSE) hybrid functional is a hybrid density functional based on a screened Coulomb potential for the exchange interaction [69]. Using a screened Coulomb potential for Hartree-Fock (HF) exchange enables fast and accurate hybrid calculations, where the Coulomb potential are decomposed into a long-range and a short range part. In practice, the splitting of the full Coulomb potential is done by means of error functions
where a is the mixing parameter and is the adjustable parameter controlling the short-range of interactions beyond which the short range interactions becomes negligible. HSE06 with standard value of and have been shown to yield good results for many systems. The PBE0 functional [70] can be obtained for = 0. is the short range exact exchange energy. and are the short and long range components of the PBE exchange energy, respectively, and is the PBE correlation energy. The M06 suite of functionals [227] contain a set of four meta hybrid GGA functionals, i.e., M06-L, M06, M06-2X and M06-HF, each of which has different amount of exact exchange. The M06-L is completely local without any HF exchange, and M06, M06-2X, and M06-HF have 27%, 54%, and 100% of HF exchange, respectively. The M06 suite mainly improves one of the big deficiencies of DFT in describing the dispersion forces. Each of them has the advantages to specific systems, for example, M06-HF is suitable for TD-DFT calculations of Rydberg and charge-transfer states.
References
- Verne, J. L’Île Mystérieuse; Pierre-Jules Hetzel: Chartres, France, 1874. [Google Scholar]
- Campbell, C.J.; Laherrére, J.H. The End of Cheap Oil. Sci. Am. 1998, 278, 60–65. [Google Scholar] [CrossRef]
- Bentley, R.; Boyle, G. Global Oil Production: Forecasts and Methodologies. Environ. Plan. B Plan. Des. 2008, 35, 609–626. [Google Scholar] [CrossRef]
- Hook, M.; Tang, X. Depletion of fossil fuels and anthropogenic climate change—A review. Energy Policy 2013, 52, 797–809. [Google Scholar] [CrossRef]
- BP Statistical Review of World Energy. 2016. Available online: http://www.bp.com/en/global/corporate/energy-economics/statistical-review-of-world-energy.html (accessed on 5 March 2017).
- Gerland, P.; Raftery, A.; Ševčíková, H.; Li, N.; Gu, D.; Spoorenberg, T.; Alkema, L.; Fosdick, B.; Chunn, J.; Lalic, N.; et al. World population stabilization unlikely this century. Science 2014, 346, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.A.; Warner, K.J. The 21st century population-energy-climate nexus. Energy Policy 2016, 93, 206–212. [Google Scholar] [CrossRef]
- Smalley, R.E. Future Global Energy Prosperity: The Terawatt Challenge. MRS Bull. 2005, 30, 412–417. [Google Scholar] [CrossRef]
- Lewis, N.S. Toward Cost-Effective Solar Energy Use. Science 2007, 315, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Armaroli, N.; Balzani, V. The Future of Energy Supply: Challenges and Opportunities. Angew. Chem. Int. Ed. 2007, 46, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Vesborg, P.C.K.; Jaramillo, T.F. Addressing the terawatt challenge: Scalability in the supply of chemical elements for renewable energy. RCS Adv. 2012, 2, 7933–7947. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [PubMed]
- Rajeshwar, K.; McConnell, R.; Harrison, K.; Licht, S. Solar Hydrogen Generation—Toward a Renewable Energy Future; Springer: Berlin, Germany, 2008. [Google Scholar]
- Herron, J.A.; Kim, J.; Upadhye, A.A.; Huber, G.W.; Maravelias, C.T. A general framework for the assessment of solar fuel technologies. Energy Environ. Sci. 2015, 8, 1754–5692. [Google Scholar] [CrossRef]
- Acar, C.; Dincer, I.; Naterer, G.F. Review of photocatalytic water-splitting methods for sustainable hydrogen production. Int. J. Energy Res. 2016, 40, 1449–1473. [Google Scholar] [CrossRef]
- Junge, H.; Rockstroh, N.; Fischer, S.; Brückner, A.; Ludwig, R.; Lochbrunner, S.; Kühn, O.; Beller, M. Light to Hydrogen: Photocatalytic Hydrogen Generation from Water with Molecularly-Defined Iron Complexes. Inorganics 2017, 5, 14. [Google Scholar] [CrossRef]
- Dura, L.; Wächtler, M.; Kupfer, S.; Kübel, J.; Ahrens, J.; Höfler, S.; Bröring, M.; Dietzek, B.; Beweries, T. Photophysics of BODIPY Dyes as Readily-Designable Photosensitisers in Light-Driven Proton Reduction. Inorganics 2017, 5, 21. [Google Scholar] [CrossRef]
- Li, C.T.; Lin, R.Y.Y.; Lin, J.T. Sensitizers for Aqueous-Based Solar Cells. Chem. Asian J. 2017, 12, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Fujisawa, J.i.; Hanaya, M. Highly-efficient dye-sensitized solar cells with collaborative sensitization by silyl-anchor and carboxy-anchor dyes. Chem. Commun. 2015, 51, 15894–15897. [Google Scholar] [CrossRef] [PubMed]
- Galliano, S.; Bella, F.; Gerbaldi, C.; Falco, M.; Viscardi, G.; Grätzel, M.; Barolo, C. Photoanode/Electrolyte Interface Stability in Aqueous Dye-Sensitized Solar Cells. Energy Technol. 2017, 5, 300–311. [Google Scholar] [CrossRef]
- Bella, F.; Galliano, S.; Falco, M.; Viscardi, G.; Barolo, C.; Gratzel, M.; Gerbaldi, C. Approaching truly sustainable solar cells by the use of water and cellulose derivatives. Green Chem. 2017, 19, 1043–1051. [Google Scholar] [CrossRef]
- Bella, F.; Galliano, S.; Falco, M.; Viscardi, G.; Barolo, C.; Gratzel, M.; Gerbaldi, C. Unveiling iodine-based electrolytes chemistry in aqueous dye-sensitized solar cells. Chem. Sci. 2016, 7, 4880–4890. [Google Scholar] [CrossRef]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef] [PubMed]
- Pinaud, B.A.; Benck, J.D.; Seitz, L.C.; Forman, A.J.; Chen, Z.; Deutsch, T.G.; James, B.D.; Baum, K.N.; Baum, G.N.; Ardo, S.; et al. Technical and economic feasibility of centralized facilities for solar hydrogen production via photocatalysis and photoelectrochemistry. Energy Environ. Sci. 2013, 6, 1983–2002. [Google Scholar] [CrossRef]
- Jafari, T.; Moharreri, E.; Amin, A.S.; Miao, R.; Song, W.; Suib, S.L. Photocatalytic Water Splitting—The Untamed Dream: A Review of Recent Advances. Molecules 2016, 21, 900. [Google Scholar] [CrossRef] [PubMed]
- Navarro Yerga, R.; Álvarez Galván, M.; del Valle, F.; Villoria de la Mano, J.; Fierro, J. Water Splitting on Semiconductor Catalysts under Visible-Light Irradiation. ChemSusChem 2009, 2, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Fang, W.Q.; Zhao, H.J.; Yang, H.G. Inorganic Photocatalysts for Overall Water Splitting. Chem. Asian J. 2012, 7, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Osterloh, F.E. Inorganic nanostructures for photoelectrochemical and photocatalytic water splitting. Chem. Soc. Rev. 2013, 42, 2294–2320. [Google Scholar] [CrossRef] [PubMed]
- Ran, J.; Zhang, J.; Yu, J.; Jaroniec, M.; Qiao, S.Z. Earth-abundant cocatalysts for semiconductor-based photocatalytic water splitting. Chem. Soc. Rev. 2014, 43, 7787–7812. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Kubota, J.; Domen, K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem. Soc. Rev. 2014, 43, 7520–7535. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37. [Google Scholar] [CrossRef] [PubMed]
- Rajeshwar, K. Fundamentals of Semiconductor Electrochemistry and Photoelectrochemistry. In Encyclopedia of Electrochemistry; Wiley-VCH: Berlin, Germany, 2007. [Google Scholar]
- Van de Krol, R.; Grätzel, M. Photoelectrochemical Hydrogen Production; Springer: Berlin, Germany, 2012. [Google Scholar]
- Lewerenz, H.J.; Peter, L. Photoelectrochemical Water Splitting: Materials, Processes and Architectures; Royal Society of Chemistry: Cambridge, UK, 2013. [Google Scholar]
- Ni, M.; Leung, M.K.; Leung, D.Y.; Sumathy, K. A review and recent developments in photocatalytic water-splitting using for hydrogen production. Renew. Sustain. Energy Rev. 2007, 11, 401–425. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Dau, H.; Limberg, C.; Reier, T.; Risch, M.; Roggan, S.; Strasser, P. The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem 2010, 2, 724–761. [Google Scholar] [CrossRef]
- Zhang, X.; Bieberle-Hütter, A. Modeling and Simulations in Photoelectrochemical Water Oxidation: From Single Level to Multiscale Modeling. ChemSusChem 2016, 9, 1223–1242. [Google Scholar] [CrossRef] [PubMed]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 3. [Google Scholar] [CrossRef]
- Iandolo, B.; Wickman, B.; Zoric, I.; Hellman, A. The rise of hematite: Origin and strategies to reduce the high onset potential for the oxygen evolution reaction. J. Mater. Chem. A 2015, 3, 16896–16912. [Google Scholar] [CrossRef]
- Montoya, J.H.; Seitz, L.C.; Chakthranont, P.; Vojvodic, A.; Jaramillo, T.F.; Norskov, J.K. Materials for solar fuels and chemicals. Nat. Mater. 2017, 16, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Bak, T.; Nowotny, J.; Rekas, M.; Sorrell, C. Photo-electrochemical hydrogen generation from water using solar energy. Materials-related aspects. Int. J. Hydrog. Energy 2002, 27, 991–1022. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Jones, R.O. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87, 897–923. [Google Scholar] [CrossRef]
