One-Pot Synthesis of Heavier Group 14 N-Heterocyclic Carbene Using Organosilicon Reductant


Indian Institute of Science Education and Research Pune, Dr. Homi Bhabha Road, Pashan, Pune-411008, India
*
Author to whom correspondence should be addressed.
Inorganics 2018, 6(3), 69; https://doi.org/10.3390/inorganics6030069
Submission received: 6 June 2018
/
Revised: 25 June 2018
/
Accepted: 30 June 2018
/
Published: 12 July 2018
(This article belongs to the Special Issue Coordination Chemistry of Silicon)
Abstract
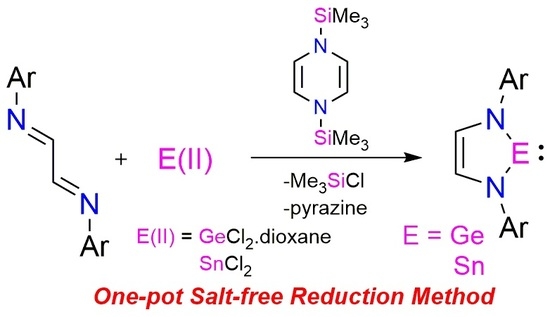
:Syntheses of heavier Group 14 analogues of “Arduengo-type” N-heterocyclic carbene majorly involved the use of conventional alkali metal-based reducing agents under harsh reaction conditions. The accompanied reductant-derived metal salts and chances of over-reduced impurities often led to isolation difficulties in this multi-step process. In order to overcome these shortcomings, we have used 1,4-bis-(trimethylsilyl)-1,4-diaza-2,5-cyclohexadiene as a milder reducing agent for the preparation of N-heterocyclic germylenes (NHGe) and stannylenes (NHSn). The reaction occurs in a single step with moderate yields from the mixture of N-substituted 1,4-diaza-1,3-butadiene, E(II) (E(II) = GeCl2·dioxane, SnCl2) and the organosilicon reductant. The volatile byproducts trimethylsilyl chloride and pyrazine could be removed readily under vacuum. No significant over reduction was observed in this process. However, N-heterocyclic silylene (NHSi) could not be synthesized using an even stronger organosilicon reductant under thermal and photochemical conditions.
{kind=link}
{kind=link}
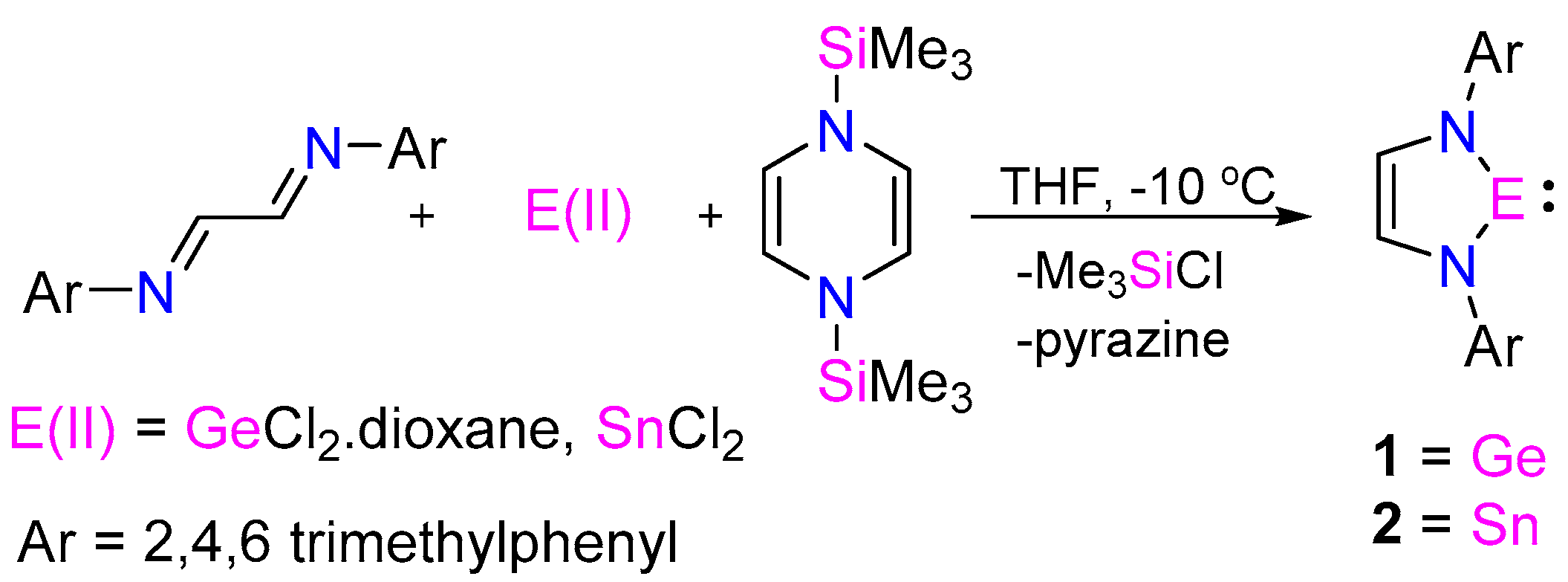{kind=link}
{kind=link}
1. Introduction
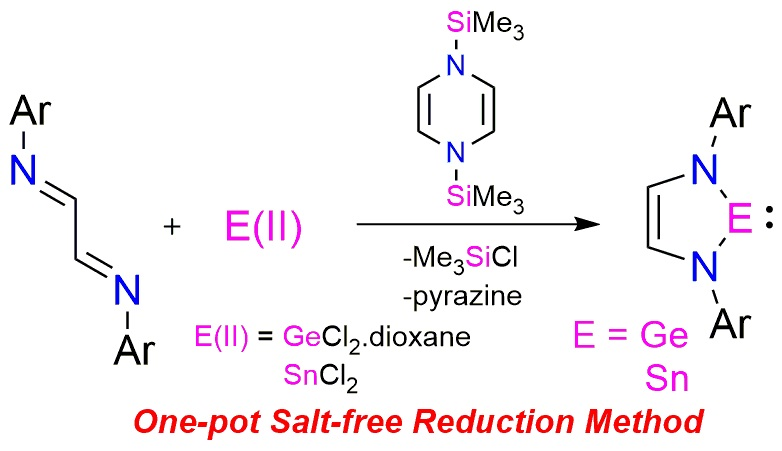
