S–H Bond Activation in Hydrogen Sulfide by NHC-Stabilized Silyliumylidene Ions
by
 , and
, and
Amelie Porzelt
1, 
Julia I. Schweizer
2,
Ramona Baierl
1,
Philipp J. Altmann
1,
Max C. Holthausen
2 and
Shigeyoshi Inoue
1,* 1
WACKER-Institute of Silicon Chemistry and Catalysis Research Center, Technische Universität München, Lichtenbergstraße 4, 85748 Garching bei München, Germany
2
Institut für Anorganische Chemie, Goethe-Universität, Max-von-Laue-Straße 7, 60438 Frankfurt/Main, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2018, 6(2), 54; https://doi.org/10.3390/inorganics6020054
Submission received: 24 April 2018
/
Revised: 14 May 2018
/
Accepted: 17 May 2018
/
Published: 24 May 2018
(This article belongs to the Special Issue Coordination Chemistry of Silicon)
Abstract
:Reactivity studies of silyliumylidenes remain scarce with only a handful of publications to date. Herein we report the activation of S–H bonds in hydrogen sulfide by mTer-silyliumylidene ion A (mTer = 2,6-Mes2-C6H3, Mes = 2,4,6-Me3-C6H2) to yield an NHC-stabilized thiosilaaldehyde B. The results of NBO and QTAIM analyses suggest a zwitterionic formulation of the product B as the most appropriate. Detailed mechanistic investigations are performed at the M06-L/6-311+G(d,p)(SMD: acetonitrile/benzene)//M06-L/6-311+G(d,p) level of density functional theory. Several pathways for the formation of thiosilaaldehyde B are examined. The energetically preferred route commences with a stepwise addition of H2S to the nucleophilic silicon center. Subsequent NHC dissociation and proton abstraction yields the thiosilaaldehyde in a strongly exergonic reaction. Intermediacy of a chlorosilylene or a thiosilylene is kinetically precluded. With an overall activation barrier of 15 kcal/mol, the resulting mechanistic picture is fully in line with the experimental observation of an instantaneous reaction at sub-zero temperatures.
1. Introduction
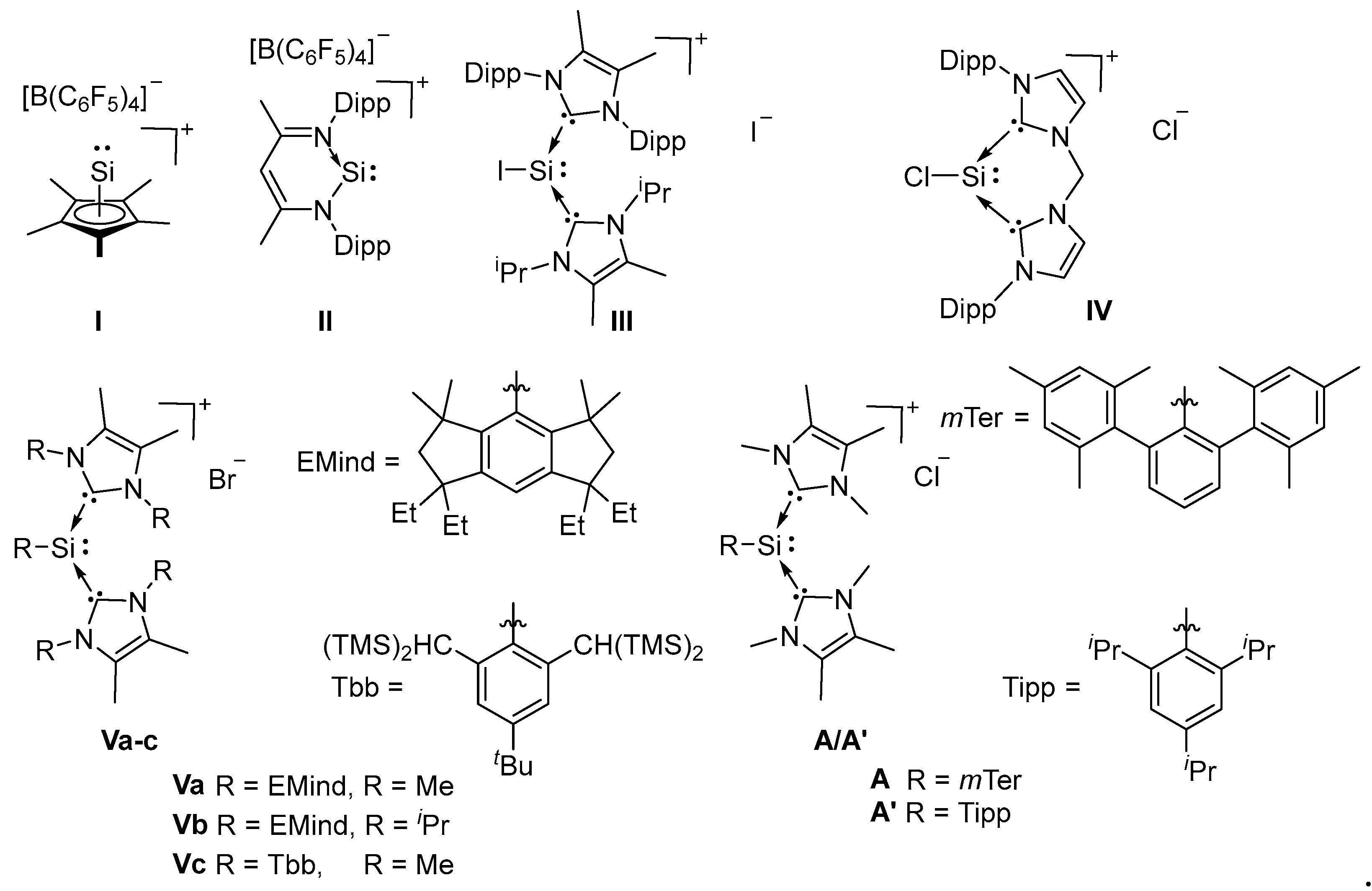
Low-valent main group chemistry is a rapidly developing field and the wealth of new structural motifs, which have been isolated in the past two decades, have increasingly gained interest in using these species for the activation of small molecules and, potentially, for catalysis (for representative reviews see [1,2,3,4,5,6,7]). Key to these developments have been the usage of suitable synthetic methodologies in combination with thermodynamic and kinetic stabilization by appropriately chosen ligands. In particular, for the heavier carbon analogue silicon, a plethora of studies reported new low-valent compounds in recent years [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25] and the chemistry of silylene base adducts has already been carefully developed [14,26,27,28,29,30,31,32,33,34,35,36]. Before these findings, silyliumylidene ions, cationic Si(II) species were found to be promising as similar versatile Lewis amphiphiles [37,38].
In 2004, Jutzi initiated the chemistry of silyliumylidene ions taking advantage of the stabilizing effects of the η5-coordinated pentamethyl-cyclopentadienyl ligand to prepare hypercoordinate silyliumylidene ion I (Figure 1) [39]. Driess and coworkers isolated the two coordinated silyliumylidene ion II, stabilized by aromatic 6π-electron delocalization as well as by intramolecular donation of the sterically encumbered β-diketiminate ligand [40]. N-heterocyclic carbenes (NHCs) represent another ligand class, widely used in modern main group chemistry. As NHCs are strong σ donors, their application in main group chemistry enabled the isolation of a large variety of low coordinate and low-valent main group compounds [41,42]. The first NHC-stabilized silyliumylidenes, III and IV were synthesized by Filippou and coworkers via a three-step protocol from SiI4 [28], and by Driess and coworkers through the reaction of Roesky’s NHC-stabilized dichlorosilylene with their bridged bis-carbene ligand [23].
Sasamori, Matsuo, Tokitoh and coworkers obtained the bulky aryl-substituted silyliumylidenes Va-c by treatment of the corresponding diaryldibromodisilene with the carbenes ImMe4 (1,3,4,5-Me4-imidazol-2-ylidene) or ImiPr2Me2 (1,3-iPr2-4,5-Me2-imidazol-2-ylidene) [43]. Around the same time our group reported the mTer- and Tipp-substituted silyliumylidenes A and A’ (mTer = 2,6-Mes2-C6H3, Mes = 2,4,6-Me3-C6H2, Tipp = 2,4,6-iPr3-C6H2) [44]. Different to all other known silyliumylidenes, A and A’ are accessible via an easy one-step synthesis: the addition of 3 equivalents of ImMe4 to the corresponding Si(IV) aryldichlorosilanes to give the silyliumylidenes via HCl removal by ImMe4 and nucleophilic substitution of chloride. The same approach was recently used by the group of Matsuo, obtaining Va via addition of ImMe4 to a solution of (EMind)dichlorosilane (EMind = 1,1,7,7-tetraethyl-3,3,5,5-tetramethyl-s-hydrindacen-4-yl) [45]. It should be noted that the corresponding iodosilyliumylidene stabilized by one NHC and one cAAC (cyclic (alkyl)aminocarbene) moiety have been reported by So and coworkers [46], as well as the parent silyliumylidene [HSi+] stabilized by two ImMe4 moieties [47].
Although a handful of silyliumylidenes have been reported in the last few years, reactivity studies are limited to the activation of elemental sulfur [24,48], the synthesis of a stable silylone from IV [23], and the catalytic application of I in the degradation of ethers [49]. For comparison, the neutral silylene derivative of silyliumylidene II (Figure 1) has been applied in the activation of several small molecules such as NH3, H2S, H2O, AsH3 and PH3 [50,51,52]. A theoretical assessment of the observed divergent reactivity was provided by Szilvási and coworkers, revealing a unique insertion step to form the 1,4 adducts, followed by varying pathways towards the products [53]. The NHC-stabilized arylchlorosilylene corresponding to A has already been published by Filippou and coworkers in 2010, as well as the chlorosilylene with sterically more demanding mTeriPr ligand (mTeriPr = 2,6-Tipp2C6H3) [54]. Subsequently, the mTeriPr-chlorosilylene was employed as the precursor for the preparation of a silylidyne complex Cp(CO)2Mo≡Si(mTeriPr) [55]. The conversion of those chlorosilylenes with lithium diphenylphosphine and LiPH2 to the corresponding phosphinosilylene and 1,2-dihydrophosphasilene reported by Driess and coworkers in 2015 [56,57] as well as the reaction towards diazoalkanes and azides presented by Filippou and coworkers [58] remain as the only reports regarding the reactivity of this species.