- Hasnip, P.J.; Refson, K.; Probert, M.I.J.; Yates, J.R.; Clark, S.J.; Pickard, C.J. Density functional theory in the solid state. Philos. Trans. R. Soc. Lond. A Math. Phys. Eng. Sci. 2014, 372, 20130270. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Dreizler, R.M.; Gross, E.K.U. Density-Functional Theory: An Approach to the Quantum Many-Body Problem; Springer: Berlin, Germany, 1990. [Google Scholar]
- Fiolhais, C.; Nogueira, F.; Marques, M. A Primer in Density Functional Theory; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Curtarolo, S.; Hart, G.L.W.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The high-throughput highway to computational materials design. Nat. Mater. 2013, 12, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Shin, Y.; Persson, K.A. Machine learning bandgaps of double perovskites. Nat. Rev. Mater. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Dai, H.L.; Ho, W. Spectroscopy and Photochemistry on Metal Surfaces; World Scientific: Singapore, Singapore, 1995. [Google Scholar]
- Liebsch, A. Electronic Excitations at Metal Surfaces; Plenum Press: Berlin, Germany, 1997. [Google Scholar]
- Nozik, A.J. Spectroscopy and hot electron relaxation dynamics in semiconductor quantum wells and quantum dots. Annu. Rev. Phys. Chem. 2001, 52, 193–231. [Google Scholar] [CrossRef] [PubMed]
- Brongersma, M.L.; Halas, N.J.; Nordlander, P. Plasmon-induced hot carrier science and technology. Nat. Nano 2015, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.R.M.; Asenjo-Garcia, A.; García de Abajo, F.J. Hot-Electron Dynamics and Thermalization in Small Metallic Nanoparticles. ACS Photonics 2016, 3, 1637–1646. [Google Scholar] [CrossRef]
- Ogawa, T.; Yanai, N.; Monguzzi, A.; Kimizuka, N. Highly Efficient Photon Upconversion in Self-Assembled Light-Harvesting Molecular Systems. Sci. Rep. 2015, 5, 10882. [Google Scholar] [CrossRef] [PubMed]
- Godby, R.W.; Schlüter, M.; Sham, L.J. Trends in self-energy operators and their corresponding exchange-correlation potentials. Phys. Rev. B 1987, 36, 6497–6500. [Google Scholar] [CrossRef]
- Godby, R.W.; Schlüter, M.; Sham, L.J. Self-energy operators and exchange-correlation potentials in semiconductors. Phys. Rev. B 1988, 37, 10159–10175. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Kuisma, M.; Ojanen, J.; Enkovaara, J.; Rantala, T.T. Kohn-Sham potential with discontinuity for band gap materials. Phys. Rev. B 2010, 82, 115106. [Google Scholar] [CrossRef]
- Hedin, L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. 1965, 139, A796–A823. [Google Scholar] [CrossRef]
- Castelli, I.E.; Hüser, F.; Pandey, M.; Li, H.; Thygesen, K.S.; Seger, B.; Jain, A.; Persson, K.A.; Ceder, G.; Jacobsen, K.W. New Light-Harvesting Materials Using Accurate and Efficient Bandgap Calculations. Adv. Energy Mater. 2015, 5, 1400915. [Google Scholar] [CrossRef]
- Pilania, G.; Mannodi-Kanakkithodi, A.; Uberuaga, B.P.; Ramprasad, R.; Gubernatis, J.E.; Lookman, T. Machine learning bandgaps of double perovskites. Sci. Rep. 2016, 6, 19375. [Google Scholar] [CrossRef] [PubMed]
- Gallino, F.; Pacchioni, G.; Valentin, C.D. Transition levels of defect centers in ZnO by hybrid functionals and localized basis set approach. J. Chem. Phys. 2010, 133, 144512. [Google Scholar] [CrossRef] [PubMed]
- Lany, S.; Zunger, A. Assessment of correction methods for the band-gap problem and for finite-size effects in supercell defect calculations: Case studies for ZnO and GaAs. Phys. Rev. B 2008, 78, 235104. [Google Scholar] [CrossRef]
- De Walle, C.G.V.; Neugebauer, J. First-principles calculations for defects and impurities: Applications to III-nitrides. J. Appl. Phys. 2004, 95, 3851–3879. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into Current Limitations of Density Functional Theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Liechtenstein, A.I.; Anisimov, V.I.; Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467–R5470. [Google Scholar] [CrossRef]
- Makov, G.; Payne, M.C. Periodic boundary conditions in ab initio calculations. Phys. Rev. B 1995, 51, 4014–4022. [Google Scholar] [CrossRef]
- Moss, T.S. The Interpretation of the Properties of Indium Antimonide. Proc. Phys. Soc. Sect. B 1954, 67, 775. [Google Scholar] [CrossRef]
- Burstein, E. Anomalous Optical Absorption Limit in InSb. Phys. Rev. 1954, 93, 632–633. [Google Scholar] [CrossRef]
- Vanpoucke, D.E.P.; Bultinck, P.; Cottenier, S.; Van Speybroeck, V.; Van Driessche, I. Aliovalent doping of CeO2: DFT study of oxidation state and vacancy effects. J. Mater. Chem. A 2014, 2, 13723–13737. [Google Scholar] [CrossRef]
- Celotti, G.; Nobili, D.; Ostoja, P. Lattice parameter study of silicon uniformly doped with boron and phosphorus. J. Mater. Sci. 1974, 9, 821–828. [Google Scholar] [CrossRef]
- Denton, A.R.; Ashcroft, N.W. Vegard’s law. Phys. Rev. A 1991, 43, 3161–3164. [Google Scholar] [CrossRef] [PubMed]
- Birch, F. Finite Elastic Strain of Cubic Crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The Compressibility of Media under Extreme Pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Asahi, R.; Morikawa, T.; Ohwaki, T.; Aoki, K.; Taga, Y. Visible-Light Photocatalysis in Nitrogen-Doped Titanium Oxides. Science 2001, 293, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, S.; Paganini, M.C.; Giamello, E.; Selloni, A.; Di Valentin, C.; Pacchioni, G. Origin of Photoactivity of Nitrogen-Doped Titanium Dioxide under Visible Light. J. Am. Chem. Soc. 2006, 128, 15666–15671. [Google Scholar] [CrossRef] [PubMed]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Reduced and n-Type Doped TiO2: Nature of Ti3+ Species. J. Phys. Chem. C 2009, 113, 20543–20552. [Google Scholar] [CrossRef]
- Kamisaka, H.; Adachi, T.; Yamashita, K. Theoretical study of the structure and optical properties of carbon-doped rutile and anatase titanium oxides. J. Chem. Phys. 2005, 123, 084704. [Google Scholar] [CrossRef] [PubMed]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Theory of Carbon Doping of Titanium Dioxide. Chem. Mater. 2005, 17, 6656–6665. [Google Scholar] [CrossRef]
- Liu, G.; Zhao, Y.; Sun, C.; Li, F.; Lu, G.; Cheng, H.M. Synergistic Effects of B/N Doping on the Visible-Light Photocatalytic Activity of Mesoporous TiO2. Angew. Chem. Int. Ed. 2008, 47, 4516–4520. [Google Scholar] [CrossRef] [PubMed]
- Long, R.; English, N.J. First-Principles Calculation of Synergistic (N,P)-Codoping Effects on the Visible-Light Photocatalytic Activity of Anatase TiO2. J. Phys. Chem. C 2010, 114, 11984–11990. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Synergistic Effects of Bi/S Codoping on Visible Light-Activated Anatase TiO2 Photocatalysts from First Principles. J. Phys. Chem. C 2009, 113, 8373–8377. [Google Scholar] [CrossRef]
- Pelaez, M.; Nolan, N.T.; Pillai, S.C.; Seery, M.K.; Falaras, P.; Kontos, A.G.; Dunlop, P.S.; Hamilton, J.W.; Byrne, J.; O’Shea, K.; et al. A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl. Catal. B Environ. 2012, 125, 331–349. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.Z.; Nagpure, S.; Kim, D.Y.; Rankin, S.E. Synthesis and Catalytic Applications of Non-Metal Doped Mesoporous Titania. Inorganics 2017, 5, 15. [Google Scholar] [CrossRef]
- Meng, X.Y.; Qin, G.W.; Li, S.; Wen, X.H.; Ren, Y.P.; Pei, W.L.; Zuo, L. Enhanced photoelectrochemical activity for Cu and Ti doped hematite: The first principles calculations. Appl. Phys. Lett. 2012, 98, 112104. [Google Scholar] [CrossRef]
- Liang, W.Y. Excitons. Phys. Educ. 1970, 5, 226. [Google Scholar] [CrossRef]
- Dember, H. Über eine photoelektronische Kraft in Kupferoxydul-Kristallen (Photoelectric E.M.F. in Cuprous-Oxide Crystals). Z. Phys. 1931, 32, 554. [Google Scholar]
- Sze, S.M. Semiconductor Devices, Physics and Technology; Wiley: Hoboken, NJ, USA, 1985. [Google Scholar]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Umezawa, H.; Ramanujam, K.; ichi Shikata, S. Thermally Stable Schottky Barrier Diode by Ru/Diamond. Appl. Phys. Express 2009, 2, 011202. [Google Scholar] [CrossRef]
- Kwon, S.; Lee, S.J.; Kim, S.M.; Lee, Y.; Song, H.; Park, J.Y. Probing the nanoscale Schottky barrier of metal/semiconductor interfaces of Pt/CdSe/Pt nanodumbbells by conductive-probe atomic force microscopy. Nanoscale 2015, 7, 12297–12301. [Google Scholar] [CrossRef] [PubMed]
- Durcan, C.A.; Balsano, R.; LaBella, V.P. Nanoscale mapping of the W/Si(001) Schottky barrier. J. Appl. Phys. 2014, 116, 023705. [Google Scholar] [CrossRef]