Carbene chemistry kick-started with the ground-breaking discovery of the bottle-able N-heterocyclic carbene (NHC) by Arduengo in 1991 [1]. Since this seminal work, the versatile NHCs have replaced the classical phosphine-based ligands for transition-metal catalysts [2]. Subsequently, the isolation of heavier Group 14 analogues of ‘Arduengo type’ carbene NHE (E = Si, Ge, and Sn) have been the subject of intense study, both for fundamental interests and potential applications in transition metal catalysis, similar to the NHCs. To date, a handful of NHEs have been synthesized and structurally characterized [3,4]. Obviously, the heavier analogues possess distinct electronic features compared to the NHCs, and hence exhibit different reactivities [3,4]. While N-heterocyclic silylenes (NHSi) have been engaged as ancillary ligands in numerous homogeneous catalysis [5,6,7], N-heterocyclic germylenes (NHGe) serve as a precursor for polymerization chemistry [8,9,10] and also for chemical vapour depositions [11].
Typically, the synthesis of the five-membered N-heterocyclic tetrylenes involves the reaction between N-substituted 1,4-diaza-1,3-butadiene, Group 14 halides, and the harsh alkali metal based reducing agents (Scheme 1) [3,4]. In the case of NHGe and NHSn, the initial step involves the reduction of N-substituted 1,4-diaza-1,3-butadiene by lithium metal, followed by cyclization of the dianion with the corresponding Group 14 E(II) halides [12,13,14,15]. While in the case of NHSi, the precursor cyclic diaminodichlorosilane was obtained by the cyclization of the dilithiated diazabutadiene with SiCl4 [16,17,18]. West et al. reported the first synthesis of NHSi by the reduction of this cyclic diaminodichlorosilane with potassium metal in tetrahydrofuran [18]. In these approaches, the choice of reductant play crucial role in the reductive dehalogenation step [12,13,14,15,16,17,18]. Moreover, the syntheses suffer from involvement of multiple steps, associated metal salts as byproduct and hence product isolation difficulties, and sometimes cases of over reduction. Certainly, these shortcomings urge a careful revisit into the synthetic methodology involved and finding an alternate milder reducing agent.
