Oxidation of Benzylic Alcohols and Lignin Model Compounds with Layered Double Hydroxide Catalysts

1
Department of Chemistry, University of Kentucky, Lexington, KY 40506, USA
2
Center for Applied Energy Research, University of Kentucky, Lexington, KY 40511, USA
3
Paul Laurence Dunbar High School, Lexington, KY 40513, USA
*
Author to whom correspondence should be addressed.
Inorganics 2018, 6(3), 75; https://doi.org/10.3390/inorganics6030075
Submission received: 12 June 2018
/
Revised: 13 July 2018
/
Accepted: 27 July 2018
/
Published: 31 July 2018
(This article belongs to the Special Issue Recent Breakthroughs with Layered Double Hydroxides)
Abstract
:Alcohol oxidation to carbonyl compounds is one of the most commonly used reactions in synthetic chemistry. Herein, we report the use of base metal layered double hydroxide (LDH) catalysts for the oxidation of benzylic alcohols in polar solvents. These catalysts are ideal reagents for alcohol oxidations due to their ease of synthesis, tunability, and ease of separation from the reaction medium. LDHs synthesized in this study were fully characterized by means of X-ray diffraction, NH3-temperature programmed desorption (TPD), pulsed CO2 chemisorption, N2 physisorption, electron microscopy, and elemental analysis. LDHs were found to effectively oxidize benzylic alcohols to their corresponding carbonyl compounds in diphenyl ether, using O2 as the terminal oxidant. LDH catalysts were also applied to the oxidation of lignin β-O-4 model compounds. Typically, for all catalysts, only trace amounts of the ketone formed from benzylic alcohol oxidation were observed, the main products comprising benzoic acids and phenols arising from β-aryl ether cleavage. This observation is consistent with the higher reactivity of the ketones, resulting from weakening of the Cβ–O4 bond that was shown to be aerobically cleaved at 180 °C in the absence of a catalyst.
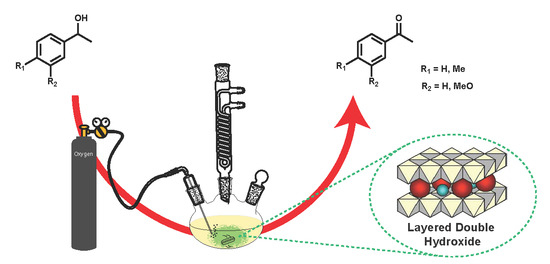
1. Introduction
The oxidation of alcohols to carbonyl compounds is one of the most widely used reactions in chemistry [1,2]. There are a variety of common reagents that are well-known for their ability to perform such transformations at the laboratory scale. Unfortunately, the use of stoichiometric reagents for alcohol oxidation typically requires toxic and/or environmentally harmful Cr(VI) [3,4,5,6] species and permanganate salts [7], or unpleasant activated DMSO [8,9,10,11,12,13,14,15]. Alternatively, catalytic systems exist that are reasonably effective for this transformation. However, in many cases, these systems utilize commercially available homogeneous catalysts such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) [16] or (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) [17,18,19], which are difficult to separate from products, or expensive noble metal catalysts (such as Pd, Pt, Rh, or Ru) [20]. Herein, we describe alcohol oxidations using easily prepared and inexpensive base metal layered double hydroxide (LDH) catalysts (also known as hydrotalcite or hydrotalcite-like compounds).
Our interest in these catalysts was piqued by a report by Choudary et al. [21], who found that Ni-Al-LDH catalysts were effective for alcohol oxidations. This was rather unexpected since Ni is rarely reported as an oxidation catalyst. Further exploration in this field showed that there are a variety of LDH catalysts that are effective for this transformation [22]. Indeed Kawabata et al. studied the use of Ni substituted Mg-Al-LDHs, finding that Mg-Ni-Al-LDH (2.5:0.5:1) produced the highest yield of benzaldehyde (ca. 51%) from benzyl alcohol of the catalysts tested [23]. Additionally, Choudhary and coworkers tested a variety of LDH catalysts, including Ni-Cr and Ni-Fe (3:1), under solvent-free conditions for the oxidation of benzyl alcohol to benzaldehyde [24]. However, although LDHs have been reported to work well in non-polar or solvent-free conditions, the use of polar solvents has yet to be thoroughly explored. While Choudhary found toluene to be an effective solvent for the oxidation of a multitude of benzylic and allylic alcohols, when 4-nitrobenzyl alcohol was oxidized in polar solvents, such as methanol and acetonitrile, only 5% and 30% of the substrate was converted to 4-nitrobenzaldehyde, respectively. Likewise, other researchers have noted decreased activity for alcohol oxidation in polar solvents using LDH catalysts [23,25]. The observed decrease in catalytic activity in polar solvent is likely the result of competitive coordination to active sites by the solvent, preventing reactant binding.
LDHs are highly flexible with regard to their tolerance for metal substitution. Indeed, the properties of LDH catalysts can be easily tuned via simple substitution of the divalent and trivalent metal ions in the interlamellar region. These anionic clays can support most first row transition metals with ionic radii similar to Mg2+ provided the trivalent metal ratio, χ (Equation (1)), is 0.2 ≤ χ ≤ 0.4 [26,27]. Catalyst preparation is simple, involving co-precipitation of the metal nitrate species under alkaline conditions (pH 8–10), followed by centrifugation and drying. Given the generally poor oxidation properties of nickel, we set out to find a transition metal substituted LDH catalyst with greater activity than the aforementioned Ni-Al-LDH. Moreover, given that most LDH catalysts are only reported to work well in non-polar solvents, thereby limiting their application, we screened a series of polar solvents for use with these catalyst systems.
The ability to function in polar solvents is particularly useful for the application of LDHs to the oxidation and depolymerization of lignin. Other researchers have applied LDHs to the oxidative depolymerization of lignin model compounds with a limited degree of success; however, the full potential of LDHs for this application has yet to be explored. Indeed, Corma and Bolm used a vanadate-containing Cu-hydrotalcite (Cu-HT) for the oxidation of lignin and lignin models [28]. While the catalyst was highly active for the oxidation of the lignin β-O-4 model, the active species was found to be homogenous in nature. Additionally, Baguc et al. studied the use of Ru/HT (i.e., Ru supported on HT) for the oxidation of the lignin model monomer veratryl alcohol with O2, finding the catalyst to yield the corresponding aldehyde in near quantitative yield [29]. Beckham et al. used Ni/HT for the depolymerization of a lignin model β-O-4 compound under thermolytic conditions, resulting in complete conversion in 1 h at 270 °C [30]. Against this background, we set out to develop a catalyst system that not only functioned well in polar solvents, but also had the potential to act as a lignin depolymerization catalyst. To this end, the synthesis, characterization, and evaluation of a variety of LDH catalysts for the oxidation of benzylic alcohols, as well as their application to models of the bio-polymer lignin, was studied.
2. Results and Discussion
2.1. Catalyst Characterization
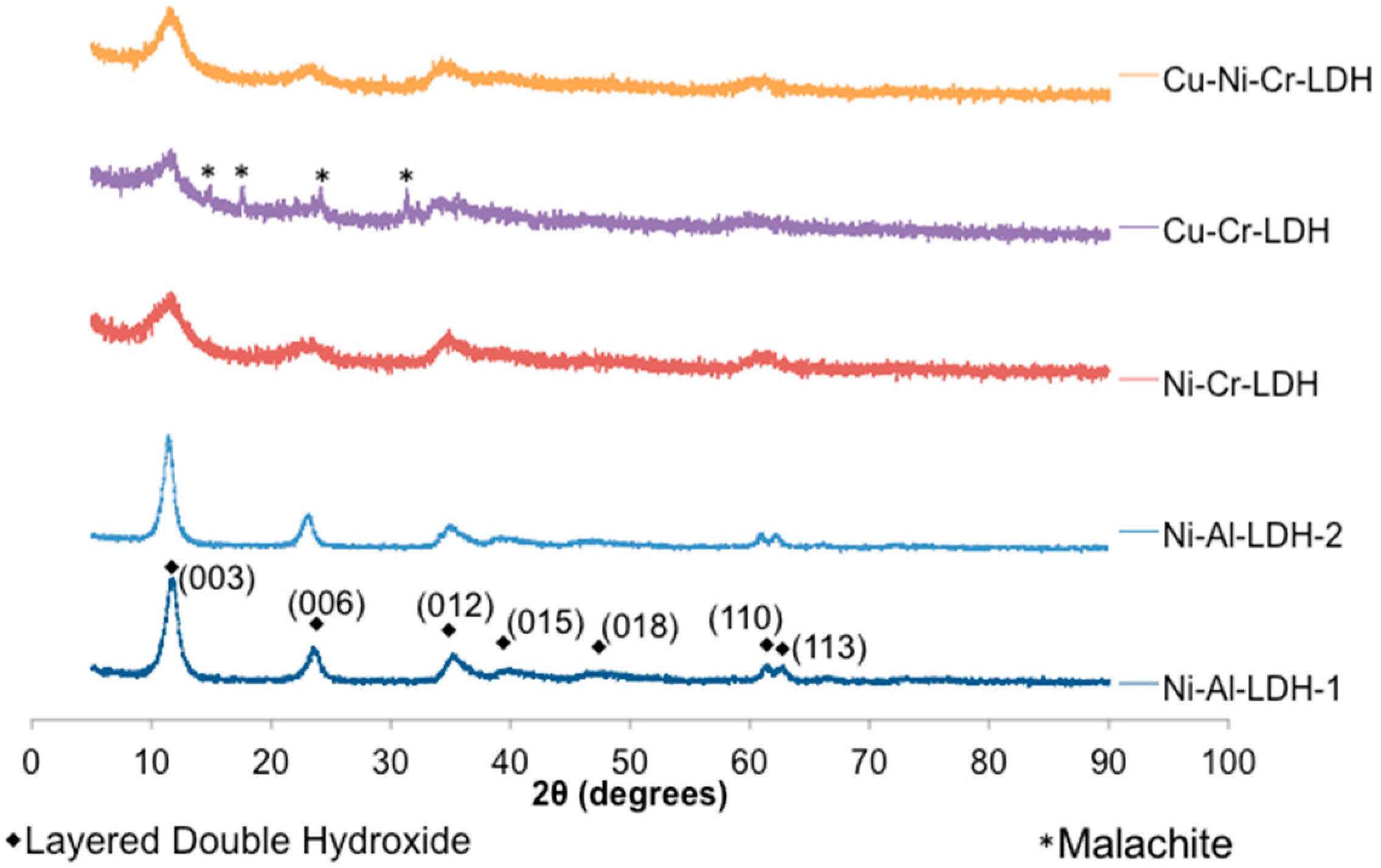
Generally, LDHs are expected to form under basic conditions as long as the metal cations (M2+ and M3+) have an ionic radius similar to Mg2+ and the trivalent metal ratio (χ) is between 0.2 and 0.4 as noted above [26]. In this study all catalysts were synthesized with the theoretical χ value within the aforementioned limits. The metal ratios of prepared catalysts and their physical properties are summarized in Table 1. Elemental analysis revealed that the catalysts returned a similar molar metal ratio to that of the solutions used for LDH synthesis. Scanning electron microscopy (SEM) images of Ni-Al-LDH-1 and Ni-Cr-LDH are shown in Figures S2 and S3, and illustrate typical LDH platelet morphology. FT-IR analysis (Figure S4) revealed that all catalysts displayed bending and stretching bands characteristic of LDH structures. Specifically, bands corresponding to the bending mode of interlayer water and asymmetric carbonate stretching were observed at ca. 1634 cm−1 and ca. 1346 cm−1, respectively.