- Durcan, C.A.; Balsano, R.; LaBella, V.P. Time dependent changes in Schottky barrier mapping of the W/Si(001) interface utilizing ballistic electron emission microscopy. J. Appl. Phys. 2015, 117, 245306. [Google Scholar] [CrossRef]
- Tung, R.T. Electron transport at metal-semiconductor interfaces: General theory. Phys. Rev. B 1992, 45, 13509–13523. [Google Scholar] [CrossRef]
- Tung, R.T. Recent advances in Schottky barrier concepts. Mater. Sci. Eng. R Rep. 2001, 35, 1–138. [Google Scholar] [CrossRef]
- Tung, R.T. The physics and chemistry of the Schottky barrier height. Appl. Phys. Rev. 2014, 1, 011304. [Google Scholar]
- Jiao, Y.; Hellman, A.; Fang, Y.; Gao, S.; Käll, M. Schottky barrier formation and band bending revealed by first- principles calculations. Sci. Rep. 2015, 5, 11374. [Google Scholar] [CrossRef] [PubMed]
- Austin, I.; Mott, N. Polarons in crystalline and non-crystalline materials. Adv. Phys. 1969, 18, 41–102. [Google Scholar] [CrossRef]
- Shluger, A.L.; Stoneham, A.M. Small polarons in real crystals: Concepts and problems. J. Phys. Condens. Matter 1993, 5, 3049. [Google Scholar] [CrossRef]
- Bosman, A.; van Daal, H. Small-polaron versus band conduction in some transition-metal oxides. Adv. Phys. 1970, 19, 1–117. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Oxidation—Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Hellman, A.; Razaznejad, B.; Lundqvist, B.I. Potential-energy surfaces for excited states in extended systems. J. Chem. Phys. 2004, 120, 4593–4602. [Google Scholar] [CrossRef] [PubMed]
- Gavnholt, J.; Olsen, T.; Engelund, M.; Schiøtz, J. ΔSelf-consistent field method to obtain potential energy surfaces of excited molecules on surfaces. Phys. Rev. B 2008, 78, 075441. [Google Scholar] [CrossRef]
- Zawadzki, P.; Laursen, A.B.; Jacobsen, K.W.; Dahl, S.; Rossmeisl, J. Oxidative trends of TiO2-hole trapping at anatase and rutile surfaces. Energy Environ. Sci. 2012, 5, 9866–9869. [Google Scholar] [CrossRef]
- Lany, S.; Zunger, A. Polaronic hole localization and multiple hole binding of acceptors in oxide wide-gap semiconductors. Phys. Rev. B 2009, 80, 085202. [Google Scholar] [CrossRef]
- Alexandrov, V.; Neumann, A.; Scherer, M.M.; Rosso, K.M. Electron Exchange and Conduction in Nontronite from First-Principles. J. Phys. Chem. C 2013, 117, 2032–2040. [Google Scholar] [CrossRef]
- Maxisch, T.; Zhou, F.; Ceder, G. Ab initio study of the migration of small polarons in olivine LixFePO4 and their association with lithium ions and vacancies. Phys. Rev. B 2006, 73, 104301. [Google Scholar] [CrossRef]
- Chen, H.; Umezawa, N. Hole localization, migration, and the formation of peroxide anion in perovskite SrTiO3. Phys. Rev. B 2014, 90, 035202. [Google Scholar] [CrossRef]
- Carvalho, A.; Alkauskas, A.; Pasquarello, A.; Tagantsev, A.K.; Setter, N. A hybrid density functional study of lithium in ZnO: Stability, ionization levels, and diffusion. Phys. Rev. B 2009, 80, 195205. [Google Scholar] [CrossRef]
- Zawadzki, P.; Jacobsen, K.W.; Rossmeisl, J. Electronic hole localization in rutile and anatase TiO2- Self-interaction correction in Δ-SCF {DFT}. Chem. Phys. Lett. 2011, 506, 42–45. [Google Scholar] [CrossRef]
- Deskins, N.A.; Rousseau, R.; Dupuis, M. Localized Electronic States from Surface Hydroxyls and Polarons in TiO2(110). J. Phys. Chem. C 2009, 113, 14583–14586. [Google Scholar] [CrossRef]
- Deskins, N.A.; Dupuis, M. Intrinsic Hole Migration Rates in TiO2 from Density Functional Theory. J. Phys. Chem. C 2009, 113, 346–358. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Holstein, T. Studies of Polaron Motion: Part I. The Molecular-Crystal Model. Ann. Phys. 2000, 8, 706–724. [Google Scholar] [CrossRef]
- Holstein, T. Studies of Polaron Motion: Part II. The “Small” Polaron. Ann. Phys. 2000, 281, 725–773. [Google Scholar] [CrossRef]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Electronic Structure of Defect States in Hydroxylated and Reduced Rutile TiO2(110) Surfaces. Phys. Rev. Lett. 2006, 97, 166803. [Google Scholar] [CrossRef] [PubMed]
- Hurum, D.C.; Agrios, A.G.; Gray, K.A.; Rajh, T.; Thurnauer, M.C. Explaining the Enhanced Photocatalytic Activity of Degussa P25 Mixed-Phase TiO2 Using EPR. J. Phys. Chem. B 2003, 107, 4545–4549. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, B.; Luo, Y. Location of Trapped Hole on Rutile-TiO2(110) Surface and Its Role in Water Oxidation. J. Phys. Chem. C 2012, 116, 7863–7866. [Google Scholar] [CrossRef]
- Kleiman-Shwarsctein, A.; Huda, M.N.; Walsh, A.; Yan, Y.; Stucky, G.D.; Hu, Y.S.; Al-Jassim, M.M.; McFarland, E.W. Electrodeposited Aluminum-Doped α-Fe2O3 Photoelectrodes: Experiment and Theory. Chem. Mater. 2010, 22, 510–517. [Google Scholar] [CrossRef]
- Liao, P.; Carter, E.A. Hole transport in pure and doped hematite. J. Appl. Phys. 2012, 112, 013701. [Google Scholar] [CrossRef]
- Adelstein, N.; Neaton, J.B.; Asta, M.; De Jonghe, L.C. Density functional theory based calculation of small-polaron mobility in hematite. Phys. Rev. B 2014, 89, 245115. [Google Scholar] [CrossRef]
- Pacchioni, G. Modeling doped and defective oxides in catalysis with density functional theory methods: Room for improvements. J. Chem. Phys. 2008, 128, 182505. [Google Scholar] [CrossRef] [PubMed]
- Shluger, A.L.; McKenna, K.P.; Sushko, P.V.; Ramo, D.M.; Kimmel, A.V. Modelling of electron and hole trapping in oxides. Model. Simul. Mater. Sci. Eng. 2009, 17, 084004. [Google Scholar] [CrossRef]
- Zou, Z.; Ye, J.; Sayama, K.; Arakawa, H. Direct splitting of water under visible light irradiation with an oxide semiconductor photocatalyst. Nature 2001, 414, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, S.; Abdullah, H. Electron transport study of indium oxide as photoanode in DSSCs: A review. Renew. Sustain. Energy Rev. 2016, 63, 245–255. [Google Scholar] [CrossRef]
- Frank, A.J.; Kopidakis, N.; van de Lagemaat, J. Electrons in nanostructured TiO2 solar cells: Transport, recombination and photovoltaic properties. Coord. Chem. Rev. 2004, 248, 1165–1179. [Google Scholar] [CrossRef]
- Riss, A.; Elser, M.J.; Bernardi, J.; Diwald, O. Stability and Photoelectronic Properties of Layered Titanate Nanostructures. J. Am. Chem. Soc. 2009, 131, 6198–6206. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yang, P.; Sun, Y.; Wu, Y.; Mayers, B.; Gates, B.; Yin, Y.; Kim, F.; Yan, H. One-Dimensional Nanostructures: Synthesis, Characterization, and Applications. Adv. Mater. 2003, 15, 353–389. [Google Scholar] [CrossRef]
- Anta, J.A.; Nelson, J.; Quirke, N. Charge transport model for disordered materials: Application to sensitized TiO2. Phys. Rev. B 2002, 65, 125324. [Google Scholar] [CrossRef]
- Göpel, W.; Rocker, G.; Feierabend, R. Intrinsic defects of TiO2(110): Interaction with chemisorbed O2, H2, CO, and CO2. Phys. Rev. B 1983, 28, 3427–3438. [Google Scholar] [CrossRef]
- Wickman, B.; Bastos Fanta, A.; Burrows, A.; Hellman, A.; Wagner, J.B.; Iandolo, B. Iron Oxide Films Prepared by Rapid Thermal Processing for Solar Energy Conversion. Sci. Rep. 2017, 7, 40500. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Z.; Li, W.; Zhang, X.; He, D.; Xiao, X. Ag Nanoparticles Located on Three-Dimensional Pine Tree-Like Hierarchical TiO2 Nanotube Array Films as High-Efficiency Plasmonic Photocatalysts. Nanoscale Res. Lett. 2017, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Antony, A.C.; Akhade, S.A.; Liang, T.; Janik, M.J.; Maranas, J.K.; Sinnott, S.B. Simulating an Applied Voltage in Molecular Dynamics Using Charge Optimized Many Body (COMB3) Potentials. ECS Trans. 2015, 69, 103–105. [Google Scholar] [CrossRef]
- Van de Lagemaat, J.; Park, N.G.; Frank, A.J. Influence of Electrical Potential Distribution, Charge Transport, and Recombination on the Photopotential and Photocurrent Conversion Efficiency of Dye-Sensitized Nanocrystalline TiO2 Solar Cells: A Study by Electrical Impedance and Optical Modulation Techniques. J. Phys. Chem. B 2000, 104, 2044–2052. [Google Scholar] [CrossRef]
- De Jongh, P.E.; Vanmaekelbergh, D. Trap-Limited Electronic Transport in Assemblies of Nanometer-Size TiO2 Particles. Phys. Rev. Lett. 1996, 77, 3427–3430. [Google Scholar] [CrossRef] [PubMed]
- Vanmaekelbergh, D.; de Jongh, P.E. Electron transport in disordered semiconductors studied by a small harmonic modulation of the steady state. Phys. Rev. B 2000, 61, 4699–4704. [Google Scholar] [CrossRef]
- Anderson, A.B.; Albu, T.V. Ab Initio Determination of Reversible Potentials and Activation Energies for Outer-Sphere Oxygen Reduction to Water and the Reverse Oxidation Reaction. J. Am. Chem. Soc. 1999, 121, 11855–11863. [Google Scholar] [CrossRef]
