Regium-π vs Cation-π Interactions in M2 and MCl (M = Cu, Ag and Au) Complexes with Small Aromatic Systems: An ab Initio Study



Department of Chemistry, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca (Baleares), Spain
*
Authors to whom correspondence should be addressed.
Inorganics 2018, 6(3), 64; https://doi.org/10.3390/inorganics6030064
Submission received: 5 June 2018
/
Revised: 18 June 2018
/
Accepted: 21 June 2018
/
Published: 24 June 2018
(This article belongs to the Special Issue Novel Non-Covalent Interactions)
Abstract
:In this study we have theoretically analyzed (RI-MP2/def2-TZVP) the ability of metal moieties involving elements from group IB (Cu, Ag and Au) to establish either regium–π or cation–π interactions with π-systems of different electronic nature. More precisely, we have used M2 (oxidation state = 0) and MCl (oxidation state = +1) molecules where M = Cu, Ag and Au. On the other hand, we have used benzene, trifluorobenzene and hexafluorobenzene as aromatic rings. Furthermore, we have used Bader’s theory of “Atoms in Molecules” as well as NBO (Natural Bonding Orbital) calculations to further investigate and characterize the regium–π and cation–π complexes described herein. We believe our findings may be important when describing and characterizing both interactions in a chemical context, as well as to further explore the nature of the recently uncovered regium–π bond.
1. Introduction
Noncovalent interactions have been recognized as a key working tool in various research fields of modern chemistry, such as supramolecular chemistry [1], molecular recognition [2] and materials science [3]. The impact of hydrogen and (more recently) halogen bonding interactions in chemical and biological media has been widely recognized by the scientific community [4,5,6]. In addition, it has been demonstrated that σ-holes can also be located in positive electrostatic potential regions of covalently bond atoms of groups IV (tetrels) to VIII (aerogens) [7,8,9], which is crucial for the understanding of σ-hole interactions. In this regard, several theoretical research groups have focused their attention on the study of their physical nature [10,11,12,13,14], concluding that it is basically understood by the interaction of a Lewis base (electron donor) with a σ-hole (electron acceptor), becoming sister interactions of hydrogen and halogen bonds [4,15]. Consequently, a vast amount of theoretical and experimental studies devoted to these “unconventional” noncovalent interactions have been carried out, showing interesting applications in solid state chemistry [16,17,18,19], biological systems [20,21,22,23,24], crystal engineering [25,26], catalytic processes [27,28,29] and supramolecular chemistry [30,31].
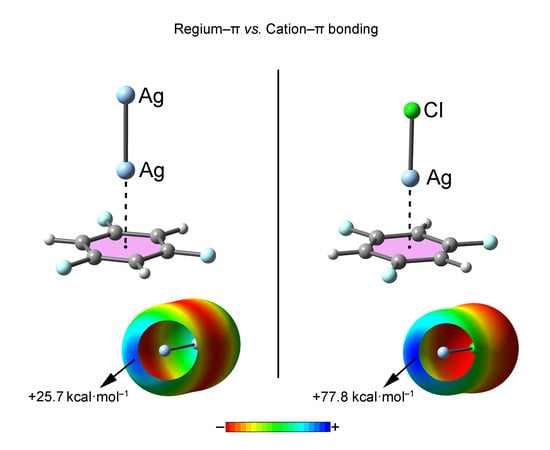
In this context, a recent report from the group of Stenlid and coworkers [32,33] introduced the concept “regium bond” to describe the interaction between a noble metal NP (Nanoparticle) and an electron rich moiety. More in detail, they demonstrated that transition metal clusters are capable of exhibiting non-spherical electron density distributions over the metal atoms due to the overlap of singly occupied s-orbitals at low coordinated atoms (σ-holes), thus behaving as Lewis acid moieties. They also found a correlation between the location of the σ-holes in Pt and Au NPs and their catalytic activity. Another important study worthy of highlighting is the recent work reported by Joy and collaborators [34] where they carried out an inspection of the CSD (Cambridge Structural Database) supported with theoretical calculations, evidencing the participation of penta-coordinated transition-metal complexes in noncovalent Cl–M···Y (M = Co, Rh and Ir, Y = electron rich atom) interactions. Other interesting theoretical studies encompass the analysis and evaluation of the interaction between transition and alkali metals and π-donors [35,36,37] as well as to dihydrogen bonding interactions involving the H2 and MF (M =Cu, Ag and Au) species [38]. Finally, our group has recently coined the term regium–π bond [39] to describe the interaction between molecular clusters formed by elements of group IB (Cu, Ag and Au) and aromatic moieties, which may be involved in processes of substrate absorption and desorption by the NP, similarly to that found for tetrel bonding interactions [40].
Although the expansion of the σ-hole concept to noble metals is on its mere naissance, it might have a great impact in a wide variety of chemical and industrial processes [41,42,43], more specifically in fields such as organic catalysis [44,45,46,47,48], biomedicine [49,50] and energy harvesting [51,52,53] where the use of noble metal nanoclusters, more concretely Cu, Ag and Au NPs, has been widespread among the scientific community. At this point, we believe it is important to properly differentiate between regium–π and cation–π interactions to avoid confusion, since transition metal atoms are able to establish both types of interactions owing to their diversity of oxidation states.
In this study, we aim to analyze and evaluate the interaction between noble metal moieties with two different oxidation states (OSs) and aromatic systems. In order to reach that goal, we have used M2 (OS = 0) and MCl (OS = +1) molecules where M = Cu, Ag and Au and several aromatic rings of different electronic nature including benzene, trifluorobenzene and hexafluorobenzene. Furthermore, we have used Bader’s AIM (Atoms in Molecules) theory as well as NBO (Natural Bonding Orbital) calculations to further describe and characterize the noncovalent interactions studied herein.