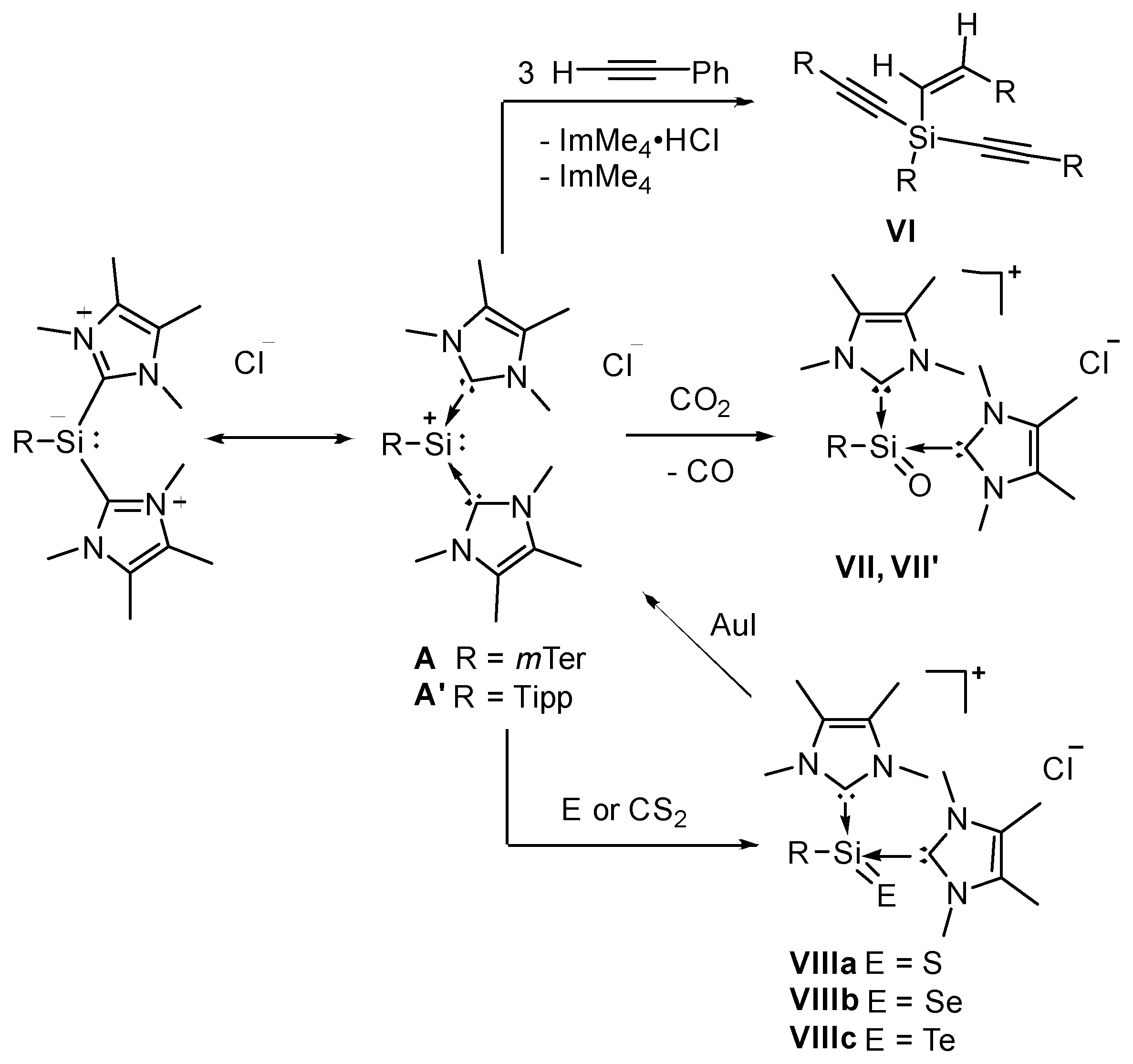
In any case, we consider silyliumylidene ions as promising candidates for small molecule activation as they possess two different reactive sites: an electron lone pair, and two empty p-orbitals at the silicon center. The electrophilicity of A and A’ is moderately mitigated by N-heterocyclic carbene coordination to the silicon center (Figure 2). Moreover, the zwitterionic representation of A/A’ (Figure 2) emphasizes the view of a silyl-anion, which appears useful further below.
We have already presented the silylene-like reactivity of A in the C–H activation of phenylacetylene to give the 1-alkenyl-1,1dialkynylsilane VI as the Z-isomer exclusively (Figure 2) [44]. We have also reported the application of A/A’ for the reduction of CO2 yielding the first NHC-stabilized silaacylium ions (VII/VII’) [59]. In addition, we have demonstrated the importance of kinetic stabilization by the steric bulk of the aryl ligands. In contrast to VII, the less shielded compound VII’ is kinetically labile even at sub-zero temperatures and could only be characterized spectroscopically. Very recently, we reported the synthesis of the corresponding heavier silaacylium ions VIIIa-c obtained from the reactions with CS2 or S8, Se, and Te, respectively [60]. Also, we could demonstrate the recovery of silyliumylidene A from VIIIa-c by the treatment with AuI as well as chalcogen transfer reactions.
For the last 30 years, neutral congeners of VIIIa-c, silanechalcogenones R2Si=E with E = S, Se, Te have been studied extensively [61]. In contrast, related compounds of type RHSi=E (E = S, Se, Te) are limited to the studies on intramolecularly stabilized silathioformamide by Driess and coworkers [50] and the NHC-stabilized heavier silaaldehydes by Müller and coworkers [62].
In this article, we further expand our series by the reaction of silyliumylidene A with hydrogen sulfide, yielding an NHC-stabilized thiosilaaldehyde, in a combined experimental and theoretical approach.
2. Results and Discussion
2.1. Reaction of Silyliumylidene A with H2S
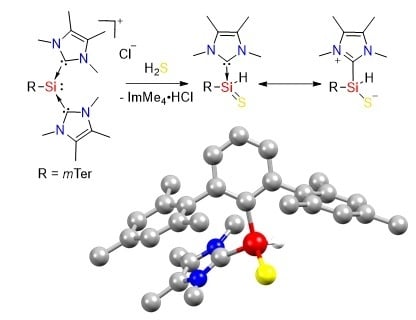
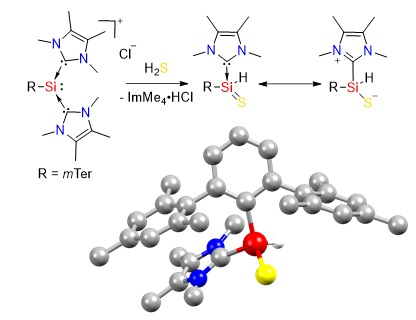
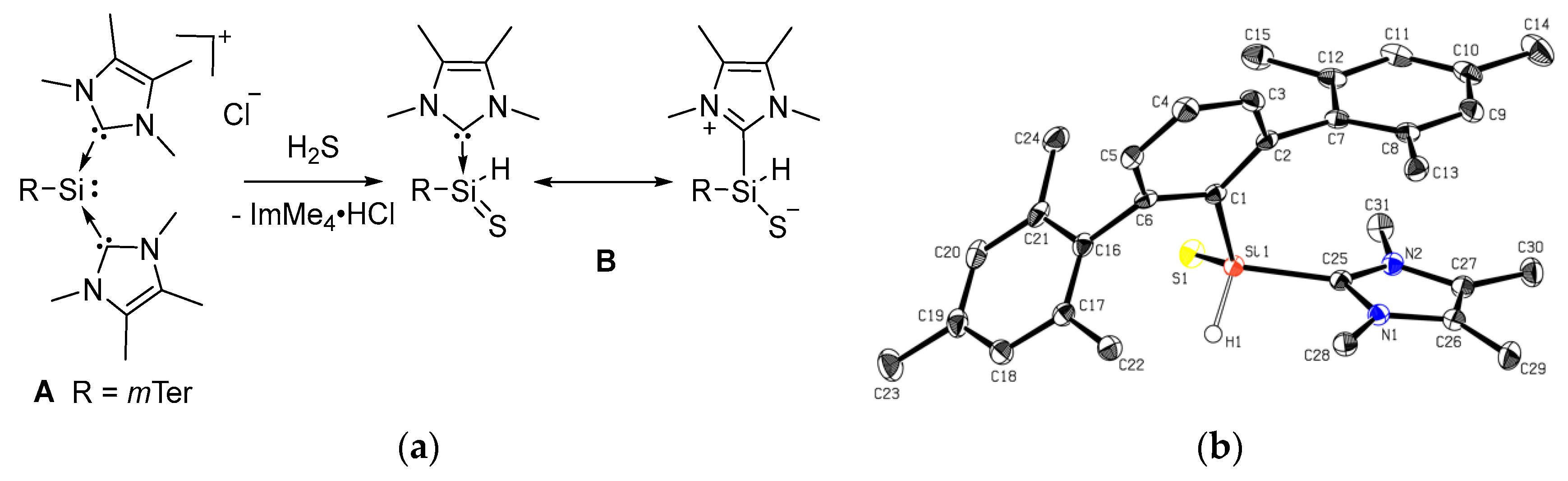
The reaction of NHC-stabilized silyliumylidene A, dissolved in acetonitrile, with 1 M H2S solution in THF proceeded rapidly even at −20 °C, and the orange color of the starting material vanished within seconds. 1H NMR spectroscopy indicated the formation of imidazolium salt, and one remaining ImMe4 coordinated to the silicon center (3.46 ppm, 6H, NCH3, ImMe4). The splitting of the signals for the ortho-methyl groups and the benzylic protons in the mesityl moieties indicated reduced symmetry in the product. A new signal at 5.35 ppm with 29Si satellites (1JSiH = 209.0 Hz) was assigned to the Si bound hydrogen atom by 1H/29Si-HMBC-NMR spectroscopy. The 29Si NMR signal was shifted down-field from −69.03 ppm in the starting material to −39.59 ppm. Single crystals were obtained after storing the reaction solution at 8 °C overnight. X-ray crystallography confirmed the formation of the NHC-stabilized thiosilaaldehyde B (Figure 3). In earlier work by Müller and coworkers, they obtained the analogous species with the bulkier mTeriPr ligand by the reaction of the NHC-stabilized hydridosilylene with elemental sulfur [62]. BmTeriPr features the same 1JSiH coupling constant (209 Hz), which is smaller compared to the one of silathioformamide (255 Hz) reported by Driess and coworkers. [50]. Compound B is stable under the inert atmosphere and shows good solubility in acetonitrile, however, in contrast to thiosilaaldehyde BmTeriPr, only a limited solubility in aromatic solvents is observed. Removal of the imidazolium byproduct from B was achieved by fractional crystallization from acetonitrile to obtain B as an analytically pure crystalline solid in 54% yield. Repetition of the experiments using A’ featured the same fast decoloring, but the attempts to isolate the corresponding B’ were not successful, most likely due to the kinetic lability of the formed product.