The milder organosilicon reductants have been well-known to efficiently reduce early transition metals without the formation of reductant-derived metal salts and over-reduced impurities [19,20]. Very recently, 2,3,5,6-tetramethyl-1,4-bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadiene have been employed for metal salt-free reduction of dibromobismuthine and dibromostibine to dibismuthine and distibine, respectively [21]. Worth mentioning, there are few discrete examples where a NHE (E = Si, Ge, and Sn) has been synthesized under metal-free conditions: Dehydrochlorination of cyclic diaminohydrochlorosilane using bulky NHC [22], dehydrogenation of dihydrogermane by frustrated Lewis pair [23], and transamination of Sn{N(SiMe3)2}2 with α-amino-aldimines [24,25], respectively. In this study, we have developed a simple one-pot synthetic route under milder conditions to synthesize NHGe and NHSn, respectively, free from reductant-derived metal salts using 1,4-bis-(trimethylsilyl)-1,4-diaza-2,5-cyclohexadiene [19,20] as the reductant.
2. Results and Discussion
The N-heterocyclic germylene (NHGe) 1 (Scheme 2) has been synthesized in one step by the low temperature addition of GeCl2·dioxane to a mixture of N1,N2-dimesitylethane-1,2-diimine and 1,4-bis-(trimethylsilyl)-1,4-diaza-2,5-cyclohexadiene in tetrahydrofuran. The volatile byproducts trimethylsilyl chloride and pyrazine, were easily removable under vacuum (Figures S1–S3). Since small amounts of insoluble solids appeared upon hexane addition, compound 1 was isolated as yellow solid from its hexane extract in an acceptable yield of 80%. Compound 1 was characterized by NMR study (Figures S4 and S5). Single crystals of 1 were grown from hexane at −40 °C, and its structure was determined using single crystal X-ray crystallography (Figure S23 and Table S1). This method is also applicable for other aromatic substituents, such as 2,6-diisopropylphenyl (Figures S6 and S7).
A similar method has been employed for the synthesis of N-heterocyclic stannylene (NHSn) 2 (Scheme 2). Formation of metallic tin was observed in the reaction mixture, which was removed by filtration. This arises due to the thermolabile nature of NHSn [24,25], leading to difficulties in acquiring NMR data (Figures S8 and S9). The presence of additional peaks in the 119Sn NMR (Figure S10) of the reaction mixture may be attributed to the formation of Sn(IV) compounds [26,27]. Single crystals of compound 2 were obtained from hexane extract at −40 °C in a yield of 35% (Figures S11–S13 and S24, Table S2). Notably, Gudat et al. reported the first synthesis of NHSn by transamination of Sn{N(SiMe3)2}2 with α-amino-aldimines [25].