Cavani et al. [27] noted that in order to incorporate copper (II) into LDH structures, it must be present with another bivalent metal in a Cu2+/M2+ ratio of less than or equal to 1. This empirical rule is attributed to the tendency of Cu2+ compounds to undergo Jahn-Teller distortions causing elongation of the octahedral coordination sphere. Therefore, Cu2+ ions must be accompanied by another M2+ metal such that the coordination sphere is undistorted in the brucite-like LDH structure. Whereas the Cu2+/M2+ rule was followed for the synthesis of Cu-Ni-Cr-LDH, the rule was disregarded in the synthesis of Cu-Cr-LDH. Unsurprisingly, in the Cu-Cr-LDH, a minor amount of a crystalline phase, which was identified as the mineral malachite, was apparent in addition to the LDH phase (Figure 1).
In addition, acidity and basicity measurements were conducted on all catalysts (Table 1 and Figures S5–S9). Notably, the Ni-Al-LDHs had the highest number of base sites on a weight basis, adsorbing 157.4 μmol CO2·g−1 and 79.2 μmol CO2·g−1 for Ni-Al-LDH-2 and Ni-Al-LDH-1, respectively. According to NH3-TPD experiments, Ni-Cr-LDH possessed the highest number of acid sites of the samples analyzed. While Mg-Al-LDH-1 had relatively few acid sites, it contained a higher relative proportion of strong acid sites (NH3 desorbed >450 °C, Figure S7). While both Ni-Al-LDH-1 and Ni-Al-LDH-2 had a similar number of total acid sites, Ni-Al-LDH-2 had a higher proportion of medium (NH3 desorbed at 250–450 °C) and strong acid sites (Figures S5 and S6). Indeed, strong and medium acid sites were virtually absent in Ni-Al-LDH-1.
2.2. Catalytic Oxidation of Lignin Model Compounds
2.2.1. Solvent Screening
Inspired by the work of Choudary et al. [21], we endeavored to find a solvent system that would be suitable for the oxidation of lignin model compounds. Given the polar nature of alcohols, including the lignin macromolecule, it is crucial to find a polar solvent that is effective for alcohol oxidations using LDH catalysts. Therefore, utilizing Ni-Al-LDH-1, which has a similar composition to the Ni-Al-LDH used by Choudary et al. [21], a series of solvents varying in polarity were screened for the oxidation of 1-phenyl ethanol, 1. As shown in Table 2, only limited conversions of 1 were obtained in most polar solvents (Table 2, entries 4–9). Acetophenone, 1a, was obtained in near quantitative yield in toluene (Table 2, entry 3), while reaction in 1,4-dioxane (Table 2, entry 7) yielded only 8% of 1a. In an attempt to amalgamate the polar ether properties of 1,4-dioxane with the electron-rich, aromatic character of toluene, phenyl ether (Table 2, entry 1) was trialed, resulting in 49% conversion of 1 to 1a.
2.2.2. Oxidation of 1 over LDH Catalysts
Having identified a suitable solvent for benzylic alcohol oxidation, LDH catalysts containing metals that are traditionally used in oxidation chemistry (copper and chromium) were synthesized (Section 3.1) and screened for activity in the oxidation of 1. As shown in Table 3, Ni-Cr-LDH (entry 6), Ni-Al-LDH-1 (thermally pretreated at 175 °C for 3 h, entry 4), and Ni-Al-LDH-2 (entry 5) gave high yields of 1a (>90%). Exploratory experiments (not shown) with Ni-Al-LDH-1 showed that light thermal pretreatment (175 °C for 3 h) appears to be optimal for catalytic performance. This is presumably since the temperature is high enough to remove H2O and/or CO2 coordinated to otherwise coordinatively unsaturated Ni2+ species that are proposed active sites (see Section 2.2.7 below). Unfortunately, thermal pretreatment with catalysts other than Ni-Al-LDH-1 did not follow the same trend. Notably, Mg-Al-LDH-1 and Mg-Al-LDH-2 showed relatively little conversion of 1, indicating that activity is not the result of catalyst basicity. Likewise, conversion of model compound 1 did not trend with acidity. Indeed, Ni-Cr-LDH, which had the highest number of acid sites, did not show the highest conversion of 1 to 1a. Moreover, while Cu-Cr-LDH had neither the most acid or base sites, it demonstrated the highest conversion of 1 to 1a, suggesting a reaction mechanism not related simply to the acidity or basicity of the catalyst.
2.2.3. Catalyst Loading Study
In order to elucidate the optimal amount of catalyst needed, a catalyst loading study was performed using Ni-Cr-LDH. The amount of catalyst was incrementally increased while keeping the amount of starting material constant at 2 mmol. As can be seen from Table 4, using 0.5 g of Ni-Cr-LDH for every 2 mmol of starting material proved to be optimal (entry 4). While this is a large amount of catalyst, it is not uncommon in the literature [25,29]. The need for a large amount of catalyst relative to starting material suggests that the active site corresponds to defect sites that are present in low concentration on the catalyst surface.
2.2.4. Leaching Study
In order to determine whether conversion was the result of leached metal in the solution, a hot filtration experiment was performed in which the Ni-Cr-LDH catalyst was hot filtered from the reaction mixture after 1 h. A sample was taken, after which the filtrate was transferred to a fresh flask and allowed to react for an additional 23 h at 150 °C. Analysis of the reaction mixture indicated a 39% conversion at 1 h with no additional conversion post-filtration, suggesting that catalysis occurred on the LDH surface and not via free metal species in solution. Elemental analysis of the reaction mixture at 24 h post-filtration did not reveal significant amounts of metal leached into solution (<1 ppm Cr and 3 ppm Ni).
2.2.5. Oxidation of Compounds 2 and 3 over LDH Catalysts
In order to determine the efficacy of these catalysts with electron-rich substrates, model compound 2 was used as a starting material. Catalysts that returned a yield of >80% in the conversion of 1 to 1a were screened for catalytic activity in the oxidation of 2. Additionally, Ni-Al-LDH-1 was also used in the reaction for comparison purposes [21]. Oxidation of 2 with the aforementioned catalysts afforded two products, the expected ketone, 2a, as well as the alcohol elimination product, 2b. As can be seen from Table 5, catalysts containing nickel yielded higher amounts of the dehydration product 2a. Dehydration was found to be most prominent for Ni-Al-LDH-2. On the other hand, copper-containing catalysts tended to be more selective towards the ketone product, 2a. In an attempt to elucidate the relationship between catalyst functionality and alcohol dehydration, acidity and basicity measurements were compared to catalyst performance. No absolute trend between acidity or basicity and alkene yield was elucidated, although the highest yields of 2b and 3b (see Table 5 and Table 6) were obtained for the LDH catalysts possessing the highest numbers of acid and base sites (Ni-Al-LDH-1, Ni-Al-LDH-2, and Ni-Cr-LDH). Mechanistically this may occur via an E2-type mechanism in which the metal alkoxide is formed on the catalyst surface, followed by deprotonation of the β-carbon and subsequent elimination of the metal oxide to form the alkene. Although both Ni-Al-LDH catalysts both performed well, overall, oxidation with Ni-Cr-LDH resulted in the highest yield of 2a. In the case of Cu-Cr-LDH, near quantitative conversion of 2 was observed. However, the selectivity to 2a (ca. 50%), which was the only identifiable compound by GC/MS, was significantly lower than other LDH catalysts (entries 2–6, Table 5), presumably due to the conversion of 2 to unidentifiable products, evidenced by the darkening of the reaction mixture.