The tetracoordinate NHC-stabilized thiosilaaldehyde B (Figure 3) exhibits a distorted tetrahedral coordination around the silicon atom with a π-stacking of the NHC and a mesityl group of the terphenyl ligand. The Si–S bond length was 2.0227(9) Å, which is slightly longer than in compound VIIa (2.013(1) Å and 2.018(1) Å for the two independent molecules) [60], as well as the intramolecular, stabilized thiosilaaldehyde by Driess and coworkers (1.9854(9) Å) [50]. It is closer to the covalent double bond radii of sulphur and silicon (2.01 Å) than to the sum of single bond radii (2.19 Å) [63]. The Si–CNHC bond (1.934(2) Å) and the Si–CmTer bond (1.902(2) Å) are shortened compared to A (1.9481(19) Å and 1.9665(19) Å/1.9355(19) Å). The structural parameters are close to those of BmTeriPr [62].
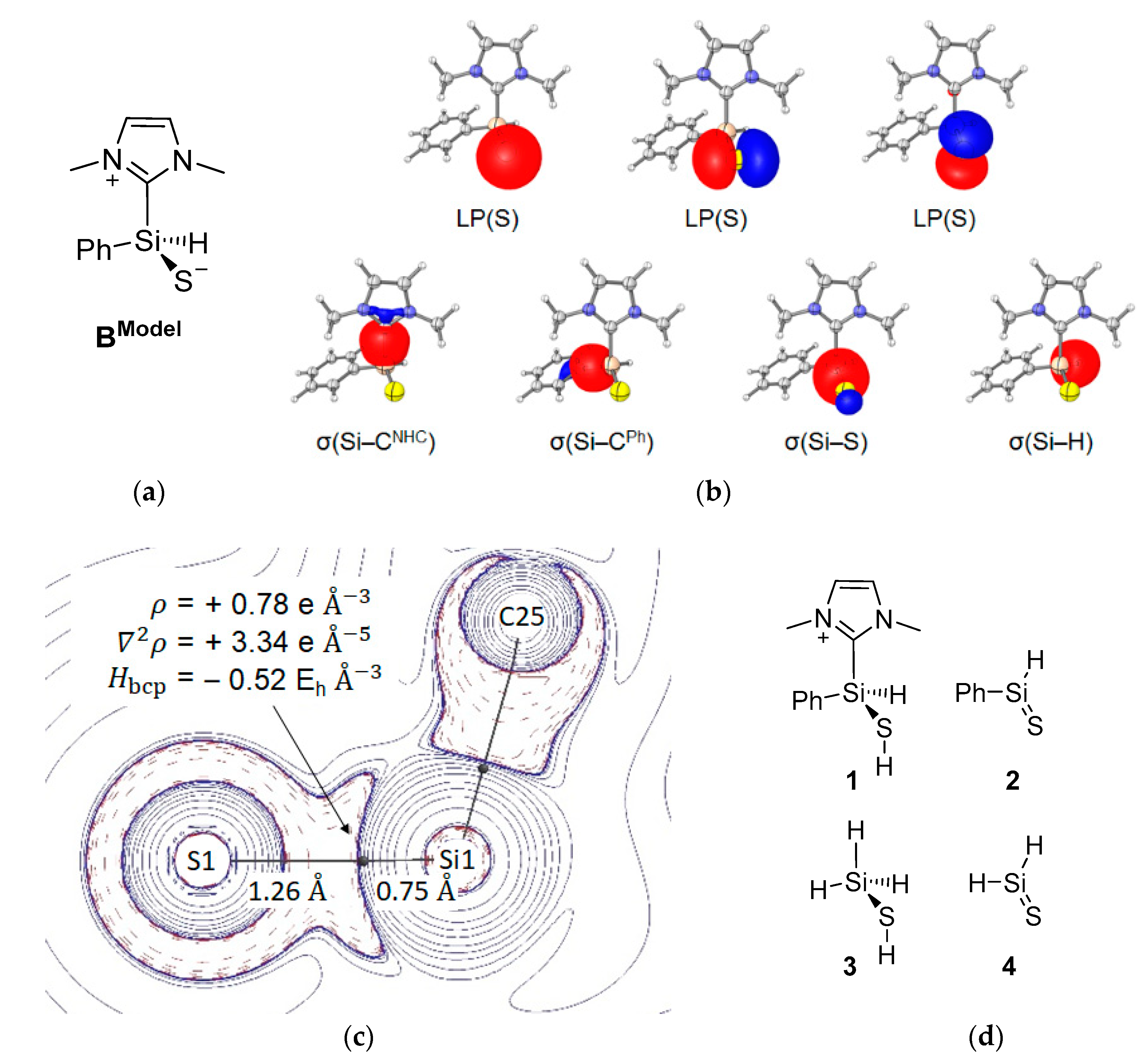
Further insight into the nature of the Si–S bond in B is provided by density functional theory (DFT) computations at the M06-L/6-311++G(2d,2p)//M06-L/6-31+G(d,p) level. For all bonding analyses, we chose a truncated molecular model replacing the mTer ligand by phenyl and ImMe4 by ImMe2H2 (1,3-Me2-imidazol-2-ylidene). The computed structural parameters of BModel agreed well with the experimental molecular structure obtained from X-ray diffraction (see Table S3). Natural bond orbital (NBO) analysis reveal natural localized molecular orbitals (NLMOs, Figure 4b) corresponding to Si–H, Si–CNHC, Si–CmTer and Si–S single bonds as well as three NLMOs representing the electron lone pairs at sulfur. This zwitterionic representation of BModel is also the dominant Lewis resonance structure within the natural resonance theory (NRT) formalism (Figure 4a). In line with analysis by Müller and co-workers [62], the short Si–S bond and the Wiberg bond index of 1.38 can be rationalized by negative hyperconjugation [64,65] of the sulphur lone pairs into the σ*(Si–R) orbitals: the occupancy of the LP(S) NBOs is significantly decreased (1.81 e, 1.76 e), while the NBOs for the anti-bonding σ*-orbitals are partly populated (Si–H: 0.11 e, Si–CNHC: 0.14 e, Si–CmTer: 0.12 e). Topological analysis of the computed electron density, by means of Bader’s quantum theory of atoms in molecules (QTAIM) [66,67], characterizes the Si–S bond as a strongly polar covalent interaction as indicated by a marked shift of the bond-critical point (bcp) towards the more electropositive Si site, a relatively large electron density , a positive Laplacian ∇2ρbcp as well as a negative total energy density Hbcp at the bcp (Figure 4c) [68,69].
For a classification of further characteristics at the Si–S bcp, we analyzed related species containing a Si–S single bond (1 and parent compound 3) or a Si=S double bond (2 and parent compound 4, Figure 4d). The results of the corresponding QTAIM analyses are summarized in Table 1. All molecular graphs display a characteristic shift of the Si–S bcp towards the more electropositive silicon site. In line with expectation, the values of ρbcp and ∇2ρbcp are higher for double bonded compounds 2 and 4 compared to single bonded compounds 1 and 3. The electron density and its Laplacian for the Si–S bond in BModel are located in between, suggesting the presence of a partial double bond [70,71]. Also, the delocalization gradient δSi,S, i.e., the number of electron pairs shared between two atoms, lies between the values for the single and double bonded species. However, the bond ellipticity εbcp, a measure of ρbcp anisotropy indicating the π character of a bond, is rather small with a value of 0.01. Nevertheless, the Si–S bond shortening, as well as the decrease in ellipticity compared to εbcp for the Si–S single bonds in 1 and 3, agree with the presence of negative hyperconjugation in BModel [72], which was already observed within the NBO framework.
These results support a zwitterionic nature of B with the partial double bond character of the Si–S bond due to negative hyperconjugation. This enhanced interaction between silicon and sulphur is reflected experimentally by the short bond length found in single crystal XRD analysis.
2.2. Mechanistic Investigations on the Reaction of Silyliumylidene A with H2S
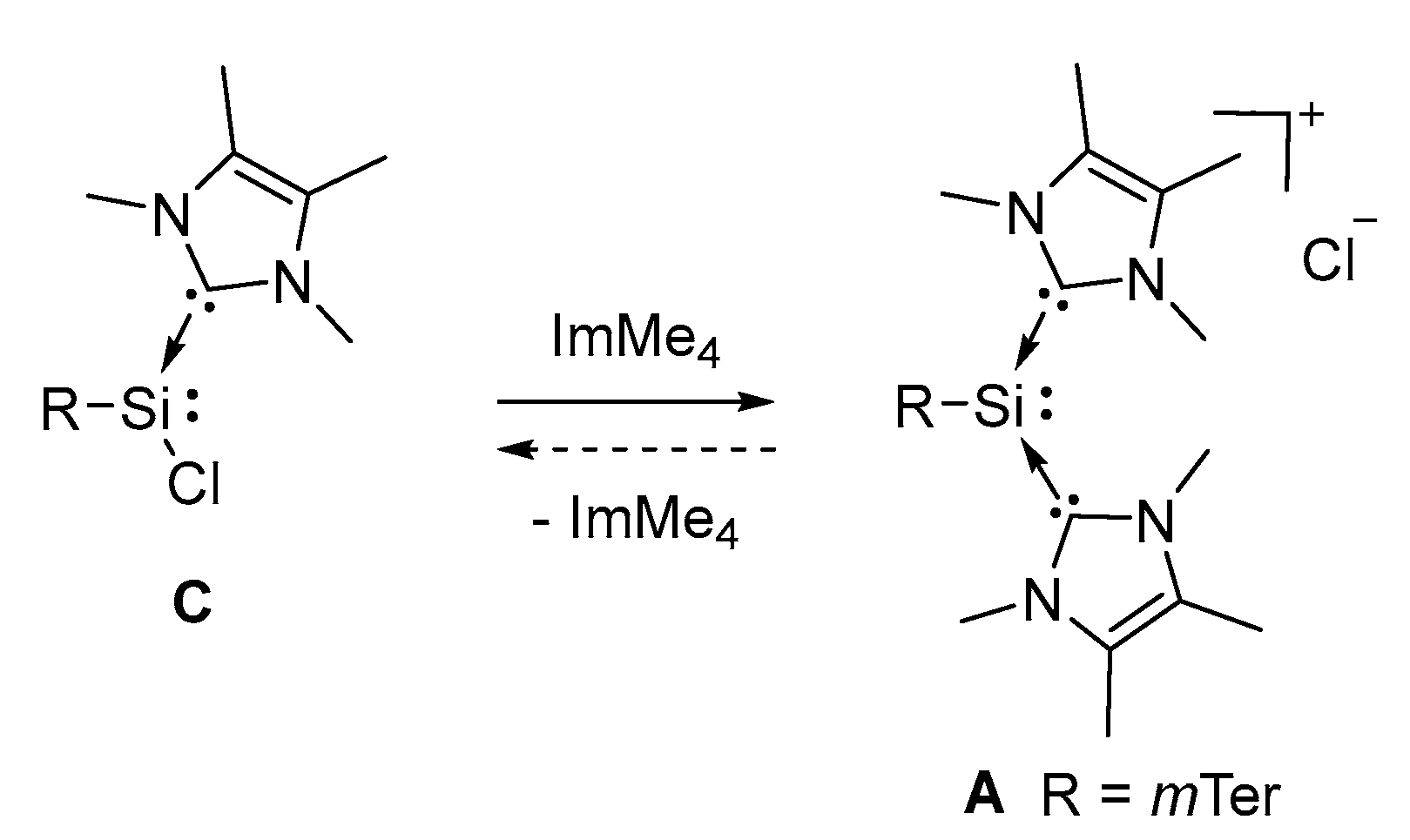
The reaction of H2S and A proceeds instantaneously, preventing the NMR detection of intermediates to gain further information on this conversion. To rule out the formation of chlorosilylene C as reaction intermediate, we investigated the interconversion of C and A in a combined experimental and theoretical approach (Figure 5 and Figure 6).