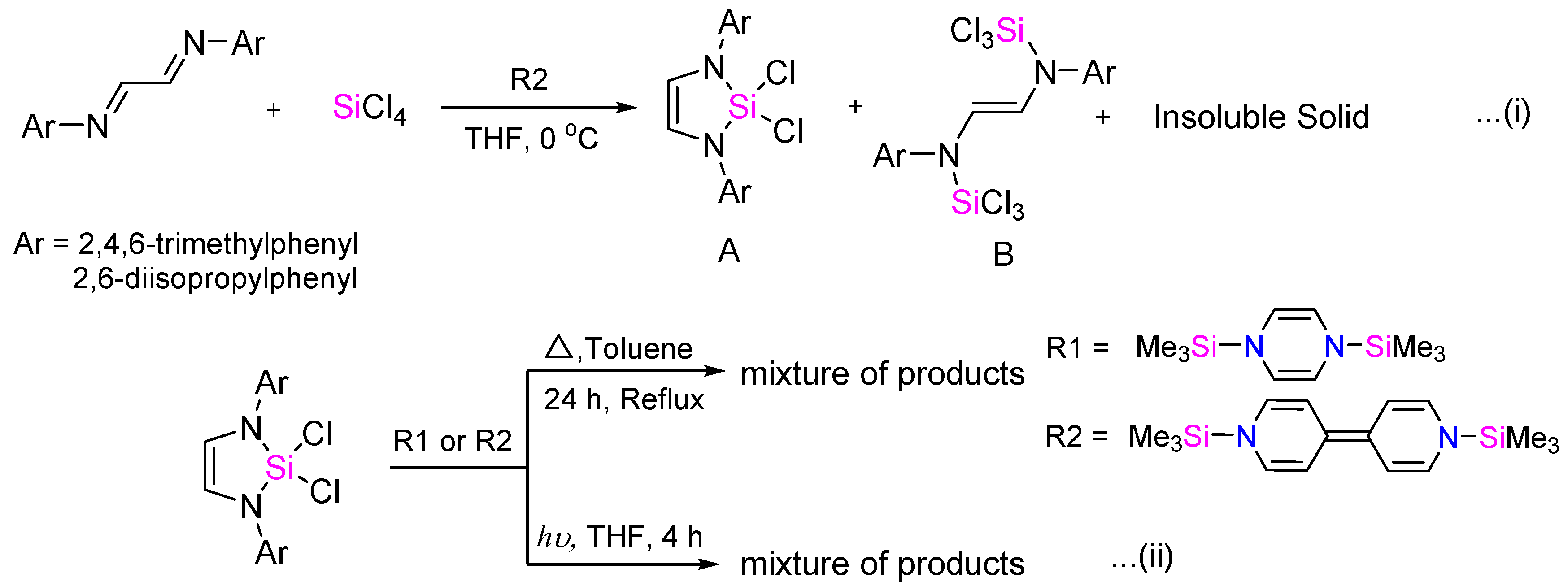
Despite several trials, we have been unsuccessful in synthesizing N-heterocyclic silylene (NHSi). Low temperature one-pot reaction (Scheme 3(i)) of N1,N2-dimesitylethane-1,2-diimine, SiCl4, and two equivalents of stronger reducing agent 1,1′-bis(trimethylsilyl)-1,1′-dihydro-4,4′-bipyridine (R2) led to a mixture of cyclic diaminodichlorosilane (A) and the disilylated product (B), along with a large amount of insoluble solid (Figures S14 and S15). Notably, the organosilicon reducing agents do not reduce diazabutadiene (Figure S22), while they react with Group 14 halides. Subsequently, we attempted to reduce the synthesized cyclic diaminodichlorosilane [16] using both 1,4-bis-(trimethylsilyl)-1,4-diaza-2,5-cyclohexadiene (R1) and also the stronger reductant 1,1′-bis(trimethylsilyl)-1,1′-dihydro-4,4′-bipyridine (R2) (Scheme 3(ii)) under thermal conditions. The NMR data always reflected the presence of unreacted cyclic diaminodichlorosilane, along with other unidentifiable products (Figures S16–S19). On a related context, 1,4-bis(trimethylsilyl)-substituted 1,4-dihydropyrazine has been reported to be capable of organosilyl group exchange reactions [28]. We have also tried reductions under UV irradiation conditions, anticipating enhanced Si–Cl bond cleavage [29]. However, each time NMR led to a mixture of unidentifiable products, in addition to the unreacted cyclic diaminodichlorosilane (Figures S20 and S21). Probably, the relatively lower reduction potentials of these organosilicon reductants do not allow the reduction of diaminodichlorosilanes.