In order to further increase lignin-like functionality on the substrate and explore functional group sensitivity, the phenolic model compound 3 was chosen. As shown in Table 6, the use of Ni-containing catalysts for the oxidation of 3 favored the formation of the dehydration product 3b. Unfortunately, poor mass balances were obtained for the oxidation of compound 3 due to suspected polymerization (chromatographically immobile material). This suggests that phenolic groups may need to be protected, e.g., by benzylation, prior to benzylic oxidation. Unexpectedly, a small amount of benzaldehyde 3c was also formed during the oxidation of 3 as a result of Cα–Cβ bond cleavage. Aldehyde formation was most prevalent when the Ni-Cu-Cr-LDH and Ni-Al-LDH-1 were used as catalysts. Moreover, Cu-Cr-LDH was active in the oxidation of 3 but did not yield identifiable products. The production of 3b likely results in phenolic or styrenic coupling reactions leading to high molecular weight polymers.
2.2.6. Catalyst Reusability Study
Catalyst reusability was studied using Ni-Al-LDH-1 and Ni-Cr-LDH in the oxidation of 1 (Table 7). After the reaction, the catalysts were filtered and washed with THF and hexanes, and then dried in a vacuum oven prior to re-use. Re-usability tests for Ni-Al-LDH-1 and Ni-Cr-LDH demonstrated a significant decrease in activity upon successive use. The X-ray diffractogram of the spent Ni-Cr-LDH (Figure S10) displayed similar peaks to the fresh catalyst with the exception of a new highly crystalline peak corresponding to chromium (III) oxide, while N2 physisorption analysis revealed a significant decrease in surface area (27.8 m2·g−1 post reaction), which is believed to be the result of phase segregation in the LDH, in addition to adsorbed organics blocking pores. Similarly, Ni-Al-LDH-1 displayed the characteristic LDH diffraction pattern but also a highly crystalline peak corresponding to Al(OH)3 (Figure S11). From these observations, it is evident that the LDH structure of the catalysts was largely retained after use, although limited segregation of the M(OH)3 phase occurs (which decomposes, in the case of Cr, to Cr2O3). Similar results were obtained for LDHs tested with other substrates (see, for example, Figure S12).
Other researchers have reported that full activity of LDH catalysts for the oxidation of benzyl alcohol is regained upon washing LDH catalysts with aqueous sodium carbonate [29,30]. After washing Ni-Cr-LDH with Na2CO3, a small amount of activity was regained. We believe that inability to completely regenerate the Ni-Cr-LDH catalyst may be related to the phase segregation observed by X-ray diffraction (Figure S10). The effect of carbonate washing was more pronounced in the case of Ni-Al-LDH-1. Indeed, Ni-Al-LDH-1 showed no activity for the oxidation of compound 1 after the first use. However, after washing with carbonate solution, activity was completely regained. In fact, conversion increased from 72% to 100%, possibly due to an increase in the number of defect sites after reconstitution with Na2CO3.
Other workers have reported that LDH anions may play an integral role in alcohol oxidation as evidenced by the reduced catalyst activity when anions are absent or substituted by another anion in the LDH [21,25]. In order to ascertain whether carbonate acts as a stoichiometric base, the amount of CO32− present in each LDH was calculated based on the idealized LDH formula [M2+1−χM3+χ(OH)2]χ+(An−)χ/n, where χ is the trivalent metal ratio, A is the anionic species, and n is the charge of the anionic species. The water content was purposefully ignored as this can vary between LDHs [26]. As can be seen in Table 8, CO32− was present in a sub-stoichiometric quantity compared to the substrate 1; hence, carbonate did not act as a stoichiometric base in these oxidation reactions.
2.2.7. Mechanistic Considerations
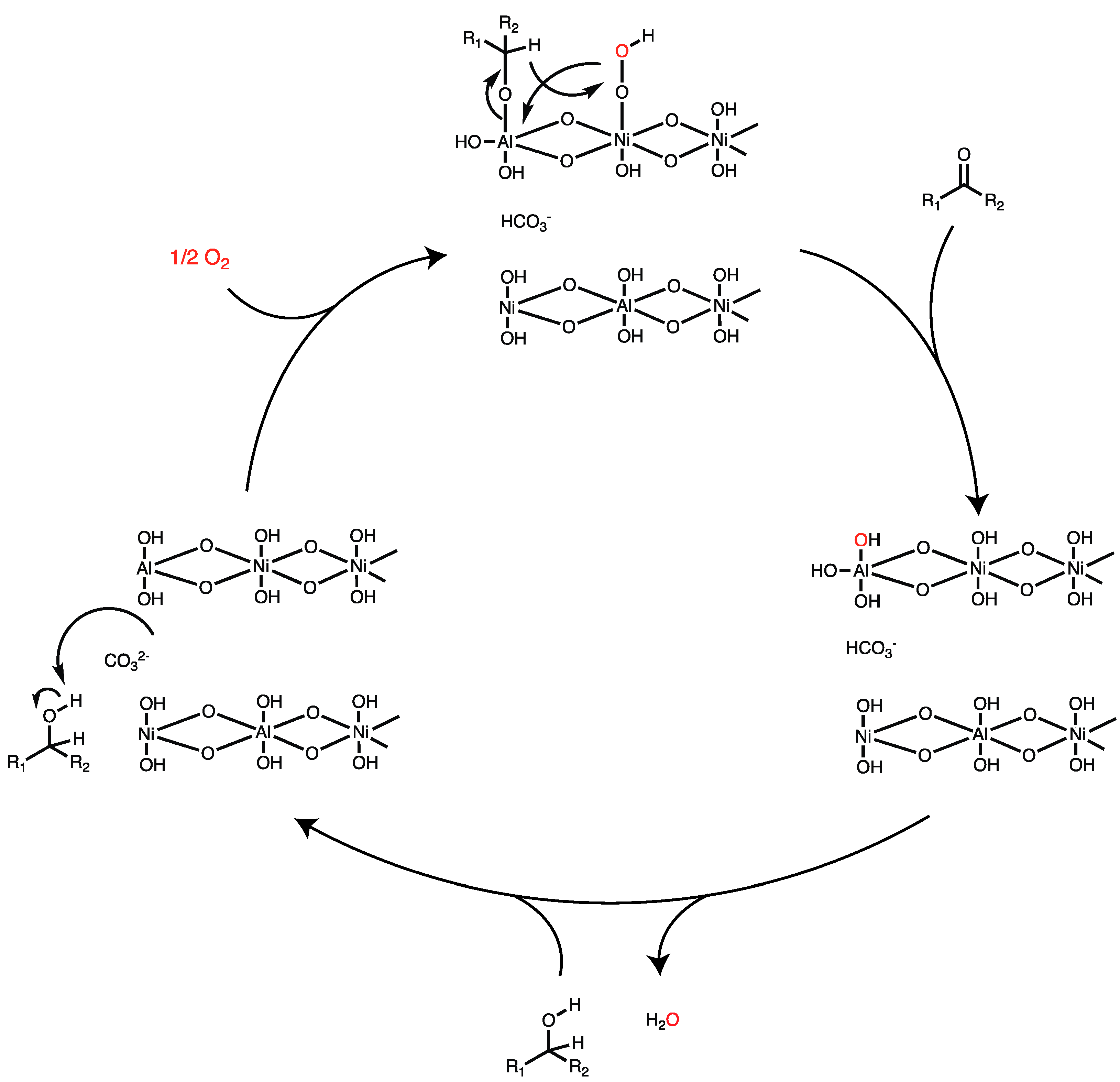
In order to determine whether oxidation proceeds via a two-electron or radical pathway, 1-cyclopropy-1-phenylcarbinol was used as a probe molecule. If oxidation proceeds via a benzylic radical then the highly strained cyclopropyl ring would open, yielding a linear propyl chain. On the other hand, if the reaction proceeds through a hydride shift (as shown in Figure 2) the cyclopropyl ring would remain after benzylic oxidation. Analysis of the reaction mixture post-oxidation revealed the presence of cyclopropyl phenyl ketone in 64% yield, with no evidence of the ring-opening product (Scheme 1). This suggests that the oxidation of benzylic alcohols to ketones proceeds through a two-electron pathway.
Given that catalytic activity is regained and even enhanced after washing with sodium carbonate, it follows that catalysis likely occurs on the catalyst edge sites (110 plane) in LDHs or an equivalent site where interlamellar carbonate anions are exposed to the reactants. Figure 2 shows a plausible mechanism, which is a modified version of that proposed by Tang et al. [30], in which the alcohol is first deprotonated by carbonate to form an alkoxide, which coordinates to an unsaturated metal site. Hydroperoxide oxidation of the metal alkoxide with a concomitant hydride shift from the alkoxide to the hydroperoxide results in net alcohol oxidation and the regeneration of metal hydroxide. Deprotonation of bicarbonate by the metal hydroxide forms water and regenerates a coordinatively unsaturated metal site.
2.2.8. Oxidation of Lignin Model Dimer Compounds over LDH Catalysts
Lignin is an amorphous biopolymer synthesized in planta in the secondary cell walls via oxidative radical condensation of three monolignols (sinapyl, coniferyl, and p-coumaryl alcohol) [31]. As such, it is composed of a variety of linkages, the most abundant of which is the β-O-4 linkage, which can compose as much as 60% of the linkages in hardwood species [31]. Moreover, several recent reports on the depolymerization of lignin have focused on the benzylic oxidation of the β-O-4 linkage, followed by a secondary cleavage step [14,16,17,32,33].