Addition of one further equivalent of ImMe4 to a solution of chlorosilylene C in benzene at RT lead to no change in color or 1H-NMR. Heating of the reaction solution to 40 °C resulted in slow darkening of the solution to orange. After several hours the formation of orange crystals in the lower part of the Schlenk tube was observed, yielding silyliumylidene A in 58% isolated yield after a prolonged reaction time of 18 h. The reverse reaction could not be demonstrated experimentally due to the limited stability of chlorosilylene C and ImMe4 in MeCN, the sole solvent in which A is soluble and stable.
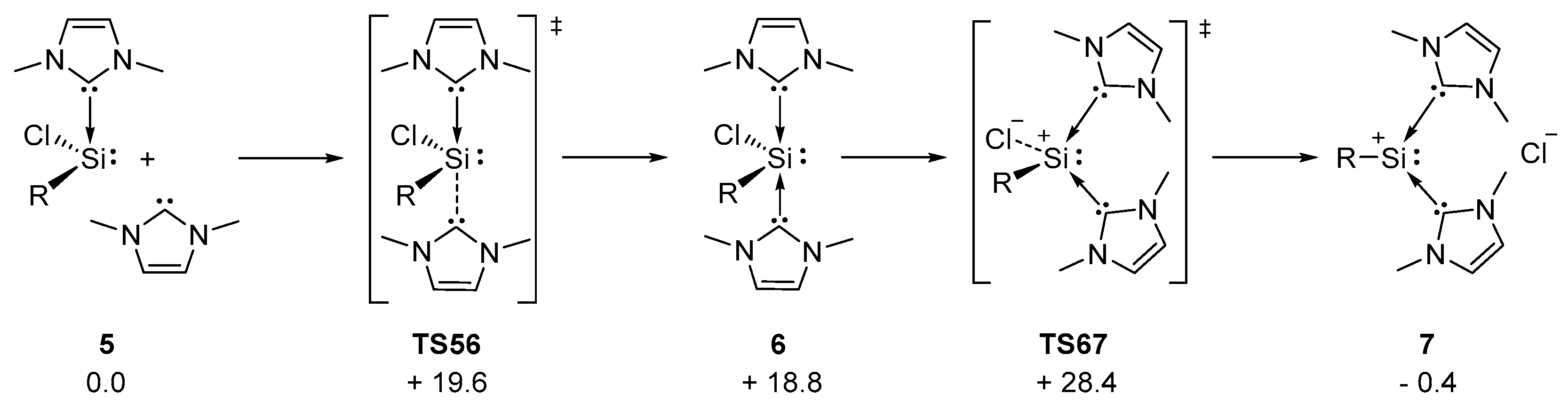
DFT calculations on the interconversion of C and A were performed at the M06-L/6-311+G(d,p)(SMD = benzene)//M06-L/6-31+G(d,p) level of theory with a marginally reduced molecular model (mTer reduced to 2,6-diphenyl-C6H3 and ImMe4 replaced by ImMe2H2). Silyliumylidene 7 is only slightly lower in energy than chlorosilylene 5 (Figure 6). This is in accordance with a report on the related silyliumylidene ions Vb and Vc, for which a substituent-dependent shift in relative stabilities was observed [43]. We further investigated the potential energy surface of the interconversion: NHC addition to chlorosilylene 5 via TS56 (Δ‡G = 19.6 kcal/mol) leads to tetracoordinate 6, which is located as an unstable intermediate (18.8 kcal/mol). En route to silyliumylidene ion 7, a substantial effective barrier of 28.4 kcal/mol for the chloride dissociation in TS67 is found. This is in line with interconversion of chlorosilylene C to silyliumylidene A taking place at elevated temperatures but clearly incompatible with the H2S activation that takes place at −20 °C. Based on our combined experimental and theoretical studies, we thus conclude that formation of thiosilaaldehyde B does not involve intermediacy of chlorosilylene C.
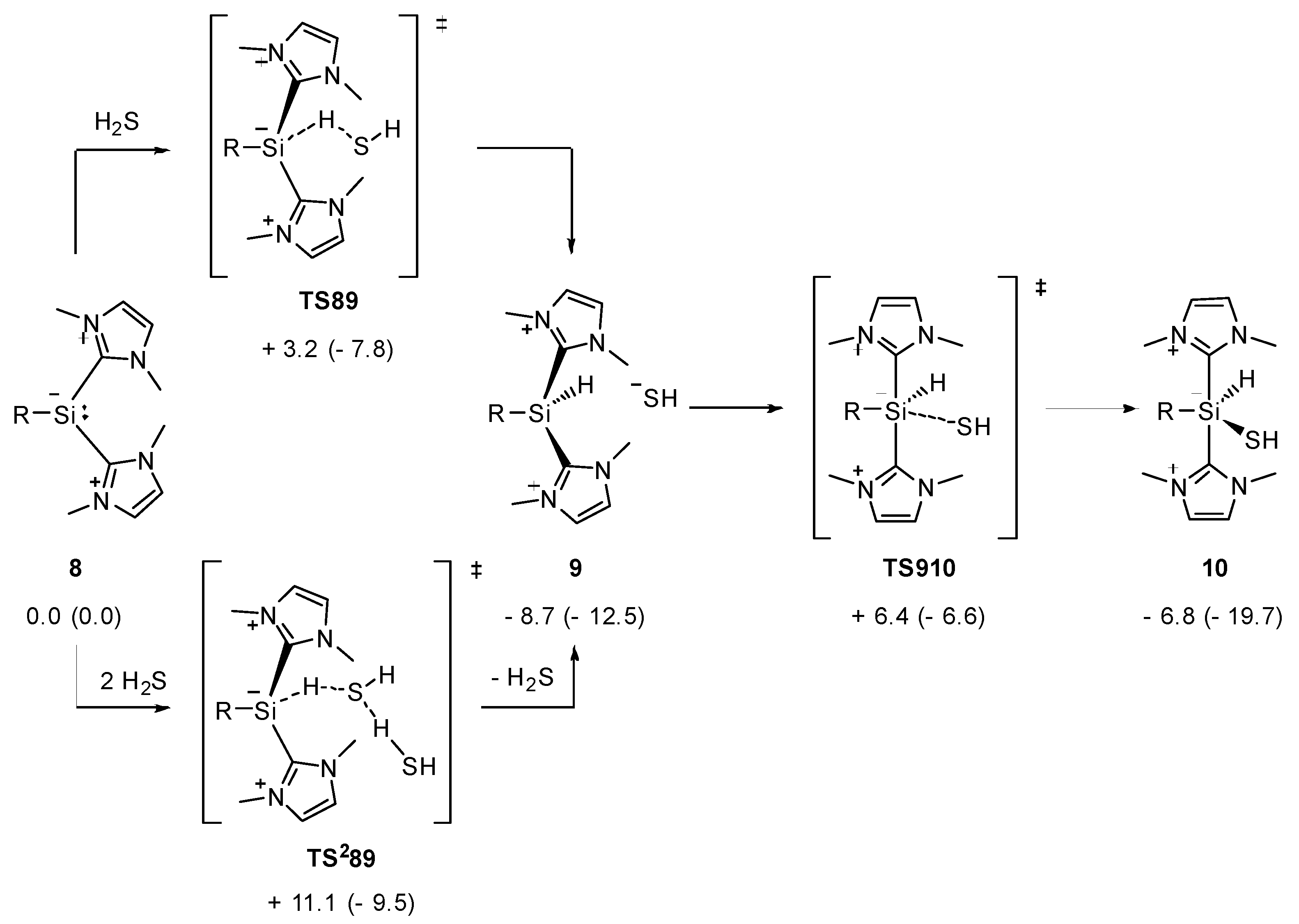
The reaction of 8 with hydrogen sulfide commences with a proton transfer via TS89 to give intermediate 9 as an ion pair in an exergonic step (Figure 7) (M06-L/6-311+G(d,p)(SMD:acetonitrile)//M06-L/6-31+G(d,p) level of theory). Subsequently, the SH moiety adds to the silicon center to yield 10, a pentacoordinate intermediate, with an effective barrier of 15.1 kcal/mol. The assistance of a second H2S molecule in TS289 is entropically disfavored compared to TS89. The NHC-stabilized silyliumylidene 8 acts as a nucleophile in the reaction with hydrogen sulfide, as its electrophilicity is saturated by the presence of two coordinating NHCs. Accordingly, the zwitterionic representation of 8 in the following best emphasizes its nucleophilic character.
