Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View


A.E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center, Russian Academy of Sciences, Arbuzov str. 8, 420088 Kazan, Russian Federation
*
Author to whom correspondence should be addressed.
Inorganics 2017, 5(4), 70; https://doi.org/10.3390/inorganics5040070
Submission received: 18 September 2017
/
Revised: 11 October 2017
/
Accepted: 12 October 2017
/
Published: 16 October 2017
(This article belongs to the Special Issue Revealing Reaction Mechanisms in Homogeneous Transition Metal Catalysis)
Abstract
:This review generalizes and specifies the oxidizing ability of a number of oxidants used in palladium (Pd)-catalyzed aromatic C–H functionalizations. The redox potentials have been analyzed as the measure of oxidant strength and applied to the reasoning of the efficiency of known reactions where catalytic cycles include cyclometalated palladium complexes (and other organopalladium key intermediates).
1. Introduction
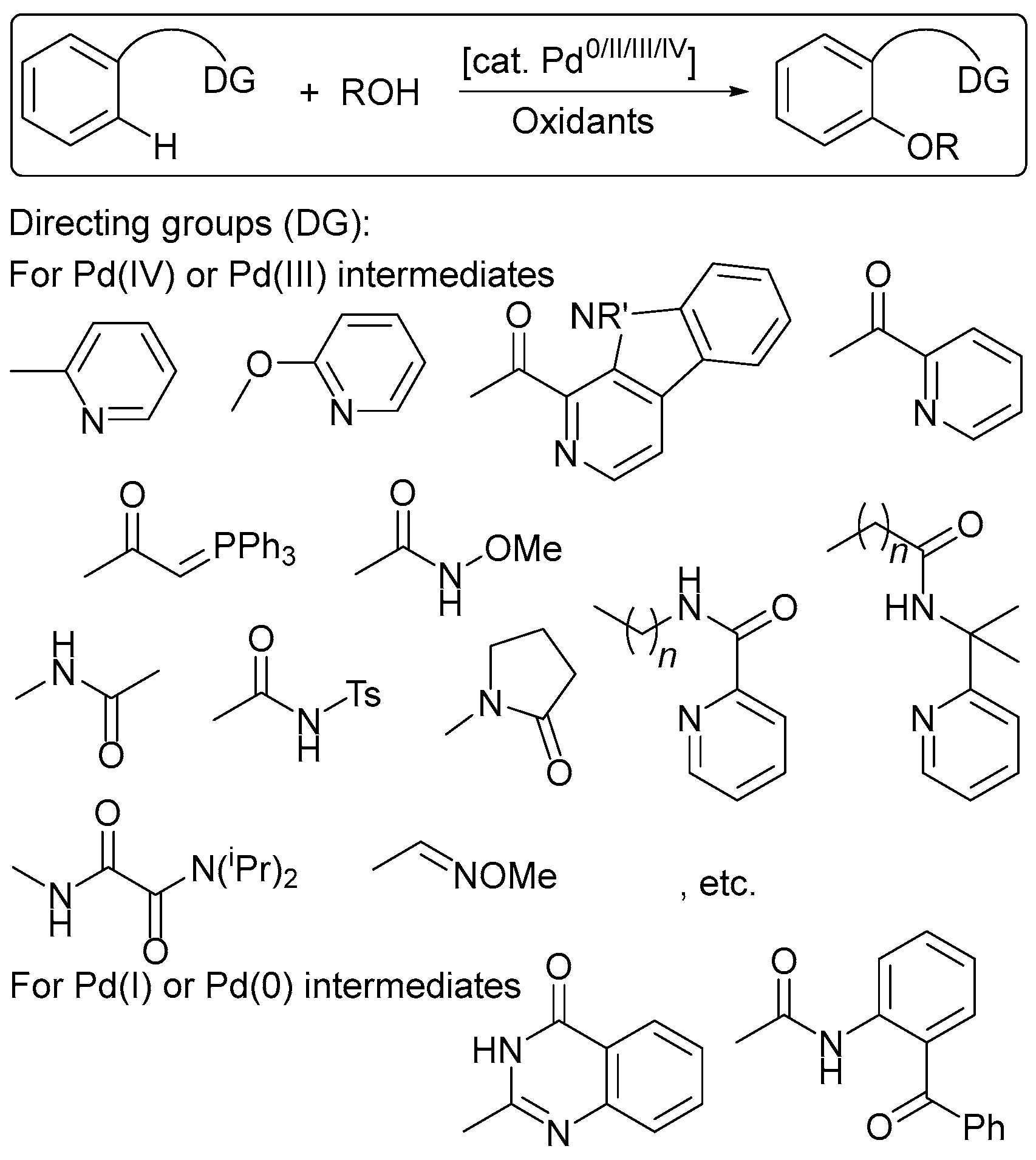
Transition-metal-catalyzed direct C–H functionalizations open a new road for diverse C–C, C–P, and C–N bond construction in a one-step and atom economical way, without the requirement of prefunctionalized C–H coupling partners [1,2,3,4,5,6]. The cleavage of high energy C–H bonds (E ~ 110 kcal·mol−1 for C(aryl)–H bonds) typically requires harsh reaction conditions that result in limited substrate scope and low functional group tolerance. The driving force of the current research is the development of the next generation of C–H transformations, which will help make these processes truly useful synthetic tools and develop reactions that proceed under milder conditions. A greater understanding of the mechanistic aspects of C–H functionalization is essential in order to choose the optimal catalytic conditions.
C–H functionalization reactions might be redox-neutral, and hence do not require the presence of an external oxidant. However, in numerous transformations, such as dehydrogenative coupling, reoxidation of the catalyst is necessary. The role of the oxidant in many cases is not only to regenerate the catalyst active form, but also to promote the reductive elimination of organometallic intermediates, which affords a product with the desired C–C (C–N, C–P) bonds, and to generate reactive radicals capable of functionalizing the desired C–H bonds. An appropriate choice of the oxidant of the appropriate force determines the success of the entire catalytic reaction.
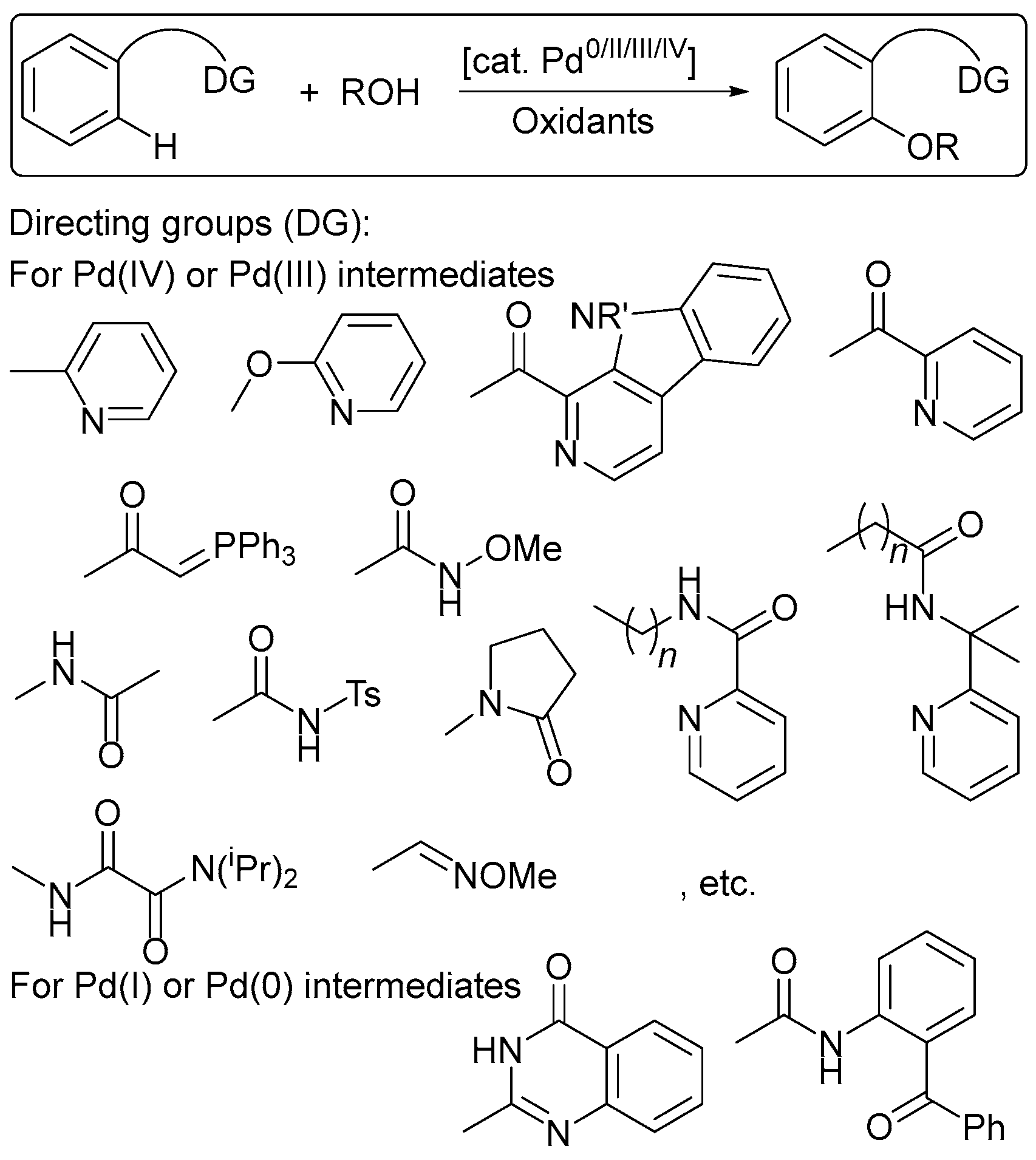
The chemistry of palladium (Pd) compounds in higher oxidation states is extremely useful in oxidative functionalizations, in which a C–H bond can be catalytically converted into another functional group [1,6,7,8,9,10,11,12]. One of the major problems in Pd(II)/Pd(III)/Pd(IV) chemistry, however, is that the co-oxidants used are either expensive to purchase (silver and other metal salts are often used), or to separate from the product mixture (particularly in the case of high molecular weight organic oxidants). Moreover, every time a new ligand or substrate or reaction condition is used, screenings must be performed in order to identify the optimal oxidant; that is, in general, the oxidants are not selective. Therefore, the choice of the optimal oxidant is one of the main difficulties of these reactions. In most cases, however, oxidative C–H transformations still rely on the presence of super-stoichiometric amounts of oxidants, which can be problematic in terms of functional group tolerance, waste generation, and cost. Electrochemical oxidation can be a cheaper, greener, and more controllable alternative to these oxidative reactions [13,14,15,16,17,18,19,20,21,22,23,24,25].
Controlling the “strength” or oxidizing ability of the oxidant is important in order to achieve high yields of products. Namely, the stage of selective oxidation of organometallic intermediates, including metallacycles affording high-valent species, is generally the key step in C–H functionalization reactions, while the catalyst regeneration stage is much less capricious and less sensitive to the nature of the oxidant. Thus, the relevant oxidant is the one that redox potential corresponds to, or the one that is higher than the oxidation potential of the key organometallic intermediate. Since intermediate compounds have not been determined and are only assumed for the most known reactions of this type, our observations reveal only some redox trends, the relationship of oxidant strength, and the redox properties of the organometallic key intermediates. It should be noted that in a number of cases, the mechanisms of aromatic C–H functionalization processes are described even without the participation of Pd(III)/Pd(IV) intermediates, but rather as Pd(II)/Pd(0) transformations, although there is no convincing evidence refuting the high-valent pathways.
In the literature, there are no summarizing publications to compare the redox properties (the “strength”) of various oxidants and the redox properties of organometallic intermediates, including palladium (Pd) complexes, to purposefully select the oxidizing conditions for successful synthesis. This review analyzes quantitative data on the redox properties of known oxidants of various natures in connection with their activity in Pd-catalyzed C–H functionalization reactions proceeding through metalacyclic or non-cyclic organometallic intermediates.
2. Oxidant Screening in Palladium Catalysis
To date, many C–H functionalization reactions are known where oxidant screening is performed and a significant effect of oxidant on the yield of product is observed. A few examples are considered below.
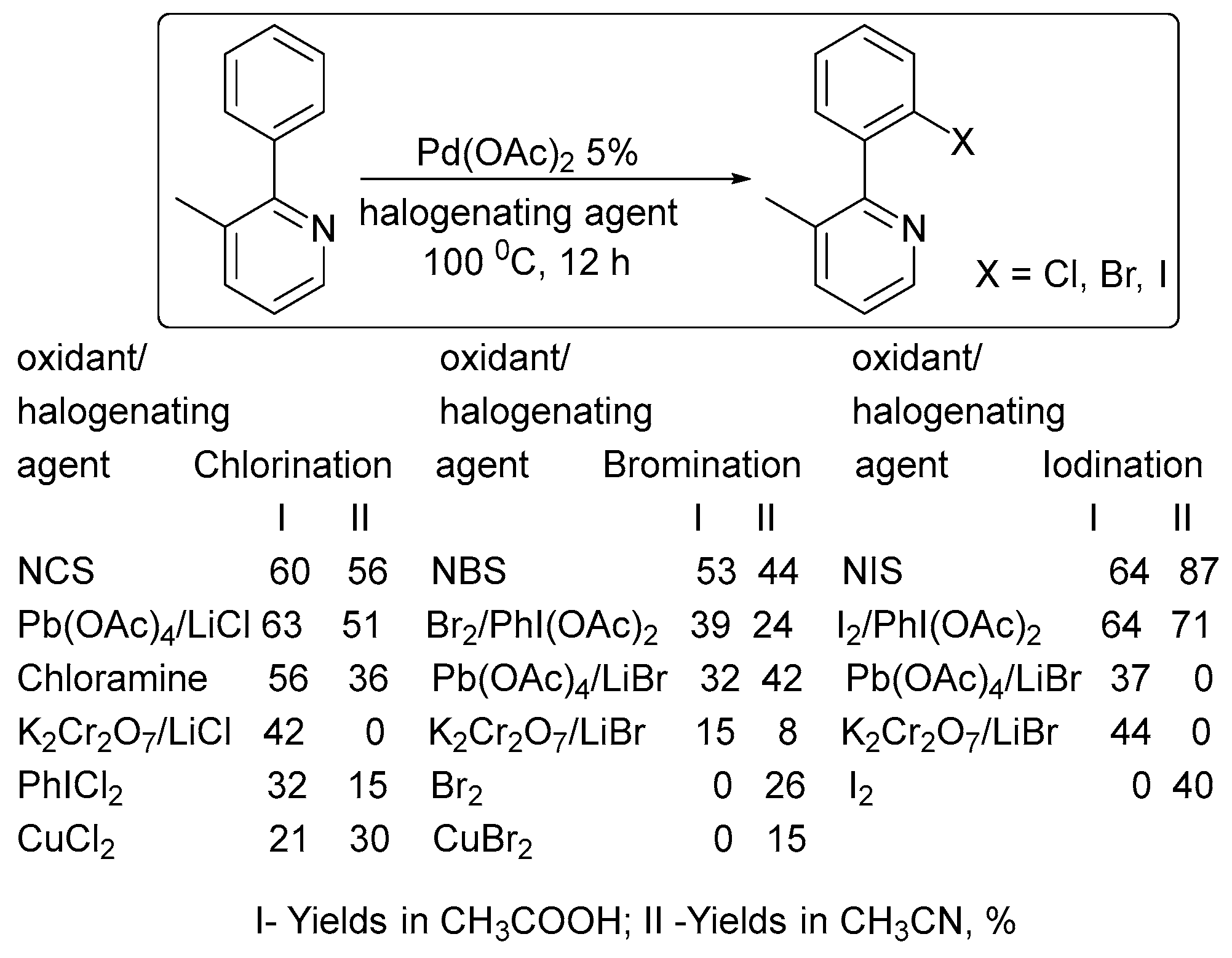
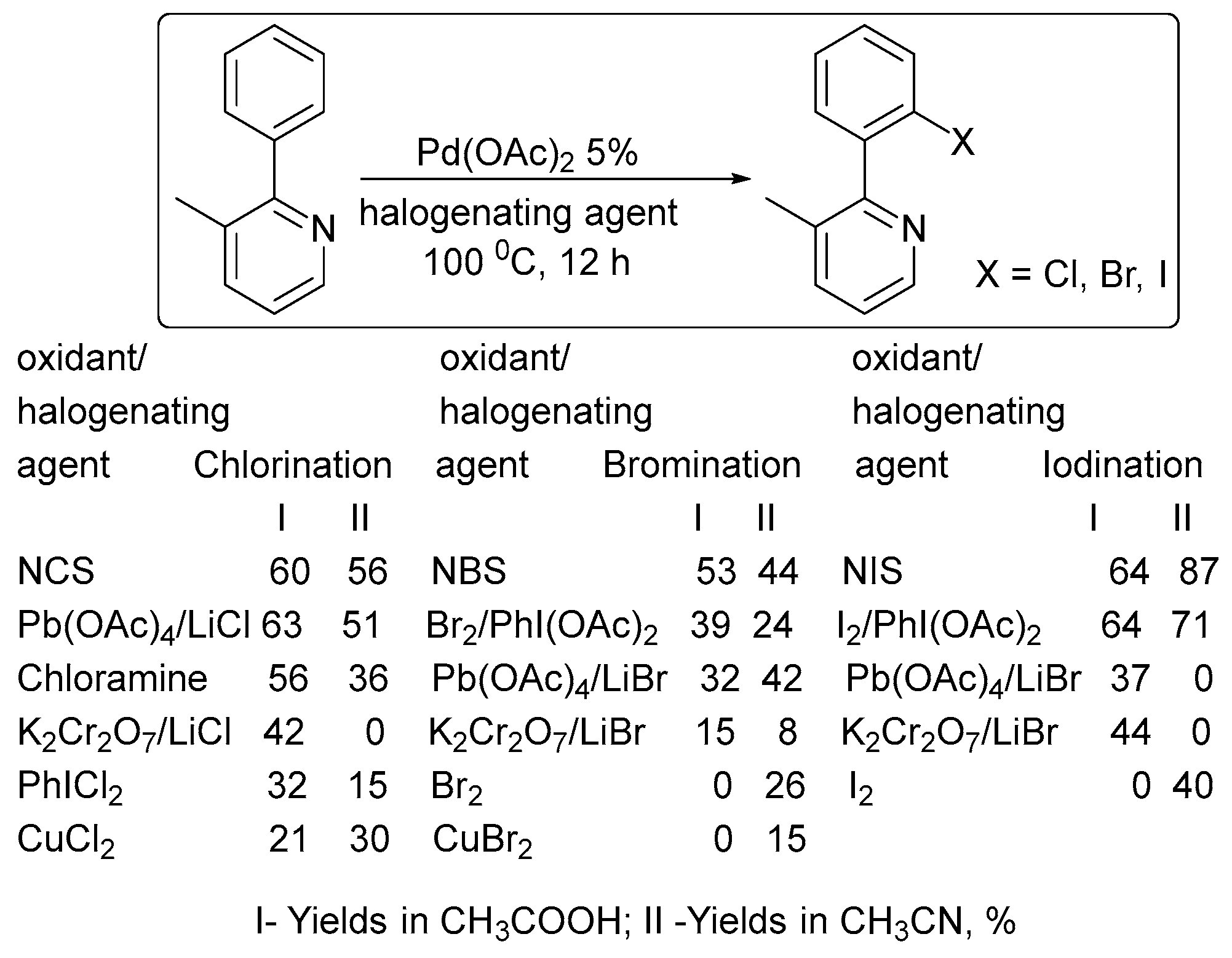
Among the other oxidants examined in ligand-directed halogenation (Scheme 1), N-halosuccinimides proved to be the best oxidants for these Pd-catalyzed reactions, providing the highest isolated yields of chlorinated (65%), brominated (56%), and iodinated (79%) products [26].