2. Results and Discussion
2.1. Preliminary MEP Analysis
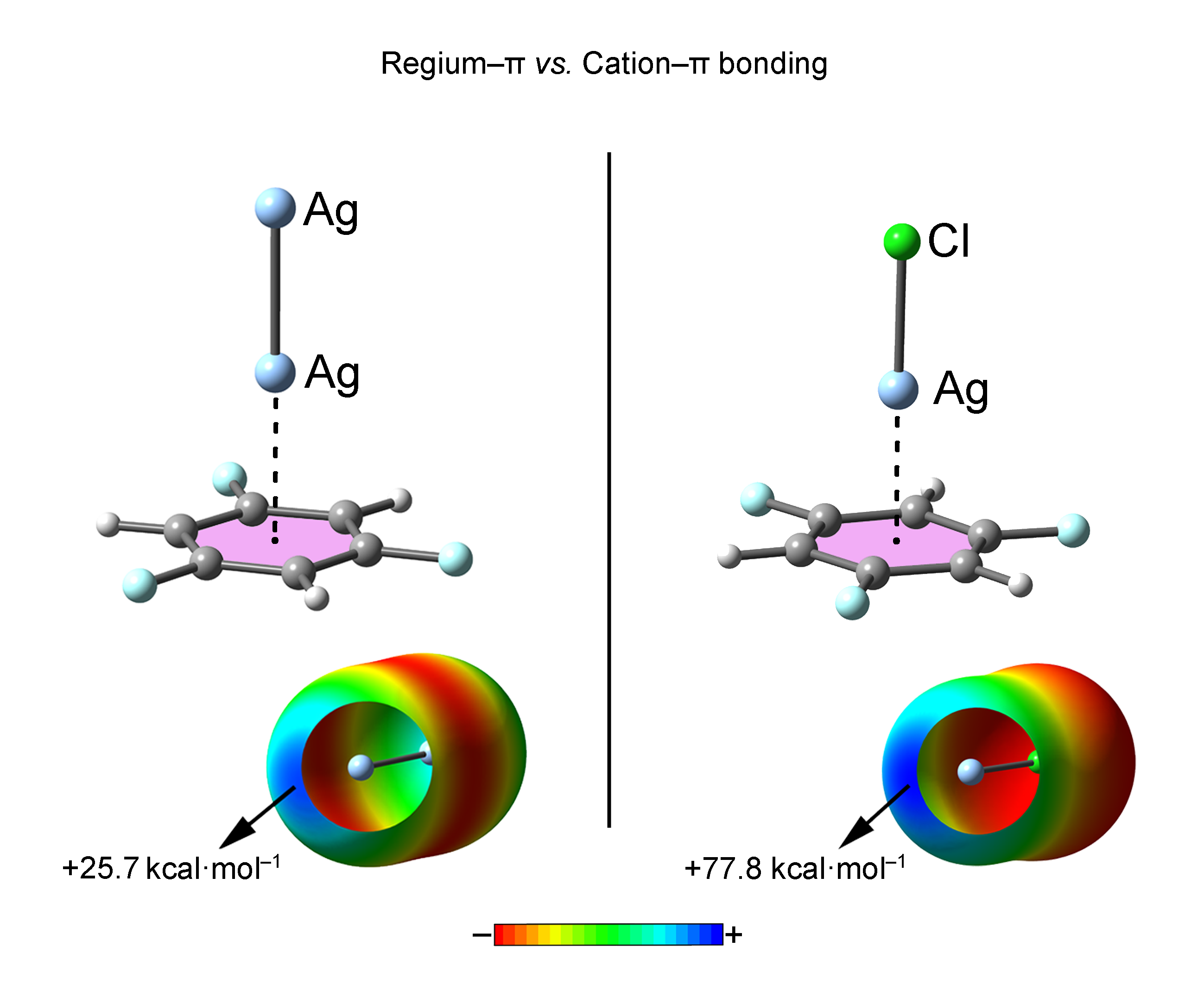
As a starting point we have computed the MEP (Molecular Electrostatic Potential) surfaces of compounds 1 to 9 used in this study (see Figure 1). First, among M2 compounds 1 to 6, a positive electrostatic potential region located at the outermost region of the M (M = Cu, Ag and Au) atom can be observed (named σ-hole). The presence of this region ensures an attractive interaction energy value with electron rich π-systems. More in detail, the MEP values at the σ-hole for compounds 1, 3, 4 and 6 are more positive than the ones obtained for compounds 2 and 5 involving Ag. Consequently, stronger interaction energy values for complexes involving Cu and Au atoms are expected compared to Ag, contrary to common forms of behavior regarding σ-hole interactions [7]. In addition, the MEP values of the Cu (1 and 3) and Au (4 and 6) derivatives are very close to each other, thus expecting a similar interaction energy strength from an electrostatic perspective. We have also computed the MEP surfaces of the aromatic compounds used in the study (compounds 7 to 9, see also Figure 1). As noted, the MEP values over the ring surface increase ongoing from benzene (7) to hexafluorobenzene (9), as it is commonly understood [54]. Thus, σ-hole complexes involving a benzene ring are expected to achieve stronger interaction energy values than those involving trifluorobenzene and hexafluorobenzene moieties. Finally, due to the almost negligible MEP value observed for trifluorobenzene, other energy contributions, such as polarization and dispersion components may play a relevant role upon complexation.