While promising results were obtained when benchtop reactions were performed on non-phenolic compounds 1 and 2, only the dehydration product (4c, 100% yield) was observed when lignin model dimer 4 was subjected to optimized reaction conditions (100% O2, 0.5 g Ni-Al-LDH-1, 150 °C, in DPE). Thus, in order to determine if higher temperatures would enhance the rate of oxidation rather than dehydration, a pressurized reaction system was used. Indeed, when lignin model dimer compounds were reacted under slightly elevated temperatures (i.e., 180 °C) using 8% O2/N2 (50 bar) significant conversion was observed (Table 9, Table 10 and Table 11). It should be noted that the use of pressurized oxygen significantly increases safety concerns for reactions in organic media. Indeed, Stahl and coworkers [34] recently reported limiting oxygen concentrations (LOC) for nine organic solvents, finding that ca. 8% O2 counter-balanced with inert gas was generally non-combustible. Thus, in this study 8% oxygen counter-balanced with nitrogen was used, which provides a nearly stoichiometric amount of oxygen (ca. 3 equiv.) for the oxidation of lignin model dimers. In addition to addressing safety concerns, the use of near stoichiometric amounts of oxygen limits over-oxidation to dicarboxylic acids, which are common products of aromatic ring over-oxidation [35].
Although modest amounts of the ketone resulting from benzylic oxidation were detected, small molecules resulting from cleavage of the model linkages were observed in more substantial yields (Table 9). Wang et al. [36] recently reported that lignin β-O-4 models oxidized at the benzylic position are more easily fragmented than the benzylic alcohol analogue, due to the weakening of the Cβ–O4 bond by approximately 87 kJ/mol. Thus, it follows that the modest yields of 4d are explained by the ready cleavage of the Cβ–O4 bond, as indicated by the observation of the phenol 4b. Product 4a, which likely also results from oxidative cleavage of 4d, was generally present in higher yield than 4b presumably due to phenol polymerization; while the mechanism for oxidative cleavage of 4d is unclear, it is presumed to undergo a similar route as that observed by Mottweiler et al. [37]. Indeed, Mottweiler et al. noted that the use of a Cu-V-LDH in the presence of O2 resulted in a large amount of the A-ring acid and aldehyde. In addition, enol ether product 4c was observed, resulting from benzylic alcohol dehydration. Product 4c is particularly interesting because enol ethers are known to undergo hydrolysis under acidic conditions [38]. This production of enol ethers in lignin would result in an easily hydrolysable linkage. Additionally, product 4e, that likely results from non-oxidative cleavage of 4d, was observed as a minor co-product. Products such as 4e are commonly observed in heterogeneous oxidation reactions of lignin model compounds [39,40,41,42]. Given the tendency of enol ethers to undergo metal-catalyzed cleavage reactions under oxidizing conditions, one can speculate that benzoic acid 4a is produced from the enol ether (4c and 5c) [43]. Indeed, when 4c was used as the feedstock in a control experiment (using Cu-Cr-LDH as catalyst), 4a was produced in low yield (7%) after 24 h.
Surprisingly, the Ni-Cr-LDH catalyst, which successfully oxidized compounds 1–3, produced modest conversions in the cases of dimer models 4–6. This may be due, in part at least, to the catalyst’s small average pore diameter (2.9 nm). Unlike compounds 1–3, which are relatively small, lignin dimer model compounds 4–6 (ca. 1.5 nm) [44] approach the pore diameter of Ni-Cr-LDH (Table 1). Moreover, other catalysts with larger pore diameters showed increased conversion of compounds 4–5. Indeed, Ni-Al-LDH-1, with a pore diameter of 7.3 nm, was found to be the most active catalyst for conversion of the lignin model compounds used in this study, resulting in >99% conversion of models 4 and 5 (Table 9 and Table 10). Unfortunately, yields of individual products were low (<20%).
In these experiments phenols resulting from β-aryl ether cleavage were recovered in low yields. As commonly reported for oxidation of lignin model compounds, phenols are often converted into unidentifiable products [16,17,36]. In order to investigate the stability of phenols under the reaction conditions, guaiacol (4b) was subjected to the same conditions in the absence of catalyst. After 16 h at 180 °C under 8% O2/N2 (720 psi), guaiacol was converted (76%) to unidentifiable products and evident darkening of the reaction solution was observed. This leads us to the conclusion that polymerization of phenols in the presence of oxygen was responsible for their low yields.
While models 4 and 5 serve as sufficiently complex models to establish reactivity trends, they do not accurately represent lignin linkages. Consequently, to better reflect native and technical lignins, model complexity was increased by the addition of a γ-carbinol group (compound 6; Table 11). The addition of a γ-carbinol provides another alcohol that can be oxidized. Once oxidized at the α or γ-position, 6 can undergo retro-aldol reactions further complicating the product mixture. Indeed, a retro-aldol reaction at the oxidized γ-position produces 4 via loss of formaldehyde, while oxidation at the α-position produces a ketone that can also undergo further oxidation.
The oxidative fragmentation of model 6 was investigated using Ni-Al-LDH-1, Cu-Cr-LDH, and Ni-Cr-LDH. Unexpectedly, Ni-Al-LDH-1 catalyzed oxidation resulted in only 34% conversion (Table 11), whereas Cu-Cr-LDH afforded similar conversion as for models 4 and 5 (80–90%). In contrast, Ni-Cr-LDH, which showed similar reactivity trends for models 4 and 5, showed relatively lower conversion for 6 (23%). The benzoic acid (4a) resulting from cleavage of the Cα–Cβ bond was observed in low yield (<7%) for all three catalysts. Furthermore, compounds similar to those produced after the oxidation of 4 and 5 (e.g., 6c) were not observed when 6 was subjected to the same reaction conditions.
The difference in the reactivity of 6 compared to 4 and 5 may have been due to diverging reaction pathways. It is hypothesized that reaction intermediates included both benzylic ketones formed via oxidation and enol ethers via dehydration. Basic sites were likely responsible for the deprotonation of the benzylic carbinol for catalysts such as Ni-Al-LDH-1. After the substrate was coordinated to the catalyst surface, base sites likely deprotonated the β-carbon. Thus, it stands to reason that substitution at the β-carbon by a γ-carbinol likely made deprotonation of the β-carbon more difficult, as suggested by the absence of 6c in the product mixture.
2.2.9. Oxidation of Model Ketone Compounds
To investigate the reason for the low yields of the ketone resulting from benzylic oxidation, models 4d–6d were subjected to the same reaction conditions as 4–6 (Table 9, Table 10 and Table 11) in the absence of a catalyst. As shown in Table 12, oxidized lignin model dimers 4d–6d were converted near quantitatively; this indicates that lignin models, once oxidized, were easily cleaved under oxidative conditions at 180 °C. Moreover, 4a was obtained in good yields from 4d–6d, indicating that the primary pathway for the production of 4a from 4–6 began with benzylic oxidation. The hypothesis that 4d–6d were converted to 4a as a result of oxidation by molecular oxygen was confirmed by the finding that in the absence of O2, 5d and 6d were not converted to identifiable products. Evidently, oxygen was a strong enough oxidant at elevated temperatures to cleave the Cβ–O bond in the ketone form of β-O-4 model compounds, indicating that oxidation of the benzylic alcohol group in β-O-4 models is the more challenging step in terms of the kinetics.
3. Experimental
3.1. Catalyst Preparation
Catalysts were prepared by co-precipitation under conditions of low supersaturation. In general, two solutions, one containing metal nitrates and the other containing a mixture of NaOH and Na2CO3, were added simultaneously and stirred while maintaining a constant pH (generally 9–10, with the exception of Cu-containing LDHs which were precipitated at lower pH). The concentration of the metal nitrate solution used was typically ca. 1.5 M (total metals), while the base solution contained Na2CO3 (ca. 1.0 M) and the calculated amount of NaOH (ca. 3 M) required for complete reaction with the divalent and trivalent metal ions. The solutions were mixed at room temperature at an addition rate of ca. 3 mL·min−1, with vigorous mechanical stirring. Unless otherwise stated, the precipitate was aged in the synthesis solution overnight at 70 °C and isolated by a cycle of centrifuging/decanting/washing with deionized water until the washings reached a neutral pH. The resulting solid was dried at 60 °C in vacuo. Additional synthetic details can be found in the supporting information. All catalysts were stored under atmospheric conditions. Unless otherwise specified, catalysts were used without further pretreatment.
3.2. Catalyst Characterization
Surface area, average pore diameter, and pore volume were determined using a Tristar 3000 porosity system (Micromeritics, Norcross, GA, USA) or a Gemini VII analyzer (Micromeritics, Norcross, GA, USA) using the Brunauer–Emmett–Teller (BET) method by N2 adsorption at −196 °C. Samples were outgassed under vacuum for at least 6 h at 160 °C prior to measurement. Note that powder X-ray diffraction (XRD) measurements confirmed that the LDH structure was retained after this pre-treatment (see Figure S1). XRD measurements were performed on a X’Pert system (PANanalytical, Almelo, The Netherlands) using Cu Kα radiation (λ = 1.5406 Å) and a step size of 0.02°. Elemental analysis was performed on a 720-ES inductively coupled plasma-optical emission spectrometer (Varian/Agilent, Santa Clara, CA, USA). Scanning electron microscopy (SEM) was performed on a S-2700 instrument equipped with a PGT EDS analyzer (Hitachi, Dallas, TX, USA) with a thin window detector and a LaB6 electron gun. FT-IR spectroscopy was performed on a Nicolet 6700 FT-IR instrument (ThermoFisher Scientific, Waltham, MA, USA) equipped with a smart iTR diamond attenuated total reflection (ATR) attachment. In all cases 32 scans were taken with a resolution of 4 cm−1. Details of the pulsed CO2 chemisorption and NH3-TPD measurements are given in the Supporting Information.