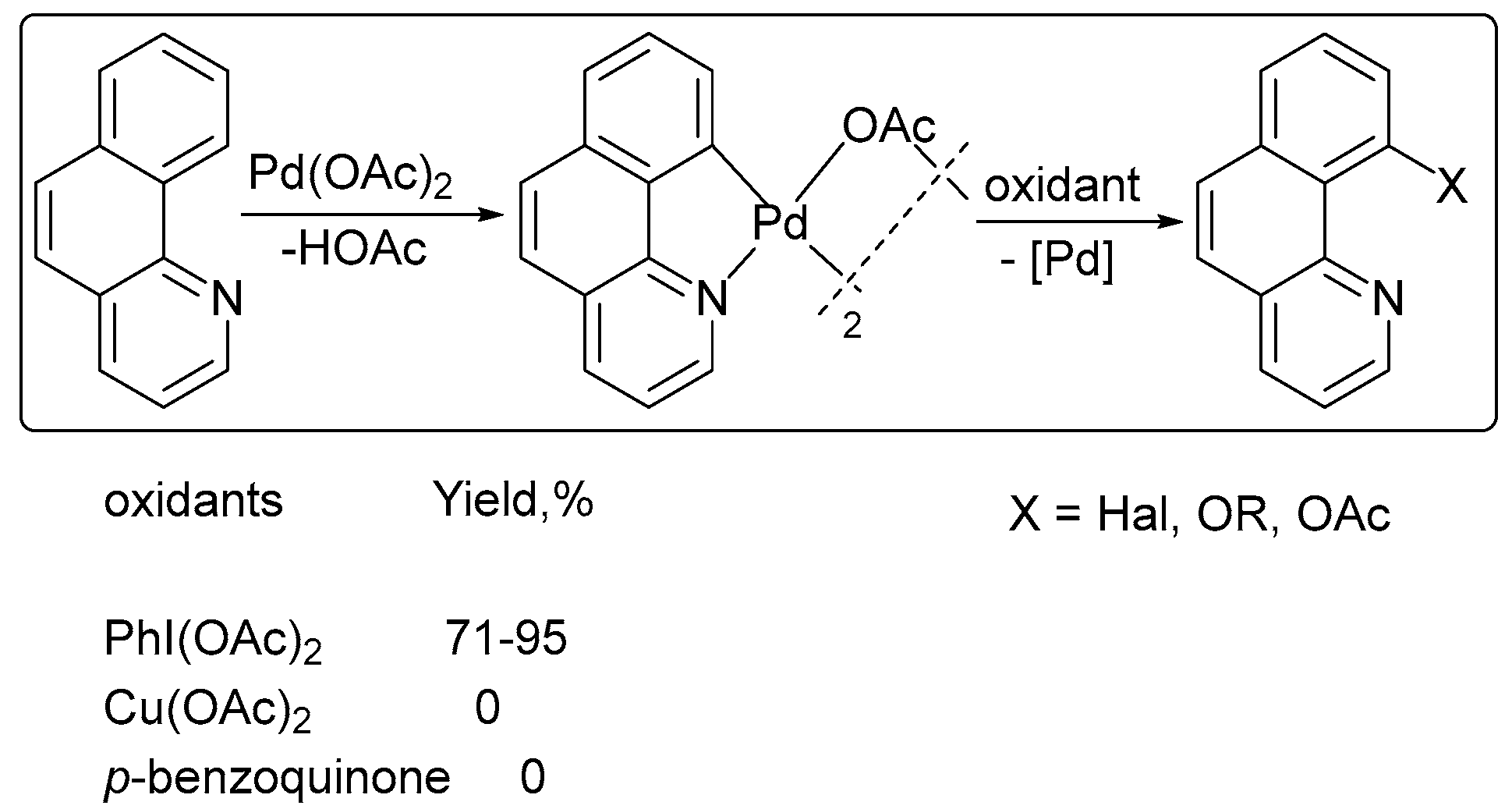
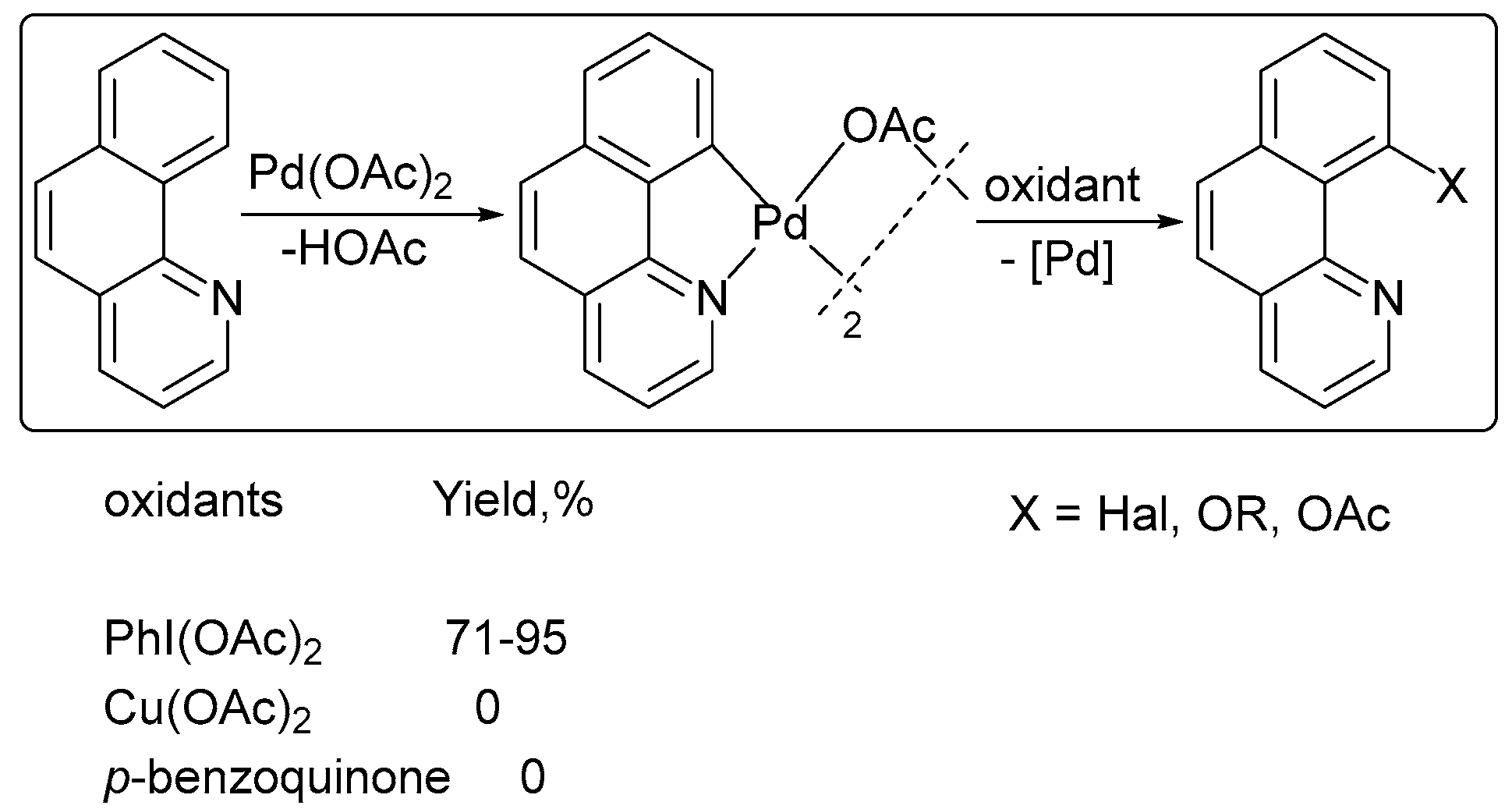
The chelate-directed oxidative functionalization of sp2 C–H bonds was reported in the presence of PhI(OAc)2 (Scheme 2) [27]. No product was formed when PhI(OAc)2 was replaced by Cu(OAc)2 or 1,4-benzoquinone.
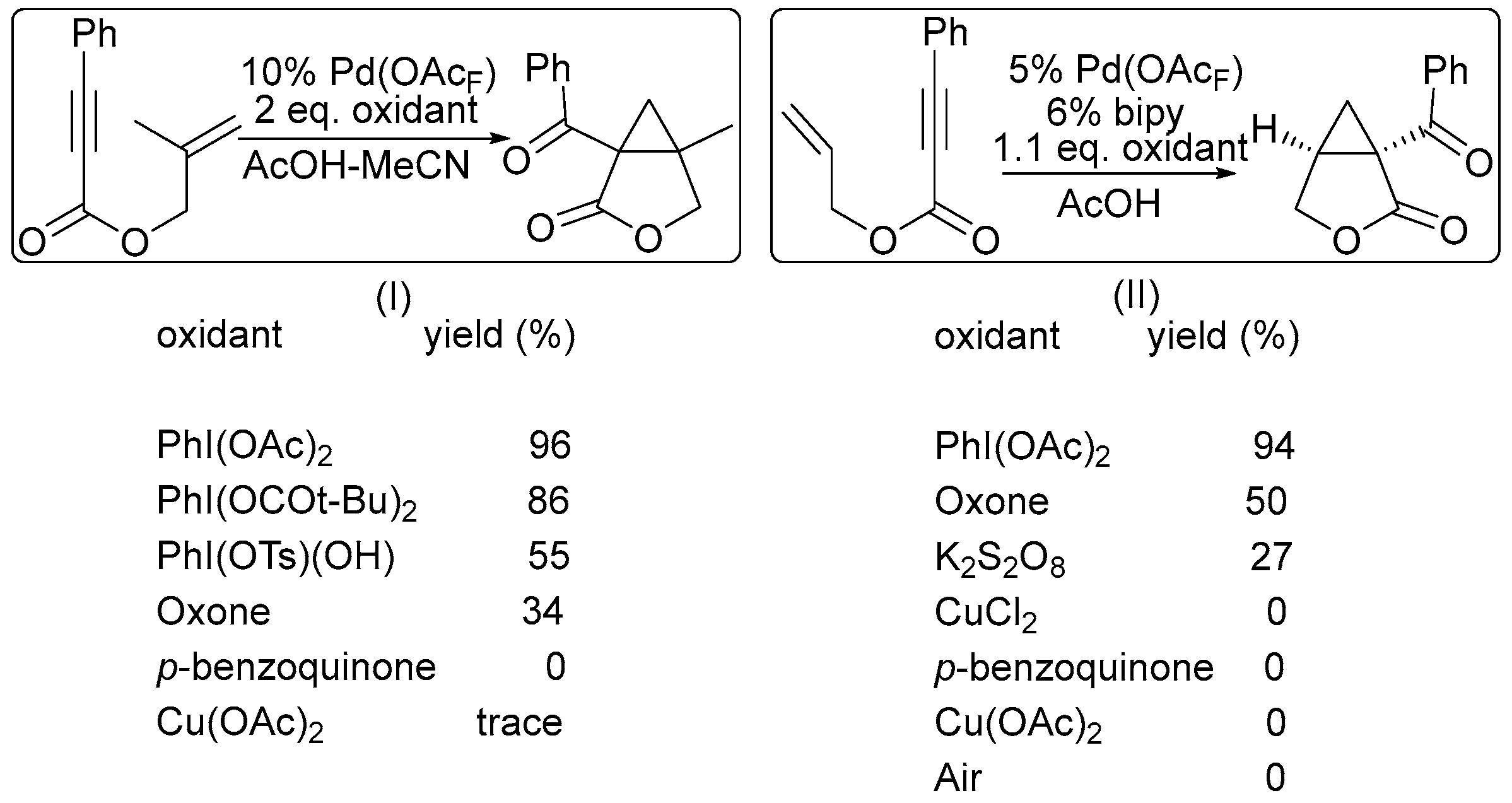
A more powerful oxidant PhI(OAc)2 promoted the desired oxidation process, producing cyclopropane (Scheme 3). The key stage of the cycle is the oxidation of a Pd(II) intermediate into a Pd(IV) complex, suppressing the side reaction of its decomposition via β-H elimination from alkyl-Pd intermediate [28,29].