3. Materials and Methods
3.1. General Information
All manipulations were carried out under a protective atmosphere of argon, applying standard Schlenk techniques or in a dry box. Tetrahydrofuran, toluene, and hexane were refluxed over sodium/benzophenone. All solvents were distilled and stored under argon and degassed prior to use. C6D6 was purchased from Sigma Aldrich (Sigma Aldrich Co., St. Louis, MO, USA) and dried over potassium. All chemicals were used as purchased. Diazabutadiene [30] ligand, and the reducing agents 1,4-bis-(trimethylsilyl)-1,4-diaza-2,5-cyclohexadiene, 1,1′-bis(trimethylsilyl)-1,1′-dihydro-4,4′-bipyridine [19,20] were synthesized according to reported literature procedure. Photochemical reactions were performed in Peschl Photoreactorsystem (Peschl Ultraviolet GmbH, Mainz, Germany). 1H, 13C{1H}, and 29Si{1H} NMR spectra were referenced to external SiMe4 using the residual signals of the deuterated solvent (1H) or the solvent itself (13C). 119Sn NMR was referenced to SnCl4 as the external standard. Melting points were determined under argon in closed NMR tubes and are uncorrected. Elemental analyses were performed on Elementar vario EL analyzer (Elementar Analysensysteme GmbH, Langenselbold, Germany). Single crystal data were collected on a Bruker SMART APEX four-circle diffractometer equipped with a CMOS photon 100 detector (Bruker Systems Inc., Fällanden, Switzerland) with a Cu Kα radiation (1.5418 Å). Data were integrated using Bruker SAINT software and absorption correction using SADABS. Structures were solved by Intrinsic Phasing module of the direct methods (SHELXS) [31] and refined using the SHELXL 2014 [32] software suite. All hydrogen atoms were assigned using AFIX instructions, while all other atoms were refined anisotropically.
3.2. Experimental Detail
Synthesis of compound 1: GeCl2 Dioxane (0.087 g. 0.38 mmol) dissolved in THF was added drop-wise to a schlenk flask containing N1,N2-dimesitylethane-1,2-diimine (0.11 g, 0.38 mmol) and 1,4-bis(trimethylsilyl)-1,4-dihydropyrazine (0.084 g, 0.38 mmol) in THF kept at −10 °C. The reaction mixture was slowly warmed to room temperature and stirred overnight. The volatiles were removed under vacuum and extracted in hexane. The solvent was removed completely to give compound 1 as a yellow solid ((0.110 g, % yield = 80), which was further bulk crystallized from hexane at −40 °C with a crystallization yield of (0.097 g, % yield = 71) (Decomp. 115–118 °C). 1H NMR (400 MHz, C6D6, TMS) δ = 6.87 (s, 4H, ArH); 6.57 (s, 2H, NCH); 2.23 (s, 12H, o-CH3); 2.19 (s, 6H, p-CH3) ppm; 13C{1H} NMR (101 MHz, C6D6, TMS ) δ = 142.41 (i-ArC); 134.87 (o-ArC); 133.61 (p-ArC); 128.88 (m-ArC); 125.16 (NCH); 20.60 (CH3); 18.06 (CH3) ppm. Elemental Analysis: Calcd. For C20H24GeN2: C, 65.80; H, 6.63; N, 7.67. Found: C, 65.82; H, 6.68; N, 7.62.
Synthesis of compound 2: SnCl2 (0.324 g, 1.71 mmol) dissolved in THF was added drop-wise to a schlenk flask containing N1,N2-dimesitylethane-1,2-diimine (0.5 g, 1.71 mmol) and 1,4-bis(trimethylsilyl)-1,4-dihydropyrazine (0.386 g, 1.71 mmol) in THF kept at −10 °C. The reaction mixture was stirred for 10 min, maintaining the same temperature. Subsequently, the volatiles were removed under vacuum. Hexane was added to the red-brown residue and the product was extracted in hexane. The red filtrate of hexane was concentrated and kept at −40 °C to give red crystals of 2 (0.247 g, crystallization yield = 35%). (Decomp. 145–147 °C). 1H NMR (400 MHz, Toluene-D8, 248 K) δ = 6.92 (s, 4H, m-CH); 6.91 (s, 2H, NCH); 2.33 (s, 12H, CH3); 2.29 (s, 6H, CH3) ppm. 13C NMR (101 MHz, Toluene-D8, 248 K) δ = 145.91 (i-ArC); 133.97 (o-ArC); 133.01 (p-ArC); 129.01 (m-ArC); 128.32 (NCH); 20.82 (CH3); 18.48 (CH3) ppm. 119Sn NMR (149.74 MHz, Toluene-D8, 248 K) δ = 250 ppm. Elemental Analysis: Calcd. For C20H24SnN2: C, 58.43; H, 5.88; N, 6.81. Found: C, 58.46; H, 5.95; N, 6.87.