3.3. General Procedure for Oxidation of 1-Phenyl Ethanol Derivatives
In a typical reaction, the alcohol compound (2 mmol), solvent (10 mL), and catalyst (0.5 g) were added to a three-neck flask equipped with an oxygen bubbler, a reflux condenser, and a glass stopper. The reaction mixture was stirred at 150 °C for 24 h, after which it was cooled to room temperature and dichloromethane (ca. 10 mL) was added. The reaction mixture was then filtered through Whatman 1 filter paper. The catalyst was washed with dichloromethane or tetrahydrofuran and the washings added to the filtrate. When compound 3 was used as the substrate, 1,4-dimethoxybenzene (0.25 g, 1.8 mmol) was added to the reaction mixture prior to reaction as an internal standard. Conversion, selectivity, and yield were determined using GC (for details see Supplementary Materials).
3.4. General Procedure for Oxidation of Lignin Model Dimer Compounds
Reactions were performed in batch mode using a Parr reactor (50 mL, Hastelloy body) equipped with a magnetic stirrer. The catalyst (0.5 g), solvent (15 mL), lignin model compound (2 mmol), and dodecane (0.25 g, internal standard) were added prior to sealing the reactor. Before each run, the system was purged three times with the reaction gas (ca. 50 bar). After cooling, the reaction mixture was filtered through Whatman 1 filter paper and washed with tetrahydrofuran. The filtrate and washings were then analyzed by GC/MS.
3.5. Derivatization Procedure and Analytical Method for Oxidation Products of Lignin Model Compounds
Following reaction, an aliquot of the reaction mixture (1 g) was derivatized with N,O-bis(trimethysilyl) trifluoroacetamide (0.1 g) with pyridine (0.1 g) as a catalyst at 60 °C for 30 min. The resulting products were analyzed by GC/MS using dodecane as the internal standard.
A 7890B GC System (Agilent, Santa Clara, CA, USA) equipped with an Agilent 5977A Extractor Mass Selective Detector (MSD) and a Flame Ionization Detector (FID) was used for analyses. The multimode inlet (MMI), containing a helix liner, was run in split mode (split ratio 15:1; split flow 48 mL/min) using an initial temperature of 100 °C. Immediately upon injection, the inlet temperature was increased at a rate of 8 °C/min to a final temperature of 380 °C, which was maintained for the duration of the analysis. Similarly, the oven temperature (initially 40 °C) was increased immediately upon injection at a rate of 4 °C/min to 325 °C, followed by a ramp of 10 °C/min to a final temperature of 400 °C, which was maintained for 12.5 min. The total run time was 91.25 min. A J&W VF-5ht column (30 m × 250 µm × 0.1 µm; 450 °C max., Agilent, Santa Clara, CA, USA) was used as the primary column. Column eluents were directed to a Siltek MXT™ Connector (Restek, Bellefonte, PA, USA), which split the flow into two streams: one leading to the MSD (J&W Ultimetal Plus Tubing, 11 m × 0.25 mm ID) and one leading to the FID (J&W Ultimetal Plus Tubing, 5 m × 0.25 mm ID). MS zone temperatures—including those of the MS source (230 °C) and quadrupole (150 °C)—remained constant for the duration of the analysis. The FID was set to 390 °C. Further details can be found in a previous contribution [28].
3.6. Synthesis of Enol Ether Intermediate
To a reaction flask containing 4-methoxyacetophenone (3.0 g, 20.3 mmol) dissolved in ethyl acetate (120 mL) was added pyridinium tribromide (90% technical grade, 7.1 g, 20.0 mmol) at 0 °C. The reaction mixture was stirred for ca. 2 h at room temperature and then quenched with saturated NaHCO3 (200 mL). The organic fraction was then removed and the aqueous fraction was extracted with dichloromethane (33 mL). The combined organic layers were washed with 1 M HCl (35 mL, ×2) and brine (33 mL) and dried over anhydrous Na2SO4. The combined organic layers were then concentrated in vacuo. The crude brominated product (4.47 g, 19.5 mmol, assuming 100% conversion) was dissolved in acetone and guaiacol (2.24 mL, 20.3 mmol) was added along with K2CO3 (11.12 g, 80.47 mmol) and NaI (0.18 g, 1.21 mmol). The reaction mixture was stirred at 80 °C for ca. 3 h. The reaction mixture was then cooled and concentrated via rotary evaporation. The dried product mixture was reconstituted in EtOAc (200 mL) and deionized water (75 mL). The organic fraction was washed with 1 M HCl (75 mL) and brine (40 mL) and dried over anhydrous Na2SO4 and concentrated in vacuo. The carbonyl product was then isolated via recrystallization from hot/cold ethanol (4.073 g, 73.7% yield). The isolated carbonyl compound (2 g, 7.35 mmol) was added to sodium borohydride (0.28 g, 7.38 mmol) in THF/MeOH (60 mL, 5:1) and stirred at room temperature for 90 min. The solution was concentrated via rotary evaporation and the dried mixture was reconstituted in EtOAc (300 mL) and 1 M hydrochloric acid (200 mL). The organic fraction was then washed with 1 M HCl (200 mL) and brine (200 mL) and dried over Na2SO4. The product was concentrated in vacuo (1.91 g, 1.94 mmol, 94.5% yield). The alcohol product (1.34 g, 4.87 mmol) was then subjected to dehydration with methanesulfonic anhydride (0.96 g, 5.52 mmol) and Et3N (1.48 mL, 10.60 mmol) in dichloromethane. The solution was stirred at 0 °C for 30 min and then allowed to reach room temperature overnight. The reaction mixture was diluted with deionized water (74 mL) and extracted with CH2Cl2 (35 mL, ×2). The combined organic fractions were washed with 1 M hydrochloric acid (100 mL) and brine (50 mL) and dried over Na2SO4 and concentrated in vacuo. The cis-product was isolated via flash column chromatography (100 g SiO2) using 0→15% EtOAc/hexanes as the eluent over 20 column volumes (0.48 g, 1.87 mmol, 38.4% yield). 1H NMR (500 MHz, CDCl3, 7.26 ppm): δ 7.68–7.66 (d, 2H, Ar2,6, J = 8.86 Hz), δ 7.1–7.06 (m, 2H, Ar), δ 6.98–6.97 (dd, 1H, Ar, J = 8.13, 1.50 Hz), δ 6.95–6.92 (ddd, 1H, Ar, J = 7.61, 1.5 Hz), δ 6.88–6.87 (d, 2H, Ar3,5, J = 8.91 Hz), δ 6.47–6.45 (d, 1H, Cα, J = 6.80 Hz), δ 5.57–5.55 (d, 1H, Cβ, J = 6.82 Hz), δ 3.90 (s, MeO, 3H), δ 3.81 (s, MeO, 3H). 13C NMR (125 MHz, CDCl3, 77.16 ppm): δ 158.27, 150.24, 146.91, 141.40, 130.11, 128.00, 124.08, 121.07, 117.81, 113.84, 112.84, 109.82, 56.27, 55.38. GC/MS: m/z 256.1 (100%), 121.1 (72%), 77.1 (33%).
4. Conclusions
LDH materials containing a variety of first row transition metal ions were found to be active catalysts for the oxidation of benzylic alcohols and lignin model dimer compounds using phenyl ether as solvent and O2 as the terminal oxidant. Upon repeated use, catalyst activity declined, although washing the spent catalyst (i.e., Ni-Al-LDH-1 and Ni-Cr-LDH) with aqueous Na2CO3 was found to restore activity in the oxidation of 1; this suggests that carbonate species play an essential role in the oxidation reaction. In the conversion of 2 and 3, Ni-containing LDH catalysts were found to show activity for alcohol dehydration, in parallel to alcohol oxidation. Moreover, in the case of phenyl ethanol derivative 3, the formation of significant amounts of unidentifiable products suggests that phenol protection is a necessity in order to prevent the occurrence of polymerization reactions. Typically, for all catalysts only trace amounts of the ketone resulting from benzylic alcohol oxidation were observed for the β-O-4 model compounds. Rather, monomeric products arising from β-aryl ether cleavage were formed. This observation is consistent with the higher reactivity of the ketones, resulting from weakening of the Cβ–O4 bond that was shown to be aerobically cleaved at 180 °C in the absence of catalyst.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/6/3/75/s1, details of catalyst preparation, catalyst leaching study, catalyst reusability, synthesis of lignin model compounds, gas chromatography analysis, catalyst acidity and basicity measurements, Figure S1: X-ray diffractogram of Ni-Al-LDH-1 pre-treated at 160 °C, Figure S2: Scanning electron micrograph of Ni-Al-LDH-1, Figure S3: Scanning electron micrograph of Ni-Cr-LDH, Figure S4: FT-IR analysis of LDH catalysts, Figure S5: NH3-TPD of Ni-Al-LDH-1, Figures S6–S9: NH3-TPD of Ni-Al-LDH-2, Mg-Al-LDH-1, Ni-Cr-LDH, and Ni-Cu-Cr-LDH, Figure S10: X-ray diffractogram of Ni-Cr-LDH after 3 cycles of use in the oxidation of 1, Figure S11: X-ray diffractogram of Ni-Al-LDH-1 after 2 cycles of use in the oxidation of 1, Figure S12: X-ray diffractogram of Ni-Al-LDH-2 after 1 cycle of use in the oxidation of 2. References [45,46,47,48,49,50,51,52] are cited in the supplementary materials.