3. Mechanistic Considerations
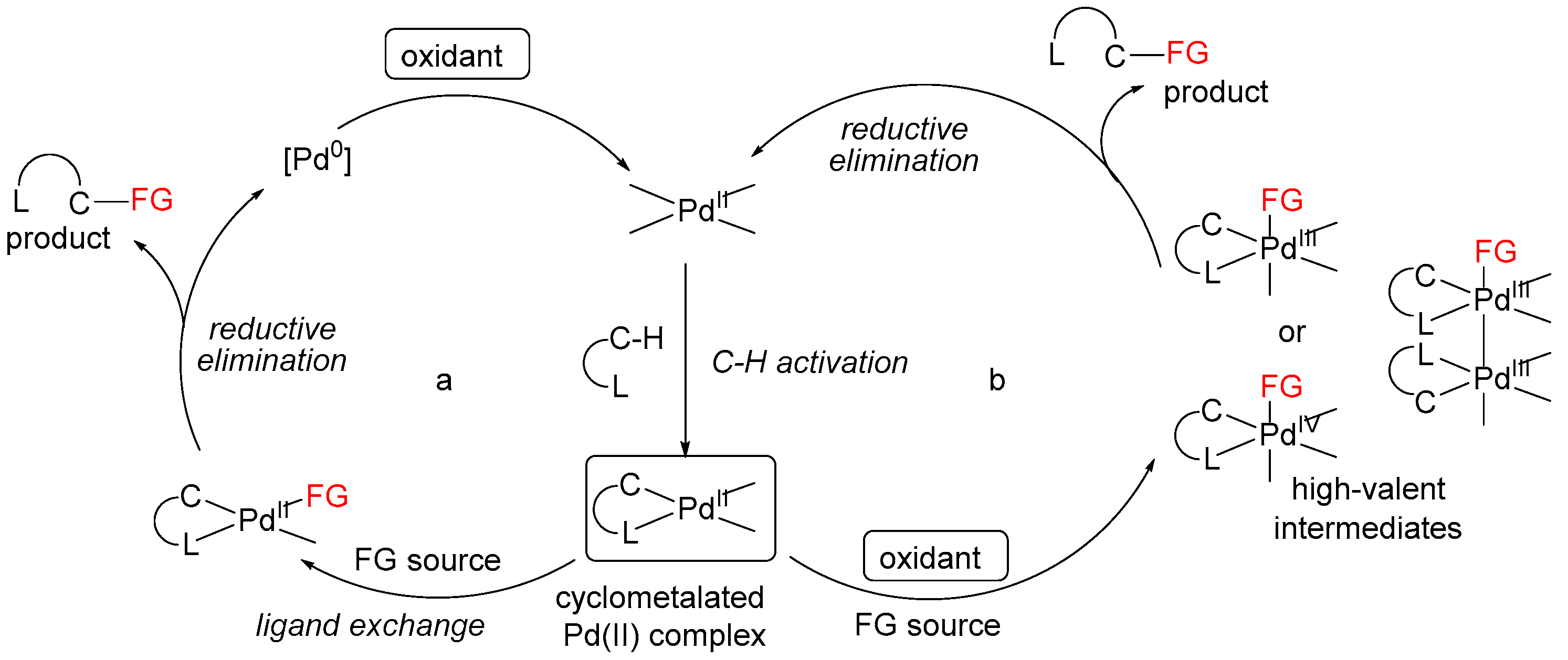
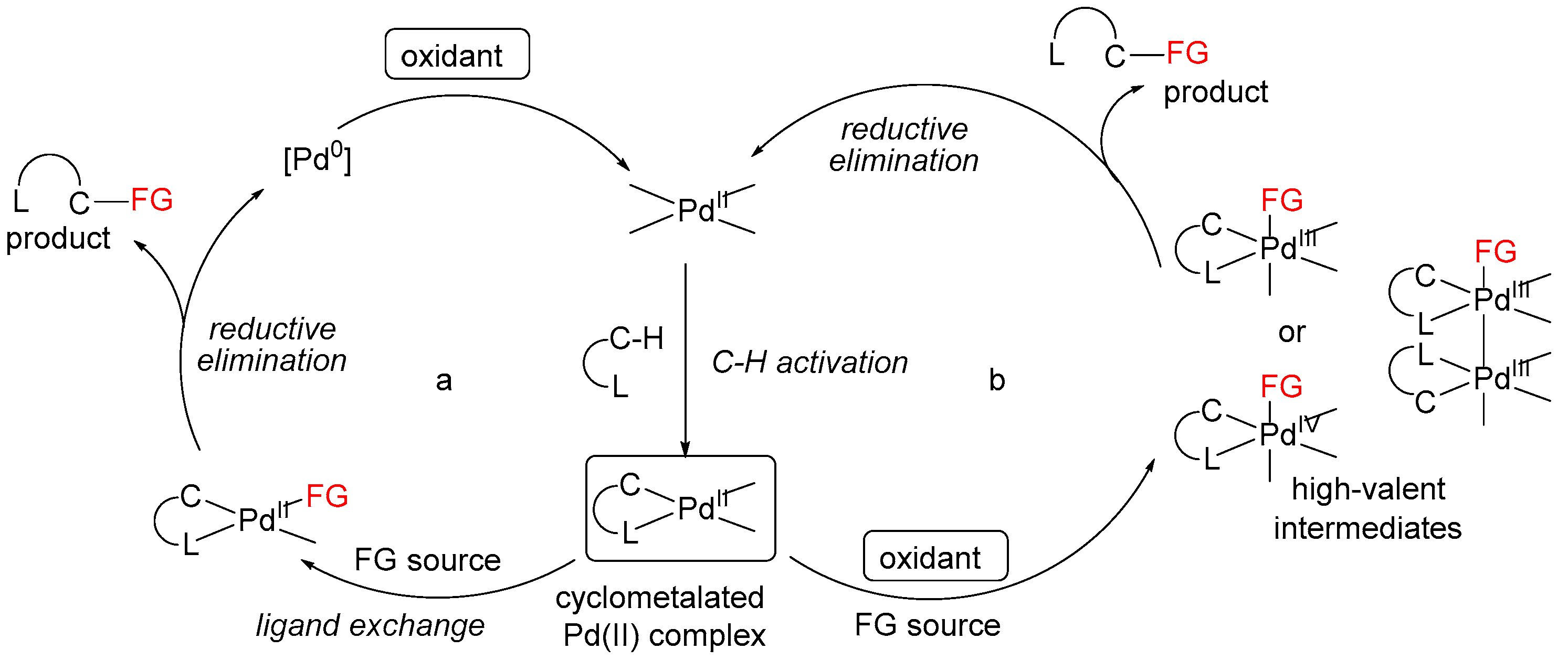
Pd-catalyzed reactions can be basically separated into two groups depending on the high- or low-valent organopalladium intermediates involved in the catalytic cycle [3]. However, the exact nature of those mechanisms is still under discussion; in some cases, catalytic cycles based on Pd(II)/Pd(III) or Pd(II)/Pd(0) redox shuttles are proposed [11,12,31]. In both cases, the reactions start from ligand-directed C–H activation at Pd(II) centers affording cyclopalladated intermediates, which then undergo functionalization by two distinct pathways. The first is based on a Pd(II)/(0) redox shuttle (Scheme 5a), where functionalization occurs via a reductive process (typically reductive elimination or β-hydride elimination/deprotonation) to release the product. The resulting Pd(0) intermediate is then oxidized to regenerate the Pd(II) catalyst. The second pathway involves the functionalization of the cyclometalated Pd(II) intermediate with an electrophilic reagent (Scheme 5b). Such reagents can react by several distinct mechanisms, including direct electrophilic cleavage of the Pd–C bond without a change in the oxidation state at the metal center, or one- or two-electron oxidation of the palladacycle generating high-valent Pd(III) or Pd(IV) intermediates, depending on the nature of the ancillary ligands at the metal center.
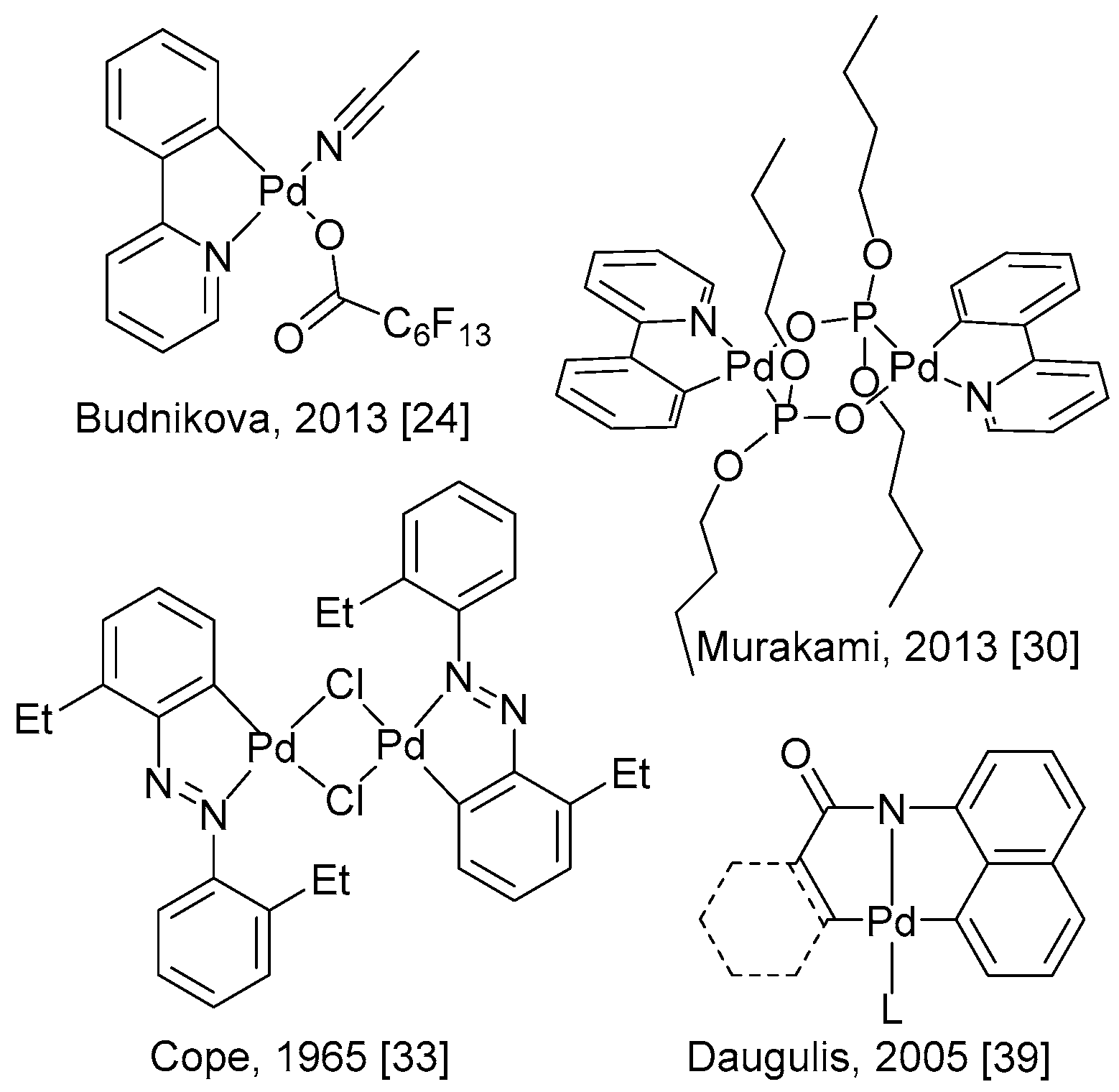
The ability of Pd(II) to activate C–H bonds through chelation-assisted processes has been known since the 1960s. Nitrogen-containing directing groups were found to promote the insertion of palladium into an ortho-C–H bond, which led to the formation of stable and isolable cyclopalladated complexes from Pd(II) starting material (Scheme 6) [25,31,32,33,34,35,36,37,38,39,40].
Despite the large literature devoted to the synthesis of various palladacycles and studies of their reactivity, their redox properties, which are necessary to describe and understand the mechanisms of catalytic reactions involving them, are elucidated only in few works [11,15,24,25,41,42,43,44]. However, the electrochemical properties of cyclometalated Pd(II) complexes (reduction/oxidation potentials) determine the mechanism of the reactions.
4. Electrode Potentials as the Measure of Oxidant/Reductant Strength
Knowledge of the oxidation potential of these important intermediates, and a comparison study of redox trends of cyclopalladated complexes depending on the ligand environment, can simplify the control over the catalytic reactions. This is very important in processes where several possible intermediates with different oxidation potentials are present. The adequate choice of the oxidation potential (or a chemical oxidant) minimizes or even prevents the formation of byproducts in these reactions.
In order to facilitate discussion, Connelly and Geiger [45] proposed to define categories of reagent strength on the basis of their formal potentials. Recognizing that the limits are subjective, the ranges in Table 1 are the basis for discussing a reagent as a weak, mild, strong, or very strong oxidant or reductant.
Table 2 shows the potentials of the most common oxidants used for catalytic C–H functionalizations; meanwhile, data for some palladium derivatives, possible, or proven intermediates in such reactions, are given in Table 3. Redox properties are not described for all palladacycles and other palladium derivatives with established structures, so only data where this estimate exists is considered for discussion. Electrochemical data for palladacycles, as established intermediates for many ligand-directed C–H-substitution reactions, are given in [25]. The potential range of the second oxidation peak of palladacycles, for the Pd(III)/Pd(IV) stage [23,24,25], is taken from supporting information data [25]. It is noteworthy that this work revealed a regularity of the complexes’ oxidation potential on the metal–metal distance in the complexes: the larger the Pd–Pd distance, the higher the oxidation potentials; in particular, monomeric palladacycles are more difficult to oxidize than dimeric ones.
A fundamental quantity providing an indication of the relative reactivity of oxidants reagents is their electrochemical standard potential, E°. Most of the oxidants used in organic chemistry for the C–H bonds functionalizations are reduced reversibly under voltammetry conditions (Table 2), and in this case E° is defined as the half-wave potential E1/2. If a redox agent undergoes chemically irreversible electron transfer, the standard potential E° can be approximately elucidated as peak potential EDPV from Differential Pulse Voltammogram (DPV), onset potential, or peak potential from slow cyclic voltammetry experiment. Nevertheless, the irreversible potentials (usually peak potentials from slow cyclic voltammetry experiments) listed in Table 2 may be used as approximations of the formal potential.
Such data (at specified experimental conditions) have proved useful in providing some measure of the relative reactivity of reagents. Studies of the reactivity of reagents in terms of their redox properties were previously carried out, for electrophilic NF fluorinating agents [71], for example, and showed good agreement between the electrochemical data and the observed experimental efficiency of the reagents.