- Anderson, A.B.; Neshev, N.M.; Sidik, R.A.; Shiller, P. Mechanism for the electrooxidation of water to {OH} and O bonded to platinum: Quantum chemical theory. Electrochim. Acta 2002, 47, 2999–3008. [Google Scholar] [CrossRef]
- Jinnouchi, R.; Anderson, A.B. Aqueous and Surface Redox Potentials from Self-Consistently Determined Gibbs Energies. J. Phys. Chem. C 2008, 112, 8747–8750. [Google Scholar] [CrossRef]
- Anderson, A.B. Insights into electrocatalysis. Phys. Chem. Chem. Phys. 2012, 14, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.B. Theory at the electrochemical interface: Reversible potentials and potential-dependent activation energies. Electrochim. Acta 2003, 48, 3743–3749. [Google Scholar] [CrossRef]
- Anderson, A.B. Theories for Predicting Reversible Potentials of Reactions on Electrode Surfaces from Internal and Gibbs Energies: Applications to ORR. ECS Trans. 2010, 28, 1–17. [Google Scholar]
- Taylor, C.D.; Kelly, R.G.; Neurock, M. A first-principles analysis of the chemisorption of hydroxide on copper under electrochemical conditions: A probe of the electronic interactions that control chemisorption at the electrochemical interface. J. Electroanal. Chem. 2007, 607, 167–174. [Google Scholar] [CrossRef]
- Taylor, C.; Kelly, R.G.; Neurock, M. First-Principles Calculations of the Electrochemical Reactions of Water at an Immersed Ni (111)/H2O Interface. J. Electrochem. Soc. 2006, 153, E207–E214. [Google Scholar] [CrossRef]
- Taylor, C.D.; Kelly, R.G.; Neurock, M. First-Principles Prediction of Equilibrium Potentials for Water Activation by a Series of Metals. J. Electrochem. Soc. 2007, 154, F217–F221. [Google Scholar] [CrossRef]
- Janik, M.J.; Taylor, C.D.; Neurock, M. First-Principles Analysis of the Initial Electroreduction Steps of Oxygen over Pt(111). J. Electrochem. Soc. 2009, 156, B126–B135. [Google Scholar] [CrossRef]
- Taylor, C.; Kelly, R.G.; Neurock, M. Theoretical Analysis of the Nature of Hydrogen at the Electrochemical Interface Between Water and a Ni(111) Single-Crystal Electrode. J. Electrochem. Soc. 2007, 154, F55–F64. [Google Scholar] [CrossRef]
- Skúlason, E.; Tripkovic, V.; Björketun, M.E.; Gudmundsdóttir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jónsson, H.; Nørskov, J.K. Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the Basis of Density Functional Theory Calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar] [CrossRef]
- Bondarenko, A.S.; Stephens, I.E.L.; Hansen, H.A.; Pérez-Alonso, F.J.; Tripkovic, V.; Johansson, T.P.; Rossmeisl, J.; Nørskov, J.K.; Chorkendorff, I. The Pt(111)/Electrolyte Interface under Oxygen Reduction Reaction Conditions: An Electrochemical Impedance Spectroscopy Study. Langmuir 2011, 27, 2058–2066. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Nørskov, J.K.; Taylor, C.D.; Janik, M.J.; Neurock, M. Calculated Phase Diagrams for the Electrochemical Oxidation and Reduction of Water over Pt(111). J. Phys. Chem. B 2006, 110, 21833–21839. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Rossmeisl, J.; Hellman, A.; Nørskov, J.K. Theoretical Trends in Particle Size Effects for the Oxygen Reduction Reaction. Z. Phys. Chem. 2007, 221, 1209–1220. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Nielsen, M.; Björketun, M.E.; Hansen, M.H.; Rossmeisl, J. Towards first principles modeling of electrochemical electrode–electrolyte interfaces. Surf. Sci. 2015, 613, 2–7. [Google Scholar] [CrossRef]
- Valdés, Á.; Qu, Z.-W.; Kroes, G.-J.; Rossmeisl, J.; Nørskov, J.K. Oxidation and Photo-Oxidation of Water on TiO2 Surface. J. Phys. Chem. C 2008, 112, 9872–9879. [Google Scholar] [CrossRef]
- Valdés, Á.; Kroes, G.J. First principles study of the photo-oxidation of water on tungsten trioxide (WO3). J. Chem. Phys. 2009, 130, 114701. [Google Scholar] [CrossRef] [PubMed]
- Valdés, Á.; Kroes, G.J. Cluster Study of the Photo-Oxidation of Water on Rutile Titanium Dioxide (TiO2). J. Phys. Chem. C 2010, 114, 1701–1708. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Koper, M.T. First-principles computational electrochemistry: Achievements and challenges. Electrochim. Acta 2012, 84, 3–11. [Google Scholar] [CrossRef]
- Karlberg, G.S.; Rossmeisl, J.; Norskov, J.K. Estimations of electric field effects on the oxygen reduction reaction based on the density functional theory. Phys. Chem. Chem. Phys. 2007, 9, 5158–5161. [Google Scholar] [CrossRef] [PubMed]
- Fenter, P.; Sturchio, N.C. Mineral-water interfacial structures revealed by synchrotron X-ray scattering. Prog. Surf. Sci. 2004, 77, 171–258. [Google Scholar] [CrossRef]
- Sato, N. Electrochemistry at Metal and Semiconductor Electrodes; Elsevier: Oxford, UK, 1998. [Google Scholar]
- Björneholm, O.; Hansen, M.H.; Hodgson, A.; Liu, L.M.; Limmer, D.T.; Michaelides, A.; Pedevilla, P.; Rossmeisl, J.; Shen, H.; Tocci, G.; et al. Simulating an Applied Voltage in Molecular Dynamics Using Charge Optimized Many Body (COMB3) Potentials. Chem. Rev. 2016, 116, 7698–7726. [Google Scholar] [CrossRef] [PubMed]
- Skulason, E.; Karlberg, G.S.; Rossmeisl, J.; Bligaard, T.; Greeley, J.; Jonsson, H.; Norskov, J.K. Density functional theory calculations for the hydrogen evolution reaction in an electrochemical double layer on the Pt(111) electrode. Phys. Chem. Chem. Phys. 2007, 9, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.Y.; Wasileski, S.A.; Janik, M.J. Electronic structure models of oxygen adsorption at the solvated, electrified Pt(111) interface. Phys. Chem. Chem. Phys. 2009, 11, 10108–10117. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.Y.; Janik, M.J.; Maranas, J.K. Molecular dynamics simulations of an electrified water/Pt(111) interface using point charge dissociative water. Electrochim. Acta 2013, 101, 308–325. [Google Scholar] [CrossRef]
- Hansen, M.H.; Jin, C.; Thygesen, K.S.; Rossmeisl, J. Finite Bias Calculations to Model Interface Dipoles in Electrochemical Cells at the Atomic Scale. J. Phys. Chem. C 2016, 120, 13485–13491. [Google Scholar] [CrossRef]
- Zhang, C.; Sprik, M. Finite field methods for the supercell modeling of charged insulator/electrolyte interfaces. Phys. Rev. B 2016, 94, 245309. [Google Scholar] [CrossRef]
- Russo, D.; Teixeira, J.; Kneller, L.; Copley, J.R.D.; Ollivier, J.; Perticaroli, S.; Pellegrini, E.; Gonzalez, M.A. Vibrational Density of States of Hydration Water at Biomolecular Sites: Hydrophobicity Promotes Low Density Amorphous Ice Behavior. J. Am. Chem. Soc. 2011, 133, 4882–4888. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Neogi, S.; Kent, P.R.C.; Bandura, A.V.; Kubicki, J.D.; Wesolowski, D.J.; Cole, D.; Sofo, J.O. Hydrogen Bonds and Vibrations of Water on (110) Rutile. J. Phys. Chem. C 2009, 113, 13732–13740. [Google Scholar] [CrossRef]
- English, N.J.; Kavathekar, R.S.; MacElroy, J. Hydrogen bond dynamical properties of adsorbed liquid water monolayers with various TiO2 interfaces. Mol. Phys. 2012, 110, 2919–2925. [Google Scholar] [CrossRef]
- Kavathekar, R.S.; Dev, P.; English, N.J.; MacElroy, J. Molecular dynamics study of water in contact with the TiO2 rutile-110, 100, 101, 001 and anatase-101, 001 surface. Mol. Phys. 2011, 109, 1649–1656. [Google Scholar] [CrossRef]
- Iandolo, B.; Hellman, A. The Role of Surface States in the Oxygen Evolution Reaction on Hematite. Angew. Chem. Int. Ed. 2014, 53, 13404–13408. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Pala, R.G.S. First-Principles Study of Photoinduced Water-Splitting on Fe2O3. J. Phys. Chem. C 2011, 115, 12901–12907. [Google Scholar] [CrossRef]
- Hellman, A.; Iandolo, B.; Wickman, B.; Grönbeck, H.; Baltrusaitis, J. Electro-oxidation of water on hematite: Effects of surface termination and oxygen vacancies investigated by first-principles. Surf. Sci. 2015, 640, 45–49. [Google Scholar] [CrossRef]
- Hansen, H.A.; Rossmeisl, J.; Norskov, J.K. Surface Pourbaix diagrams and oxygen reduction activity of Pt, Ag and Ni(111) surfaces studied by DFT). Phys. Chem. Chem. Phys. 2008, 10, 3722–3730. [Google Scholar] [CrossRef] [PubMed]
- Persson, K.A.; Waldwick, B.; Lazic, P.; Ceder, G. Prediction of solid-aqueous equilibria: Scheme to combine first-principles calculations of solids with experimental aqueous states. Phys. Rev. B 2012, 85, 235438. [Google Scholar] [CrossRef]
- Todorova, M.; Neugebauer, J. Extending the Concept of Defect Chemistry from Semiconductor Physics to Electrochemistry. Phys. Rev. Appl. 2014, 1, 014001. [Google Scholar] [CrossRef]
- Huang, L.F.; Rondinelli, J.M. Electrochemical phase diagrams for Ti oxides from density functional calculations. Phys. Rev. B 2015, 92, 245126. [Google Scholar] [CrossRef]
- Zeng, Z.; Chan, M.K.Y.; Zhao, Z.J.; Kubal, J.; Fan, D.; Greeley, J. Towards First Principles-Based Prediction of Highly Accurate Electrochemical Pourbaix Diagrams. J. Phys. Chem. C 2015, 119, 18177–18187. [Google Scholar] [CrossRef]