4. Conclusions
We have established a salt-free reductive route for the synthesis of N-heterocyclic germylene and stannylene in one step. The easily removable volatile byproducts of the reaction leads to easy isolation of N-heterocyclic tetrylenes in acceptable yields. However, we have not been able to synthesize N-heterocyclic silylene using organosilicon reductants under thermal or photochemical conditions. The potential of this benign salt-free reduction method in the synthesis of other interesting low-valent main-group compounds is a much coveted area to explore.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/6/3/69/s1, detailed synthetic trials for preparing NHSi, NMR Spectra Figures, Crystallography Tables, ORTEP Figures. Figures S1 and S2: 1H and 13C NMR of Compound 1 (crude reaction mixture, before crystallization), Figure S3: 1H NMR in CDCl3 of the hexane insoluble solid residue from the crude reaction mixture of compound 1, Figures S4 and S5: 1H NMR, 13C NMR of Compound 1, Figure S6: 1H NMR, Figure S7: 13C NMR, Figures S8–S10: 1H, 13C, and 119Sn NMR of Compound 2 (crude reaction mixture, before crystallization), Figure S11–S13: 1H, 13C, and 119Sn NMR of Compound 2, Figures S14 and S15: 1H and 13C NMR plot of Trial 1 (* = 4,4’-Bipyridine, ’ = A, ’’ = B), Figures S16 and S17: 1H and 29Si NMR of Trial 2, Figures S18 and S19: 1H and 29Si of Trial 3, Figures S20 and S21: 1H and 29Si NMR of Trial 4, Figure S22: 1H NMR study for the reaction between diazabutadiene and organosilicon reductant, Figure S23: Molecular structure of 1 in the solid state (thermal ellipsoids at 30%, H atoms omitted for clarity). Selected bond lengths [Å] and bond angle [°]: Ge1–N1 = 1.8679 (18) Å, Ge1–N2 = 1.8786 (18); N1–Ge–N2 = 83.62 (8), Figure S24: Molecular structure of 2 in the solid state (thermal ellipsoids at 30%, H atoms omitted for clarity). Selected bond lengths [Å] and bond angle [°]: Sn1–N1 = 2.089 (4) Å, Sn1–N2 = 2.096 (4); N1–Sn–N2 = 77.95 (16), Table S1: Crystal data and structure refinement for Compound 1, Table S2: Crystal data and structure refinement for Compound 2.
Author Contributions
M.M. conceptualized the work, designed the experiments and wrote the paper; R.K.R. performed most of the experiments; S.F.A. and P.S. performed the photochemical reactions; V.K. carried out the NMR studies.
Funding
This work has been financially supported by the Department of Science and Technology (DST), India (EMR/2015/001135) and Council of Scientific and Industrial Research (CSIR), India No. 01/(2877)/17/EMR-II.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arduengo, A.J., III; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Peris, E. Smart N-heterocyclic carbene ligands in catalysis. Chem. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Asay, M.; Jones, C.; Driess, M. N-Heterocyclic carbene analogues with low-valent group 13 and group 14 elements: Syntheses, structures, and reactivities of a new generation of multitalented ligands. Chem. Rev. 2011, 111, 354–396. [Google Scholar] [CrossRef] [PubMed]
- Mizuhata, Y.; Sasamori, T.; Tokitoh, N. Stable Heavier Carbene Analogues. Chem. Rev. 2009, 109, 3479–3511. [Google Scholar] [CrossRef] [PubMed]
- Raoufmoghaddam, S.; Zhou, Y.-P.; Wang, Y.; Driess, M. N-Heterocyclic Silylenes as powerful steering ligands in catalysis. J. Organomet. Chem. 2017, 829, 2–10. [Google Scholar] [CrossRef]