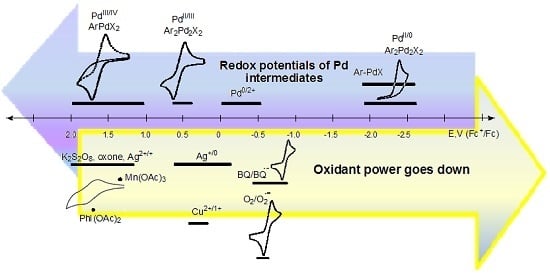
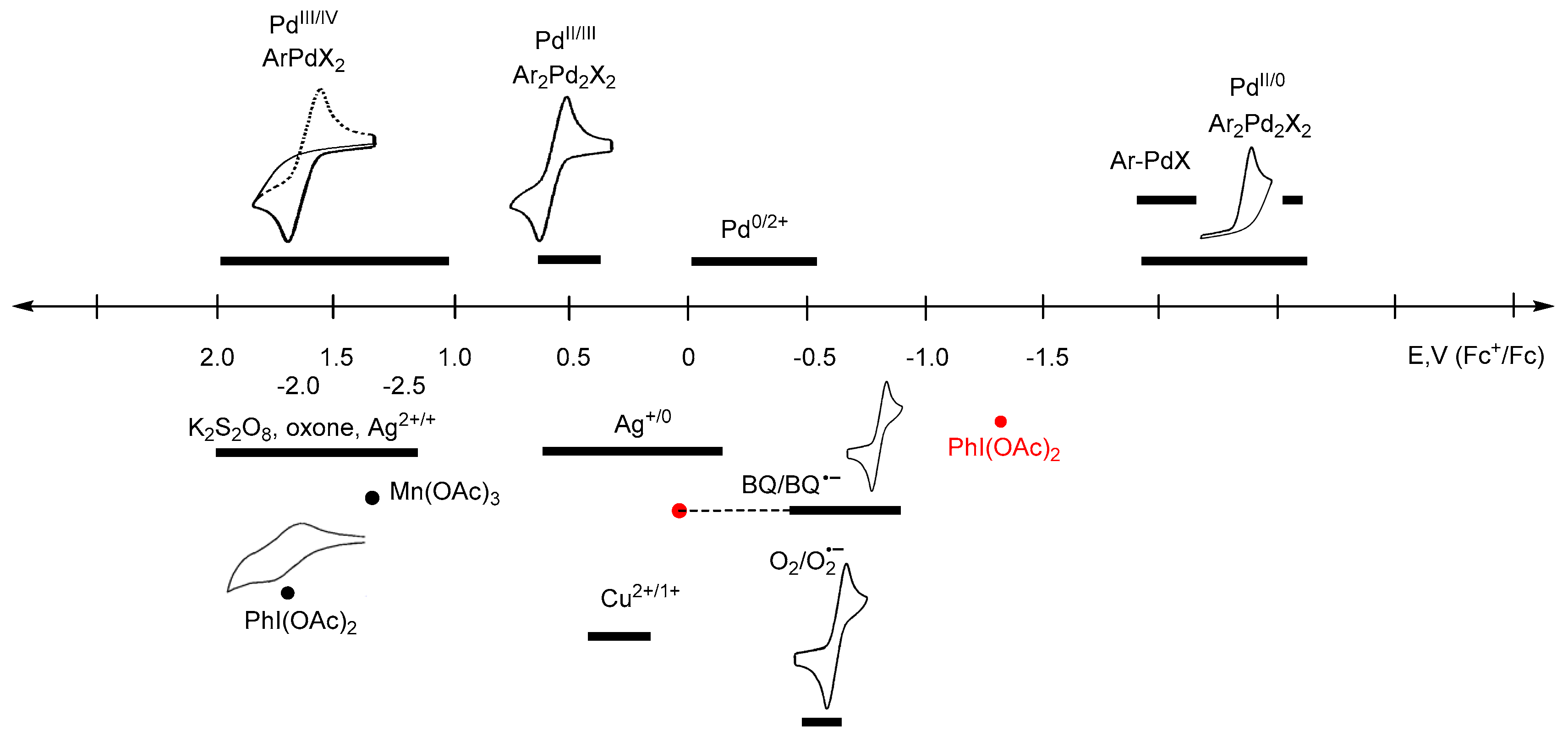
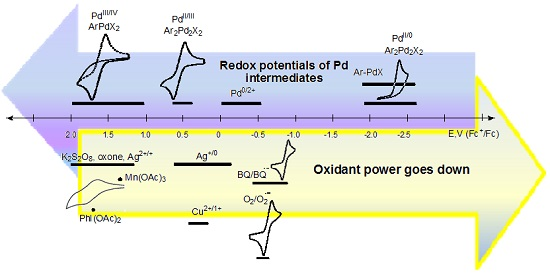
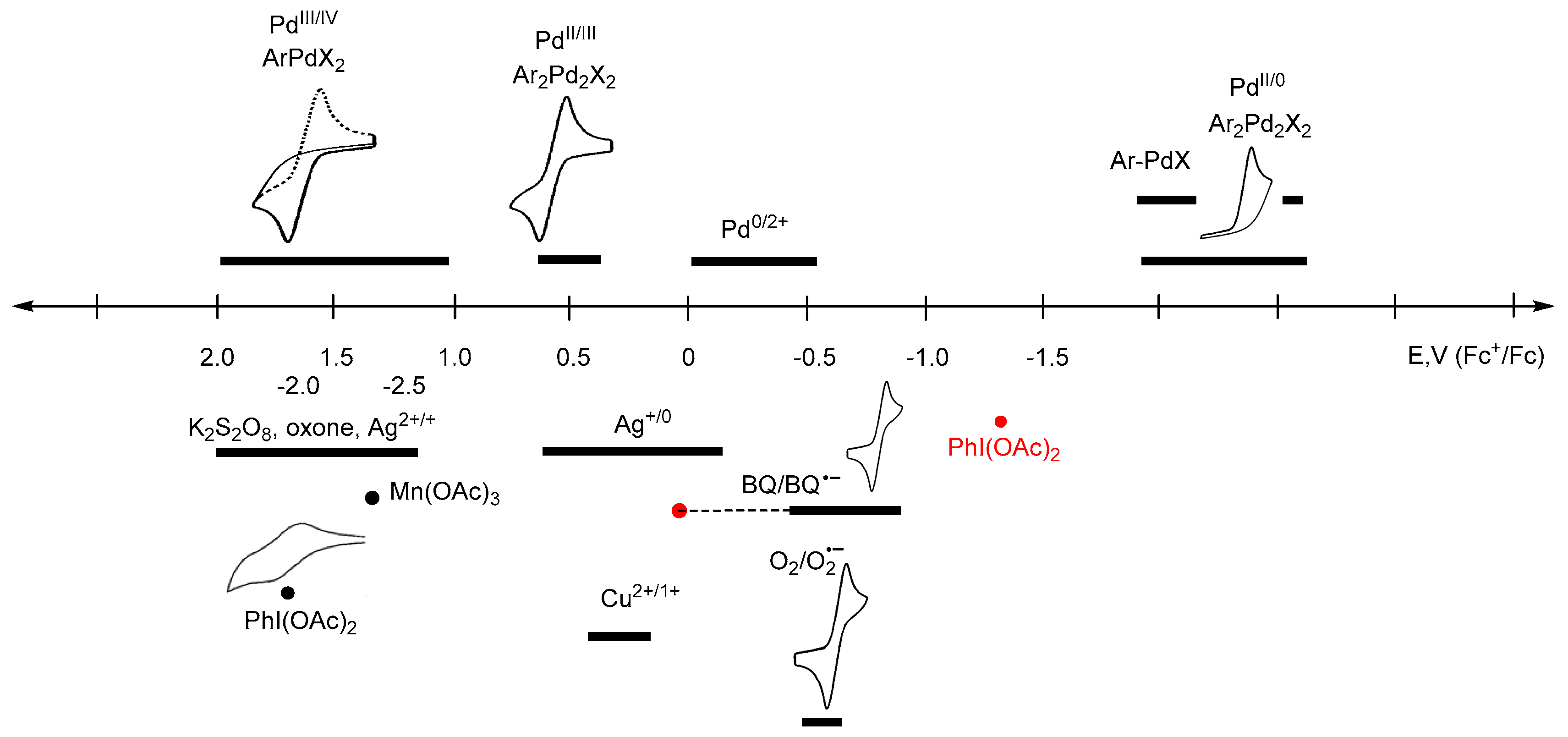
In Figure 1, the approximate ranges of standard redox potentials (or close to them) are given for some of the most commonly used oxidants and possible palladium intermediates. Certainly, the range of potentials attributed to each participant in the redox cycle (oxidant, metallocycle, or other organometallic intermediate) is not constant, and can vary depending on the process conditions, the composition of the reaction medium, and the nature of aromatic substrates. The diagram shows the known values of redox potentials published in the literature to date, and refined in this paper, and also shows when there was a discrepancy between the literature data or the data was absurd (for example, for PhI(OAc)2). Unreproducible data is shown in red.
Based on the literature data [58], where the reduction potential of PhI(OAc)2 is −1.29 V (vs. Fc+/Fc) or −0.91 V (vs. SCE) (Table 2), it should be recognized as a very weak oxidant or even a mild reductant (Table 1). This contradicts the results of plenty of oxidative transformations, where it works perfectly. That is, this data [58], which then was quoted in [43], is obviously erroneous. Perhaps the error is due to the instability of PhI(OAc)2 under the selected conditions used in the voltammetric measurements (acetonitrile with Et4ClO4 and LiClO4 elecrolytes), which prompted the authors to conduct kinetic studies of these first-order decomposition reactions [58]. To get reliable data and avoid possible side reactions, we investigated both the commercial (Acros Organics) and the PhI(OAc)2 obtained by the procedure [72], which we found to be more efficient than what [73] used in previous studies [58]. Cyclic voltammograms were taken both in acetonitrile solution (Bu4NBF4 electrolyte), and in solid state when PhI(OAc)2 crystals were added to the carbon paste electrode, according to the previously described procedure [62]. The first reduction peak potential of PhI(OAc)2 was found to occur equally both in the CH3CN solution and in the carbon paste electrode (1.70 V vs. Fc+/Fc) as a two-electron process. These redox properties are in good agreement with the experimental data on their high oxidative ability, as hypervalent iodine oxidants have been used for the two-electron oxidations of numerous Pd(II) to Pd(IV) cyclometallated complexes [3,7,8,10,11,12,26,27,28,29,31].
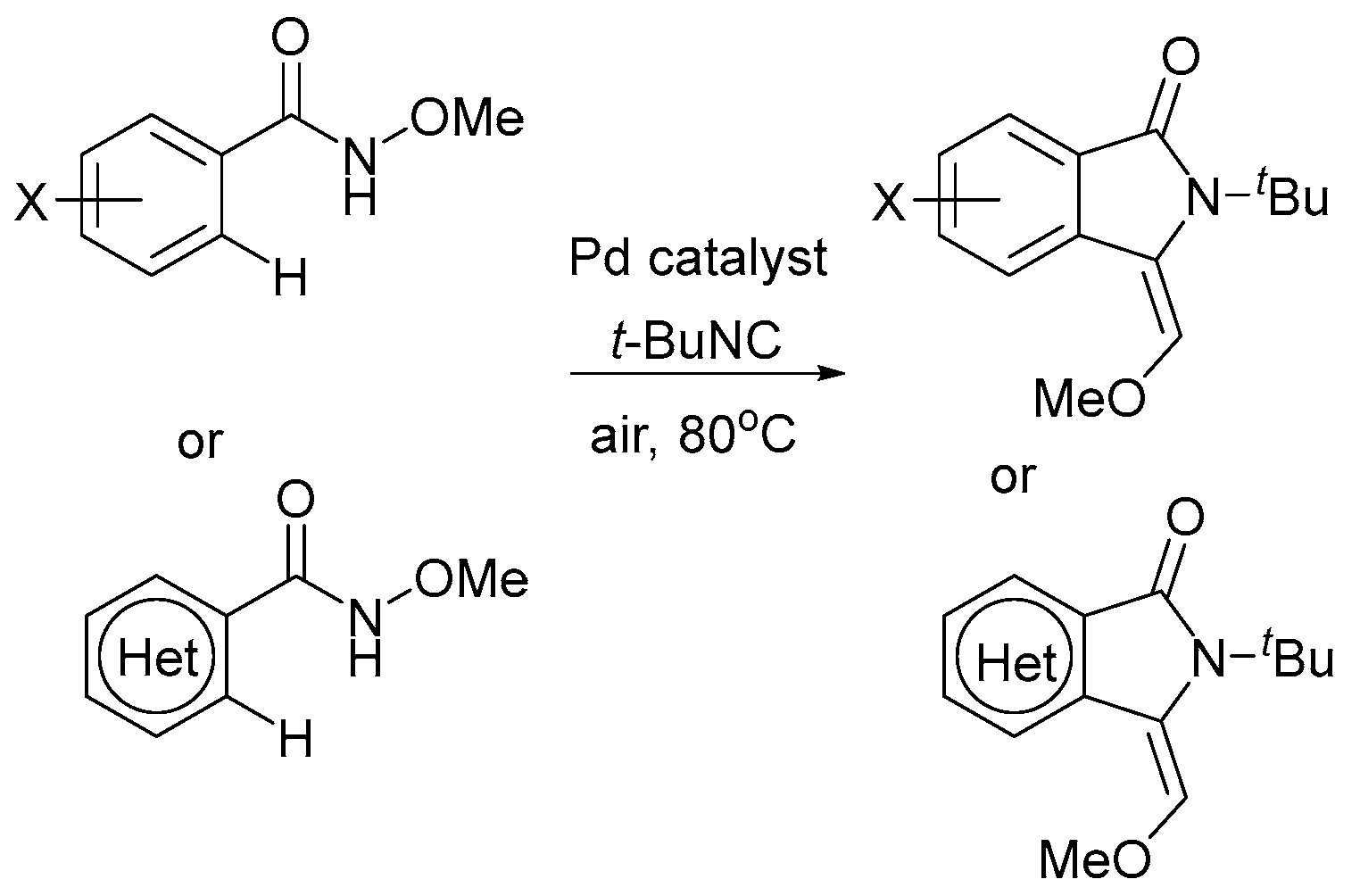
The recently reported work [31] analyzes the oxidant dependence and radical participation in Pd-catalyzed dehydrogenative alkoxylation of arenes (Scheme 7), as well as the efficiency of the procedures and key intermediates proposed in the literature. The latter are mainly Pd(IV) complexes for reactions with PhI(OAc)2, K2S2O8, or oxone, which seems to be fair and corresponds to the strength of these oxidants (the strongest). Only two Pd(II) intermediates and a Pd(II)/Pd(0) cycle were proposed for these reactions [31]. In 2011/2012, Reddy’s team proposed a Pd(II)/Pd(0) catalytic cycle for N-(2-benzoylphenyl)benzamides [74] and 2-arylquinazolin-4(3H)-ones substrates [75], but nevertheless proposed a Pd(IV) intermediate for the alkoxylation of 1-aryl-3,4-dihydroisoquinolines [76]. However, no evidence was provided. Considering the unexpected stability of the known palladacycles(II) to reduce [25], this path Pd(II)/Pd(0) seems to be unbelievable, since the Pd(II)/Pd(0) transfer occurs at high negative potentials (−1.8 to −2.6V). To form the products under reductive conditions, the palladacycles need to be treated with strong reducing agents, rather than strong oxidants, which actually work here (PhI(OAc)2, K2S2O8, or oxone). Some other unproven assumptions about the possible pathways that are based on Pd(II)/Pd(0) shuttle [77] in the presence of strong oxidants, such as PhI(OAc)2, also seem to be unlikely.
Sometimes, TEMPO (2,2,6,6-tetramethylpiperidyl-1-oxyl) is used as an oxidant or a radical trap. However, in oxidative conditions, conclusions should be drawn with caution. For example, to clarify the reaction mechanism of the pd-catalyzed direct ortho-C–O bond construction of azoxybenzenes with carboxylic acids and alcohols [78], 4.0 equiv. of TEMPO was added to the reactions under the optimized conditions, and no desirable products were detected. Therefore, it was suggested that these reactions involve a free radical pathway. So, a Pd(II) intermediate was reacted with the alkoxyl radical or carbonyl oxygen radicals to form a Pd(III) or Pd(IV) complex. However, the failure of the reaction in the presence of TEMPO can be explained; the latter is easily oxidized itself by the applied oxidant PhI(OAc)2 (Table 2). Moreover, if TEMPO was taken in two-fold excess over the oxidant, the entire oxidant was consumed by the side reaction of TEMPO oxidation, which is easily oxidized to about 0 V [60,61], but not by the oxidation of more difficultly oxidized palladacycles. It is known that oxammonium salts are effective oxidant species derived from TEMPO [79].