- Ulissi, Z.W.; Singh, A.R.; Tsai, C.; Nørskov, J.K.; Todorova, M.; Neugebauer, J. Automated Discovery and Construction of Surface Phase Diagrams Using Machine Learning. J. Phys. Chem. Lett. 2016, 7, 3931–3935. [Google Scholar] [CrossRef] [PubMed]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd ed.; National Association of Corrosion Engineers: Houston, TX, USA, 1974. [Google Scholar]
- Nguyen, M.T.; Piccinin, S.; Seriani, N.; Gebauer, R. Photo-Oxidation of Water on Defective Hematite(0001). ACS Catal. 2015, 5, 715–721. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Bahn, S.; Hansen, L.B.; Bollinger, M.; Bengaard, H.; Hammer, B.; Sljivancanin, Z.; Mavrikakis, M.; et al. Universality in Heterogeneous Catalysis. J. Catal. 2002, 209, 275. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Seriani, N.; Gebauer, R. Water adsorption and dissociation on α-Fe2O3(0001): PBE+U calculations. J. Chem. Phys. 2013, 138, 194709. [Google Scholar] [CrossRef] [PubMed]
- Toroker, M.C. Theoretical Insights into the Mechanism of Water Oxidation on Nonstoichiometric and Titanium-Doped Fe2O3(0001). J. Phys. Chem. C 2014, 118, 23162–23167. [Google Scholar] [CrossRef]
- Zeng, Z.; Hansen, M.H.; Greeley, J.P.; Rossmeisl, J.; Björketun, M.E. Ab Initio Thermodynamic Modeling of Electrified Metal–Oxide Interfaces: Consistent Treatment of Electronic and Ionic Chemical Potentials. J. Phys. Chem. C 2014, 118, 22663–22671. [Google Scholar] [CrossRef]
- Chan, K.; Nørskov, J.K. Electrochemical Barriers Made Simple. J. Phys. Chem. Lett. 2015, 6, 2663–2668. [Google Scholar] [CrossRef] [PubMed]
- Abild-Pedersen, F.; Greeley, J.; Studt, F.; Rossmeisl, J.; Munter, T.R.; Moses, P.G.; Skúlason, E.; Bligaard, T.; Nørskov, J.K. Scaling Properties of Adsorption Energies for Hydrogen-Containing Molecules on Transition-Metal Surfaces. Phys. Rev. Lett. 2007, 99, 016105. [Google Scholar] [CrossRef] [PubMed]
- Bligaard, T.; Nørskov, J.; Dahl, S.; Matthiesen, J.; Christensen, C.; Sehested, J. The Brønsted–Evans–Polanyi relation and the volcano curve in heterogeneous catalysis. J. Catal. 2004, 224, 206–217. [Google Scholar] [CrossRef]
- Norskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Man, I.C.; Su, H.Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- García-Mota, M.; Vojvodic, A.; Metiu, H.; Man, I.C.; Su, H.Y.; Rossmeisl, J.; Nørskov, J.K. Tailoring the Activity for Oxygen Evolution Electrocatalysis on Rutile TiO2(110) by Transition-Metal Substitution. ChemCatChem 2011, 3, 1607–1611. [Google Scholar] [CrossRef]
- Montoya, J.H.; Garcia-Mota, M.; Norskov, J.K.; Vojvodic, A. Theoretical evaluation of the surface electrochemistry of perovskites with promising photon absorption properties for solar water splitting. Phys. Chem. Chem. Phys. 2015, 17, 2634–2640. [Google Scholar] [CrossRef] [PubMed]
- Calle-Vallejo, F.; Martinez, J.I.; Rossmeisl, J. Density functional studies of functionalized graphitic materials with late transition metals for oxygen reduction reactions. Phys. Chem. Chem. Phys. 2011, 13, 15639–15643. [Google Scholar] [CrossRef] [PubMed]
- Dogutan, D.K.; Bediako, D.K.; Teets, T.S.; Schwalbe, M.; Nocera, D.G. Efficient Synthesis of Hangman Porphyrins. Org. Lett. 2010, 12, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- McGuire R., Jr.; Dogutan, D.K.; Teets, T.S.; Suntivich, J.; Shao-Horn, Y.; Nocera, D.G. Oxygen reduction reactivity of cobalt(II) hangman porphyrins. Chem. Sci. 2010, 1, 411–414. [Google Scholar] [CrossRef]
- Rosenthal, J.; Nocera, D. Oxygen activation chemistry of Pacman and Hangman porphyrin architectures based on xanthene and dibenzofuran spacers. In Progress in Inorganic Chemistry; Wiley: Hoboken, NJ, USA, 2007; Volume 55, pp. 483–544. [Google Scholar]
- Dogutan, D.K.; Stoian, S.A.; McGuire, R.; Schwalbe, M.; Teets, T.S.; Nocera, D.G. Hangman Corroles: Efficient Synthesis and Oxygen Reaction Chemistry. J. Am. Chem. Soc. 2011, 133, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chng, L.L.; Nocera, D.G. Proton-Coupled O–O Activation on a Redox Platform Bearing a Hydrogen-Bonding Scaffold. J. Am. Chem. Soc. 2003, 125, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Baran, J.D.; Grönbeck, H.; Hellman, A. Analysis of Porphyrines as Catalysts for Electrochemical Reduction of O2 and Oxidation of H2O. J. Am. Chem. Soc. 2014, 136, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Que, L.; Tolman, W.B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Shook, R.L.; Borovik, A.S. Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg. Chem. 2010, 49, 3646–3660. [Google Scholar] [CrossRef] [PubMed]
- Wang, B. Electronic Structure and Optical Properties of Solar Energy Materials. Ph.D. Dissertation, Royal Institute of Technology, KTH, Stockholm, Sweden, 2014. [Google Scholar]
- Martin, R.M. Electronic Structure : Basic Theory and Practical Methods; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Slater, J.C. A Simplification of the Hartree-Fock Method. Phys. Rev. 1951, 81, 385–390. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef]
- Csonka, G.I.; Perdew, J.P.; Ruzsinszky, A.; Philipsen, P.H.T.; Lebègue, S.; Paier, J.; Vydrov, O.A.; Ángyán, J.G. Assessing the performance of recent density functionals for bulk solids. Phys. Rev. B 2009, 79, 155107. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Tests of a ladder of density functionals for bulk solids and surfaces. Phys. Rev. B 2004, 69, 075102. [Google Scholar] [CrossRef]
- Perdew, J.P. Accurate Density Functional for the Energy: Real-Space Cutoff of the Gradient Expansion for the Exchange Hole. Phys. Rev. Lett. 1985, 55, 1665–1668. [Google Scholar] [CrossRef] [PubMed]
- Langreth, D.C.; Mehl, M.J. Easily Implementable Nonlocal Exchange-Correlation Energy Functional. Phys. Rev. Lett. 1981, 47, 446–450. [Google Scholar] [CrossRef]
- Gunnarson, O.; Lundqvist, B.; Lundqvist, S. Screening in a spin-polarized electron liquid. Solid State Commun. 1972, 11, 149–153. [Google Scholar] [CrossRef]
- Beck, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Armiento, R.; Mattsson, A.E. Functional designed to include surface effects in self-consistent density functional theory. Phys. Rev. B 2005, 72, 085108. [Google Scholar] [CrossRef]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta? Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Constantin, L.A.; Sun, J. Workhorse Semilocal Density Functional for Condensed Matter Physics and Quantum Chemistry. Phys. Rev. Lett. 2009, 103, 026403. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2008, 4, 1849–1868. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A new mixing of Hartree Fock and local density functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Kim, K.; Jordan, K.D. Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem. 1994, 98, 10089–10094. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
Figure 1.
A schematic energy diagram for a photoanode (n-type semiconductor). Several important steps are illustrated, namely: (i) light absorption ; (ii) charge transport ; and (iii) surface chemical reactions . Photons with energy equal to or larger than the band-gap E are absorbed with efficiency . The built-in electric field drives holes towards the photoanode/electrolyte interface and electrons to the cathode (usually via a charge collector and an external circuit). is the fraction of photogenerated charges reaching the respective solid/liquid interface. Holes are transferred to the electrolyte with efficiency and take part in the oxygen evolution reaction (OER). An external bias/PV device supplies electrons with sufficient energy so that they take part in the hydrogen evolution reaction (HER). Reproduced with permission from ref. [40], published by The Royal Society of Chemistry.
Figure 1.
A schematic energy diagram for a photoanode (n-type semiconductor). Several important steps are illustrated, namely: (i) light absorption ; (ii) charge transport ; and (iii) surface chemical reactions . Photons with energy equal to or larger than the band-gap E are absorbed with efficiency . The built-in electric field drives holes towards the photoanode/electrolyte interface and electrons to the cathode (usually via a charge collector and an external circuit). is the fraction of photogenerated charges reaching the respective solid/liquid interface. Holes are transferred to the electrolyte with efficiency and take part in the oxygen evolution reaction (OER). An external bias/PV device supplies electrons with sufficient energy so that they take part in the hydrogen evolution reaction (HER). Reproduced with permission from ref. [40], published by The Royal Society of Chemistry.