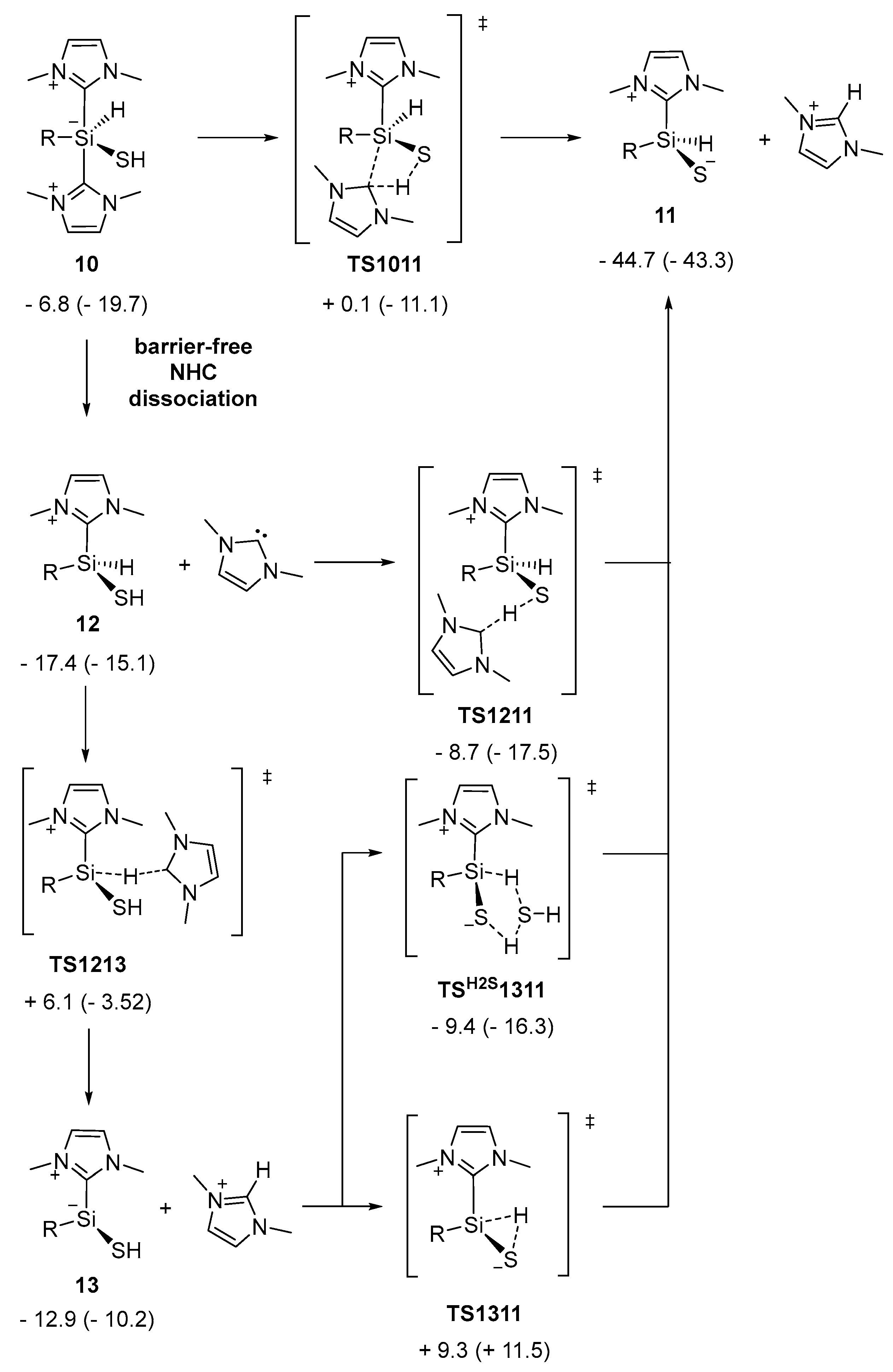
Starting from 10, different pathways to NHC-stabilized thiosilaaldehyde 11 were examined (Figure 8). The concerted NHC dissociation and S–H proton abstraction in transition state TS1011 is connected with a barrier of 6.9 kcal/mol and directly yield 11 in a strongly exergonic reaction. Alternatively, dissociation of one NHC ligand from 10 to 12 was thermodynamically favored and proceeded barrierlessly, as indicated by relaxed potential energy surface scans along the Si–CNHC bonds (see Figures S7 and S8). The NHC liberated subsequently abstracts, with clear kinetic preference, the S–H proton in 12 (TS1211: Δ‡G = 8.7 kcal/mol). The alternative route for Si–H hydride abstraction via TS1213 is connected with a substantially higher activation barrier (Δ‡G = 23.5 kcal/mol), which renders this path to 11 kinetically irrelevant. The atomic charges obtained by natural population analysis of 10 (HSi: −0.17 e, HS: 0.18 e) supported the view that the increased activation barrier goes back to the additional charge transfer occuring in the course of the hydride abstraction. Overall, the addition of H2S to silyliumylidene 8 via TS910 is rate-limiting with an effective activation barrier of 15 kcal/mol. Subsequent isomerization to thiosilaaldehyde 11 is initiated by barrierless NHC dissociation and accomplished by abstraction of the S–H proton by the free carbene. Concerted proton abstraction and NHC dissociation (TS1011) is kinetically disfavored.
In conclusion, we have presented the activation of hydrogen sulfide by silyliumylidene ion A to give the thiosilaaldehyde B. Its nucleophilicity is best rationalized by assuming a zwitterionic character. Combined experimental and theoretical investigations reveal that the thiosilaaldehyde formation does not involve intermediacy of chlorosilylene C or thiosilylene 13. The NHC-stabilized silyliumylidene A adds H2S in a stepwise reaction sequence followed by NHC dissociation. Proton abstraction by the latter yields thiosilaaldehyde in a strongly exergonic reaction. With an overall activation barrier of 15 kcal/mol, the resulting mechanistic picture is fully in line with the experimental observation of an instantaneous reaction at sub-zero temperatures.
3. Materials and Methods
3.1. General Methods and Instruments
All manipulations were carried out under the argon atmosphere using standard Schlenk or glovebox techniques. Glassware was heat-dried under vacuum prior to use. Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich (Steinheim, Germany) and used as received. Benzene, n-hexane, and acetonitrile were refluxed over standard drying agents (benzene/hexane over sodium and benzophenone, acetonitrile over CaH2), distilled and deoxygenated prior to use. Deuterated acetonitrile (CD3CN) and benzene (C6D6) were dried by short refluxing over CaH2 (CD3CN) and/or storage over activated 3 Å molecular sieves (CD3CN and C6D6). All NMR samples were prepared under argon in J. Young PTFE tubes. mTerSiHCl2, chlorosilylene C and ImMe4 were synthesized according to procedures described in literature [54,73,74]. NMR spectra were recorded on Bruker AV-400 spectrometer (Rheinstetten, Germany) at ambient temperature (300 K). 1H, 13C, and 29Si NMR spectroscopic chemical shifts δ are reported in ppm relative to tetramethylsilane. δ(1H) and δ(13C) were referenced internally to the relevant residual solvent resonances. δ(29Si) was referenced to the signal of tetramethylsilane (TMS) (δ = 0 ppm) as the external standard. Elemental analyses (EA) were conducted with a EURO EA (HEKA tech, Wegberg, Germany) instrument equipped with a CHNS combustion analyzer. Details on XRD data are given in the supplementary materials.
3.2. Improved and Upscaled Synthesis of Silyliumylidene A
mTerSiHCl2 (1.00 g, 2.42 mmol, 1.0 eq.) and ImMe4 (901 mg, 7.26 mmol, 3.0 eq.) were each dissolved in 17.5 mL of dry benzene in two different flasks. The ImMe4 solution was added very slowly to the silane solution to generate a layer of immediately formed imidazolium hydrogenchloride salt separating both solutions without stirring. After complete addition/overlaying stirring was switched on, both solutions mixed thoroughly as fast as possible and the precipitated imidazolium hydrogenchloride salt was allowed to settle down for a short time. The supernatant dark red solution was filtered into a new flask, the residue was washed with 2 mL of dry benzene and the combined solutions were allowed to stand overnight for complete crystallization of the orange silyliumylidene. The yellow supernatant was separated from the orange crystalline solid, washed four times with 5 mL dry hexane to remove residues of white imidazolium hydrogenchloride salt and dried in vacuo. An orange crystalline product was obtained in 66% yield (1.00 g). Analytical data are the same as previously published [44].
3.3. Synthesis of Thiosilaaldehyde B
Silyliumylidene A (150 mg, 221 µmol, 1.0 eq.) was dissolved in MeCN (3.0 mL), cooled to −20 °C and an excess of H2S solution (approx. 0.8 M) in THF was added. The solution quickly turned from orange to yellow to blue-green while a white precipitate was formed. The solution was allowed to warm to RT upon which the precipitate redissolved. The solution was concentrated to halve the volume and stored in the fridge for crystallization overnight. The supernatant was filtered off and the white residue was washed with MeCN (0.5 mL) at 0 °C. The solid was dried in vacuo. B was obtained as a white crystalline solid in 54% yield (59.0 mg, 118 μmol). Storage of a crude reaction mixture at 8 °C yield single crystals of B suitable for X-ray diffraction analysis.
1H-NMR (400 MHz, 298 K, CD3CN) δ 7.45 (t, J = 7.6 Hz, 1H, C4H, C6H3), 6.98 (s, 2H, C3/5H, Mes), 6.91 (d, J = 7.6 Hz, 2H, C3,5H, C6H3), 6.76 (s, 2H, C3/5H, Mes), 5.36 (s, 1H, SiH, 1JSiH = 209.0 Hz), 3.46 (s, 6H, NCH3, ImMe4), 2.36 (s, 6H, C1/3/5CH3, Mes), 2.31 (s, 6H, C1/3/5CH3, Mes), 1.98 (s, 6H, CCH3, ImMe4), 1.93 (s, 6H, C1/3/5CH3, Mes); 13C-NMR (126 MHz, 298 K, CD3CN) δ 149.84, 148.75, 141.06, 138.76, 137.45, 137.42, 137.05, 130.08, 129.63, 128.45, 34.12 (NCH3, ImMe4), 22.21 (C2/4/6CH3, Mes), 21.76 (C2/4/6CH3, Mes), 21.23 (C2/4/6CH3, Mes), 8.68 (CCH3, ImMe4); 29Si-INEPT-NMR (99 MHz, 298 K, CD3CN) δ −39.58; EA experimental (calculated): C 74.27 (74.65), H 7.66 (7.68), N 5.61 (5.62), S 6.25 (6.43) %.