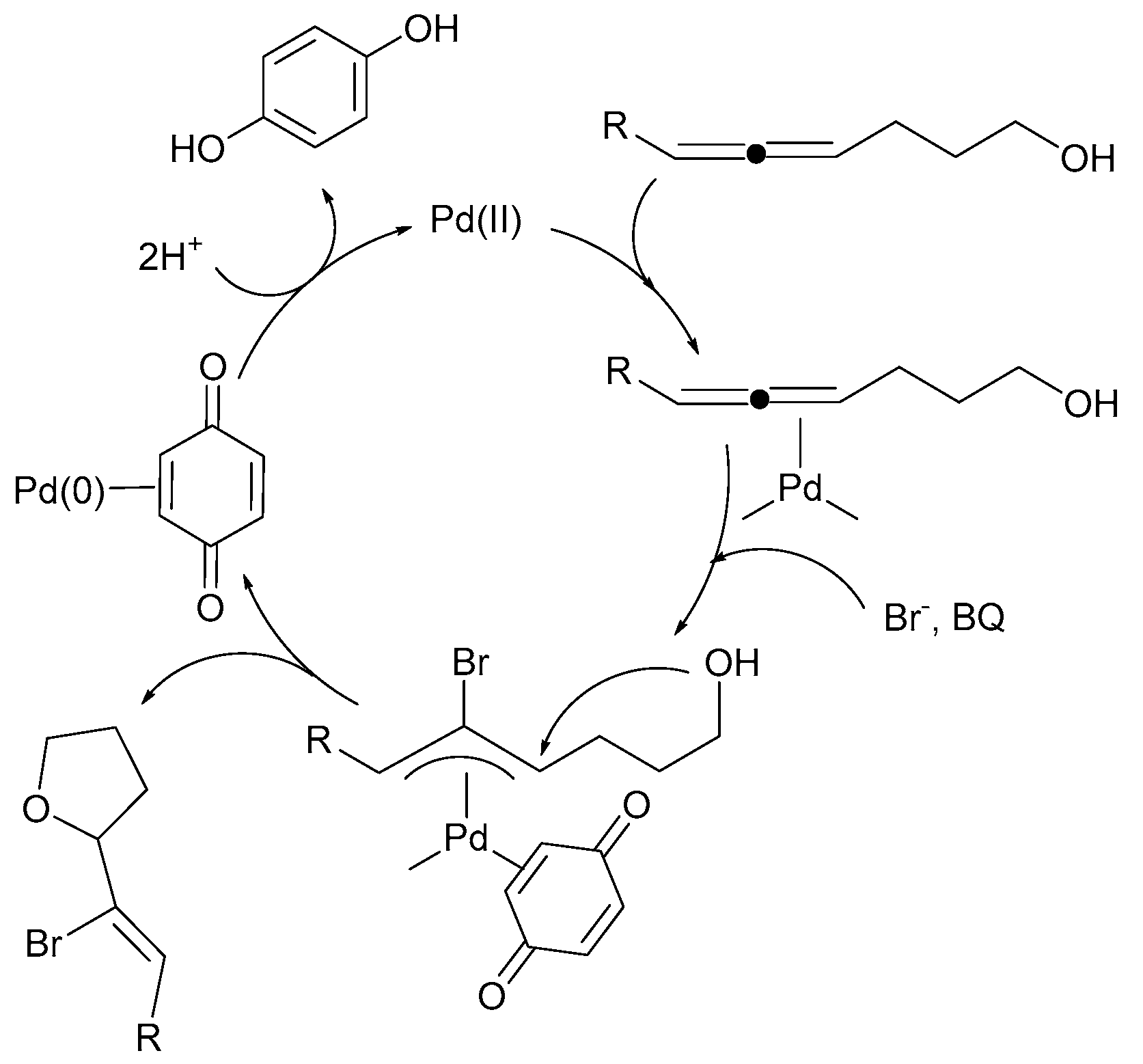
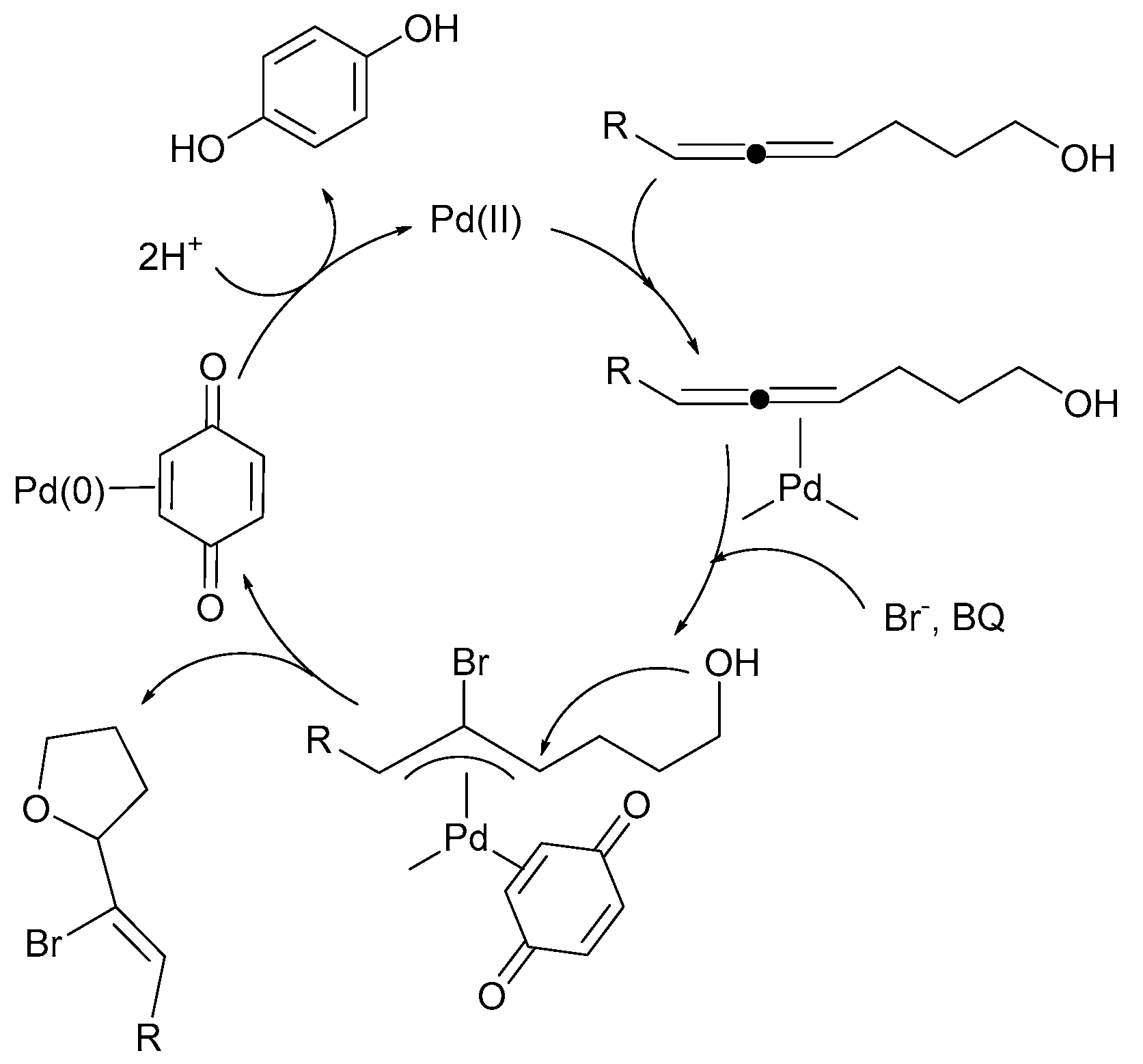
Reoxidants such as Cu(OAc)2, AgOAc, Ag2CO3, and 1,4-benzoquinone typically mediate the Pd(0)/Pd(II) catalysis of a dehydrogenative Heck reaction [80]. For example, intramolecular Pd(II) catalyzed 1,2-addition to allenes (Scheme 8). The reaction proceeds through an external nucleophilic attack of bromide anion on a (π-allene)palladium complex, which produces a (π-allyl)palladium intermediate. A subsequent intramolecular attack by the second internal nucleophile gives the product. The intermediate (π-allyl)palladium complexes were isolated and characterized [80]. Two different reoxidants for this Pd(0)/Pd(II) system (1,4-benzoquinone or Cu(OAc)2) were used.
Another example, a Pd(II)-catalyzed carboxylation of ortho-C–H bonds in anilides to form N-acyl anthranilic acids has been developed using 10 mol % Pd(OAc)2 and 1 equiv of benzoquinone (BQ) as the terminal oxidant [81]. The monomeric palladacycle containing p-toluenesulfonate as an anionic ligand is the well-characterized intermediate of the Pd(II)/Pd(0) catalytic cycle. According to the diagram (Figure 1), in this case, the oxidant is only able to reoxidize Pd(0) into a Pd(II) complex.
Therefore, in order to establish the mechanism of a reaction proceeding through Pd(IV)/Pd(II), Pd(III)/Pd(II), or Pd(II)/Pd(0) transformations, or the apparent redox-neutral pathway, one can focus on the redox properties of the process participants: key cyclopalladated intermediates and oxidants (or reducing agents). Thus, it is obvious that such weak oxidants as oxygen (−0.78 to −1.26 V), 1,4-benzoquinone (0.16 to −0.875V) are not able to oxidize many palladacycles even up to Pd(III) (~0.25 to 1.4 V) complexes, the more so to Pd(IV) (~1.0 to 1.6V) [25]. This explains the numerous failed attempts to use them in such Pd-catalysed C–H functionalizations, such as 1,4-benzoquinone [27,28,29,75,82,83,84,85], and O2 [77,82,86,87,88] for example.
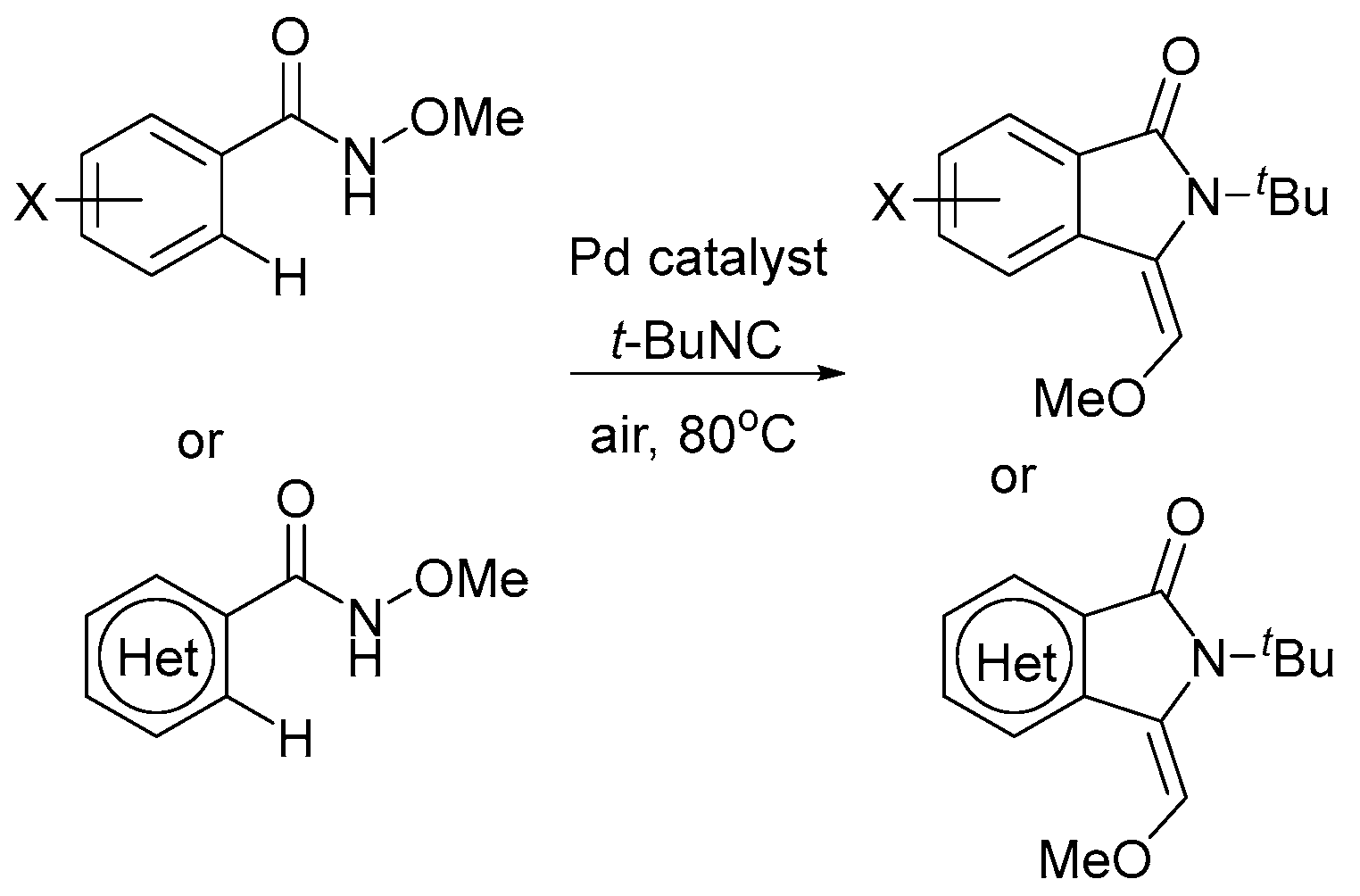
There are reactions catalyzed by Pd(II) complexes involving oxygen under mild conditions, such as the C–C couplings for example. However, in these cases, the mechanism should include Pd(0)/Pd(II) reoxidation of the catalyst, but not high-valent palladium intermediates [89,90,91]. However, as can be seen from the diagram in Figure 1, the oxidative strength of oxygen may not be enough to oxidize Pd(0) under certain conditions (the potential ranges of Pd(0) oxidation and O2 reduction overlap insignificantly, at about −0.5 V). A strategy to facilitate directed Pd-catalyzed C–H functionalizations conducted in the presence of heterocycles that relies on the relatively inefficient oxidation of Pd(0) by air was demonstrated by the groups of Dai and Yu [92] (Scheme 9). More detailed discussions on air or oxygen used as oxidants are described in recent reviews [93,94,95,96].
At the same time, a mild oxidant, AgOAc, is sufficient for the success of many Pd(OAc)2-catalyzed reactions, especially in CH2Cl2 or DCE [97,98,99], passing through Pd(III) but not Pd(IV) intermediates, for which its strength is not enough [74,75,87,100,101].
Functionalizations of aromatic C–H bonds under oxidative conditions can be controlled in two ways. Shifting the oxidation potential of the key palladacycle to less positive potentials (through using additives capable to replace counteranions in the palladacycles, changing its structure, etc.), decreases the HOMO energy and facilitates oxidation. The second way is to choose a stronger oxidant (it should be taken into account that the oxidant’s power depends on the solvent medium, and the presence of acids or bases).
Coordinating solvents capable of converting dipalladium complexes into monopalladacycles leads to an increase in the oxidation potential of palladium intermediates [24,25]; that is, their oxidation is more difficult, and stronger oxidants or more harsh conditions for their oxidation are required. For example, for a Pd(II)-catalyzed carboxylation of ortho-C–H bonds in anilides affording N-acyl anthranilic acids [81], it was demonstrated that the key intermediates are dipalladacycles in CH2Cl2 or THF, and monopalladacycles in dioxane. Correspondingly, the yield of product in the latter case is lower, and additives are required to succeed the reaction.
Many inorganic strong oxidants are poorly or completely insoluble in organic solvents; as a result, reactions with them take place under heterogeneous conditions (especially in the case of generally accepted large excesses of oxidants). The known data for the redox potentials of such inorganic oxidants are in most cases measured in aqueous solutions, and therefore are not suitable for the discussion of non-aqueous reactions. The data of solid state electrochemistry is more appropriate, and generally more correct, because such oxidants do not dissolve in organic solvents, and the oxidation stage is heterogeneous.
Aqueous media, especially when acidic, significantly increase the strength of some oxidants through shifting their reduction potentials toward the less negative region (Table 2). This effect is observed when protons of the medium participate in the oxidation/reduction reactions and shift the equilibrium potential to higher positive potentials. Such effect is a property, for example, of K2S2O8, 1,4-benzoquinone, O2.