Author Contributions
M.C., J.K.M. and J.A.J. conceived and designed the experiments; J.K.M., J.A.J., T.M. and A.K. performed the experiments; J.K.M., J.A.J. and M.C. analyzed the data; J.K.M., J.A.J. and M.C. wrote the paper.
Funding
This material is based on work supported by the National Science Foundation under Cooperative Agreement No. 1355438, NSF MRI Award No. 1531637 and NSF-EFRI-0937657 as well as the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02-07ER64494).
Acknowledgments
The authors would also like to thank Tian Li, Yaying Ji, Shelley Hopps, Mark Meier, and Gerald Thomas for their assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sheldon, R.A.; Kochi, J.K. Oxygen-containing compounds. In Metal-Catalyzed Oxidations of Organic Compounds; Academic Press, Inc.: Cambridge, MA, USA, 1981; pp. 350–386. [Google Scholar]
- Markó, I.E.; Giles, P.R.; Tsukazaki, M.; Brown, S.M.; Urch, C.J. Copper-catalyzed oxidation of alcohols to aldehydes and ketones: An efficient, aerobic alternative. Science 1996, 274, 2044–2046. [Google Scholar] [CrossRef] [PubMed]
- Cainelli, G.; Cardillo, G. Oxidation of alcohols. In Chromium Oxidations in Organic Chemistry; Springer: Berlin/Heidelberg, Germany, 1984; pp. 118–216. [Google Scholar]
- Collins, J.C.; Hess, W.W.; Frank, F.J. Dipyridine-chromium(VI) oxide oxidation of alcohols in dichloromethane. Tetrahedron Lett. 1968, 9, 3363–3366. [Google Scholar] [CrossRef]
- Corey, E.J.; Suggs, J.W. Pyridinium chlorochromate. An efficient reagent for oxidation of primary and secondary alcohols to carbonyl compounds. Tetrahedron Lett. 1975, 16, 2647–2650. [Google Scholar] [CrossRef]
- Corey, E.J.; Schmidt, G. Useful procedures for the oxidation of alcohols involving pyridinium dichromate in aprotic media. Tetrahedron Lett. 1979, 20, 399–402. [Google Scholar] [CrossRef]
- Kim, K.S.; Chung, S.; Cho, I.H.; Hahn, C.S. Selective oxidation of alcohols by manganates. Tetrahedron Lett. 1989, 30, 2559–2562. [Google Scholar] [CrossRef]
- Pfitzner, K.E.; Moffatt, J.G. Sulfoxide-carbodiimide reactions. I. A facile oxidation of alcohols. J. Am. Chem. Soc. 1965, 87, 5661–5670. [Google Scholar] [CrossRef]
- Fenselau, A.H.; Moffatt, J.G. Sulfoxide-carbodiimide reactions. III.1 mechanism of the oxidation reaction. J. Am. Chem. Soc. 1966, 88, 1762–1765. [Google Scholar] [CrossRef]
- Albright, J.D.; Goldman, L. Dimethyl sulfoxide-acid anhydride mixtures. New reagents for oxidation of alcohols. J. Am. Chem. Soc. 1965, 87, 4214–4216. [Google Scholar] [CrossRef]
- Mancuso, A.J.; Swern, D. Activated dimethyl sulfoxide: Useful reagents for synthesis. Synthesis 1981, 1981, 165–185. [Google Scholar] [CrossRef]
- Huang, S.L.; Omura, K.; Swern, D. Further studies on the oxidation of alcohols to carbonyl compounds by dimethyl sulfoxide/trifluoroacetic anhydride. Synthesis 1978, 1978, 297–299. [Google Scholar] [CrossRef]
- Omura, K.; Swern, D. Oxidation of alcohols by “activated” dimethyl sulfoxide. A preparative, steric and mechanistic study. Tetrahedron 1978, 34, 1651–1660. [Google Scholar] [CrossRef]
- Mobley, J.K.; Yao, S.G.; Crocker, M.; Meier, M. Oxidation of lignin and lignin β-O-4 model compounds via activated dimethyl sulfoxide. RSC Adv. 2015, 5, 105136–105148. [Google Scholar] [CrossRef]
- Parikh, J.R.; Doering, W.V.E. Sulfur trioxide in the oxidation of alcohols by dimethyl sulfoxide. J. Am. Chem. Soc. 1967, 89, 5505–5507. [Google Scholar] [CrossRef]
- Lancefield, C.S.; Ojo, O.S.; Tran, F.; Westwood, N.J. Isolation of functionalized phenolic monomers through selective oxidation and C–O bond cleavage of the β-O-4 linkages in lignin. Angew. Chem. Int. Ed. 2015, 54, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, A.; Azarpira, A.; Kim, H.; Ralph, J.; Stahl, S.S. Chemoselective metal-free aerobic alcohol oxidation in lignin. J. Am. Chem. Soc. 2013, 135, 6415–6418. [Google Scholar] [CrossRef] [PubMed]
- Hoover, J.M.; Stahl, S.S. A highly practical copper(I)/tempo catalyst system for chemoselective aerobic oxidation of primary alcohols. J. Am. Chem. Soc. 2011, 133, 16901–16910. [Google Scholar] [CrossRef] [PubMed]
- Hoover, J.M.; Steves, J.E.; Stahl, S.S. Copper(I)/tempo-catalyzed aerobic oxidation of primary alcohols to aldehydes with ambient air. Nat. Protoc. 2012, 7, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Kochi, J.K. Oxygen-containing compounds. In Metal-Catalyzed Oxidations of Organic Compounds; Academic Press, Inc.: Cambridge, MA, USA, 1981; pp. 189–214. [Google Scholar]
- Choudary, B.M.; Kantam, M.L.; Rahman, A.; Reddy, C.V.; Rao, K.K. The first example of activation of molecular oxygen by nickel in Ni-Al hydrotalcite: A novel protocol for the selective oxidation of alcohols. Angew. Chem. Int. Ed. 2001, 40, 763–766. [Google Scholar] [CrossRef]
- Mobley, J.K.; Crocker, M. Catalytic oxidation of alcohols to carbonyl compounds over hydrotalcite and hydrotalcite-supported catalysts. RSC Adv. 2015, 5, 65780–65797. [Google Scholar] [CrossRef]
- Kawabata, T.; Shinozuka, Y.; Ohishi, Y.; Shishido, T.; Takaki, K.; Takehira, K. Nickel containing Mg-Al hydrotalcite-type anionic clay catalyst for the oxidation of alcohols with molecular oxygen. J. Mol. Catal. A Chem. 2005, 236, 206–215. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Chaudhari, P.A.; Narkhede, V.S. Solvent-free liquid phase oxidation of benzyl alcohol to benzaldehyde by molecular oxygen using non-noble transition metal containing hydrotalcite-like solid catalysts. Catal. Commun. 2003, 4, 171–175. [Google Scholar] [CrossRef]
- Kaneda, K.; Yamashita, T.; Matsushita, T.; Ebitani, K. Heterogeneous oxidation of allylic and benzylic alcohols catalyzed by Ru-Al-Mg hydrotalcites in the presence of molecular oxygen. J. Org. Chem. 1998, 63, 1750–1751. [Google Scholar] [CrossRef]
- De Roy, A.; Forano, C.; Besse, J.P. Layered double hydroxides: Synthesis and post-synthesis modification. In Layered Double Hydroxides: Present and Future; Rives, V., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2001. [Google Scholar]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Morgan, T.; Santillan-Jimenez, E.; Huff, K.; Javed, K.R.; Crocker, M. Use of dual detection in the gas chromatographic analysis of oleaginous biomass feeds and biofuel products to enable accurate simulated distillation and lipid profiling. Energy Fuels 2017, 31, 9498–9506. [Google Scholar] [CrossRef]
- Matsushita, T.; Ebitani, K.; Kaneda, K. Highly efficient oxidation of alcohols and aromatic compounds catalysed by the Ru-Co-Al hydrotalcite in the presence of molecular oxygen. Chem. Commun. 1999, 265–266. [Google Scholar] [CrossRef]