3.4. Conversion of Chlorosilylene C to Silyliumylidene A
Chlorosilylene C (40.0 mg, 80 µmol, 1.0 eq.) and ImMe4 (10.1 mg, 80 µmol, 1.0 eq.) were dissolved in 1.5 mL of dry benzene in a Schlenk tube. The tube was placed in an oil bath and heated to 40 °C for 18 h. After this time, a large amount of orange crystals was formed with some white precipitate (ImMe4·HCl) and a slightly yellow supernatant, which was removed via the syringe. The orange crystals were washed two times with 2 mL benzene and three times with 2 mL hexane to remove the white precipitate. The crystalline material was dried in vacuo to give A in 58% yield (29.0 mg). Analytical data are the same as previously published [44].
3.5. DFT Calculations
Geometry optimizations and harmonic frequency calculations have been performed using Gaussian09 [75] employing the M06-L/6-31+G(d,p) [76,77,78] level of density functional theory. The SMD polarizable continuum model was used to account for solvent effects of acetonitrile and benzene [79]. The ‘ultrafine’ grid option was used for numerical integrations [80]. Stationary points were characterized as minima or transition states by analysis of computed Hessians. The connectivity between minima and transition states was validated by IRC calculations [81] or displacing the geometry along the transition mode, followed by unconstrained optimization. For improved energies, single point calculations were conducted at the SMD-M06-L/6-311+G(d,p) [82,83] level of theory; wave functions used for bonding analysis were obtained at the M06-L/6-311++G(2d,2p) [82,83] level. Natural bond orbital (NBO) and natural resonance theory (NRT) analyses were performed using the NBO 6.0 program [84], interfaced with Gaussian09 [85,86]. The AIMALL [87] program was used for QTAIM analyses [66,67]. Unscaled zero-point vibrational energies, as well as thermal and entropic correction terms, were obtained from Hessians computed at the M06-L/6-31+G(d,p) level using standard procedures. Pictures of molecular structures were generated with the ChemCraft [88] program.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/6/2/54/s1, Figures S1–S4: NMR spectra of B, Figures S5 and S6, Tables S1 and S2: Crystallographic details of B (CCDC 1839062), Table S3: Comparison of calc. and exp. Structures, Tables S4-S7: Details of NBO and QTAIM analyses, Figures S7–S8: Relaxed potential energy scans along the Si–CNHC bonds in 10, Table S9: Energies of all calculated compounds, Tables S10–S35: Cartesian coordinates of all calculated compounds.
Author Contributions
A.P. and R.B. performed the experiments. A.P. and J.I.S. conducted the calculations. P.J.A. measured and solved the SC-XRD data. M.C.H. and S.I. supervised the complete project. All authors discussed the results and commented on the manuscript.
Acknowledgments
We are exceptionally grateful to the WACKER Chemie AG and European Research Council (SILION 63794) for financial support. We thank Samuel Powley, Technische Universität München, for helpful discussion and proofreading the manuscript and Alexander Pöthig for advice pertaining to crystallography. Quantum-chemical calculations were performed at the Center for Scientific Computing (CSC) Frankfurt on the FUCHS and the LOEWE-CSC high-performance compute clusters and at the Leibniz Supercomputing Center of the Bavarian Academy of Science and Humanities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bayne, J.M.; Stephan, D.W. Phosphorus Lewis acids: Emerging reactivity and applications in catalysis. Chem. Soc. Rev. 2016, 45, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Hadlington, T.J.; Driess, M.; Jones, C. Low-valent group 14 element hydride chemistry: Towards catalysis. Chem. Soc. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.K.; Roesky, H.W. Group 14 Hydrides with Low Valent Elements for Activation of Small Molecules. Acc. Chem. Res. 2012, 45, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Power, P.P. Main-group elements as transition metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.M.D.; Rivard, E. Pushing Chemical Boundaries with N-Heterocyclic Olefins (NHOs): From Catalysis to Main Group Element Chemistry. Acc. Chem. Res. 2017, 50, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Saha, S.; Sen, S.S. Compounds with Low-Valent p-Block Elements for Small Molecule Activation and Catalysis. ChemCatChem 2016, 8, 486–501. [Google Scholar] [CrossRef]
- Yao, S.; Xiong, Y.; Driess, M. Zwitterionic and Donor-Stabilized N-Heterocyclic Silylenes (NHSis) for Metal-Free Activation of Small Molecules. Organometallics 2011, 30, 1748–1767. [Google Scholar] [CrossRef]
- Alvarado-Beltran, I.; Rosas-Sanchez, A.; Baceiredo, A.; Saffon-Merceron, N.; Branchadell, V.; Kato, T. A Fairly Stable Crystalline Silanone. Angew. Chem. Int. Ed. 2017, 56, 10481–10485. [Google Scholar] [CrossRef] [PubMed]
- Arz, M.I.; Geiß, D.; Straßmann, M.; Schnakenburg, G.; Filippou, A.C. Silicon(i) chemistry: The NHC-stabilised silicon(i) halides Si2X2(Idipp)2 (X = Br, I) and the disilicon(i)-iodido cation [Si2(I)(Idipp)2]+. Chem. Sci. 2015, 6, 6515–6524. [Google Scholar] [CrossRef]
- Arz, M.I.; Schnakenburg, G.; Meyer, A.; Schiemann, O.; Filippou, A.C. The Si2H radical supported by two N-heterocyclic carbenes. Chem. Sci. 2016, 7, 4973–4979. [Google Scholar] [CrossRef]
- Boehme, C.; Frenking, G. Electronic Structure of Stable Carbenes, Silylenes, and Germylenes. J. Am. Chem. Soc. 1996, 118, 2039–2046. [Google Scholar] [CrossRef]
- Burchert, A.; Müller, R.; Yao, S.; Schattenberg, C.; Xiong, Y.; Kaupp, M.; Driess, M. Taming Silicon Congeners of CO and CO2: Synthesis of Monomeric SiII and SiIV Chalcogenide Complexes. Angew. Chem. Int. Ed. 2017, 56, 6298–6301. [Google Scholar] [CrossRef] [PubMed]
- Denk, M.; Lennon, R.; Hayashi, R.; West, R.; Belyakov, A.V.; Verne, H.P.; Haaland, A.; Wagner, M.; Metzler, N. Synthesis and Structure of a Stable Silylene. J. Am. Chem. Soc. 1994, 116, 2691–2692. [Google Scholar] [CrossRef]
- Ghana, P.; Arz, M.I.; Das, U.; Schnakenburg, G.; Filippou, A.C. Si=Si Double Bonds: Synthesis of an NHC-Stabilized Disilavinylidene. Angew. Chem. Int. Ed. 2015, 54, 9980–9985. [Google Scholar] [CrossRef] [PubMed]
- Kira, M.; Ishida, S.; Iwamoto, T.; Kabuto, C. The First Isolable Dialkylsilylene. J. Am. Chem. Soc. 1999, 121, 9722–9723. [Google Scholar] [CrossRef]
- Mondal, K.C.; Roesky, H.W.; Schwarzer, M.C.; Frenking, G.; Tkach, I.; Wolf, H.; Kratzert, D.; Herbst-Irmer, R.; Niepotter, B.; Stalke, D. Conversion of a Singlet Silylene to a stable Biradical. Angew. Chem. Int. Ed. 2013, 52, 1801–1805. [Google Scholar] [CrossRef] [PubMed]
- Mondal, K.C.; Roy, S.; Dittrich, B.; Andrada, D.M.; Frenking, G.; Roesky, H.W. A Triatomic Silicon(0) Cluster Stabilized by a Cyclic Alkyl(amino) Carbene. Angew. Chem. Int. Ed. 2016, 55, 3158–3161. [Google Scholar] [CrossRef] [PubMed]
- Nieder, D.; Yildiz, C.B.; Jana, A.; Zimmer, M.; Huch, V.; Scheschkewitz, D. Dimerization of a marginally stable disilenyl germylene to tricyclic systems: Evidence for reversible NHC-coordination. Chem. Commun. 2016, 52, 2799–2802. [Google Scholar] [CrossRef] [PubMed]
- Protchenko, A.V.; Birjkumar, K.H.; Dange, D.; Schwarz, A.D.; Vidovic, D.; Jones, C.; Kaltsoyannis, N.; Mountford, P.; Aldridge, S. A Stable Two-Coordinate Acyclic Silylene. J. Am. Chem. Soc. 2012, 134, 6500–6503. [Google Scholar] [CrossRef] [PubMed]
- Rekken, B.D.; Brown, T.M.; Fettinger, J.C.; Tuononen, H.M.; Power, P.P. Isolation of a Stable, Acyclic, Two-Coordinate Silylene. J. Am. Chem. Soc. 2012, 134, 6504–6507. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Xie, Y.; Wei, P.; Schaefer, H.F., III; Schleyer, P.V.R.; Robinson, G.H. Stabilization of elusive silicon oxides. Nat. Chem. 2015, 7, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Wendel, D.; Reiter, D.; Porzelt, A.; Altmann, P.J.; Inoue, S.; Rieger, B. Silicon and Oxygen’s Bond of Affection: An Acyclic Three-Coordinate Silanone and Its Transformation to an Iminosiloxysilylene. J. Am. Chem. Soc. 2017, 139, 17193–17198. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yao, S.; Inoue, S.; Epping, J.D.; Driess, M. A Cyclic Silylone (“Siladicarbene”) with an Electron-Rich Silicon(0) Atom. Angew. Chem. Int. Ed. 2013, 52, 7147–7150. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yao, S.; Inoue, S.; Irran, E.; Driess, M. The Elusive Silyliumylidene [ClSi:]+ and Silathionium [ClSi=S]+ Cations Stabilized by Bis(Iminophosphorane) Chelate Ligand. Angew. Chem. Int. Ed. 2012, 51, 10074–10077. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Sekiguchi, A.; Driess, M. An N-Heterocyclic Carbene−Disilyne Complex and Its Reactivity toward ZnCl2. J. Am. Chem. Soc. 2010, 132, 14061–14063. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.J.; Huch, V.; Rzepa, H.S.; Scheschkewitz, D. Equilibrium between a cyclotrisilene and an isolable base adduct of a disilenyl silylene. Nat. Chem. 2013, 5, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Chernov, O.; Schnakenburg, G. SiBr2(Idipp): A Stable N-Heterocyclic Carbene Adduct of Dibromosilylene. Angew. Chem. Int. Ed. 2009, 48, 5687–5690. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Lebedev, Y.N.; Chernov, O.; Straßmann, M.; Schnakenburg, G. Silicon(II) Coordination Chemistry: N-Heterocyclic Carbene Complexes of Si2+ and SiI+. Angew. Chem. Int. Ed. 2013, 52, 6974–6978. [Google Scholar] [CrossRef] [PubMed]