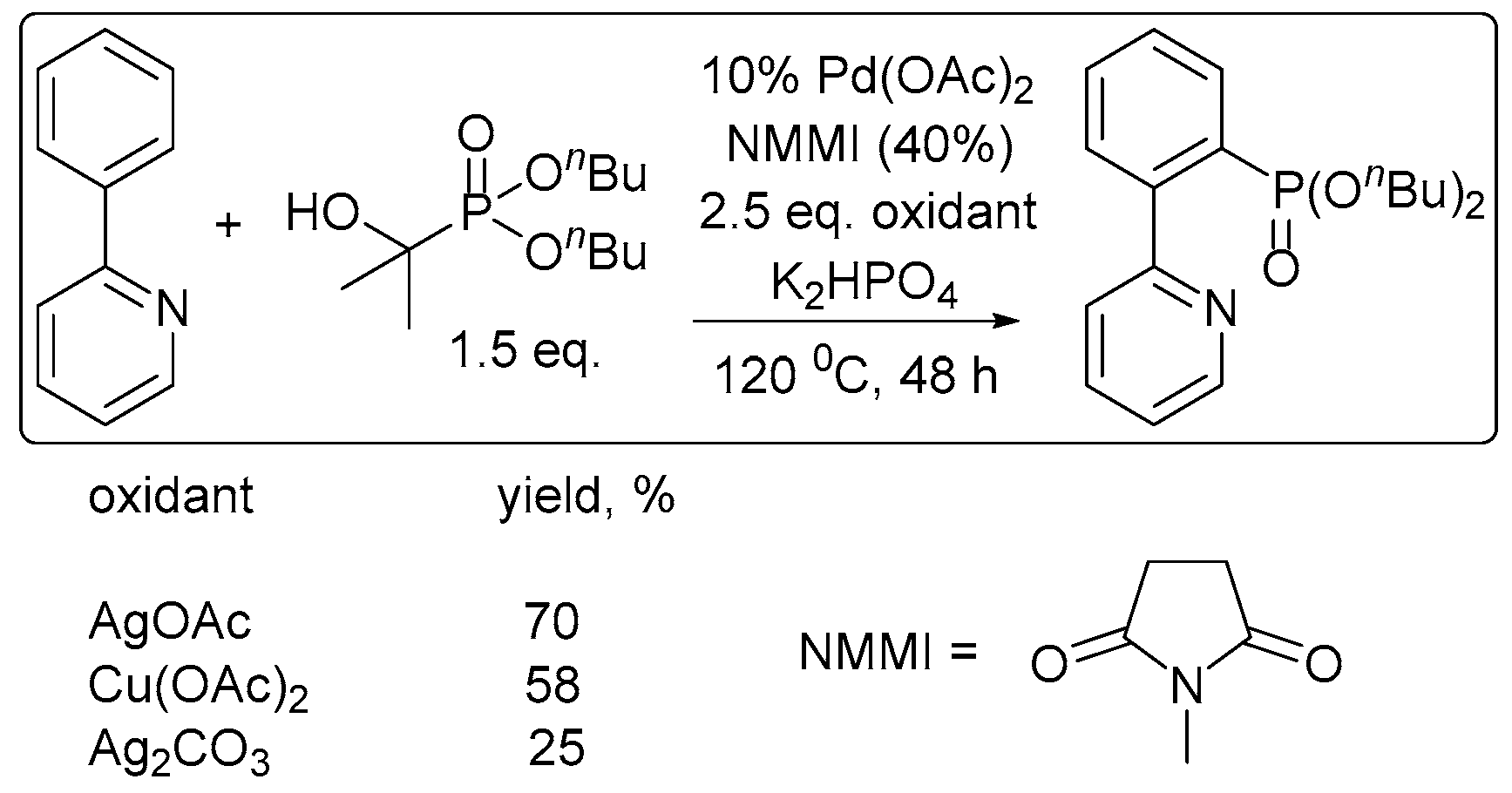
The revealed regularity in the oxidation chemistry of cyclopalladated complexes might be useful to determine conditions in ligand-directed C–H functionalization reactions. For example, as phosphonate-bridged palladacycles are significantly more difficult to oxidize than acetate-bridged ones [25], the reactivity of both complexes (key intermediates) should be considered in C–H phosphorylation reactions, such as the phosphorylation of 2-phenylpyridine with H-phosphonates catalyzed by palladium acetate [14,15,25,30]. Selective C–H phosphonation requires the correct choice of an oxidant (or electrolysis potential) to promote the oxidation of the phosphonate-bridged dipalladacycle. In the case of a “weak” oxidant (or a low electrolysis potential), the oxidation of an acetate-bridged dipalladacycle is the dominant process that results in the formation of an acetoxylated byproduct. The problem of finding an appropriate chemical oxidant likely explains the low yields and very few aromatic C–H phosphonation reactions in the literature, which are usually carried out at high temperature, with special additives (NMMI, Scheme 6 [30]).
5. Electrocatalytic Ligand-Directed Substitution of C(sp2)–H Bonds
An important aspect of electrochemical methodology is that many electrochemical processes meet standards for environmental cleanliness. From the viewpoint of implementation, electrochemical reactions have several advantages: mild conditions, high rates, selectivity, and convenient operational control through switching current density and potential. Electrochemical methods can be recommended for the preventive protection of the environment, because they do not require any special reagents [13,14,15,16,17,18,19,20,21,22,23,24,25,102,103,104].
We propose developing cheaper, greener, and more controllable electrochemical alternatives to these oxidation reactions. One can easily see the advantages of having a recyclable and much cheaper redox co-catalyst (the electrode vs. silver or organic oxidants), where you can simply dial in the potential. Scheme 3 shows an oxidative cyclization reaction where (diacetoxyiodo)benzene was employed. An example of the utility of the electrochemical alternative is the replacement of this expensive reagent with just a graphite electrode in the presence of sodium acetate, which is more than 200 times less expensive than (diacetoxyiodo)benzene. Moreover, ligand effects were noted to affect the yields and selectivity in this reaction, so bulk electrolysis could combine different ligands without worries of overpotential effects.
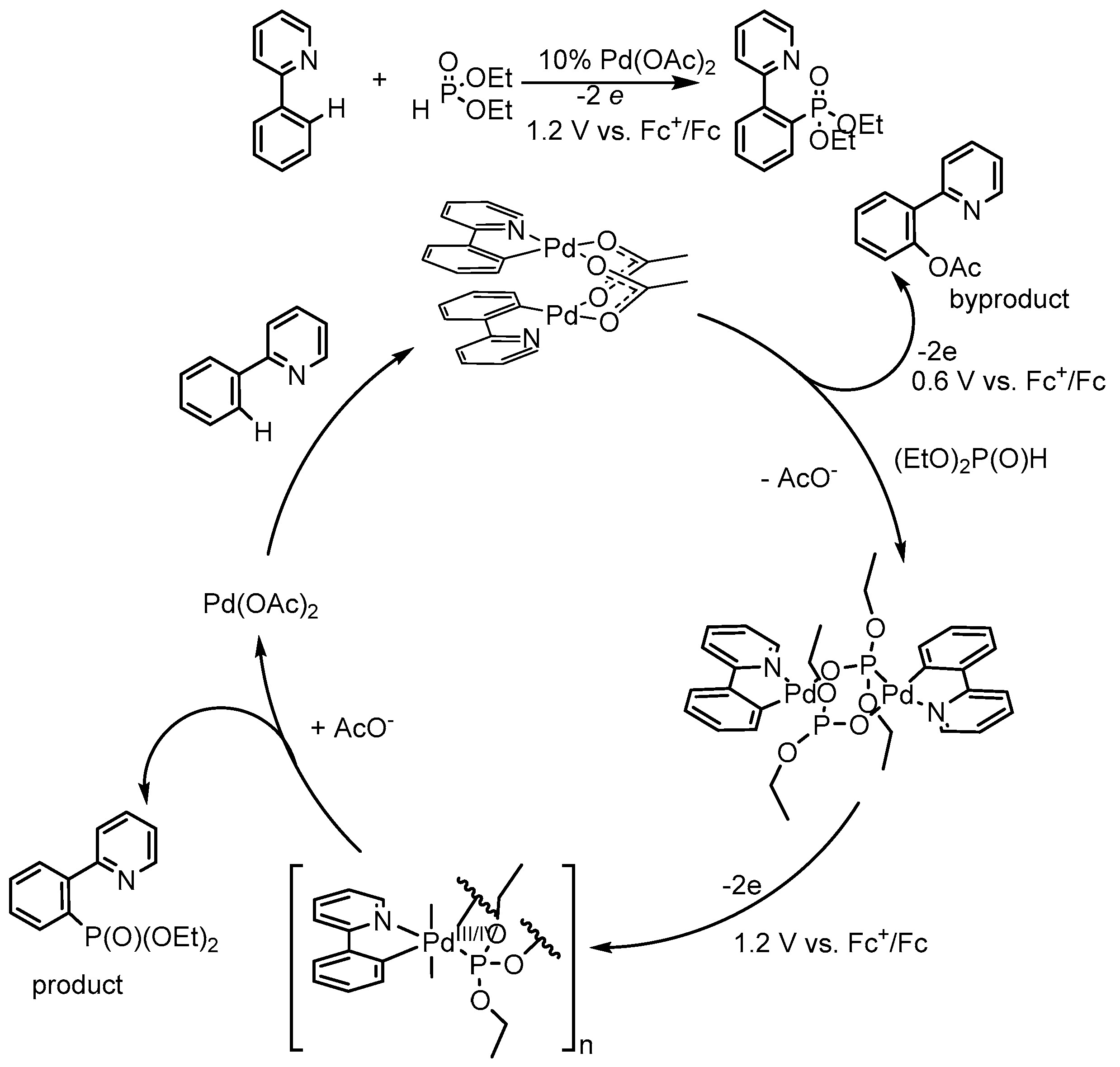
Also, ligand-directed substitution at the C–H bond in 2-phenylpyridine, which was earlier employed for acetoxylation and perfluoroalkylation under electrochemical conditions [23,24] in the presence of Pd(II) acetate, proved to be effective in the case of phosphorylation [13,14,15,25]. The introduction of the phosphonate group is based on the electrochemical oxidation of a mixture of palladium acetate, phenylpyridine, and diethyl phosphite at room temperature, with phenylpyridine orto-phosphonate being obtained without special external oxidants (Scheme 10).
In order to empower the ligand-directed phosphonation of aromatic C–H bonds, a series of diphosphonate-bridged dipalladacycles (with 2-phenylpyridine, benzo[h]quinoline, and 1-phenylpyrazole) was prepared and characterized by NMR spectroscopy and cyclic voltammetry in acetonitrile solutions and in carbon paste electrode [14,15]. A comparative quantitative study on the redox properties of structurally related diphosphonate-bridged dipalladacycles under the same conditions, their stability in high oxidation states, the influence of aromatic ligand nature on the potentials of Pd(II)/Pd(III) and Pd(III)/Pd(IV) transfers and electrochemical synthesis of arylphosphonates from these complexes was performed [14,15,25].
The electrochemical oxidation of diphosphonate-bridged palladacycles was explored in a divided electrolyser in CH3CN solution and monitored by 31P NMR spectroscopy. It was found that the amount of electricity required for the complete conversion of the complex into the product is 2 e per each atom of palladium, so, Pd(II)/Pd(IV) pair runs. It should also be noted that the mononuclear arylphosphonate palladium complex was detected as reaction intermediate. Thus, phosphonate palladacycles are oxidized irreversibly and quantitatively, thus affording corresponding arylphosphonates.
Therefore, this method of oxidation may be applied to the synthesis of new arylphosphonates from different arenes in ligand-directed aromatic C–H phosphonation reactions. Advantages of the method are the mild conditions (room temperature), and absence of specially added chemical oxidants.
6. Conclusions
Selective oxidation reactions have extraordinary value in organic chemistry, ranging from the conversion of petrochemical feedstocks into industrial chemicals and polymer precursors to the introduction of heteroatom functional groups into pharmaceutical and agrochemical intermediates. The search for the optimal oxidant for these transformations is very important. The selective choice of a suitable oxidizing agent should be based on the redox properties of both the oxidants themselves, including their reduction potentials, the number of electrons (one, two or more electron oxidants), and the substrates and intermediates of the catalytic conversions. The series of potentials used herein involving possible participants of such Pd-catalysed C–H transformations can be further extended to other oxidation reactions with other metals, if the potentials of organometallic intermediates are known, or to induce researchers to obtain such useful electrochemical data ahead of planning new costly syntheses, and only after that to choose more effective catalytic conditions.
Acknowledgments
This work was supported by the Russian Science Foundation (grant No. 14-23-00016).