2.2. Energetic Study
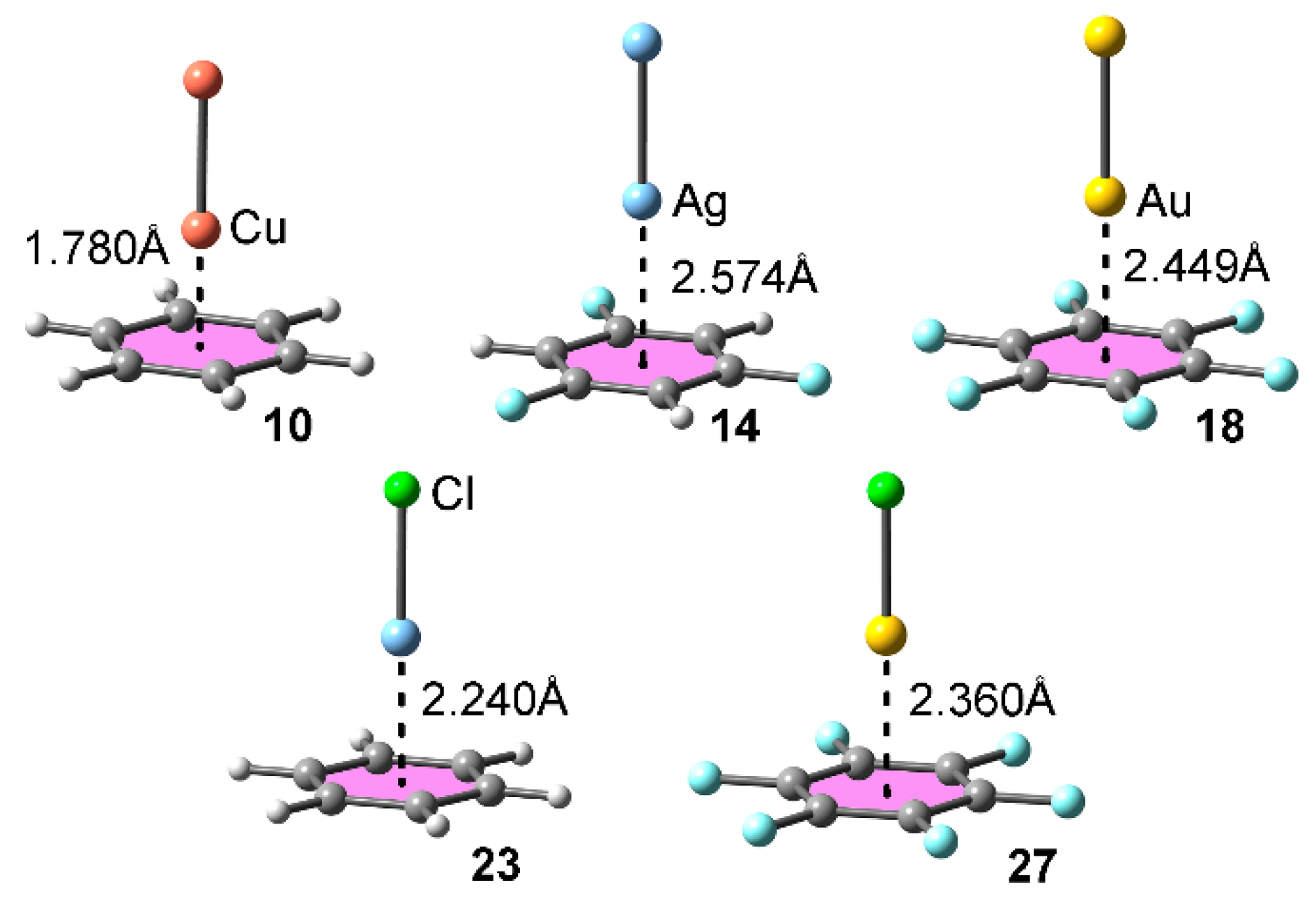
As previously reported by us [39], the main feature that differentiates regium–π and cation–π interactions is that the former implies a metal oxidation state of 0, while cation–π interactions involve a positive oxidation state of the metal atom. Bearing this concept in mind, Table 1 gathers the interaction energies and equilibrium distances of the optimized complexes 10 to 27 (see Scheme 1) computed at the RI-MP2/def2-TZVP level of theory. The optimized geometries of some complexes are shown in Figure 2. From the analysis of the results, several points are worthy of discussion. First, in all cases with the exception of complex 16 attractive and moderately strong interaction energy values were obtained, ranging from −21.0 to −2.2 kcal·mol−1. Second, cation–π complexes 19 to 27 obtained larger interaction energy values and shorter equilibrium distances than the analogous regium–π, due to the more favored electrostatic and polarization contributions. Third, complex 19 involving CuCl and benzene obtained the largest interaction energy value of the study, in agreement with the MEP analysis discussed above.
Among regium–π complexes 10 to 18, those involving Au2 (12, 15 and 18) achieved more favorable interaction energy values than their corresponding Cu and Ag analogous (10, 11, 13, 14, 16 and 17), also in agreement with the MEP analysis shown above and indicating that polarization and dispersion contributions are important when analyzing regium–π interactions involving Au. In addition, those complexes involving benzene as aromatic system (10 to 12) obtained the largest interaction energy values of the set (−9.7, −6.8 and −12.7 kcal·mol−1, respectively). Among Cu2 and Ag2 complexes, those involving the latter achieved larger interaction energies (complex 15, −7.8 kcal·mol−1 and complex 17, −2.2 kcal·mol−1) with the exception of complex 10 involving Cu2 and benzene as aromatic moiety (−9.7 kcal·mol−1). With respect to complex 16 involving Cu2 and hexafluorobenzene, it exhibits a slightly positive (unfavorable) interaction energy (+0.5 kcal·mol−1), thus suggesting that the electrostatic repulsion between the positive regions is not totally compensated by the other favorable terms like dispersion and polarization. In this particular case, we have re-computed the interaction energy using a larger basis set (def2-TZVPD) and we have obtained an almost negligible interaction energy value (−0.1 kcal·mol−1), thus confirming the delicate balance between the different forces that contribute to the binding energy in this complex.
On the other hand, in the case of cation–π complexes 19 to 27, those involving benzene as the aromatic system (19 to 21) achieved the largest interaction energy values of the set, in agreement to that observed for the M2 complexes. In addition, complexes involving AuCl and trifluorobenzene and hexafluorobenzene rings (24 and 27) obtained larger interaction energy values (−12.9 and −8.1 kcal·mol−1, respectively) that their corresponding CuCl and AgCl analogous. On the other hand, in case of benzene ring, complex 19 involving CuCl achieved a more favorable binding energy value (−21.0 kcal·mol−1) than AgCl (18) and AuCl (20) complexes (−15.1 and −20.1 kcal·mol−1, respectively), in agreement with the MEP analysis discussed above. Finally, among CuCl and AgCl complexes (19, 20, 22, 23, 25 and 26), the former achieved larger interaction energy values in all three aromatic systems.