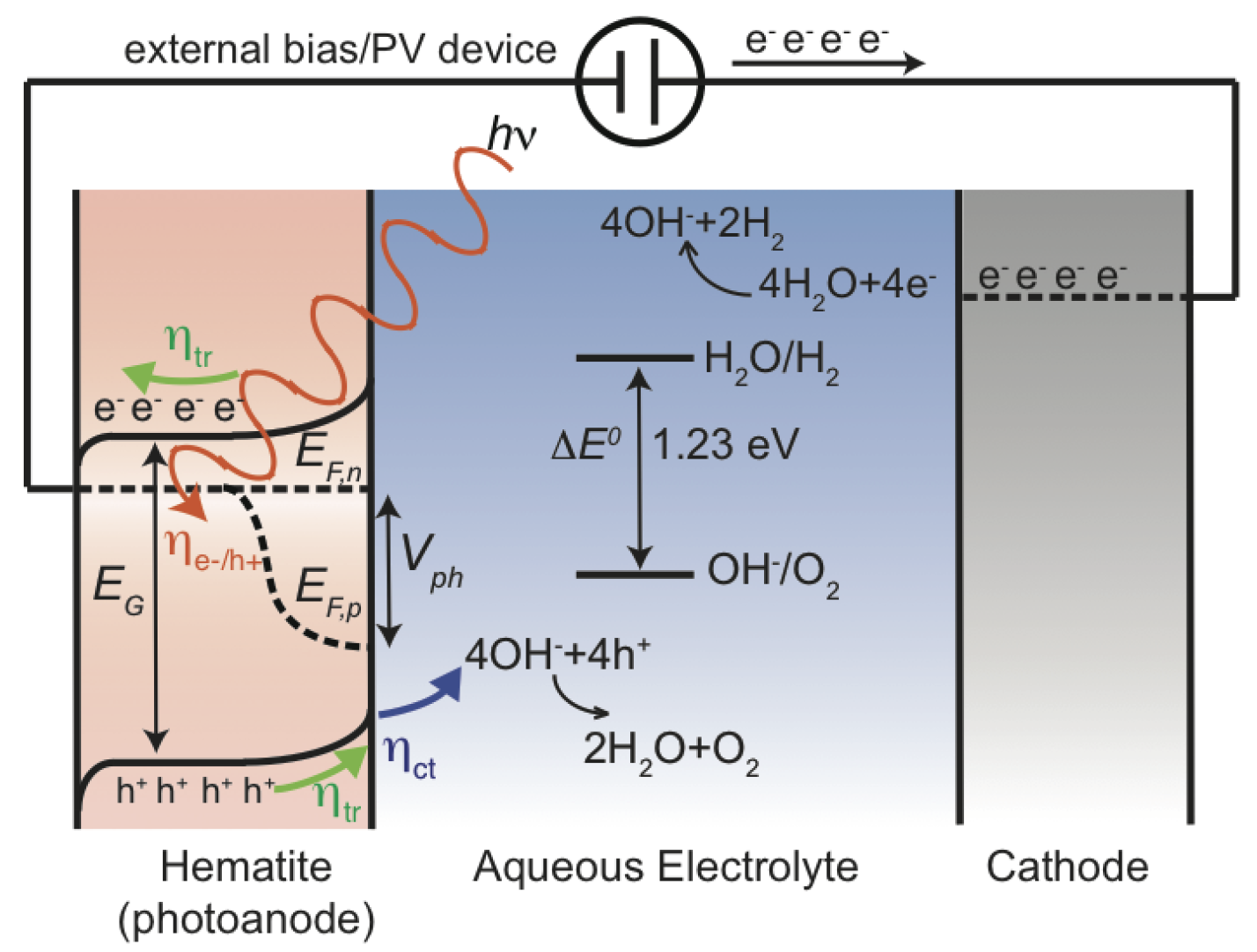
Figure 2.
PEC performance gap between a state-of-the-art photoanode and the ideal photoanode. The arrows indicate the main performance limiting factors for increasingly anodic potential: first charge transfer at the photoanode/electrolyte interface and the small photovoltage, then charge transport within photoanode and finally light absorption in the semiconductor. Reproduced with permission from ref. [40], published by The Royal Society of Chemistry.
Figure 2.
PEC performance gap between a state-of-the-art photoanode and the ideal photoanode. The arrows indicate the main performance limiting factors for increasingly anodic potential: first charge transfer at the photoanode/electrolyte interface and the small photovoltage, then charge transport within photoanode and finally light absorption in the semiconductor. Reproduced with permission from ref. [40], published by The Royal Society of Chemistry.

Figure 3.
The relaxation process of hot electron-hole pairs. There exist several pathways for dissipation of the energy of the electron-hole pairs. More details of these are given in the text. Reprinted by permission from Nature Nanotechnology, copyright 2015 [54].
Figure 3.
The relaxation process of hot electron-hole pairs. There exist several pathways for dissipation of the energy of the electron-hole pairs. More details of these are given in the text. Reprinted by permission from Nature Nanotechnology, copyright 2015 [54].
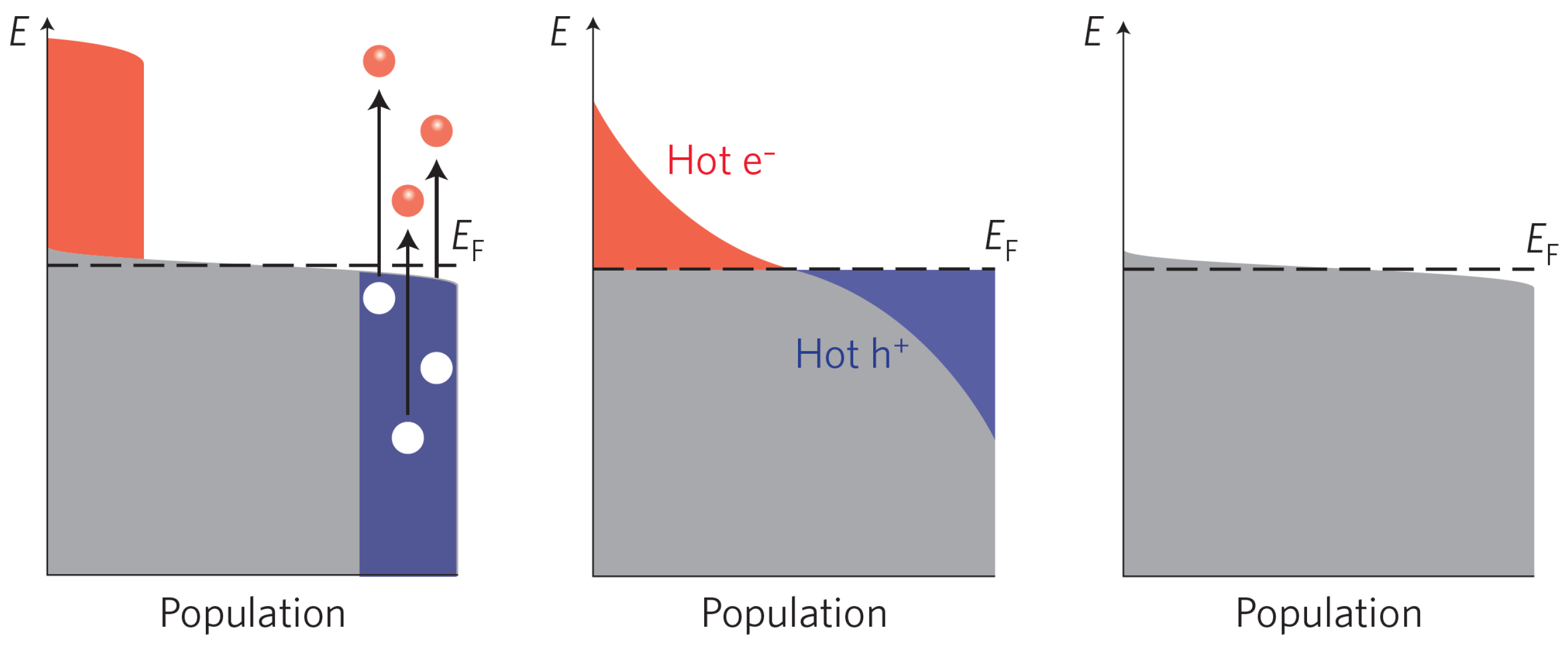
Figure 4.
Schematic picture showing the electronic energy levels at the interface between an n-type semiconductor and an electrolyte containing a redox couple. The four cases indicated are: (a) flat band potential, where no space-charge layer exists in the semiconductor; (b) accumulation layer, where excess electrons have been injected into the solid producing a downward bending of the conduction and valence band towards the interface; (c) depletion layer, where electrons have moved from the semiconductor to the electrolyte, producing an upward bending of the bands; and (d) inversion layer, where the electrons have been depleted below their intrinsic level, enhancing the upward band bending and rendering the semiconductor p-type at the surface [96]. Reprinted by permission from Macmillan Publishers Ltd.: Nature 414 338, copyright 2001.
Figure 4.
Schematic picture showing the electronic energy levels at the interface between an n-type semiconductor and an electrolyte containing a redox couple. The four cases indicated are: (a) flat band potential, where no space-charge layer exists in the semiconductor; (b) accumulation layer, where excess electrons have been injected into the solid producing a downward bending of the conduction and valence band towards the interface; (c) depletion layer, where electrons have moved from the semiconductor to the electrolyte, producing an upward bending of the bands; and (d) inversion layer, where the electrons have been depleted below their intrinsic level, enhancing the upward band bending and rendering the semiconductor p-type at the surface [96]. Reprinted by permission from Macmillan Publishers Ltd.: Nature 414 338, copyright 2001.
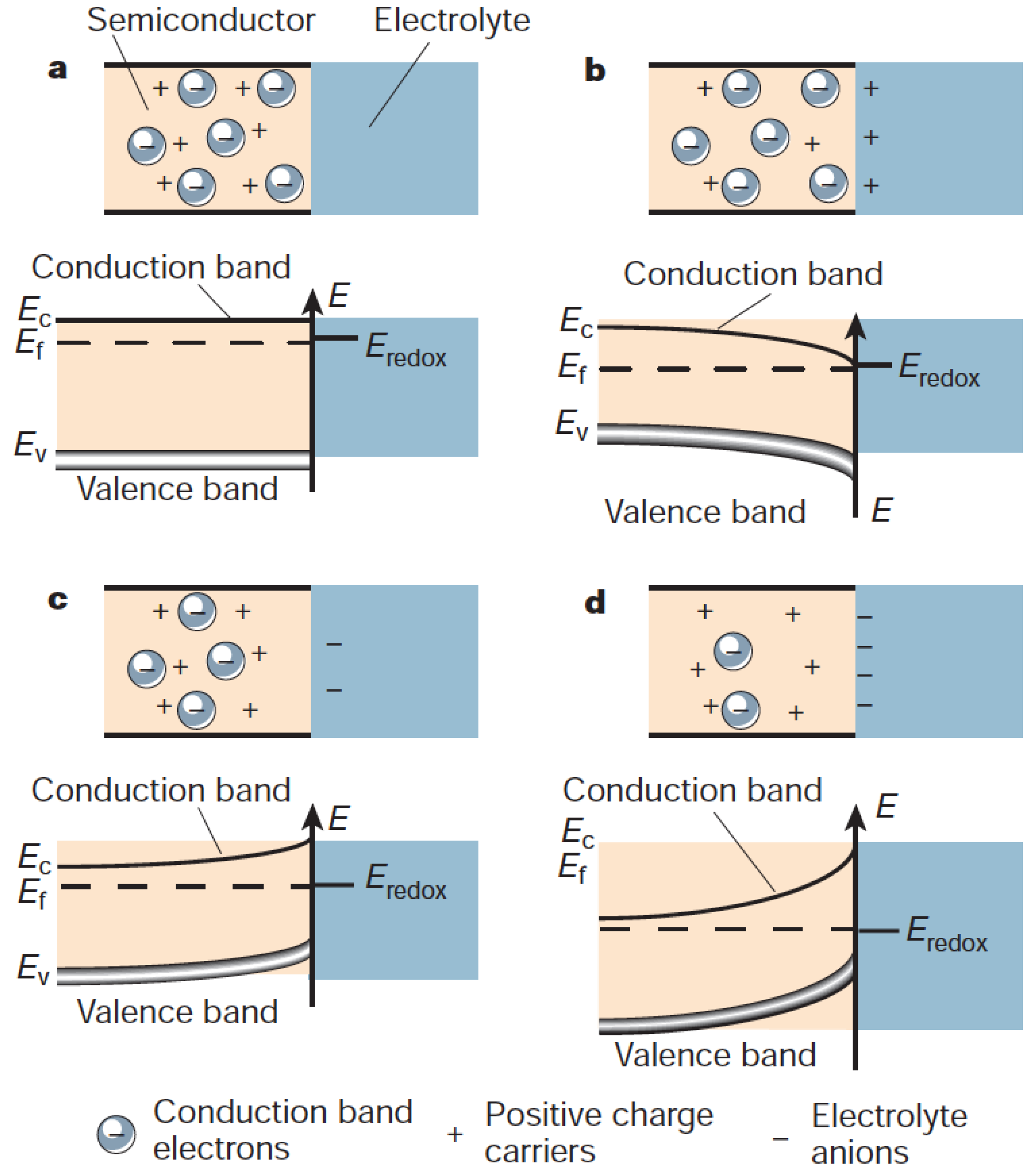
Figure 5.