Author Contributions
Yulia H. Budnikova generated the basic idea of the review, summarized and discussed the results of individual studies, wrote the paper; Yulia B. Dudkina conceived and designed the experiments and wrote the paper; Mikhail N. Khrizanforov performed the experiments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gensch, T.; Hopkinson, M.N.; Glorius, F.; Wencel-Delord, J. Mild metal-catalyzed C–H activation: Examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Shi, Z.; Yuan, Y. Transition-metal-catalyzed Chelation-assisted C–H Functionalization of Aromatic Substrates. Chem. Rec. 2016, 16, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium-Catalyzed Ligand-Directed C−H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Kim, J.G.; Carr, K.J.T.; Bellina, F.; Soulé, J.-F.; Wang, G.-W.; Bruneau, C.; Dana, S.; Li, J.; Ackermann, L.; et al. C–H Bond Activation and Catalytic Functionalization I; Dixneuf, P.H., Doucet, H., Eds.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Ilies, L.; Nakamura, E.; Chatani, N.; Hirano, K.; Jones, W.; Sustac Roman, D.; Zhou, T.; Dailler, D.; Samantaray, M.K.; Bézier, D.; et al. C–H Bond Activation and Catalytic Functionalization II; Dixneuf, P.H., Doucet, H., Eds.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A. Oxidative Coupling between Two Hydrocarbons: An Update of Recent C–H Functionalizations. Chem. Rev. 2015, 115, 12138–12204. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-M.; Li, B.-J.; Yang, Z.; Shi, Z.-J. Organopalladium(IV) chemistry. Chem. Soc. Rev. 2010, 39, 712–733. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, P.; Taylor, R.J.K.; Fairlamb, I.J.S. Emergence of Palladium(IV) Chemistry in Synthesis and Catalysis. Chem. Rev. 2010, 110, 824–889. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Mei, T.-S.; Wang, X.; Yu, J.-Q. Bystanding F+ Oxidants Enable Selective Reductive Elimination from High-Valent Metal Centers in Catalysis. Angew. Chem. Int. Ed. 2011, 50, 1478–1491. [Google Scholar] [CrossRef] [PubMed]
- Hickman, A.J.; Sanford, M.S. High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.C.; Geibel, M.A.L.; Klein, J.E.M.N.; Ritter, T. Bimetallic Palladium Catalysis: Direct Observation of Pd(III)−Pd(III) Intermediates. J. Am. Chem. Soc. 2009, 131, 17050–17051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deprez, N.R.; Sanford, M.S. Synthetic and Mechanistic Studies of Pd-Catalyzed C−H Arylation with Diaryliodonium Salts: Evidence for a Bimetallic High Oxidation State Pd Intermediate. J. Am. Chem. Soc. 2009, 131, 11234–11241. [Google Scholar] [CrossRef] [PubMed]
- Budnikova, Y.H.; Gryaznova, T.V.; Grinenko, V.V.; Dudkina, Y.B.; Khrizanforov, M.N. Eco-efficient electrocatalytic C–P bond formation. Pure Appl. Chem. 2017, 89, 311–330. [Google Scholar] [CrossRef]
- Grayaznova, T.V.; Dudkina, Y.B.; Islamov, D.R.; Kataeva, O.N.; Sinyashin, O.G.; Vicic, D.A.; Budnikova, Y.H. Pyridine-directed palladium-catalyzed electrochemical phosphonation of C(sp2)–H bond. J. Organomet. Chem. 2015, 785, 68–71. [Google Scholar] [CrossRef]
- Gryaznova, T.; Dudkina, Y.; Khrizanforov, M.; Sinyashin, O.; Kataeva, O.; Budnikova, Y. Electrochemical properties of diphosphonate-bridged palladacycles and their reactivity in arene phosphonation. J. Solid State Electrochem. 2015, 19, 2665–2672. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Strekalova, S.O.; Kholin, K.V.; Khrizanforova, V.V.; Kadirov, M.K.; Gryaznova, T.V.; Budnikova, Y.H. Novel approach to metal-induced oxidative phosphorylation of aromatic compounds. Catal. Today 2017, 279, 133–141. [Google Scholar] [CrossRef]
- Grinenko, V.V.; Khrizanforov, M.N.; Strekalova, S.O.; Khrizanforova, V.V.; Kholin, K.V.; Gryaznova, T.V.; Budnikova, Y.H. Electrooxidative phosphorylation of coumarins by bimetallic catalytic systems Ni(II)/Mn(II) or Co(II)/Mn(II). Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1660–1661. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Strekalova, S.O.; Grinenko, V.V.; Khrizanforova, V.V.; Gryaznova, T.V.; Budnikova, Y.H. Various ways of C–P bonds formation via selective electrochemical phosphorylation of aromatic C–H bonds. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1491–1493. [Google Scholar] [CrossRef]
- Gryaznova, T.V.; Khrizanforov, M.N.; Strekalova, S.O.; Budnikova, Y.H.; Sinyashin, O.G. Electrochemical oxidative phosphonation of azoles. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1658–1659. [Google Scholar] [CrossRef]
- Strekalova, S.O.; Khrizanforov, M.N.; Shamsieva, A.V.; Grinenko, V.V.; Gryaznova, T.V.; Musina, E.I.; Karasik, A.A.; Budnikova, Y.H. Direct phosphorylation of pyridine in the presence of Ni(BF4)2bpy and CoCl2bpy metal complexes. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1545–1546. [Google Scholar] [CrossRef]
- Khrizanforova, V.V.; Khrizanforov, M.N.; Gryaznova, T.V.; Budnikova, Y.H. Electrochemical pathway to CH/PH functionalization of diphenylphosphine oxide. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1602–1603. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Strekalova, S.O.; Kholin, K.V.; Khrizanforova, V.V.; Grinenko, V.V.; Gryaznova, T.V.; Budnikova, Y.H. One-stage Synthesis of FcP(O)(OC2H5)2 from Ferrocene and α-Hydroxyethylphosphonate. RSC Adv. 2016, 6, 42701–42707. [Google Scholar] [CrossRef]
- Dudkina, Y.B.; Mikhaylov, D.Y.; Gryaznova, T.V.; Sinyashin, O.G.; Vicic, D.A.; Budnikova, Y.H. MII/MIII-Catalyzed ortho-Fluoroalkylation of 2-Phenylpyridine. Eur. J. Org. Chem. 2012, 2114–2117. [Google Scholar] [CrossRef]
- Dudkina, Y.B.; Mikhaylov, D.Y.; Gryaznova, T.V.; Tufatullin, A.I.; Kataeva, O.N.; Vicic, D.A.; Budnikova, Y.H. Electrochemical Ortho Functionalization of 2-Phenylpyridine with Perfluorocarboxylic Acids Catalyzed by Palladium in Higher Oxidation States. Organometallics 2013, 32, 4785–4792. [Google Scholar] [CrossRef]
- Dudkina, Y.B.; Kholin, K.V.; Gryaznova, T.V.; Islamov, D.R.; Kataeva, O.N.; Rizvanov, I.K.; Levitskaya, A.I.; Fominykh, O.D.; Balakina, M.Y.; Sinyashin, O.G.; et al. Redox Trends in Cyclometalated Palladium(II) Complexes. Dalton Trans. 2017, 46, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, D.; Dick, A.R.; Anani, W.Q.; Sanford, M.S. Scope and selectivity in palladium-catalyzed directed C–H bond halogenation reactions. Tetrahedron 2006, 62, 11483–11498. [Google Scholar] [CrossRef]
- Dick, A.R.; Hull, K.L.; Sanford, M.S. A Highly Selective Catalytic Method for the Oxidative Functionalization of C–H Bonds. J. Am. Chem. Soc. 2004, 126, 2300–2301. [Google Scholar] [CrossRef] [PubMed]
- Tsujihara, T.; Takenaka, K.; Onitsuka, K.; Hatanaka, M.; Sasai, H. PdII/PdIV Catalytic Enantioselective Synthesis of Bicyclo[3.1.0]hexanes via Oxidative Cyclization of Enynes. J. Am. Chem. Soc. 2009, 131, 3452–3453. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium(II/IV) catalyzed cyclopropanation reactions: Scope and mechanism. Tetrahedron 2009, 65, 3211–3221. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yano, T.; Ishida, N.; Murakami, M. Pyridine-directed palladium-catalyzed phosphonation of C(sp2)–H bonds. Angew. Chem. Int. Ed. 2013, 52, 9801–9804. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, J.; Muzart, J. Dehydrogenative (Hetero)arene Alkoxylations Triggered by PdII-Catalyzed C(sp2)–H Activation and Coordinating Substituent: PdII,III or PdIV Complex as Key Intermediate? Eur. J. Org. Chem. 2017, 3528–3548. [Google Scholar] [CrossRef]
- Hartwell, G.E.; Lawrence, R.V.; Smas, M.J. The formation of palladium(II)– and platinum(II)–carbon bonds by proton abstraction from benzo[h]quinoline and 8-Methylquinoline. J. Chem. Soc. Chem. Commun. 1970, 912. [Google Scholar] [CrossRef]
- Cope, A.C.; Siekman, R.W. Formation of Covalent Bonds from Platinum or Palladium to Carbon by Direct Substitution. J. Am. Chem. Soc. 1965, 87, 3272–3273. [Google Scholar] [CrossRef]
- Cope, A.C.; Friedrich, E.C. Electrophilic aromatttic substitution reactions by platinum(II) and palladium(II) chlorides on N,N-dimethylbenzylamines. J. Am. Chem. Soc. 1968, 90, 909–913. [Google Scholar] [CrossRef]
- Constable, A.G.; McDonald, W.G.; Sawkins, L.C.; Shaw, B.L. Palladation of dimethylhydrazones, oximes, and oxime O-allyl ethers: Crystal structure of [Pd3(ON=CPriPh)6]. J. Chem. Soc. Chem. Commun. 1978, 1061–1062. [Google Scholar] [CrossRef]
- Fuchita, Y.; Hiraki, K.; Uchiyama, T. Metallation of aliphatic carbon atoms. Part 1. Synthesis and characterization of the cyclopalladated complexes of 2-neopentylpyridine. J. Chem. Soc. Dalton Trans. 1983, 897–899. [Google Scholar] [CrossRef]
- Alsters, P.L.; Engel, P.F.; Hogerheide, M.P.; Copijn, M.; Spek, A.L.; van Koten, G. Rigid five- and six-membered C,N,N’-bound aryl-, benzyl-, and alkylorganopalladium complexes: sp2 vs. sp3 carbon-hydrogen activation during cyclopalladation and palladium(IV) intermediates in oxidative addition reactions with dihalogens and alkyl halides. Organometallics 1993, 12, 1831–1844. [Google Scholar] [CrossRef]
- Rouquet, G.; Chatani, N. Catalytic Functionalization of C(sp2)–H and C(sp3)–H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef] [PubMed]
- Zaitsev, V.G.; Shabashov, D.; Daugulis, O. Highly Regioselective Arylation of sp3 C−H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13154–13155. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C–H Functionalization Reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.G.; Powers, D.C.; Raynaud, J.; Graham, M.J.; Xie, P.; Lee, E.; Ritter, T. Synthesis and structure of solution-stable one-dimensional palladium wires. Nat. Chem. 2011, 3, 949–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bercaw, J.E.; Durrell, A.C.; Gray, H.B.; Green, J.C.; Hazari, N.; Labinger, J.A.; Winkler, J.R. Electronic Structures of PdII Dimers. Inorg. Chem. 2010, 49, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.N.; Adrio, L.A.; Albrecht, T.; White, A.J.P.; Newton, M.A.; Nachtegaal, M.; Figueroa, S.J.A.; Hii, K.K.M. Electronic structures of cyclometalated palladium complexes in the higher oxidation states. Dalton Trans. 2015, 44, 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Fabre, I.; von Wolff, N.; Le Duc, G.; Flegeau, E.F.; Bruneau, C.; Dixneuf, P.H.; Jutand, A. Autocatalytic Intermolecular versus Intramolecular Deprotonation in C–H Bond Activation of Functionalized Arenes by Ruthenium(II) or Palladium(II) Complexes. Chem. Eur. J. 2013, 19, 7595–7604. [Google Scholar] [CrossRef] [PubMed]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef] [PubMed]
- Wacławek, S.; Grübel, K.; Cěrník, M. Simple spectrophotometric determination of monopersulfate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 149, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Block, P.A.; Brown, R.A.; Robinson, D. Novel Activation Technologies for Sodium Persulfate in Situ Chemical Oxidation. In Proceedings of the Fourth International Conference on Remediation of Chlorinated and Recalcitrant Compounds, Monterey, CA, USA, 24–27 May 2004. [Google Scholar]
- Haynes, W.M. CRC Handbook of Chemistry and Physics, 93rd ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Wulfsberg, G. Inorganic Chemistry; University Science Books: Sausalito, CA, USA, 2000. [Google Scholar]
- Nayak, B.; Dash, U.N. The silver/silver acetate electrode Part I. Standard potential in formamide at 25 °C. J. Electroanal. Chem. 1973, 41, 323–328. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Mebi, C.A.; Petro, B.J.; Vannucci, A.K.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Review of electrochemical studies of complexes containing the Fe2S2 core characteristic of [FeFe]-hydrogenases including catalysis by these complexes of the reduction of acids to form dihydrogen. J. Organomet. Chem. 2009, 694, 2681–2699. [Google Scholar] [CrossRef]
- Bour, J.R.; Camasso, N.M.; Meucci, E.A.; Kampf, J.W.; Canty, A.J.; Sanford, M.S. Carbon–carbon bond-forming reductive elimination from isolated Nickel(III) complexes. J. Am. Chem. Soc. 2016, 138, 16105–16111. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Reduction Potentials of One-Electron Couples Involving Free Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef]
- Prieto-Simón, B.; Fàbregas, E. Comparative study of electron mediators used in the electrochemical oxidation of NADH. Biosens. Bioelectron. 2004, 19, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kitagawa, T. Solvent effects of 1,4-benzoquinone and its anion radicals probed by resonance Raman and absorption spectra and their correlation with redox potentials. J. Raman Spectrosc. 1998, 29, 773–780. [Google Scholar] [CrossRef]
- Jin, B.-K.; Li, L.; Huang, J.-L.; Zhang, S.-Y.; Tian, Y.-P.; Yang, J.-X. IR Spectroelectrochemical Cyclic Voltabsorptometry and Derivative Cyclic Voltabsorptometry. Anal. Chem. 2009, 81, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Steckhan, E. Indirect Electroorganic Syntheses—A Modern Chapter of Organic Electrochemistry. Angew. Chem. Int. Ed. 1986, 28, 683–701. [Google Scholar] [CrossRef]
- Kokkinidis, G.; Papadopoulou, M.; Varvoglis, A. Electrochemical reduction of [bis(acyloxy)iodo]arenes. Electrochim. Acta 1989, 34, 133–139. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J. Electrocatalytic Oxygen Reduction Reaction. In PEM Fuel Cell Electrocatalysts and Catalyst Layers. Fundamentals and Applications; Zhang, J., Ed.; Springer-Verlag: London, UK, 2008. [Google Scholar]
- Jeena, V.; Robinson, R.S. Convenient photooxidation of alcohols using dye sensitised zinc oxide in combination with silver nitrate and TEMPO. Chem. Commun. 2012, 48, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Rychnosky, S.D.; Vaidyanathan, R.; Beauchamp, T.; Lin, R.; Farmer, P.J. AM1-SM2 Calculations Model the Redox Potential of Nitroxyl Radicals Such as TEMPO. J. Org. Chem. 1999, 64, 6745–6749. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Arkhipova, D.M.; Shekurov, R.P.; Gerasimova, T.P.; Ermolaev, V.V.; Islamov, D.R.; Miluykov, V.A.; Kataeva, O.N.; Khrizanforova, V.V.; Sinyashin, O.G.; et al. Novel paste electrodes based on phosphonium salt room temperature ionic liquids for studying the redox properties of insoluble compounds. J. Sol. State Electrochem. 2015, 19, 2883–2890. [Google Scholar] [CrossRef]
- Favier, I.; Duñach, E. New protic salts of aprotic polar solvents. Tetrahedron Lett. 2004, 45, 3393–3395. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A.; Lemaître, F.; Ricard, J.L.; Kozuch, S.; Shaik, S. Formation of anionic palladium(0) complexes ligated by the trifluoroacetate ion and their reactivity in oxidative addition. J. Organomet. Chem. 2004, 689, 3728–3734. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A.; Khalil, F. Neutral palladium(0) complexes from Pd(OAc)2 and tri-2-furylphosphine and their reactivity in oxidative addition of iodobenzene. ARKIVOC 2006, 38–48. [Google Scholar] [CrossRef]
- Diculescu, V.C.; Chiorcea-Paquim, A.-M.; Corduneanu, O.; Oliveira-Brett, A.M. Palladium nanoparticles and nanowires deposited electrochemically: AFM and electrochemical characterization. J. Solid State Electrochem. 2007, 11, 887–898. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods. Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2001; ISBN 978-0-471-04372-0. [Google Scholar]