2.3. AIM and NBO Analyses
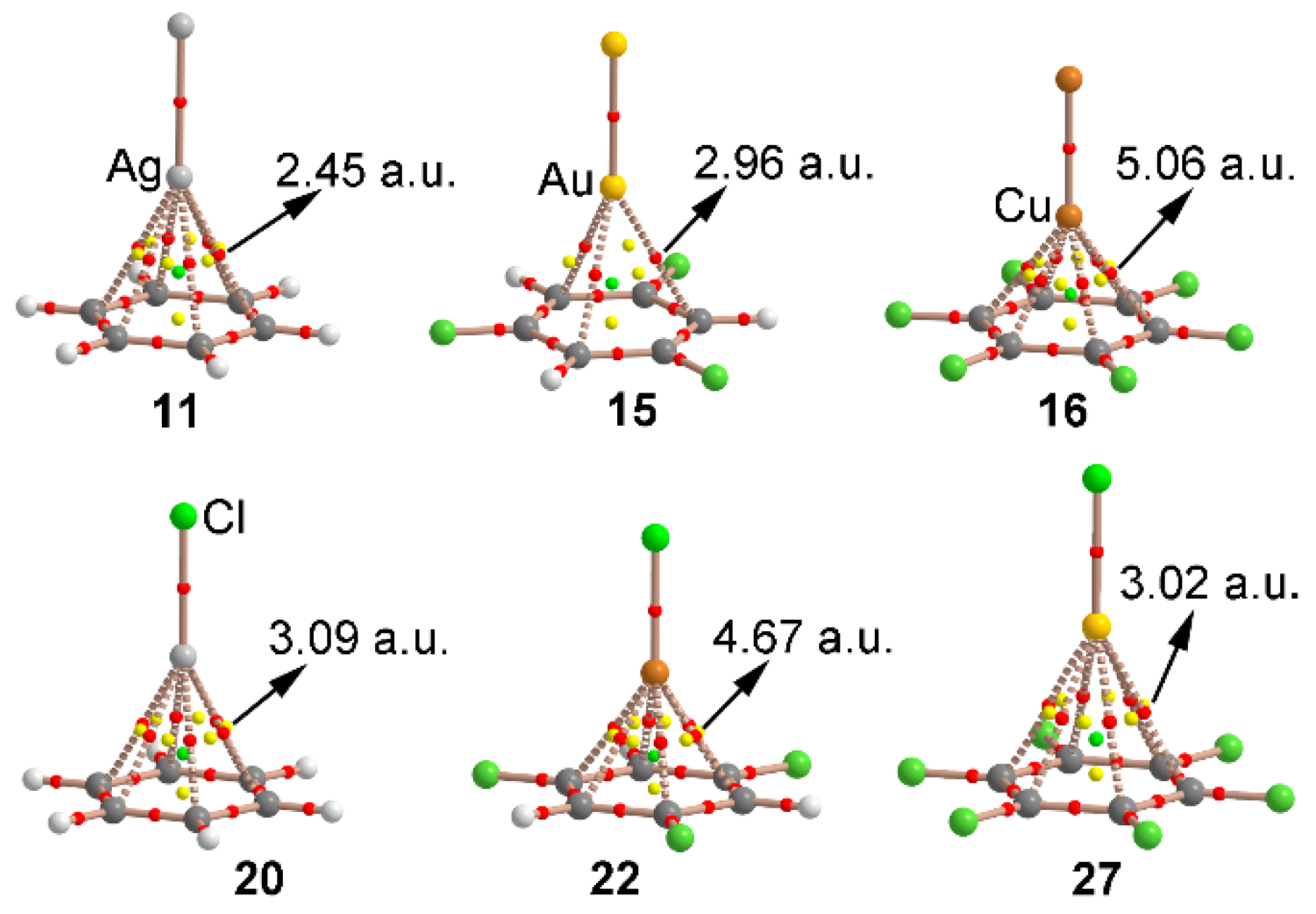
We have used the Bader’s AIM theory to characterize the noncovalent interactions present in complexes 10–27. A bond critical point (CP) and a bond path provide valuable structural information by indicating which atoms interact in any given system. The AIM distribution of critical points and bond paths computed for some complexes is show in Figure 3. As noted, in all cases the regium–π (11, 15 and 16) and cation–π (20 and 22) interactions are characterized by the presence of six symmetrically distributed bond CPs that connect the metal atom and the carbon atoms of the aromatic moiety. In addition, for complexes 11, 16, 20 and 22 involving Ag and Cu atoms, six ring CPs emerge due to the formation of several supramolecular rings. In case of complex 15 involving Au only three ring CPs are observed. Furthermore, in all cases a cage critical point is observed, which further characterizes the interaction. Finally, the values of the laplacian are in all cases positive, as it is common in closed shell calculations.
In order to study if orbital contributions are important to explain the regium–π and cation–π complexes described above, we have performed NBO calculations focusing our attention on the second order perturbation analysis, due to its utility in the analysis of donor-acceptor interactions [55]. The results for some representative examples are summarized in Table 2. From the inspection of the results some interesting issues arise.
First, for regium–π complexes (10 to 18) the main orbital contribution comes from the interaction between a bonding (BD) C–C orbital and an antibonding (BD*) M–M (M = Cu, Ag and Au) orbital. In addition, the magnitude of this orbital contribution in complexes involving Ag2 and the aromatic systems of benzene and trifluorobenzene (11 and 14) is lower than for their Cu2 (10 and 13) and Au2 (12 and 15) analogous. However, the opposite behavior is observed in case of the hexafluorobenzene moiety, where the orbital interaction increases ongoing from lighter to heavier metal atoms (complexes 16 to 18), in agreement with the interaction energy values obtained.