- Tang, Q.; Wu, C.; Qiao, R.; Chen, Y.; Yang, Y. Catalytic performances of Mn–Ni mixed hydroxide catalysts in liquid-phase benzyl alcohol oxidation using molecular oxygen. Appl. Catal. A Gen. 2011, 403, 136–141. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, A.; Ulbrich, A.; Coon, J.J.; Stahl, S.S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 2014, 515, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.D.; Yao, S.G.; Meier, M.S.; Mobley, J.K.; Crocker, M. Selective cleavage of the Cα–Cβ linkage in lignin model compounds via Baeyer–Villiger oxidation. Org. Biomol. Chem. 2015, 13, 3243–3254. [Google Scholar] [CrossRef] [PubMed]
- Osterberg, P.M.; Niemeier, J.K.; Welch, C.J.; Hawkins, J.M.; Martinelli, J.R.; Johnson, T.E.; Root, T.W.; Stahl, S.S. Experimental limiting oxygen concentrations for nine organic solvents at temperatures and pressures relevant to aerobic oxidations in the pharmaceutical industry. Org. Process Res. Dev. 2015, 19, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Vardon, D.R.; Franden, M.A.; Johnson, C.W.; Karp, E.M.; Guarnieri, M.T.; Linger, J.G.; Salm, M.J.; Strathmann, T.J.; Beckham, G.T. Adipic acid production from lignin. Energy Environ. Sci. 2015, 8, 617–628. [Google Scholar] [CrossRef]
- Wang, M.; Lu, J.; Zhang, X.; Li, L.; Li, H.; Luo, N.; Wang, F. Two-step, catalytic C–C bond oxidative cleavage process converts lignin models and extracts to aromatic acids. ACS Catal. 2016, 6, 6086–6090. [Google Scholar] [CrossRef]
- Mottweiler, J.; Puche, M.; Rauber, C.; Schmidt, T.; Concepcion, P.; Corma, A.; Bolm, C. Copper- and vanadium-catalyzed oxidative cleavage of lignin using dioxygen. Chemsuschem 2015, 8, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Deng, W.; Zhang, H.; Wu, X.; Li, R.; Zhang, Q.; Wang, Y. Oxidative conversion of lignin and lignin model compounds catalyzed by CeO2-supported Pd nanoparticles. Green Chem. 2015, 17, 5009–5018. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, J.; Chen, X.; Ma, D.; Yan, N. A metal-free, carbon-based catalytic system for the oxidation of lignin model compounds and lignin. ChemPlusChem 2014, 79, 825–834. [Google Scholar] [CrossRef]
- Sturgeon, M.R.; O’Brien, M.H.; Ciesielski, P.N.; Katahira, R.; Kruger, J.S.; Chmely, S.C.; Hamlin, J.; Lawrence, K.; Hunsinger, G.B.; Foust, T.D.; et al. Lignin depolymerisation by nickel supported layered-double hydroxide catalysts. Green Chem. 2014, 16, 824–835. [Google Scholar] [CrossRef]
- Kruger, J.S.; Cleveland, N.S.; Zhang, S.; Katahira, R.; Black, B.A.; Chupka, G.M.; Lammens, T.; Hamilton, P.G.; Biddy, M.J.; Beckham, G.T. Lignin depolymerization with nitrate-intercalated hydrotalcite catalysts. ACS Catal. 2016, 6, 1316–1328. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Fomova, N.A.; Kuznetsov, B.N.; Ivanchenko, N.M.; Kudryashev, A.V. On the mechanism of vanillin formation in the catalytic oxidation of lignin with oxygen. React. Kinet. Catal. Lett. 1995, 55, 161–170. [Google Scholar] [CrossRef]
- Jennings, J.A.; Parkin, S.; Munson, E.; Delaney, S.P.; Calahan, J.L.; Isaacs, M.; Hong, K.; Crocker, M. Regioselective Baeyer–Villiger oxidation of lignin model compounds with tin beta zeolite catalyst and hydrogen peroxide. RSC Adv. 2017, 7, 25987–25997. [Google Scholar] [CrossRef] [Green Version]
- Dawange, M.; Galkin, M.V.; Samec, J.S.M. Selective Aerobic Benzylic Alcohol Oxidation of Lignin Model Compounds: Route to Aryl Ketones. ChemCatChem 2015, 7, 401–404. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, M.; Kim, I. Palladium-Catalyzed α-Arylation of Aryloxyketones for the Synthesis of 2,3-Disubstituted Benzofurans. J. Org. Chem. 2014, 79, 6153–6163. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Yan, M.; Wang, J.; Zhang, Z.C.; Yao, K. Selective Reductive Cleavage of Inert Aryl C–O Bonds by an Iron Catalyst. Angew. Chem. In. Ed. 2013, 52, 12674–12678. [Google Scholar] [CrossRef] [PubMed]
- Tien Thao, N.; Kim Huyen, L.T. Catalytic oxidation of styrene over Cu-doped hydrotalcites. Chem. Eng. J. 2015, 279, 840–850. [Google Scholar] [CrossRef]
- Ross, G.J.; Kodama, H. Properties of a Synthetic Magnesium–Aluminum Carbonate Hydroxide and its Relationship to Magnesium–Aluminum Double Hydroxide, Manasseite and Hydrotalcite. Am. Mineral. 1967, 52, 1036–1047. [Google Scholar]
- Kloprogge, J.T.; Frost, R.L. Fourier Transform Infrared and Raman Spectroscopic Study of the Local Structure of Mg-, Ni-, and Co-Hydrotalcites. J. Solid State Chem 1999, 146, 506–515. [Google Scholar] [CrossRef]
- Ehlsissen, K.T.; Delahaye-Vidal, A.; Genin, P.; Figlarz, M.; Willmann, P. Preparation and characterization of turbostratic Ni/Al layered double hydroxides for nickel hydroxide electrode applications. J. Mater. Chem. 1993, 3, 883–888. [Google Scholar] [CrossRef]
- Kooli, F.; Rives, V.; Ulibarri, M.A. Preparation and Study of Decavanadate-Pillared Hydrotalcite-like Anionic Clays Containing Transition Metal Cations in the Layers. 2. Samples containing Magnesium-Chromium and Nickel-Chromium. Inorg. Chem. 1995, 34, 5122–5128. [Google Scholar] [CrossRef]
Figure 1.
X-ray diffraction (XRD) analysis of synthesized LDH catalysts.

Scheme 1.
Oxidation of 1-cyclopropyl 1-phenylcarbinol.

Figure 2.
Plausible mechanism for LDH-catalyzed aerobic alcohol oxidation.


{kind=link}
{kind=link}
{kind=link}
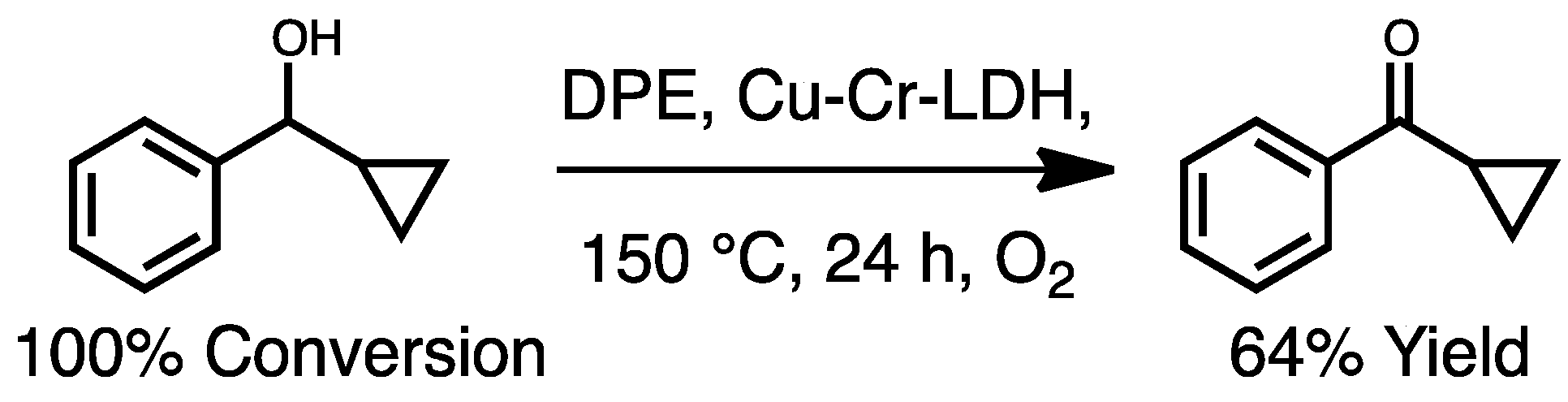{kind=link}
Table 1.
Elemental analysis and gas adsorption data for LDH catalysts.
| Entry | Catalyst | Target Formula | Actual Formula | χ | BET SA (m2·g−1) | Avg. Pore Volume (cm3·g−1) | Avg. Pore Diameter (nm) | Basicity a (μmol CO2 ads./g Catalyst) | Acidity b (μmol NH3 ads./g Catalyst) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Mg-Al-LDH-1 | Mg0.75Al0.25 | Mg0.68Al0.32 | 0.32 | 100.5 | 0.429 | 17.1 | 71.6 | 136.0 |
| 2 | Mg-Al-LDH-2 | Mg0.80Al0.20 | Mg0.73Al0.27 | 0.27 | 36.4 | 0.211 | 23.2 | 40.5 | 75.3 |
| 3 | Ni-Al-LDH-1 | Ni0.67Al0.33 | Ni0.65Al0.35 | 0.35 | 136.6 | 0.249 | 7.3 | 79.2 | 281.4 |
| 4 | Ni-Al-LDH-2 | Ni0.75Al0.25 | Ni0.73Al0.27 | 0.27 | 127.8 | 0.286 | 9.0 | 157.4 | 271.1 |
| 5 | Ni-Cr-LDH | Ni0.67Cr0.33 | Ni0.68Cr0.32 | 0.32 | 76.6 | 0.055 | 2.9 | 51.8 | 383.1 |
| 6 | Ni-Cu-Cr-LDH | Ni0.34Cu0.32Cr0.34 | Ni0.35Cu0.33Cr0.33 | 0.33 | 103.2 | 0.251 | 9.5 | 14.3 | 39.7 |
| 7 | Cu-Cr-LDH | Cu0.67Cr0.33 | Cu0.68Cr0.32 | 0.32 | 134.2 | 0.177 | 5.1 | 27.8 | 51.5 |
a Determined by pulsed CO2 chemisorption; b Evaluated by ammonia temperature programmed desorption (NH3-TPD).
Table 2.