- Blom, B.; Gallego, D.; Driess, M. N-Heterocyclic Silylene Complexes in Catalysis: New frontiers in an emerging field. Inorg. Chem. Front. 2014, 1, 134–148. [Google Scholar] [CrossRef]
- Blom, B.; Stoelzel, M.; Driess, M. New Vistas in N-Heterocyclic Silylene (NHSi) Transition-Metal Coordination Chemistry: Syntheses, Structures and Reactivity towards Activation of Small Molecules. Chem. Eur. J. 2013, 19, 40–62. [Google Scholar] [CrossRef] [PubMed]
- Shoda, S.-I.; Iwata, S.; Yajima, K.; Yagi, K.; Ohnishi, Y.; Kobayashi, S. Synthesis of germanium enolate polymers from germylene monomers. Tetrahedron 1997, 53, 15281–15295. [Google Scholar] [CrossRef]
- Shoda, S.-I.; Iwata, S.; Kim, H.J.; Hiraishi, M.; Kobayashi, S. Poly(germanium thiolate): A new class of organometallic polymers having a germanium-sulfur bond in the main chain. Macromol. Chem. Phys. 1996, 197, 2437–2445. [Google Scholar] [CrossRef]
- Kobayashi, S.; Iwata, S.; Hiraishi, M. Novel 2:1 periodic copolymers from cyclic germylenes and p-benzoquinone derivatives. J. Am. Chem. Soc. 1994, 116, 6047–6048. [Google Scholar] [CrossRef]
- Veprek, S.; Prokop, J.; Glatz, F.; Merica, R.; Klingan, F.R.; Herrmann, W.A. Organometallic chemical vapour deposition of germanium from a cyclic germylene, 1,3-Di-tert-butyl-1,3,2-diazagermolidin-2-ylidene. Chem. Mater. 1996, 8, 825–831. [Google Scholar] [CrossRef]
- Hermann, W.A.; Denk, M.; Behm, J.; Scherer, W.; Klingan, F.; Bock, H.; Solouki, B.; Wagner, M. Stable cyclic germanediyls (“cyclogermylenes”): Synthesis, structure, metal complexes and thermolysis. Angew. Chem. Int. Ed. 1992, 31, 1485–1488. [Google Scholar] [CrossRef]
- Baker, R.J.; Jones, C.; Mills, D.P.; Pierce, G.A.; Waugh, M. Investigation into the preparation of groups 13-15 N-heterocyclic carbene analogues. Inorg. Chim. Acta 2008, 361, 427–435. [Google Scholar] [CrossRef]
- Piskunov, A.V.; Aivaz’yan, I.A.; Cherkasov, V.K.; Abakumov, G.A. New paramagnetic N-heterocyclic stannylenes: An EPR study. J. Organomet. Chem. 2006, 691, 1531–1534. [Google Scholar] [CrossRef]
- Veith, M. Cyclic nitrogen derivatives of tetra- and divalent tin. Angew. Chem. Int. Ed. 1975, 14, 263–264. [Google Scholar] [CrossRef]
- Park, P.; Schäfer, A.; Mitra, A.; Haase, D.; Saak, W.; West, R.; Müller, T. Synthesis and reactivity of N-aryl substituted N-heterocyclic silylenes. J. Organomet. Chem. 2010, 695, 398–408. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, J.; Song, H.; Cui, C. N-Aryl substituted heterocyclic silylenes. Dalton Trans. 2009, 5444–5446. [Google Scholar] [CrossRef] [PubMed]
- Denk, M.; Lennon, R.; Hayashi, R.; West, R.; Belyakov, A.V.; Verne, H.P.; Haaland, A.; Wagner, M.; Metzler, N. Synthesis and structure of a stable silylene. J. Am. Chem. Soc. 1994, 116, 2691–2692. [Google Scholar] [CrossRef]
- Saito, T.; Nishiyama, H.; Tanahashi, H.; Kawakita, K.; Tsuragi, H.; Mashima, K. 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes as strong salt-free reductants for generating low-valent early transition metals with electron-donating ligands. J. Am. Chem. Soc. 2014, 136, 5161–5170. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W. Effects of cyclic 8π-electron conjugation in reductively silylated nitrogen heterocycles. J. Am. Chem. Soc. 1983, 105, 707–713. [Google Scholar] [CrossRef]