On the other hand, in case of cation–π complexes (19 to 27) the main orbital contribution relies on the interaction between a bonding (BD) C–C orbital and an unfilled Rydberg orbital (Ry*) from the metal atom. In this set of complexes, the orbital contributions are larger for those involving CuCl and AuCl moieties and the aromatic rings of benzene of hexafluorobenzene (complexes 19 and 21, respectively). In case of the trifluorobenzene ring, an increase in the magnitude of the orbital interaction ongoing from lighter to heavier metal atoms (complexes 22 to 24) is observed. Furthermore, the orbital contributions involving cation–π complexes are generally larger than those involving the regium–π set (see for instance complexes 10 and 19 or 15 and 24), in agreement with the results derived from the energetic study, only in case of complexes involving Cu and trifluorobenzene moieties (13 and 22) and Au and hexafluorobenzene (18 and 27) the opposite behavior was observed.
Finally, the magnitude of the orbital contributions is remarkable in both regium–π and cation–π complexes compared to the total interaction energy (~40% for 12, ~80% for 16, ~40% for 19 and ~40% for 24), thus highlighting the impact of orbital interactions as a noticeable source of stabilization of both type of interactions studied herein.
2.4. SAPT Analysis of Ag Complexes
In Table 3 we gather the total symmetry adapted perturbation theory (SAPT) energy values computed for the Ag complexes using the density-fitting density functional theory (DF-DFT, see computational methods). The total DF-DFT-SAPT interaction energies for these complexes are overestimated compared to those obtained using the RI-MP2/def2-TZVP level of theory likely due to the large BSSE correction applied to the MP2 energies. The different energetic contributions indicate that the cation–π complexes 20, 23 and 26 exhibit larger electrostatic contribution (Epol) than the regium–π complexes 11, 14 and 17, in line with the MEP values commented above. Furthermore, the contribution of induction (Eind + Eind_exc) is significantly more favorable in the cation–π complexes because the positive metal center Ag(I) is able to induce a larger dipole than the neutral one Ag(0). This contribution is comparable to the electrostatic one in the cation–π complexes 20 and 23. Remarkably, it is the most important contribution in the complex with hexafluorobenzene (26). Finally, the contribution of dispersion (Edisp + Edisp_exc) is similar in both series of complexes varying from −13.7 to −9.0 kcal·mol−1 and comparable to the induction contribution in complexes 14 and 17.
3. Theoretical Methods
The energies of all complexes included in this study were computed at the RI-MP2/def2-TZVP level of theory. For Ag and Au, we have used ECPs (Effective Core Potentials) [56] to accelerate the calculations and to account for relativistic effects, which cannot be neglected. The calculations have been performed by using the program TURBOMOLE version 7.0 [57]. During the optimizations the C6v and C3v symmetry point groups were used. The Bader’s “Atoms in molecules” theory has been used to study the interactions discussed herein by means of the AIMall calculation package [58]. The calculations for the wavefunction (MP2/def2-TZVP) and NBO (HF/def2-TZVP) analyses have been carried out using Gaussian 09 software [59]. The partitioning of the interaction energies into the individual electrostatic, induction, dispersion and exchange-repulsion components was carried out with the symmetry adapted intermolecular perturbation theory approach DFT-SAPT at the PBE0/def2-TZVP level of theory. For Ag the def2-SVP basis set was used. The SAPT calculations have been carried out using the program MOLPRO [60,61].
4. Conclusions
In conclusion, we have demonstrated the ability of M2 and MCl (M = Cu, Ag and Au) moieties to establish regium–π and cation–π interactions with aromatic portions of different electronic natures (i.e., benzene, trifluorobenzene and hexafluorobenzene). In all cases with the exception of complex 16 between Cu2 and hexafluorobenzene, a moderately strong and attractive interaction energy value was obtained, ranging from −21.0 to −2.2 kcal·mol−1. In addition, the cation–π set of complexes achieve larger interaction energy values than their regium–π analogous, in agreement with the MEP analysis. Furthermore, complex 19 involving CuCl and benzene as aromatic portions obtained the largest interaction energy value of the study. We have also used Bader’s theory of “Atoms in Molecules” as well as NBO calculations to further describe the interactions presented herein. Related to this, orbital interactions play an important role in the stabilization of both regium–π and cation–π complexes studied. Owing to the recent discovery of regium bonds as a promising and potential tool in the fields of supramolecular chemistry and catalysis, we believe that the findings derived from this study might be useful to proper differentiate between regium–π and cation–π interactions, which often share a similar chemical context.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/6/3/64/s1, cartesian coordinates of the complexes and results from the CSD search.
Author Contributions
A.B. and A.F. conceived and designed the calculations; A.B. and A.F. analyzed the data; A.B. wrote the paper.
Funding
We thank the MINECO/AEI (projects CTQ2014-57393-C2-1-P and CTQ2017-85821-R FEDER funds) for financial support.