Conversion of 1-phenyl ethanol (1) to acetophenone (1a) in selected solvents.
| Entry | Solvent | Time (h) | Temperature (°C) | 1a Yield (%) |
|---|---|---|---|---|
| 1 | Phenyl Ether | 6 | 90 | 49 |
| 2 | Phenyl Ether | 24 | 150 | 66 |
| 3 | Toluene | 6 | 85 | 99 |
| 4 | Dimethyl Sulfoxide | 6 | 120 | 4 |
| 5 | Chloroform | 6 | 60 | 1 |
| 6 | Hexachloroacetone | 6 | 85 | <1 |
| 7 | 1,4-Dioxane | 6 | 85 | 8 |
| 8 | Benzonitrile | 24 | 90 | 1 |
| 9 | 1,2-Dichlorobenzene | 6 | 85 | <1 |
Table 3.
Conversion of 1-phenyl ethanol, 1 to acetophenone, 1a in phenyl ether with various LDH catalysts.
Table 3.
Conversion of 1-phenyl ethanol, 1 to acetophenone, 1a in phenyl ether with various LDH catalysts.

| Entry | Catalyst | Conversion (%) a | Selectivity (%) |
|---|---|---|---|
| 1 | Mg-Al-LDH-1 | 8 | >99 |
| 2 | Mg-Al-LDH-2 | 12 | >99 |
| 3 | Ni-Al-LDH-1 | 66 | >99 |
| 4 b,c | Ni-Al-LDH-1 | 91 ± 3 | >99 |
| 5 | Ni-Al-LDH-2 | 91 | >99 |
| 6 c | Ni-Cr-LDH | 92 ± 5 | >99 |
| 7 | Ni-Cu-Cr-LDH | 80 | >99 |
| 8 | Cu-Cr-LDH | >99 | >99 |
a 24 h reaction time, 10 mL of phenyl ether as solvent; b Calcined at 175 °C/3 h, reaction time 23 h; c Average of 3 reactions ± st. dev.
Table 4.
Ni-Cr-LDH catalyst loading study a.

| Entry | Catalyst Loading (g) | Conversion (%) | Selectivity (%) to 1a |
|---|---|---|---|
| 1 | 0.05 | 18 | >99 |
| 2 | 0.1 | 37 | >99 |
| 3 | 0.25 | 69 | >99 |
| 4 b | 0.5 | 92 ± 5 | >99 |
a 2 mmol of starting material, 10 mL phenyl ether; b Average of 3 reactions ± st. dev.
Table 5.
Conversion of 2 over various LDH catalysts in phenyl ether with O2.

| Entry | Catalyst | Conversion (%) | Ketone Yield (%) 2a | Alkene Yield (%) 2b |
|---|---|---|---|---|
| 1 | None | 3 | 1 | 1 |
| 2 | Ni-Al-LDH-1 | 92 | 74 | 18 |
| 3 a | Ni-Al-LDH-1 | 83 | 75 | 8 |
| 4 b | Ni-Al-LDH-2 | 99 ± 0 | 71 ± 8 | 27 ± 7 |
| 5 | Ni-Cr-LDH | 99 | 96 | 3 |
| 6 | Ni-Cu-Cr-LDH | 90 | 88 | 2 |
| 7 | Cu-Cr-LDH | >99 | 50 | 0 |
a Calcined at 175 °C for 3 h; b Average of 3 reactions ± st. dev.
Table 6.
Conversion of 3 over LDHs in phenyl ether with O2.

| Entry | Catalyst | Conversion (%) | Ketone Yield 3a (%) | Alkene 3b Yield (%) | Aldehyde 3c Yield (%) |
|---|---|---|---|---|---|
| 1 | None (Blank) | 27 | 8 | 18 | 1 |
| 2 a | Ni-Al-LDH-1 | 99 | 3 | 9 | 5 |
| 3 | Ni-Al-LDH-2 | 99 | 1 | 4 | 1 |
| 4 | Ni-Cr-LDH | 58 | 8 | 29 | 2 |
| 5 | Ni-Cu-Cr-LDH | 98 | 9 | 5 | 7 |
| 6 | Cu-Cr-LDH | >99 | 0 | 0 | 0 |
a Calcined at 175 °C for 3 h.
Table 7.
Catalyst reusability study in the oxidation of 1 (phenyl ether as solvent).
| Cycle # | Catalyst | Temperature (°C) | Time (h) | Yield of 1a (%) |
|---|---|---|---|---|
| 1 | Ni-Cr-LDH | 150 | 24 | 78 |
| 2 | Ni-Cr-LDH | 150 | 24 | 35 |
| 3 | Ni-Cr-LDH | 150 | 24 | 15 |
| 4 b | Ni-Cr-LDH | 150 | 24 | 28 |
| 1 a | Ni-Al LDH-1 | 150 | 24 | 72 |
| 2 a | Ni-Al LDH-1 | 150 | 24 | 0 |
| 3 a,b | Ni-Al LDH-1 | 150 | 24 | 100 |
a Catalyst thermally pretreated at 175 °C for 3 h; b Catalyst regeneration with Na2CO3 solution.
Table 8.
Formulae of LDH catalysts and molar ratio of carbonate to 1.
| LDH | Formula | mmol CO32−/mmol 1 |
|---|---|---|
| Mg-Al-LDH-1 | [Mg0.68Al0.32(OH)2]0.32+(CO32−)0.16 | 0.58 |
| Mg-Al-LDH-2 | [Mg0.73Al0.27(OH)2]0.27+(CO32−)0.135 | 0.50 |
| Ni-Al-LDH-1 | [Ni0.65Al0.35(OH)2]0.35+(CO32−)0.175 | 0.47 |
| Ni-Al-LDH-2 | [Ni0.73Al0.27(OH)2]0.27+(CO32−)0.135 | 0.37 |
| Ni-Cr-LDH | [Ni0.68Cr0.32(OH)2]0.32+(CO32−)0.160 | 0.43 |
| Ni-Cu-Cr-LDH | [Ni0.35 Cu0.33Cr0.33(OH)2]0.33+(CO32−)0.163 | 0.40 |
| Cu-Cr-LDH | [Cu0.68Cr0.32(OH)2]0.32+(CO32−)0.160 | 0.39 |
Table 9.
Conversion of 4 over LDH catalysts in phenyl ether with O2.

| Catalyst | Conversion (%) | Yield (%) | ||||
|---|---|---|---|---|---|---|
| 4a | 4b | 4c | 4d | 4e | ||
| - | 58 | 6 | 5 | 3 | 21 | 0 |
| Ni-Al-LDH-1 | >99 | 12 | 18 | 14 | 0 | 7 |
| Cu-Cr-LDH | 81 | 1 | 3 | 0 | 0 | 0 |
| Cu-Cr-LDH a | 93 | 3 | 1 | 0 | 0 | 0 |
| Ni-Cr-LDH | 56 | 3 | 4 | 1 | 2 | 0 |
aReaction time 24 h.
Table 10.
Conversion of 5 over LDH catalysts in phenyl ether with O2.

| Catalyst | Conversion (%) | Yield (%) | ||||
|---|---|---|---|---|---|---|
| 4a | 5b | 5c | 5d | 4e | ||
| - | 38 | 0 | 1 | 0 | 3 | 0 |
| Ni-Al LDH-1 a | >99 | 15 | 10 | 8 | 0 | 5 |
| Cu-Cr-LDH | 65 | 4 | 2 | 0 | 0 | 1 |
| Cu-Cr-LDH b | 84 | 8 | 1 | 0 | 0 | 1 |
| Ni-Cr-LDH | 41 | 2 | 1 | 0 | 0 | 0 |
a Calcined at 175 °C for 3 h; b Reaction time 24 h.
Table 11.
Conversion of 6 over LDH catalysts in phenyl ether with O2.

| Catalyst | Conversion (%) | Yield (%) | ||||
|---|---|---|---|---|---|---|
| 4a | 4b | 6c | 6d | 4e | ||
| - | 6 | 3 | 3 | 0 | 5 | 0 |
| Ni-Al-LDH-1 | 34 | 2 | 6 | 0 | 1 | 2 |
| Cu-Cr-LDH | 80 | 7 | 7 | 0 | 0 | 4 |
| Ni-Cr-LDH | 23 | 3 | 1 | 0 | 1 | 0 |
Table 12.
Conversion of 4d–6d in phenyl ether with O2.

| Model | Conversion (%) | Yield (%) | |||
|---|---|---|---|---|---|
| 4a | 4b | 5b | Anisoin | ||
| 4d | >99 | 43 | 5 | - | 0 |
| 5d | >99 | 60 | - | 22 | 0 |
| 5d a | 65 | 1 | - | 9 | 24 |
| 6d | >99 | 50 | 18 | - | 0 |
| 6d a | 82 | 1 | 1 | 6 | 0 |
a Reaction was performed under an argon atmosphere.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mobley, J.K.; Jennings, J.A.; Morgan, T.; Kiefer, A.; Crocker, M. Oxidation of Benzylic Alcohols and Lignin Model Compounds with Layered Double Hydroxide Catalysts. Inorganics 2018, 6, 75. https://doi.org/10.3390/inorganics6030075
AMA Style
Mobley JK, Jennings JA, Morgan T, Kiefer A, Crocker M. Oxidation of Benzylic Alcohols and Lignin Model Compounds with Layered Double Hydroxide Catalysts. Inorganics. 2018; 6(3):75. https://doi.org/10.3390/inorganics6030075
Chicago/Turabian StyleMobley, Justin K., John A. Jennings, Tonya Morgan, Axel Kiefer, and Mark Crocker. 2018. "Oxidation of Benzylic Alcohols and Lignin Model Compounds with Layered Double Hydroxide Catalysts" Inorganics 6, no. 3: 75. https://doi.org/10.3390/inorganics6030075
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.