- Stahl, S.S.; Alsters, P.L. Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives; Wiley-VCH: WeinHeim, Germany, 2016. [Google Scholar]
- Jutand, A. Contribution of Electrochemistry to Organometallic Catalysis. Chem. Rev. 2008, 108, 2300–2347. [Google Scholar] [CrossRef] [PubMed]
- Jutand, A.; Mosleh, A. Nickel- and Palladium-Catalyzed Homocoupling of Aryl Triflates. Scope, Limitation, and Mechanistic Aspects. J. Org. Chem. 1997, 62, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.S.; Pez, G.P.; Syvret, R.G. Electrophilic NF Fluorinating Agents. Chem. Rev. 1996, 96, 1737–1755. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.D.; Kitamura, T. Alternative, Easy Preparation of (Diacetoxyiodo)arenes from Iodoarenes Using Potassium Peroxodisulfate as the Oxidant. Synthesis 2005, 12, 1932–1934. [Google Scholar] [CrossRef]
- Varvoglis, A. Aryliodine(III) dicarboxylates. Chem. Soc. Rev. 1981, 10, 377–407. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Revathi, G.; Reddy, A.S.; Yadav, J.S. Regioselective ortho-acetoxylation/methoxylation of N-(2-benzoylphenyl)benzamides via substrate directed C–H activation. Tetrahedron Lett. 2011, 52, 5926–5929. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Narasimhulu, G.; Umadevi, N.; Yadav, J.S. Quinazolinone-Directed C–H Activation: A Novel Strategy for the Acetoxylation–Methoxylation of the Arenes. Synlett 2012, 23, 1364–1370. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Umadevi, N.; Narasimhulu, G.; Yadav, J.S. Oxidative C–H functionalization: A novel strategy for the acetoxylation/alkoxylation of arenes tethered to 3,4-dihydroisoquinolines. Tetrahedron Lett. 2012, 53, 6091–6094. [Google Scholar] [CrossRef]
- Wang, G.-W.; Yuan, T.-T. Palladium-Catalyzed Alkoxylation of N-Methoxybenzamides via Direct sp2 C−H Bond Activation. J. Org. Chem. 2010, 75, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Cui, X.; Yang, F.; Zhang, Q.; Zhu, Y.; Wu, Y. Palladium-catalyzed direct ortho C–O bond construction of azoxybenzenes with carboxylic acids and alcohols. Org. Chem. Front. 2015, 2, 951–955. [Google Scholar] [CrossRef]
- Piancatelli, G.; Leonelli, F. Oxidation of nerol to neral with iodobenzene diacetate and tempo. Org. Synth. 2006, 18–23. [Google Scholar] [CrossRef]
- Jonasson, C.; Horvath, A.; Backvall, J.E. Intramolecular Palladium(II)-Catalyzed 1,2-Addition to Allenes. J. Am. Chem. Soc. 2000, 122, 9600–9609. [Google Scholar] [CrossRef]
- Giri, R.; Lam, J.K.; Yu, J.-Q. Synthetic Applications of Pd(II)-Catalyzed C−H Carboxylation and Mechanistic Insights: Expedient Routes to Anthranilic Acids, Oxazolinones, and Quinazolinones. J. Am. Chem. Soc. 2010, 132, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Kuang, C. Palladium-Catalyzed Ortho-Alkoxylation of 2-Aryl-1,2,3-triazoles. J. Org. Chem. 2014, 79, 6105–6112. [Google Scholar] [CrossRef] [PubMed]
- Hull, K.L.; Lanni, E.L.; Sanford, M.S. Highly Regioselective Catalytic Oxidative Coupling Reactions: Synthetic and Mechanistic Investigations. J. Am. Chem. Soc. 2006, 128, 14047–14049. [Google Scholar] [CrossRef] [PubMed]
- Topczewski, J.J.; Sanford, M.S. Carbon–hydrogen (C–H) bond activation at PdIV: A Frontier in C–H functionalization catalysis. Chem. Sci. 2015, 6, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Han, J.; Chen, C.P.; Shi, D.Q.; Zhao, Y.S. Palladium-catalyzed oxygenation of C(sp2)–H and C(sp3)–H bonds under the assistance of oxalyl amide. RSC Adv. 2015, 5, 28430–28434. [Google Scholar] [CrossRef]
- Li, W.; Sun, P. Pd(OAc)2-Catalyzed Alkoxylation of Arylnitriles via sp2 C–H Bond Activation Using Cyano as the Directing Group. J. Org. Chem. 2012, 77, 8362–8366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Sun, P. Palladium-Catalyzed Direct C(sp2)–H Alkoxylation of 2-Aryloxypyridines Using 2-Pyridyloxyl as the Directing Group. J. Org. Chem. 2014, 79, 8457–8461. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kim, M.; Park, J.; Kim, M.; Kwak, J.H.; Jung, Y.H.; Oh, J.S.; Lee, Y.; Kim, I.S. Palladium-Catalyzed Direct Acylation of Ketoximes and Aldoximes from the Alcohol Oxidation Level through C–H Bond Activation. Eur. J. Org. Chem. 2013, 6656–6665. [Google Scholar] [CrossRef]
- Gandeepan, P.; Cheng, C.-H. Allylic Carbon–Carbon Double Bond Directed Pd-Catalyzed Oxidative ortho-Olefination of Arenes. J. Am. Chem. Soc. 2012, 134, 5738–5741. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Wang, D.-H.; Yu, J.-Q. Ligand-Accelerated C–H Activation Reactions: Evidence for a Switch of Mechanism. J. Am. Chem. Soc. 2010, 132, 14137–14151. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.D.; Sale, D.; Engle, K.M.; Yu, J.-Q.; Blackmond, D.G. Mechanistic Rationalization of Unusual Kinetics in Pd-Catalyzed C–H Olefination. J. Am. Chem. Soc. 2012, 134, 4600–4606. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-J.; Xu, H.; Kong, W.-J.; Shang, M.; Dai, H.-X.; Yu, J.-Q. Overcoming the limitations of directed C–H functionalizations of heterocycles. Nature 2014, 515, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.N.; Stahl, S.S. Overcoming the “Oxidant Problem”: Strategies to Use O2 as the Oxidant in Organometallic C–H Oxidation Reactions Catalyzed by Pd (and Cu). Acc. Chem. Res. 2012, 45, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Jiang, H. Palladium-Catalyzed Oxidation of Unsaturated Hydrocarbons Using Molecular Oxygen. Acc. Chem. Res. 2012, 45, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Gulzar, N.; Schweitzer-Chaput, B.; Klussmann, M. Oxidative coupling reactions for the functionalisation of C–H bonds using oxygen. Catal. Sci. Technol. 2014, 4, 2778–2796. [Google Scholar] [CrossRef]
- Baslé, O. Cross-Dehydrogenative-Coupling Reactions with Molecular Oxygen as the Terminal Oxidant. In From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling; Li, C.-J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 197–218. ISBN 978-1-84973-797-5. [Google Scholar]
- Wencel-Delord, J.; Colobert, F. A remarkable solvent effect of fluorinated alcohols on transition metal catalysed C–H functionalizations. Org. Chem. Front. 2016, 3, 394–400. [Google Scholar] [CrossRef]
- Dherbassy, Q.; Schwertz, G.; Chessé, M.; Hazra, C.K.; Wencel-Delord, J.; Colobert, F. 1,1,1,3,3,3-Hexafluoroisopropanol as a Remarkable Medium for Atroposelective Sulfoxide-Directed Fujiwara–Moritani Reaction with Acrylates and Styrenes. Chem. Eur. J. 2016, 22, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Wesch, T.; Leroux, F.R.; Colobert, F. Atropodiastereoselective C–H Olefination of Biphenyl p-Tolyl Sulfoxides with Acrylates. Adv. Synth. Catal. 2013, 355, 2139–2144. [Google Scholar] [CrossRef]
- Villuendas, P.; Serrano, E.; Urriolabeitia, E.P. Pd-catalysed ortho-alkoxylation of benzamides N-protected with an iminophosphorane functionality. New J. Chem. 2015, 39, 3077–3083. [Google Scholar] [CrossRef]
- Yu, M.; Wang, Z.; Tian, M.; Lu, C.; Li, S.; Du, H. Purinyl N3-Directed Palladium-Catalyzed C–H Alkoxylation of N9-Arylpurines: A Late-Stage Strategy to Synthesize N9-(ortho-Alkoxyl)arylpurines. J. Org. Chem. 2016, 81, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Dudkina, Y.B.; Khrizanforov, M.N.; Gryaznova, T.V.; Budnikova, Y.H. Prospects of Synthetic Electrochemistry in the Development of New Methods of Electrocatalytic Fluoroalkylation. J. Organomet. Chem. 2014, 751, 301–305. [Google Scholar] [CrossRef]
- Budnikova, Y.H. Metal complex catalysis in organic electrosynthesis. Russ. Chem. Rev. 2002, 71, 111–139. [Google Scholar] [CrossRef]
- Mikhaylov, D.Y.; Gryaznova, T.V.; Dudkina, Y.B.; Khrizanphorov, M.N.; Latypov, S.K.; Kataeva, O.N.; Vicic, D.A.; Sinyashin, O.G.; Budnikova, Y.G. Electrochemical nickel-induced fluoroalkylation: Synthetic, structural and mechanistic study. Dalton Trans. 2012, 41, 165–172. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Effect of the oxidant on the product yield in ligand-directed halogenation [26].
Scheme 1.
Effect of the oxidant on the product yield in ligand-directed halogenation [26].
Scheme 2.
Effect of the oxidant on the product yield in ligand-directed functionalizations. Reprinted with permission from American Chemical Society [27].
Scheme 2.
Effect of the oxidant on the product yield in ligand-directed functionalizations. Reprinted with permission from American Chemical Society [27].

Scheme 4.
Effect of the oxidant on the product yield in ligand-directed phosphonation [30].
Scheme 4.
Effect of the oxidant on the product yield in ligand-directed phosphonation [30].

Scheme 5.
Proposed catalytic cycle pathways.
Scheme 6.
Examples of Pd(II) palladacycles resulting from a directed C–H activation.

Figure 1.
Diagram of the redox potentials of selected oxidants and some Pd intermediates in catalytic reactions. Red points are erroneous literary data.
Figure 1.
Diagram of the redox potentials of selected oxidants and some Pd intermediates in catalytic reactions. Red points are erroneous literary data.
Scheme 7.
Directing groups used in the Pd-catalyzed dehydrogenative alkoxylation of arenes [31].
Scheme 7.
Directing groups used in the Pd-catalyzed dehydrogenative alkoxylation of arenes [31].
Scheme 8.
Intramolecular palladium(II)-catalyzed 1,2-addition to allenes. Reprinted with permission from American Chemical Society [80].
Scheme 8.
Intramolecular palladium(II)-catalyzed 1,2-addition to allenes. Reprinted with permission from American Chemical Society [80].
Scheme 9.
Directed Pd-catalyzed C–H functionalizations using air as the oxidant. Adapted with permission from Macmillan Publishers Ltd: Nature [92].
Scheme 9.
Directed Pd-catalyzed C–H functionalizations using air as the oxidant. Adapted with permission from Macmillan Publishers Ltd: Nature [92].
Scheme 10.
Electrochemical aromatic palladium-catalyzed C–H phosphorylation [14].
Scheme 10.
Electrochemical aromatic palladium-catalyzed C–H phosphorylation [14].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Suggested Categories for Strengths of Chemical Redox Agents (V vs. Fc). Reprinted with permission from American Chemical Society [45].
Table 1.
Suggested Categories for Strengths of Chemical Redox Agents (V vs. Fc). Reprinted with permission from American Chemical Society [45].
| Strength | Oxidants | Reductants |
|---|---|---|
| Very strong | >0.8 | <−2.5 |
| Strong | 0.8–0.2 | −1.5 to −2.5 |
| Mild | 0.2 to −0.5 | −0.5 to −1.5 |
| Weak | <−0.5 | >−0.5 |
Table 2.
Formal Potentials of Selected Oxidizing Agents (all potential values are recalculated to Fc+/Fc redox couple).
Table 2.
Formal Potentials of Selected Oxidizing Agents (all potential values are recalculated to Fc+/Fc redox couple).
| Oxidant | Solvent | E° (V vs. Fc+/Fc) | Reference |
|---|---|---|---|
| Oxone | H2O | 1.98 | [46] |
| K2S2O8 | H2O | 1.48 | [47] |
| H2O | 1.39 | [48] | |
| H2O + [H+] | 1.50 | [48] | |
| CPE, solid | 1.58 | a | |
| [N(C6H2Br3-2,4,6)3]+ | CH3CN | 1.36 | [45] |
| Ag2+ | H2O | 1.36 | [49] |
| CPE, solid | 1.16 | a | |
| Mn(OAc)3 | CPE, solid | 1.32 | a |
| [N(C6H3Br2-2,4)3]+ | CH3CN | 1.14 | [45] |
| H2O2 | H2O | 1.18 | [47] |
| [NO]+ | CH2Cl2 | 1.00 | [45] |
| [Ru(phen)3]3+ | CH3CN | 0.87 | [45] |
| [NO]+ | CH3CN | 0.87 | [45] |
| [N(C6H4Br-4)3]+ | CH2Cl2 | 0.70 | [45] |
| CH3CN | 0.67 | [45] | |
| Ag+ | CH2Cl2 | 0.65 | [45] |
| THF | 0.41 | [45] | |
| acetone | 0.18 | [45] | |
| AgOAc | CPE, solid | 1.16 | a |
| AgOAc | formamide | −0.198 | [50] |
| AgNO3 | CH3CN | −0.08 | [51] |
| AgBF4 | CH3CN | −0.04 | [52] |
| Ag2O | CPE, solid | −1.26 | a |
| [Fe(η-C5H4COMe)2]+ | CH2Cl2 | 0.49 | [45] |
| [CuTf2] | CH3CN | 0.40 | [45] |
| Cu(OAc)2·H2O | CPE, solid | 0.33 | a |
| [Ni(tfd)2] | CH2Cl2 | 0.33 | [45] |
| [PtCl6]2− | H2O | 0.31 | [45] |
| Cl2 | CH3CN | 0.18 | [45] |
| DDQ | CH3CN | 0.13 | [45] |
| 1,4-BQ | H2O | 0.16 | [53,54] |
| H2O | −0.535 | [55] | |
| H2O | −0.526 | b | |
| CH3CN | −0.73 | [56] | |
| CH3CN | −0.86 | [55] | |
| CH3CN | −0.88 | c | |
| CH3CN + [H+] | d | ||
| CH2Cl2 | −0.805 | [55] | |
| Acetone | −0.875 | [55] | |
| Br2 | CH3CN | 0.07 | [45] |
| (FcBF4) [FeCp2]+ | 0 | [45] | |
| I2 | CH3CN | −0.14 | [45] |
| 0.0 | [57] | ||
| I+ | 0.33 | [56] | |
| TCNE | CH3CN | −0.27 | [45] |
| TCNQ | CH3CN | −0.30 | [45] |
| [FeCp*2]+ | CH3CN | −0.59 | [45] |
| CH2Cl2 | −0.48 | [45] | |
| PhI(OAc)2 | CH3CN | −1.293 | [43,58] |
| CH3CN | 1.70 | c | |
| CPE, solid | 1.70 | a | |
| O2 | H2O | −0.78 | [59] |
| H2O | −0.81 | b | |
| DMSO | −1.16 | [59] | |
| DMF | −1.24 | [59] | |
| Py | −1.24 | [59] | |
| MeCN | −1.25 | [59] | |
| Quinoline | −1.25 | [59] | |
| EMIBF4 | −1.23 | [59] | |
| PMIBF4 | −1.20 | [59] | |
| BMIBF4 | −1.24 | [59] | |
| [bmim]HFP | −1.26 | [59] | |
| TEMPO | CH2Cl2 | 0.014 | [60,61] |
a Redox potentials were measured in CPE (carbon paste electrode) according to the reported procedure [62]; b Redox potentials were measured in H2O/0.1M NaBF4 at 20 °C, 100 mV·s−1, platinum working electrode; c Redox potentials were measured in CH3CN/0.1M Bu4NBF4 at 20 °C, 100 mV·s−1, platinum working electrode; d Redox potentials were measured in CH3CN/0.1M Bu4NBF4 at 20 °C, 100 mV·s−1, platinum working electrode, [DMF-H+]BF4 was used as a proton source [63].
Table 3.
Formal Potentials of Selected Palladium Intermediates (all potential values are recalculated to Fc+/Fc redox couple).
Table 3.
Formal Potentials of Selected Palladium Intermediates (all potential values are recalculated to Fc+/Fc redox couple).
| Palladium Complex | Solvent | E° (V vs. Fc+/Fc) | Reference |
|---|---|---|---|
| Pd2+/0 | DMF | −0.38 | [64] a |
| DMF | −0.02 | [65] b | |
| 0.1 M phosphate buffer | −0.64 | [66] | |
| −0.29 | [67] | ||
| H2O | −0.02 | [68] c | |
| PhPdIL | DMF | −0.88 reduction | [69] d |
| PdCl2(PPh3)2 | DMF | −1.29 reduction | [70] |
| Pd(OAc)2(TFP)2 | −1.34 reduction | [65] | |
| ArPdCl(PPh3) | DMF | >−2.03 reduction | [70] |
| [Pd(C^N)(OR)]2 | ACN | −2.03 to −2.37 reduction | [25] |
| 0.44–0.58 oxidation | [25] | ||
| DCM | −1.81 to −2.47 reduction | [25] | |
| 0.4 to 0.75 oxidation | [25] | ||
| [Pd(C^N)X]2 | ACN | −1.85 to −2.35 reduction | [25] |
| 0.63 to 1.00 oxidation | [25] | ||
| DCM | −2.29 to −2.43 reduction | [25] | |
| 0.73 to 0.74 oxidation | [25] | ||
| Pd(C^N)(CH3CN)ORF | ACN | −1.61 to −1.71 reduction | [25] |
| 1.19 to 1.32 oxidation | [25] |
a Pd0(PPh3)2(OAc)-HNEt3+; b Pd0(TFP)3, TFP = tri-2-furylphosphine; c in the presence of Br−; d L = AsPh3.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Budnikova, Y.H.; Dudkina, Y.B.; Khrizanforov, M.N. Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View. Inorganics 2017, 5, 70. https://doi.org/10.3390/inorganics5040070
AMA Style
Budnikova YH, Dudkina YB, Khrizanforov MN. Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View. Inorganics. 2017; 5(4):70. https://doi.org/10.3390/inorganics5040070
Chicago/Turabian StyleBudnikova, Yulia H., Yulia B. Dudkina, and Mikhail N. Khrizanforov. 2017. "Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View" Inorganics 5, no. 4: 70. https://doi.org/10.3390/inorganics5040070
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.