Acknowledgments
We thank the CTI at the Universitat de les Illes Balears for computational facilities.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| AIM | Atoms in molecules |
| MEP | Molecular electrostatic potential |
| MP2 | Second order Moller-Plesset |
| BSSE | Basis Set Superposition Error |
| NBO | Natural Bonding Orbital |
| CP | Critical point |
| MINECO | Ministerio de Economía y Competitividad |
| AEI | Agencia Española de Investigación |
| NP | Nanoparticle |
| OS | Oxidation State |
| ECP | Efective Core Potential |
| CSD | Cambridge Structural Database |
| CTI | Centre de Tecnologies de la Informació |
| SAPT | Symmetry Adapted Perturbation Theory |
References
- Schneider, H.J. Binding mechanisms in supramolecular complexes. Angew. Chem. Int. Ed. 2009, 48, 3924–3977. [Google Scholar] [CrossRef] [PubMed]
- Maharramov, A.M.; Mahmudov, K.T.; Kopylovich, M.N.; Pombeiro, A.J.L. Non-Covalent Interactions in the Synthesis and Design of New Compounds; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Vickaryous, W.J.; Herges, R.; Jonhson, D.W. Arsenic–π interactions stabilize a self-assembled As2L3 supramolecular complex. Angew. Chem. Int. Ed. 2004, 43, 5831–5833. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. What is the covalency of hydrogen bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Murrayrust, P.; Motherwell, W.D.S. Computer retrieval and analysis of molecular geometry. 4. Intermolecular interactions. J. Am. Chem. Soc. 1979, 101, 4374–4376. [Google Scholar] [CrossRef]
- Bai, Y.; Sosnick, T.R.; Mayne, L.; Englander, S.W. Protein folding intermediates: Native-state hydrogen exchange. Science 1995, 269, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Aerogen bonding interaction: A new supramolecular force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Triel bonds-complexes of boron and aluminum trihalides and trihydrides with benzene. Struct. Chem. 2017, 28, 1163–1171. [Google Scholar] [CrossRef]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Marín-Luna, M.; Alkorta, I.; Elguero, J. Cooperativity in Tetrel Bonds. J. Phys. Chem. A 2016, 120, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen bond: A sister noncovalent bond to halogen bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. π-hole aerogen bonding interactions. Phys. Chem. Chem. Phys. 2015, 17, 24748–24753. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Halogen bonding: An interim discussion. ChemPhysChem 2013, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Přáda, A.; Padělková, Z.; Pecina, A.; Macháček, J.; Lepšík, M.; Holub, J.; Růžička, A.; Hnyk, D.; Hobza, P. The dominant role of chalcogen bonding in the crystal packing of 2D/3D aromatics. Angew. Chem. Int. Ed. 2014, 53, 10139–10142. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Brand, J.; Jetter, S.E. Donor-acceptor complexes between organoamines and phosphorus tribromide. Z. Naturforsch. 2001, 56, 1163–1171. [Google Scholar] [CrossRef]
- Dai, X.; Schulz, D.L.; Braun, C.W.; Ugrinov, A.; Boudjouk, P. “Inverse sandwich” complexes of perhalogenated cyclohexasilane. Organometallics 2010, 29, 2203–2205. [Google Scholar] [CrossRef]
- Goettel, J.T.; Haensch, V.G.; Schrobilgen, J.G. Stable chloro- and bromoxenate cage anions; [X3(XeO3)3]3– and [X4(XeO3)4]4– (X = Cl or Br). J. Am. Chem. Soc. 2017, 139, 8725–8733. [Google Scholar] [CrossRef] [PubMed]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Liu, Z.; Chen, T.; Chen, T.; Wang, Z.; Tian, G.; Shi, J.; Wang, X.; Lu, Y.; Yan, X.; et al. Utilization of halogen bond in lead optimization: A case study of rational design of potent phosphodiesterase type 5 (PDE5) inhibitors. J. Med. Chem. 2011, 54, 5607–5611. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. RCH3···O interactions in biological systems: Are they trifurcated H-bonds or noncovalent carbon bonds? Crystals 2016, 6, 26. [Google Scholar] [CrossRef]
- García-Llinás, X.; Bauzá, A.; Seth, S.K.; Frontera, A. Importance of R–CF3···O tetrel bonding interactions in biological systems. J. Chem. Phys. A 2017, 121, 5371–5376. [Google Scholar] [CrossRef] [PubMed]
- Iwaoka, M.; Isozumi, N. Possible roles of S···O and S···N interactions in the functions and evolution of phospholipase A2. Biophysics 2006, 2, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, F.; Dubois, P. Halogen bonding at work: Recent applications in synthetic chemistry and materials science. CrystEngComm 2013, 15, 3058–3071. [Google Scholar] [CrossRef]
- Britvin, S.N.; Kashtanov, S.A.; Krivovichev, S.V.; Chukanov, N.V. Xenon in rigid oxide frameworks: Structure, bonding and explosive properties of layered perovskite K4Xe3O12. J. Am. Chem. Soc. 2016, 138, 13838–13841. [Google Scholar] [CrossRef] [PubMed]
- Bruckmann, A.; Pena, M.A.; Bolm, C. Organocatalysis through halogen-bond activation. Synlett 2008, 900–902. [Google Scholar] [CrossRef]
- Benz, S.; Macchione, M.; Verolet, Q.; Mareda, J.; Sakai, N.; Matile, S. Anion transport with chalcogen bonds. J. Am. Chem. Soc. 2016, 138, 9093–9096. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.R.T.; Walden, D.M.; Fallan, C.; Greenhalgh, M.D.; Cheong, P.H.-Y.; Smith, A.D. Non-bonding 1,5-S···O interactions govern chemo- and enantioselectivity in isothiourea-catalyzed annulations of benzazoles. Chem. Sci. 2016, 7, 6919–6927. [Google Scholar] [CrossRef] [PubMed]
- Aakeroy, C.B.; Rajbanshi, A.; Metrangolo, P.; Resnati, G.; Parisi, M.F.; Desper, J.; Pilati, T. The quest for a molecular capsule assembled via halogen bonds. CrystEngComm 2012, 14, 6366–6368. [Google Scholar] [CrossRef] [Green Version]
- Yang, X. Synthesis and crystal structure of tetramethylammonium fluoride octadecasil. Mater. Res. Bull. 2006, 41, 54–66. [Google Scholar] [CrossRef]
- Stenlid, J.H.; Brinck, T. Extending the σ-hole concept to metals: An electrostatic interpretation of the effects of nanostructure in gold and platinum catalysis. J. Am. Chem. Soc. 2017, 139, 11012–11015. [Google Scholar] [CrossRef] [PubMed]