The color map shows the local density of states (LDOS) along the direction normal to the interface. The Nb-dopant is marked as Nb in the inserted structure plot. The dopant state below the bottom of the conduction band traps most of the excess electron donated by Nb. The band edges of the pristine TiO are also shown (gray dashed lines). The band bending is caused by the dopant-induced charge polarization. Reprinted with permission from ref. [104].
Figure 5.
The color map shows the local density of states (LDOS) along the direction normal to the interface. The Nb-dopant is marked as Nb in the inserted structure plot. The dopant state below the bottom of the conduction band traps most of the excess electron donated by Nb. The band edges of the pristine TiO are also shown (gray dashed lines). The band bending is caused by the dopant-induced charge polarization. Reprinted with permission from ref. [104].
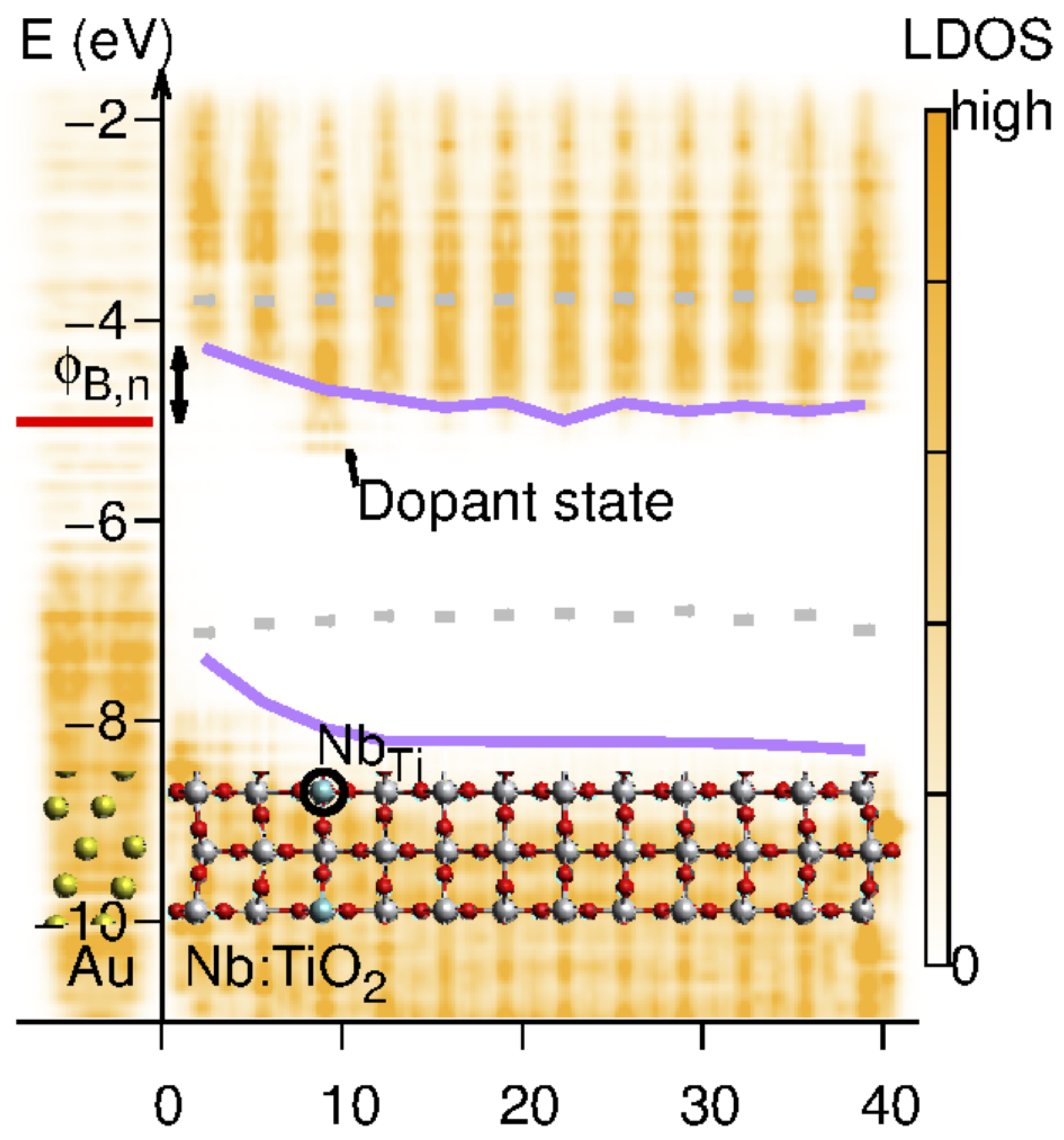
Figure 6.
Schematic density profile of interfacial water near the solid–aqueous interface. Reprinted with permission from ref. [168]. Copyright 2004 Elsevier.
Figure 6.
Schematic density profile of interfacial water near the solid–aqueous interface. Reprinted with permission from ref. [168]. Copyright 2004 Elsevier.
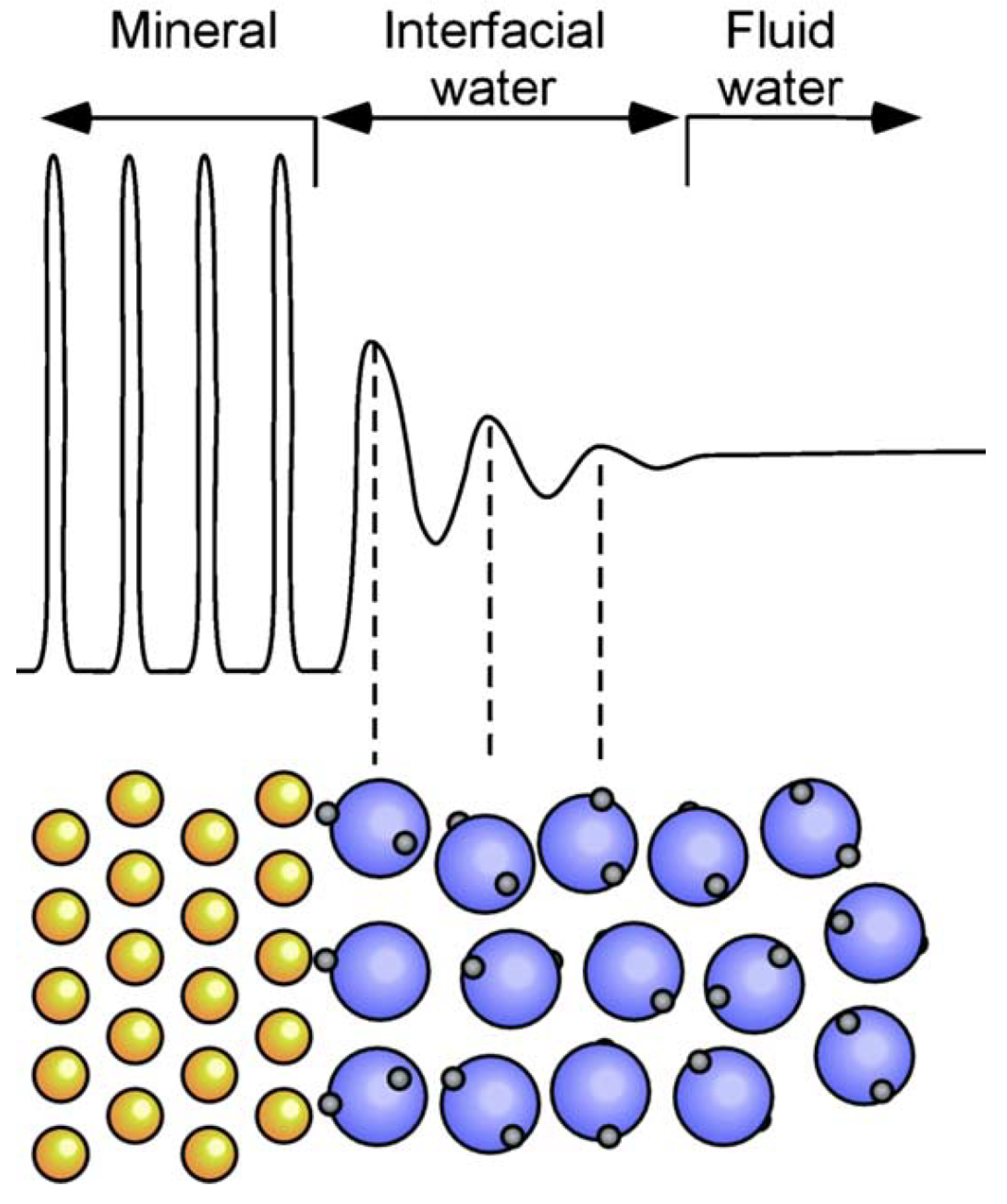
Figure 7.