- Ghadwal, R.S.; Pröpper, K.; Dittrich, B.; Jones, P.G.; Roesky, H.W. Neutral Pentacoordinate Silicon Fluorides Derived from Amidinate, Guanidinate, and Triazapentadienate Ligands and Base-Induced Disproportionation of Si2Cl6 to Stable Silylenes. Inorg. Chem. 2011, 50, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Ghadwal, R.S.; Roesky, H.W.; Merkel, S.; Henn, J.; Stalke, D. Lewis Base Stabilized Dichlorosilylene. Angew. Chem. Int. Ed. 2009, 48, 5683–5686. [Google Scholar] [CrossRef] [PubMed]
- Rivard, E. Donor-acceptor chemistry in the main group. Dalton Trans. 2014, 43, 8577–8586. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, J.I.; Meyer, L.; Nadj, A.; Diefenbach, M.; Holthausen, M.C. Unraveling the Amine-Induced Disproportionation Reaction of Perchlorinated Silanes—A DFT Study. Chem. Eur. J. 2016, 22, 14328–14335. [Google Scholar] [CrossRef] [PubMed]
- Sinhababu, S.; Kundu, S.; Paesch, A.N.; Herbst-Irmer, R.; Stalke, D.; Fernández, I.; Frenking, G.; Stückl, A.C.; Schwederski, B.; Kaim, W.; et al. A Route to Base Coordinate Silicon Difluoride and the Silicon Trifluoride Radical. Chem. Eur. J. 2018, 24, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, J.I.; Scheibel, M.G.; Diefenbach, M.; Neumeyer, F.; Würtele, C.; Kulminskaya, N.; Linser, R.; Auner, N.; Schneider, S.; Holthausen, M.C. A Disilene Base Adduct with a Dative Si–Si Single Bond. Angew. Chem. Int. Ed. 2016, 55, 1782–1786. [Google Scholar] [CrossRef] [PubMed]
- Tillmann, J.; Meyer, L.; Schweizer, J.I.; Bolte, M.; Lerner, H.W.; Wagner, M.; Holthausen, M.C. Chloride-Induced Aufbau of Perchlorinated Cyclohexasilanes from Si2Cl6: A Mechanistic Scenario. Chem. Eur. J. 2014, 20, 9234–9239. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Wegner, F.; Nadj, A.; Bolte, M.; Auner, N.; Wagner, M.; Holthausen, M.C.; Lerner, H.W. The Perchlorinated Silanes Si2Cl6 and Si3Cl8 as Sources of SiCl2. Chem. Eur. J. 2011, 17, 4715–4719. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, P.P. Learning from silylenes and supersilylenes. In Organosilicon Chemistry VI: From Molecules to Materials, 1; Auner, N., Weis, J., Eds.; Wiley-VCH: Weinheim, Germany, 2005; Volume 2, pp. 10–24. ISBN 9783527618224. [Google Scholar]
- Müller, T. Stability, Reactivity, and Strategies for the Synthesis of Silyliumylidenes, RSi:+. A Computational Study. Organometallics 2010, 29, 1277–1283. [Google Scholar] [CrossRef]
- Jutzi, P.; Mix, A.; Rummel, B.; Schoeller, W.W.; Neumann, B.; Stammler, H.-G. The (Me5C5)Si+ Cation: A Stable Derivative of HSi+. Science 2004, 305, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Driess, M.; Yao, S.; Brym, M.; van Wüllen, C. Low-Valent Silicon Cations with Two-Coordinate Silicon and Aromatic Character. Angew. Chem. Int. Ed. 2006, 45, 6730–6733. [Google Scholar] [CrossRef] [PubMed]
- Hudnall, T.W.; Ugarte, R.A.; Perera, T.A. Main group complexes with N-Heterocyclic carbenes: Bonding, stabilization and applications in catalysis. In N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools (2); The Royal Society of Chemistry: London, UK, 2017; pp. 178–237. ISBN 978-1-78262-423-3. [Google Scholar]
- Melaimi, M.; Jazzar, R.; Soleilhavoup, M.; Bertrand, G. Cyclic (Alkyl)(amino)carbenes (CAACs): Recent Developments. Angew. Chem. Int. Ed. 2017, 56, 10046–10068. [Google Scholar] [CrossRef] [PubMed]
- Agou, T.; Hayakawa, N.; Sasamori, T.; Matsuo, T.; Hashizume, D.; Tokitoh, N. Reactions of Diaryldibromodisilenes with N-Heterocyclic Carbenes: Formation of Formal Bis-NHC Adducts of Silyliumylidene Cations. Chem. Eur. J. 2014, 20, 9246–9249. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.U.; Szilvási, T.; Inoue, S. A facile access to a novel NHC-stabilized silyliumylidene ion and C-H activation of phenylacetylene. Chem. Commun. 2014, 50, 12619–12622. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, N.; Sadamori, K.; Mizutani, S.; Agou, T.; Sugahara, T.; Sasamori, T.; Tokitoh, N.; Hashizume, D.; Matsuo, T. Synthesis and Characterization of N-Heterocyclic Carbene-Coordinated Silicon Compounds Bearing a Fused-Ring Bulky Eind Group. Inorganics 2018, 6, 30. [Google Scholar] [CrossRef]
- Li, Y.; Chan, Y.-C.; Li, Y.; Purushothaman, I.; De, S.; Parameswaran, P.; So, C.-W. Synthesis of a Bent 2-Silaallene with a Perturbed Electronic Structure from a Cyclic Alkyl(amino) Carbene-Diiodosilylene. Inorg. Chem. 2016, 55, 9091–9098. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, Y.-C.; Leong, B.-X.; Li, Y.; Richards, E.; Purushothaman, I.; De, S.; Parameswaran, P.; So, C.-W. Trapping a Silicon(I) Radical with Carbenes: A Cationic cAAC–Silicon(I) Radical and an NHC–Parent-Silyliumylidene Cation. Angew. Chem. Int. Ed. 2017, 56, 7573–7578. [Google Scholar] [CrossRef] [PubMed]
- Yeong, H.-X.; Xi, H.-W.; Li, Y.; Lim, K.H.; So, C.-W. A Silyliumylidene Cation Stabilized by an Amidinate Ligand and 4-Dimethylaminopyridine. Chem. Eur. J. 2013, 19, 11786–11790. [Google Scholar] [CrossRef] [PubMed]
- Leszczyńska, K.; Mix, A.; Berger, R.J.F.; Rummel, B.; Neumann, B.; Stammler, H.-G.; Jutzi, P. The Pentamethylcyclopentadienylsilicon(II) Cation as a Catalyst for the Specific Degradation of Oligo(ethyleneglycol) Diethers. Angew. Chem. Int. Ed. 2011, 50, 6843–6846. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, A.; Inoue, S.; Präsang, C.; Driess, M. Steering S–H and N–H Bond Activation by a Stable N-Heterocyclic Silylene: Different Addition of H2S, NH3, and Organoamines on a Silicon(II) Ligand versus Its Si(II)→Ni(CO)3 Complex. J. Am. Chem. Soc. 2010, 132, 3038–3046. [Google Scholar] [CrossRef] [PubMed]
- Präsang, C.; Stoelzel, M.; Inoue, S.; Meltzer, A.; Driess, M. Metal-Free Activation of EH3 (E=P, As) by an Ylide-like Silylene and Formation of a Donor-Stabilized Arsasilene with a HSi=AsH Subunit. Angew. Chem. Int. Ed. 2010, 49, 10002–10005. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Brym, M.; van Wüllen, C.; Driess, M. From a Stable Silylene to a Mixed-Valent Disiloxane and an Isolable Silaformamide–Borane Complex with Considerable Silicon–Oxygen Double-Bond Character. Angew. Chem. Int. Ed. 2007, 46, 4159–4162. [Google Scholar] [CrossRef] [PubMed]
- Szilvási, T.; Nyíri, K.; Veszprémi, T. Unique Insertion Mechanisms of Bis-dehydro-β-diketiminato Silylene. Organometallics 2011, 30, 5344–5351. [Google Scholar] [CrossRef]
- Filippou, A.C.; Chernov, O.; Blom, B.; Stumpf, K.W.; Schnakenburg, G. Stable N-Heterocyclic Carbene Adducts of Arylchlorosilylenes and Their Germanium Homologues. Chem. Eur. J. 2010, 16, 2866–2872. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Chernov, O.; Stumpf, K.W.; Schnakenburg, G. Metal–Silicon Triple Bonds: The Molybdenum Silylidyne Complex [Cp(CO)2Mo≡Si-R]. Angew. Chem. Int. Ed. 2010, 49, 3296–3300. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Szilvási, T.; Blom, B.; Driess, M. A Persistent 1,2-Dihydrophosphasilene Adduct. Angew. Chem. Int. Ed. 2015, 54, 15060–15063. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Szilvási, T.; Blom, B.; Irran, E.; Driess, M. From an Isolable Acyclic Phosphinosilylene Adduct to Donor-Stabilized Si=E Compounds (E=O, S, Se). Chem. Eur. J. 2015, 21, 18930–18933. [Google Scholar] [CrossRef] [PubMed]
- Arz, M.I.; Hoffmann, D.; Schnakenburg, G.; Filippou, A.C. NHC-stabilized Silicon(II) Halides: Reactivity Studies with Diazoalkanes and Azides. Z. Anorg. Allg. Chem. 2016, 642, 1287–1294. [Google Scholar] [CrossRef]
- Ahmad, S.U.; Szilvási, T.; Irran, E.; Inoue, S. An NHC-Stabilized Silicon Analogue of Acylium Ion: Synthesis, Structure, Reactivity, and Theoretical Studies. J. Am. Chem. Soc. 2015, 137, 5828–5836. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Wendel, D.; Ahmad, S.U.; Szilvasi, T.; Pothig, A.; Inoue, S. Chalcogen-atom transfer and exchange reactions of NHC-stabilized heavier silaacylium ions. Dalton Trans. 2017, 46, 16014–16018. [Google Scholar] [CrossRef] [PubMed]