- Stenlid, H.; Johansson, A.J.; Brinck, T. σ-Holes and σ-lumps direct the Lewis basic and acidic interactions of noble metal nanoparticles: Introducing regium bonds. Phys. Chem. Chem. Phys. 2018, 20, 2676–2692. [Google Scholar] [CrossRef] [PubMed]
- Joy, J.; Jemmis, E.D. Contrasting behavior of the Z bonds in X–Z···Y weak interactions: Z = main group elements versus the transition metals. Inorg. Chem. 2017, 56, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; De Sarkar, A. A systematic investigation of acetylene activation and hydracyanation of the activated acetylene on Aun (n = 3–10) clusters via density functional theory. Phys. Chem. Chem. Phys. 2016, 18, 13830–13843. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, T.; Rauk, A. A theoretical study of the ethylene-metal bond in complexes between copper(1+), silver(1+), gold(1+), platinum(0) or platinum(2+) and ethylene, based on the Hartree-Fock-Slater transition-state method. Inorg. Chem. 1979, 18, 1558–1565. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Ruipérez, F. π⋅⋅⋅H+ ⋅⋅⋅π Hydrogen bonds and their lithium and gold analogues: MP2 and CASPT2 calculations. ChemPhysChem 2017, 18, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J.; Ruipérez, F. Dihydrogen bond interactions as a result of H2 cleavage at Cu, Ag and Au centres. Phys. Chem. Chem. Phys. 2016, 18, 12810–12818. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A.; Bauzá, A. Regium–π bonds: An unexplored link between noble metal nanoparticles and aromatic surfaces. Chem. Eur. J. 2018, 24, 7228–7234. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bond–σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D.; Lu, F.; Aranzaes, J.R. Nanoparticles as recyclable catalysts: The frontier between homogeneous and heterogeneous catalysis. Angew. Chem. Int. Ed. 2005, 44, 7852–7872. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.-C.; Astruc, D. Gold nanoparticles: assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Haruda, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Ali, Z.I.; Ghazy, O.A.; Meligi, G.; Saleh, H.H.; Bekhit, M. Copper nanoparticles: Synthesis, characterization and its application as catalyst for p-nitrophenol reduction. J. Inorg. Organomet. Polym. Mater. 2018, 28, 1195–1205. [Google Scholar] [CrossRef]
- Venkatesha, N.J.; Rameh, S. Citric acid-assisted synthesis of nanoparticle copper catalyst supported on an oxide system for the reduction of furfural to furfuryl alcohol in the vapor phase. Ind. Eng. Chem. Res. 2018, 57, 1506–1515. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Y.; Zhang, L. Fabrication of high efficient silver nanoparticle catalyst supported on poly(glycidyl methacrylate)–polyacrylamide. Ind. Eng. Chem. Res. 2016, 55, 12398–12406. [Google Scholar] [CrossRef]
- Schroder, F.; Sharma, U.K.; Mertens, M.; Devred, F.; Debecker, D.P.; Luque, R.; Van der Eycken, E.V. Silver-nanoparticle-catalyzed dearomatization of indoles toward 3-spiroindolenines via a 5-exo-dig spirocyclization. ACS Catal. 2016, 6, 8156–8161. [Google Scholar] [CrossRef]
- Stratakis, M.; García, H. Catalysis by supported gold nanoparticles: Beyond aerobic oxidative processes. Chem. Rev. 2012, 112, 4469–4506. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, P.; Strano, G.; Zuccarello, L.; Satriano, C. Gold and silver nanoparticles for applications in theranostics. Curr. Top. Med. Chem. 2016, 16, 3069–3102. [Google Scholar] [CrossRef]
- Kar, S.; Bagchi, B.; Kundu, B.; Bhandary, S.; Basu, R.; Nandy, P.; Das, S. Synthesis and characterization of Cu/Ag nanoparticle loaded mullite nanocomposite system: A potential candidate for antimicrobial and therapeutic applications. Biochim. Biophys. Acta 2014, 1840, 3264–3276. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; López-Puente, V.; Wang, Q.; Polavarapu, L.; Pastoriza-Santos, I.; Xu, Q.-H. Plasmon-enhanced light harvesting: Applications in enhanced photocatalysis, photodynamic therapy and photovoltaics. RSC Adv. 2015, 5, 29076–29097. [Google Scholar] [CrossRef]
- Jung, H.-Y.; Yeo, I.-S.; Kim, T.-U.; Ki, H.-C.; Gu, H.-B. Surface plasmon resonance effect of silver nanoparticles on a TiO2 electrode for dye-sensitized solar cells. Appl. Surf. Sci. 2018, 432, 266–271. [Google Scholar] [CrossRef]
- Shen, P.; Liu, Y.; Long, Y.; Shen, L.; Kang, B. High-performance polymer solar cells enabled by copper nanoparticles-induced plasmon resonance enhancement. J. Phys. Chem. C 2016, 120, 8900–8906. [Google Scholar] [CrossRef]
- Frontera, A.; Gamez, P.; Mascal, M.; Mooibroek, T.J.; Reedijk, J. Putting anion–π interactions into perspective. Angew. Chem. Int. Ed. 2011, 50, 9564–9583. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bär, M.; Hacer, M.; Horn, H.; Kömel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 13.05.06), TK Gristmill Software: Overland Park, KS, USA, 2013.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Heβelmann, A.; Jansen, G. The helium dimer potential from a combined density functional theory and symmetry-adapted perturbation theory approach using an exact exchange-correlation potential. Phys. Chem. Chem. Phys. 2003, 5, 5010–5014. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
Figure 1.
MEP surfaces of compounds 1 to 9 used in the study. Energies at selected points of the surface (0.001 a.u.) are given in kcal·mol−1.
Figure 1.
MEP surfaces of compounds 1 to 9 used in the study. Energies at selected points of the surface (0.001 a.u.) are given in kcal·mol−1.

Scheme 1.
Compounds 1 to 9 and complexes 10 to 27 used in this study.

Figure 2.
Optimized geometries of complexes 10, 14, 18, 23 and 27 at the RI-MP2/def2-TZVP level of theory.
Figure 2.
Optimized geometries of complexes 10, 14, 18, 23 and 27 at the RI-MP2/def2-TZVP level of theory.

Figure 3.
Distribution of critical points and bond paths in complexes 11, 15, 16, 20, 22 and 27. Bond, ring and cage critical points are represented by red, yellow and green spheres, respectively. The bond paths connecting bond critical points are also represented. The value of the density at the bond critical point (ρ× 02) is also indicated in a.u.