The relative stability of all considered surface terminations as a function of applied potential and at two different pH, namely pH = 0 (left) and pH = 14 (right). Reprinted with permission from [181]. Copyright 2011 American Chemical Society.
Figure 7.
The relative stability of all considered surface terminations as a function of applied potential and at two different pH, namely pH = 0 (left) and pH = 14 (right). Reprinted with permission from [181]. Copyright 2011 American Chemical Society.
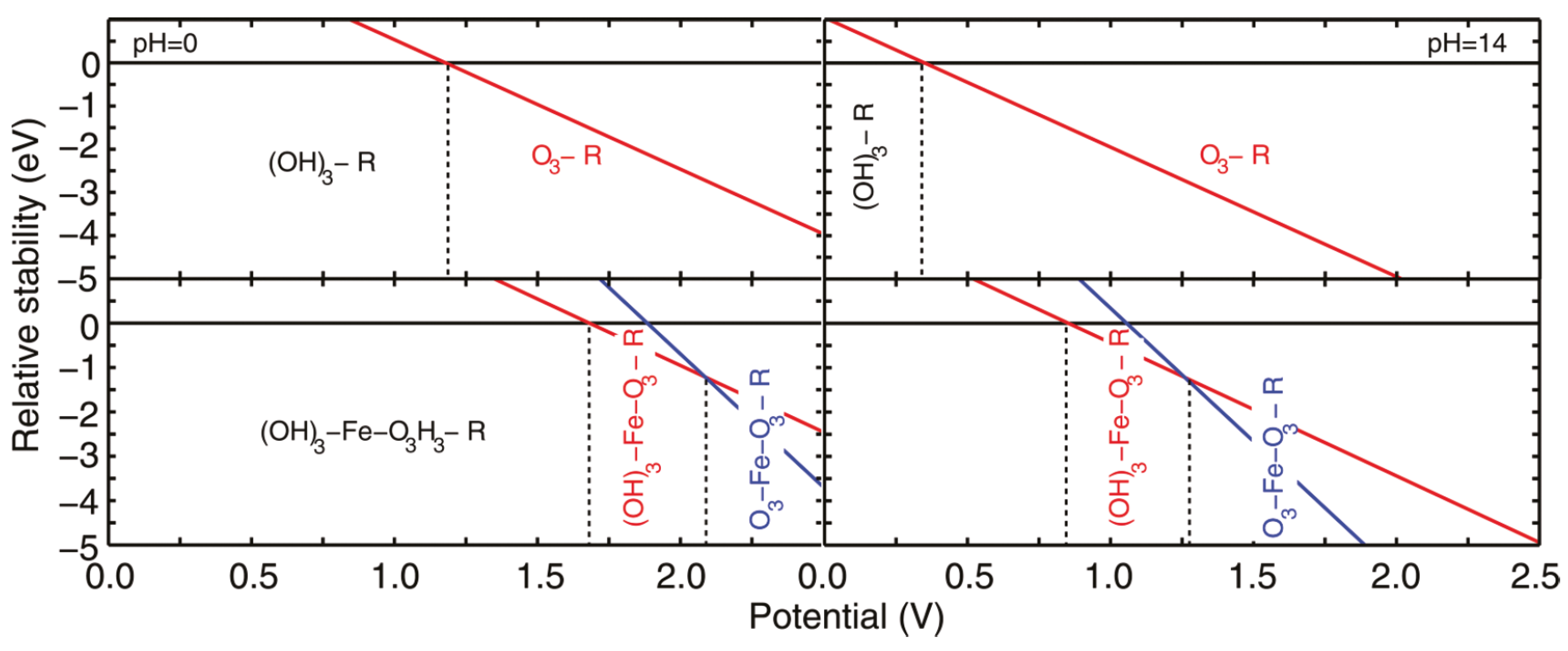
Figure 8.
The water splitting reaction at different potentials. At potentials between 0 and 0.78 V all steps in the oxygen reduction are exothermic. For potentials beyond 2.55 V all water splitting reaction steps become exothermic. This variation is obtained by varying the term eU in the free energy per electron transferred to the electrode. Reprinted from Chemical Physics 319 (2005) 178, Copyright 2005, with permission from Elsevier [158].
Figure 8.
The water splitting reaction at different potentials. At potentials between 0 and 0.78 V all steps in the oxygen reduction are exothermic. For potentials beyond 2.55 V all water splitting reaction steps become exothermic. This variation is obtained by varying the term eU in the free energy per electron transferred to the electrode. Reprinted from Chemical Physics 319 (2005) 178, Copyright 2005, with permission from Elsevier [158].

Figure 9.
(a) Adsorption energies of HOO* as a function of adsorption energies of HO* on rutile, perovskites, anatase, MnO, CoO and NiO oxides. Open and solid symbols represent the adsorption energies on clean surfaces and on high-coverage surfaces, respectively. The star represents the binding energies required for an ideal electrocatalyst. The dotted line represents the relationship between E and E for an ideal catalyst, which is given by E = E + 2.44 eV; (b) Activity trends toward oxygen evolution for rutile oxides, CoO, and MnO. The negative value of the theoretical overpotential is plotted as a function of the standard free energy of Gand G; (c) Standard free-energy diagram for the OER on O*-covered RuO at three different potentials: U = 0, 1.23, and 1.60 V. Reprinted with permission from [200].
Figure 9.
(a) Adsorption energies of HOO* as a function of adsorption energies of HO* on rutile, perovskites, anatase, MnO, CoO and NiO oxides. Open and solid symbols represent the adsorption energies on clean surfaces and on high-coverage surfaces, respectively. The star represents the binding energies required for an ideal electrocatalyst. The dotted line represents the relationship between E and E for an ideal catalyst, which is given by E = E + 2.44 eV; (b) Activity trends toward oxygen evolution for rutile oxides, CoO, and MnO. The negative value of the theoretical overpotential is plotted as a function of the standard free energy of Gand G; (c) Standard free-energy diagram for the OER on O*-covered RuO at three different potentials: U = 0, 1.23, and 1.60 V. Reprinted with permission from [200].
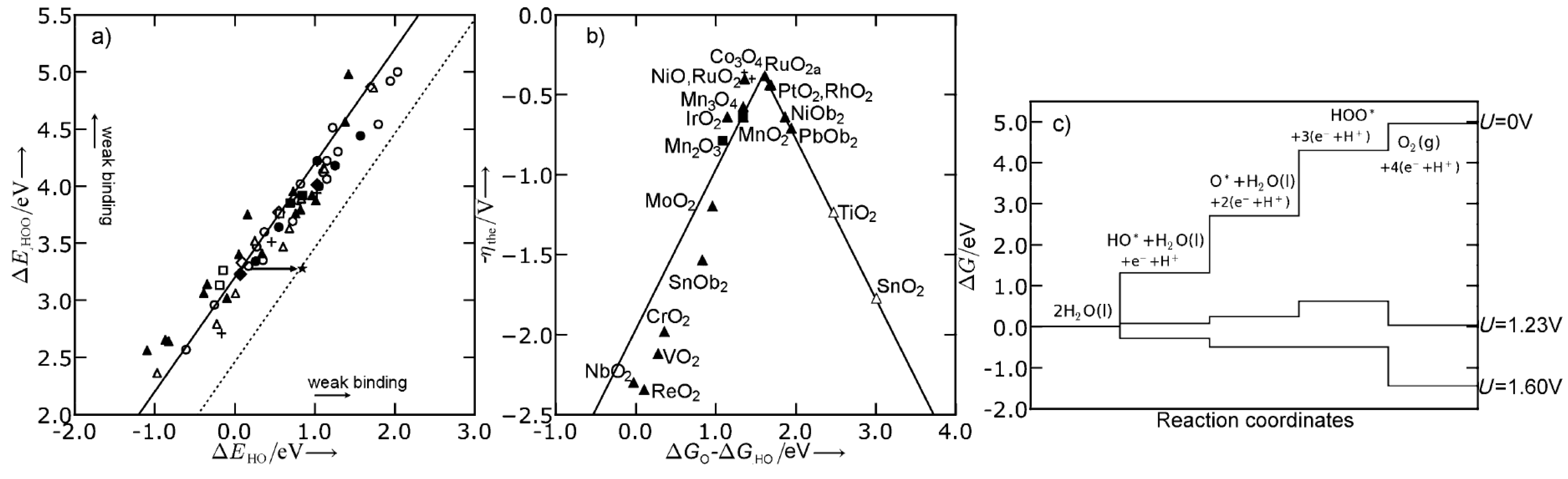
Figure 10.
Scaling relation between the Gibbs free energies of adsorption of *OH, *O, and *OOH. Squares represent metal porphyrine, triangles hangman metalloporphyrins and rectangles metaltetrafluorophenyloporphyrines molecules, respectively. The dashed lines at 0 and 4.92 eV represent energy of HO and O, respectively. The grey area separates the metals from the Group 9 and before from the 10 and after of the periodic table. Reprinted with permission from [208]. Copyright 2014 American Chemical Society.
Figure 10.
Scaling relation between the Gibbs free energies of adsorption of *OH, *O, and *OOH. Squares represent metal porphyrine, triangles hangman metalloporphyrins and rectangles metaltetrafluorophenyloporphyrines molecules, respectively. The dashed lines at 0 and 4.92 eV represent energy of HO and O, respectively. The grey area separates the metals from the Group 9 and before from the 10 and after of the periodic table. Reprinted with permission from [208]. Copyright 2014 American Chemical Society.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hellman, A.; Wang, B. First-Principles View on Photoelectrochemistry: Water-Splitting as Case Study. Inorganics 2017, 5, 37. https://doi.org/10.3390/inorganics5020037
AMA Style
Hellman A, Wang B. First-Principles View on Photoelectrochemistry: Water-Splitting as Case Study. Inorganics. 2017; 5(2):37. https://doi.org/10.3390/inorganics5020037
Chicago/Turabian StyleHellman, Anders, and Baochang Wang. 2017. "First-Principles View on Photoelectrochemistry: Water-Splitting as Case Study" Inorganics 5, no. 2: 37. https://doi.org/10.3390/inorganics5020037
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.