- Baceiredo, A.; Kato, T. Multiple Bonds to Silicon (Recent Advances in the Chemistry of Silicon Containing Multiple Bonds). In Organosilicon Compounds: Theory and Experiment (Synthesis); Lee, V.Y., Ed.; Academic Press: London, UK, 2017; pp. 533–618. ISBN 978-0-12-801981-8. [Google Scholar]
- Lutters, D.; Merk, A.; Schmidtmann, M.; Müller, T. The Silicon Version of Phosphine Chalcogenides: Synthesis and Bonding Analysis of Stabilized Heavy Silaaldehydes. Inorg. Chem. 2016, 55, 9026–9032. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P.; Atsumi, M. Molecular Double-Bond Covalent Radii for Elements Li–E112. Chem. Eur. J. 2009, 15, 12770–12779. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Webb, R.L.; McElhill, E.A. The Electrical Effect of the Trifluoromethyl Group. J. Am. Chem. Soc. 1950, 72, 408–411. [Google Scholar] [CrossRef]
- Von Ragué Schleyer, P.; Kos, A.J. The importance of negative (anionic) hyperconjugation. Tetrahedron 1983, 39, 1141–1150. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990; ISBN 0198558651. [Google Scholar]
- Matta, C.F.; Boyd, R.J. An Introduction to the Quantum Theory of Atoms in Molecule; Wiley-VCH: Weinheim, Germany, 2007; ISBN 3527610707. [Google Scholar]
- Macchi, P.; Sironi, A. Chemical bonding in transition metal carbonyl clusters: Complementary analysis of theoretical and experimental electron densities. Coord. Chem. Rev. 2003, 238, 383–412. [Google Scholar] [CrossRef]
- Macchi, P.; Sironi, A. Interactions involving metals—From ‘Chemical Categories’ to QTAIM, and Backwards. In The Quantum Theory of Atoms in Molecules; Matta, C.F., Boyd, R.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Bader, R.F.W.; Slee, T.S.; Cremer, D.; Kraka, E. Description of conjugation and hyperconjugation in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5061–5068. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E.; Slee, T.S.; Bader, R.F.W.; Lau, C.D.H.; Nguyen Dang, T.T.; MacDougall, P.J. Description of homoaromaticity in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5069–5075. [Google Scholar] [CrossRef]
- Mandado, M.; Mosquera, R.A.; Graña, A.M. On the effects of electron correlation and conformational changes on the distortion of the charge distribution in alkyl chains. Chem. Phys. Lett. 2002, 355, 529–537. [Google Scholar] [CrossRef]
- Simons, R.S.; Haubrich, S.T.; Mork, B.V.; Niemeyer, M.; Power, P.P. The Syntheses and Characterization of the Bulky Terphenyl Silanes and Chlorosilanes 2,6-Mes2C6H3SiCl3, 2,6-Trip2C6H3SiCl3, 2,6-Mes2C6H3SiHCl2, 2,6-Trip2C6H3SiHCl2, 2,6-Mes2C6H3SiH3, 2,6-Trip2C6H3SiH3and 2,6-Mes2C6H3SiCl2SiCl3. Main Group Chem. 1998, 2, 275–283. [Google Scholar] [CrossRef]
- Kuhn, N.; Kratz, T. Synthesis of Imidazol-2-ylidenes by Reduction of Imidazole-2(3H)-thiones. Synthesis 1993, 1993, 561–562. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Revision, D.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Houk, K.N. Integration Grid Errors for Meta-GGA-Predicted Reaction Energies: Origin of Grid Errors for the M06 Suite of Functionals. J. Chem. Theory Comput. 2010, 6, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Glendening Eric, D.; Landis Clark, R.; Weinhold, F. Natural bond orbital methods. WIRES Ccomput. Mol. Sci. 2011, 2, 1–42. [Google Scholar] [CrossRef]
- Glendening Eric, D.; Landis Clark, R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Chem. Theory Comput. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. AIMAll (Version 17. 01. 25); TK Gristmill Software: Overland Park, KS, USA, 2017. [Google Scholar]
- Andrienko, G.A. ChemCraf—graphical software for visualization of quantum chemistry computations. Available online: http://www.chemcraftprog.com (accessed on 3 January 2015).
Figure 1.
Selected examples of isolated silyiumylidenes.

Figure 2.
Reactions of silyliumylidenes A and A’.

Figure 3.
(a) Conversion of silyliumylidene A to B with H2S and (b) the molecular structure of B. Thermal ellipsoids are shown at the 50% probability level. Except for the H1 atom, hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] of B: S1–Si1 2.0227(9), Si1–C1 1.902(2), Si1–C25 1.934(2), Si1–H1 1.41(3), S1–Si1–H1 113.5(11), C1–Si1–S1 121.14(8), C1–Si1–H1 113.3(11).
Figure 3.
(a) Conversion of silyliumylidene A to B with H2S and (b) the molecular structure of B. Thermal ellipsoids are shown at the 50% probability level. Except for the H1 atom, hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] of B: S1–Si1 2.0227(9), Si1–C1 1.902(2), Si1–C25 1.934(2), Si1–H1 1.41(3), S1–Si1–H1 113.5(11), C1–Si1–S1 121.14(8), C1–Si1–H1 113.3(11).

Figure 4.
Results of the bonding analysis of BModel. (a) Dominant Lewis resonance structure according to NRT analysis, (b) NLMOs representing the electron lone pairs at sulphur and the Si–CNHC, Si–CPh, Si–S, and Si–H single bonds, (c) 2D plot of ∇2ρ(r) charge concentration (- - -), and depletion (―), bond path (―) and bcps (black dots) with characteristic properties and bond path lengths of the Si–S bond, (d) related compounds 1–4.
Figure 4.
Results of the bonding analysis of BModel. (a) Dominant Lewis resonance structure according to NRT analysis, (b) NLMOs representing the electron lone pairs at sulphur and the Si–CNHC, Si–CPh, Si–S, and Si–H single bonds, (c) 2D plot of ∇2ρ(r) charge concentration (- - -), and depletion (―), bond path (―) and bcps (black dots) with characteristic properties and bond path lengths of the Si–S bond, (d) related compounds 1–4.

Figure 5.
Interconversion of chlorosilylene C and silyliumylidene A at 40 °C.

Figure 6.
Computed pathway for the interconversion between 5 and 7, R = 2,6-diphenyl-C6H3 and ImMe2H2; ΔG298 in kcal/mol.
Figure 6.
Computed pathway for the interconversion between 5 and 7, R = 2,6-diphenyl-C6H3 and ImMe2H2; ΔG298 in kcal/mol.

Figure 7.
Computed pathway for the reaction from 8 to 10, R = 2,6-diphenyl-C6H3; ΔG298 (ΔH298) in kcal/mol.
Figure 7.
Computed pathway for the reaction from 8 to 10, R = 2,6-diphenyl-C6H3; ΔG298 (ΔH298) in kcal/mol.

Figure 8.
Computed pathway for the reaction from 10 to 11, R = 2,6-diphenyl-C6H3; ΔG298 (ΔH298) in kcal/mol.
Figure 8.
Computed pathway for the reaction from 10 to 11, R = 2,6-diphenyl-C6H3; ΔG298 (ΔH298) in kcal/mol.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Results of QTAIM analyses of BModel and 1–4. Selected properties of the electron density distribution of the Si–S bond: Bond path lengths dSi–S, and distances to bcps dSi–bcp and dbcp–S, the electron density ρbcp, the Laplacian of the electron density ∇2ρbcp, the total energy density Hbcp, the bond ellipticity εbcp = λ1/λ2 − 1 (derived from the two negative eigenvalues of the Hessian matrix of the electron density at the bcp with λ1 ≥ λ2), delocalization index δSi,S.
Table 1.
Results of QTAIM analyses of BModel and 1–4. Selected properties of the electron density distribution of the Si–S bond: Bond path lengths dSi–S, and distances to bcps dSi–bcp and dbcp–S, the electron density ρbcp, the Laplacian of the electron density ∇2ρbcp, the total energy density Hbcp, the bond ellipticity εbcp = λ1/λ2 − 1 (derived from the two negative eigenvalues of the Hessian matrix of the electron density at the bcp with λ1 ≥ λ2), delocalization index δSi,S.
| Compound | dSi–S [Å] | dSi–bcp [Å] | dbcp–S [Å] | ρbcp [eÅ−3] | ∇2ρbcp [eÅ−5] | Hbcp [EhÅ−3] | εbcp | δSi,S |
|---|---|---|---|---|---|---|---|---|
| BModel | 2.00 | 0.75 | 1.26 | 0.78 | 3.33 | −0.52 | 0.01 | 0.78 |
| 1 | 2.13 | 0.77 | 1.36 | 0.66 | 1.36 | −0.43 | 0.12 | 0.56 |
| 2 | 1.95 | 0.73 | 1.22 | 0.83 | 5.24 | −0.55 | 0.21 | 1.15 |
| 3 | 2.14 | 0.78 | 1.37 | 0.64 | 1.36 | −0.40 | 0.10 | 0.57 |
| 4 | 1.94 | 0.73 | 1.21 | 0.83 | 5.39 | −0.56 | 0.23 | 1.25 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Porzelt, A.; Schweizer, J.I.; Baierl, R.; Altmann, P.J.; Holthausen, M.C.; Inoue, S. S–H Bond Activation in Hydrogen Sulfide by NHC-Stabilized Silyliumylidene Ions. Inorganics 2018, 6, 54. https://doi.org/10.3390/inorganics6020054
AMA Style
Porzelt A, Schweizer JI, Baierl R, Altmann PJ, Holthausen MC, Inoue S. S–H Bond Activation in Hydrogen Sulfide by NHC-Stabilized Silyliumylidene Ions. Inorganics. 2018; 6(2):54. https://doi.org/10.3390/inorganics6020054
Chicago/Turabian StylePorzelt, Amelie, Julia I. Schweizer, Ramona Baierl, Philipp J. Altmann, Max C. Holthausen, and Shigeyoshi Inoue. 2018. "S–H Bond Activation in Hydrogen Sulfide by NHC-Stabilized Silyliumylidene Ions" Inorganics 6, no. 2: 54. https://doi.org/10.3390/inorganics6020054
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.