Figure 3.
Distribution of critical points and bond paths in complexes 11, 15, 16, 20, 22 and 27. Bond, ring and cage critical points are represented by red, yellow and green spheres, respectively. The bond paths connecting bond critical points are also represented. The value of the density at the bond critical point (ρ× 02) is also indicated in a.u.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Interaction energies with correction for basis set superposition error (ΔEBSSE in kcal·mol−1) of complexes 10 to 27 at the RI-MP2/def2-TZVP level of theory, equilibrium distances (Re in Å) and value of the density at the bond critical point (ρ × 102 in a.u.).
Table 1.
Interaction energies with correction for basis set superposition error (ΔEBSSE in kcal·mol−1) of complexes 10 to 27 at the RI-MP2/def2-TZVP level of theory, equilibrium distances (Re in Å) and value of the density at the bond critical point (ρ × 102 in a.u.).
| Complex | ΔEBSSE | Re a | 102 × ρ |
|---|---|---|---|
| 10 | −9.7 | 1.780 | 4.91 |
| 11 | −6.8 | 2.381 | 2.45 |
| 12 | −12.7 | 2.244 | 3.57 |
| 13 | −3.5 | 1.849 | 4.48 |
| 14 | −3.7 | 2.574 | 1.83 |
| 15 | −7.8 | 2.374 | 2.96 |
| 16 | +0.5 (−0.1) b | 1.785 | 5.06 |
| 17 | −2.2 | 2.674 | 1.56 |
| 18 | −5.2 | 2.449 | 2.64 |
| 19 | −21.0 | 1.767 | 5.01 |
| 20 | −15.1 | 2.240 | 3.09 |
| 21 | −20.1 | 2.230 | 3.64 |
| 22 | −11.9 | 1.829 | 4.63 |
| 33 | −7.9 | 2.349 | 2.65 |
| 24 | −12.9 | 2.316 | 3.22 |
| 25 | −5.4 | 1.813 | 4.83 |
| 26 | −2.4 | 2.416 | 2.37 |
| 27 | −8.1 | 2.360 | 3.02 |
a Distances measured from the metal atom to the ring centroid. b Interaction energy value using the def2-TZVPD basis set.
Table 2.
Donor and acceptor NBOs with indication of the second-order interaction energy E(2) (kcal·mol−1) for complexes 10–27.
Table 2.
Donor and acceptor NBOs with indication of the second-order interaction energy E(2) (kcal·mol−1) for complexes 10–27.
| Complex | Donor a | Acceptor | E(2) |
|---|---|---|---|
| 10 | BD C–C | BD* Cu–Cu | 6.96 |
| 11 | BD C–C | BD* Ag–Ag | 1.50 |
| 12 | BD C–C | BD* Au–Au | 5.46 |
| 13 | BD C–C | BD* Cu–Cu | 6.75 |
| 14 | BD C–C | BD* Ag–Ag | 1.74 |
| 15 | BD C–C | BD* Au–Au | 4.59 |
| 16 | BD C–C | BD* Cu–Cu | 0.39 |
| 17 | BD C–C | BD* Ag–Ag | 3.09 |
| 18 | BD C–C | BD* Au–Au | 6.09 |
| 19 | BD C–C | Ry* Cu | 9.18 |
| 20 | BD C–C | Ry* Ag | 7.0 |
| 21 | BD C–C | Ry* Au | 13.46 |
| 22 | BD C–C | Ry* Cu | 2.79 |
| 23 | BD C–C | Ry* Ag | 3.52 |
| 24 | BD C–C | Ry* Au | 5.97 |
| 25 | BD C–C | Ry* Cu | 5.54 |
| 26 | BD C–C | Ry* Ag | 4.04 |
| 27 | BD C–C | Ry* Au | 5.93 |
a BD, BD* and Ry* stand for bonding, anti-bonding and unfilled Rydberg orbital, respectively.
Table 3.
DFT-SAPT partition energies (electrostatic, Epol; exchange, Eex; induction Eind + Eind_exc; dispersion, Edisp + Edisp_exc and total Etotal) computed for cation–π and regium–π Ag complexes in kcal·mol−1.
Table 3.
DFT-SAPT partition energies (electrostatic, Epol; exchange, Eex; induction Eind + Eind_exc; dispersion, Edisp + Edisp_exc and total Etotal) computed for cation–π and regium–π Ag complexes in kcal·mol−1.
| Complex | Epol | Eex | Eind + Eind_exc | Edisp + Edisp_exc | Etotal |
|---|---|---|---|---|---|
| 11 | −36.4 | 55.5 | −23.3 | −13.7 | −17.8 |
| 14 | −21.3 | 35.2 | −11.7 | −10.4 | −8.2 |
| 17 | −15.1 | 28.0 | −8.4 | −9.0 | −4.5 |
| 20 | −39.4 | 59.4 | −34.2 | −13.3 | −27.4 |
| 23 | −24.8 | 44.7 | −24.8 | −11.3 | −16.2 |
| 26 | −15.8 | 37.4 | −20.6 | −10.2 | −9.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bauzá, A.; Frontera, A. Regium-π vs Cation-π Interactions in M2 and MCl (M = Cu, Ag and Au) Complexes with Small Aromatic Systems: An ab Initio Study. Inorganics 2018, 6, 64. https://doi.org/10.3390/inorganics6030064
AMA Style
Bauzá A, Frontera A. Regium-π vs Cation-π Interactions in M2 and MCl (M = Cu, Ag and Au) Complexes with Small Aromatic Systems: An ab Initio Study. Inorganics. 2018; 6(3):64. https://doi.org/10.3390/inorganics6030064
Chicago/Turabian StyleBauzá, Antonio, and Antonio Frontera. 2018. "Regium-π vs Cation-π Interactions in M2 and MCl (M = Cu, Ag and Au) Complexes with Small Aromatic Systems: An ab Initio Study" Inorganics 6, no. 3: 64. https://doi.org/10.3390/inorganics6030064
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.