- Majhi, P.K.; Ikeda, H.; Sasamori, T.; Tsuragi, H.; Mashima, K.; Tokitoh, N. Inorganic-salt-free reduction in main-group chemistry: Synthesis of a dibismuthene and a distibene. Organometallics 2017, 36, 1224–1226. [Google Scholar] [CrossRef]
- Cui, H.; Shao, Y.; Li, X.; Kong, L.; Cui, C. Dehydrochlorination to silylenes by N-heterocyclic carbenes. Organometallics 2009, 28, 5191–5195. [Google Scholar] [CrossRef]
- Jana, A.; Tavčar, G.; Roesky, H.; Schulzke, C. Facile synthesis of dichlorosilane by metathesis reaction and dehydrogenation. Dalton Trans. 2010, 39, 6217–6220. [Google Scholar] [CrossRef] [PubMed]
- Gans-Eichler, T.; Gudat, D.; Nӓttinen, K.; Nieger, M. The transfer of tin and germanium atoms from N-heterocyclic stannylenes and germylenes to diazadienes. Chem. Eur. J. 2006, 12, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Gans-Eichler, T.; Gudat, D.; Nieger, M. Tin analogues of “arduengo carbenes”: Synthesis of 1,3,2λ2-diazastannoles and transfer of Sn atoms between a 1,3,2λ2-diazastannole and a diazadiene. Angew. Chem. Int. Ed. 2002, 41, 1888–1891. [Google Scholar] [CrossRef]
- Mansell, S.M.; Russel, C.A.; Wass, D.F. Synthesis of chelating diamido Sn(IV)compounds from oxidation of Sn(II) and directly from Sn(IV) precursors. Dalton Trans. 2015, 44, 9756–9765. [Google Scholar] [CrossRef] [PubMed]
- Sarazin, Y.; Coles, S.J.; Hughes, D.L.; Hursthouse, M.B.; Bochmann, M. Cationic brønsted acids for the preparation of SnIV salts: Synthesis and characterization of [Ph3Sn(OEt2)][H2N{B(C6F5)3}2], [Sn(NMe2)3(HNMe2)2][B(C6F5)4] and [Me3Sn(HNMe2)2][B(C6F5)4]. Eur. J. Inorg. Chem. 2006, 3211–3220. [Google Scholar] [CrossRef]
- Lichtblau, A.; Eblend, A.; Hausen, H.-D.; Kaim, W. N,N’-disilylated 1,4-dihydropyrazines: Organosilyl substitution reactions, structural effects of steric hindrance, and electron exchange with C60. Chem. Ber. 1995, 128, 745–750. [Google Scholar] [CrossRef]
- Rej, S.; Pramanik, S.; Tsurugi, H.; Mashima, K. Dehalogenation of vicinal dihalo compounds by 1,1′-bis(trimethylsilyl)-1H, 1′H-4,4′-bipyridinylidene for giving alkenes and alkynes in a salt-free manner. Chem. Commun. 2017, 53, 13157–13160. [Google Scholar] [CrossRef] [PubMed]
- Koten, G.V.; Vrieze, K. 1,4-Diaza-1,3-butadiene (α-diimine) ligands: Their coordination modes and the reactivity of their metal complexes. Adv. Organomet. Chem. 1982, 21, 151–239. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
Scheme 1.
Typical synthetic route for heavier Group 14 analogue of “Arduengo-type” carbene.

Scheme 2.
One-pot synthesis of NHE (E = Ge, Sn) using a salt-free reduction method.

Scheme 3.
Attempts to synthesize N-heterocyclic silylene (NHSi) using organosilicon reductants.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Raut, R.K.; Amin, S.F.; Sahoo, P.; Kumar, V.; Majumdar, M. One-Pot Synthesis of Heavier Group 14 N-Heterocyclic Carbene Using Organosilicon Reductant. Inorganics 2018, 6, 69. https://doi.org/10.3390/inorganics6030069
AMA Style
Raut RK, Amin SF, Sahoo P, Kumar V, Majumdar M. One-Pot Synthesis of Heavier Group 14 N-Heterocyclic Carbene Using Organosilicon Reductant. Inorganics. 2018; 6(3):69. https://doi.org/10.3390/inorganics6030069
Chicago/Turabian StyleRaut, Ravindra K., Sheikh Farhan Amin, Padmini Sahoo, Vikas Kumar, and Moumita Majumdar. 2018. "One-Pot Synthesis of Heavier Group 14 N-Heterocyclic Carbene Using Organosilicon Reductant" Inorganics 6, no. 3: 69. https://doi.org/10.3390/inorganics6030069
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.