Ddpd as Expanded Terpyridine: Dramatic Effects of Symmetry and Electronic Properties in First Row Transition Metal Complexes

 ,
,
Abstract
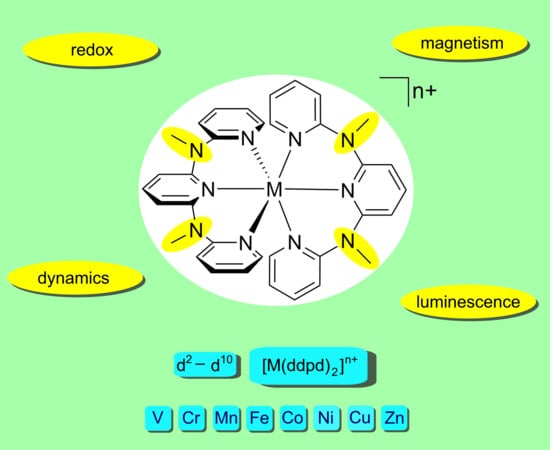
:
1. Introduction
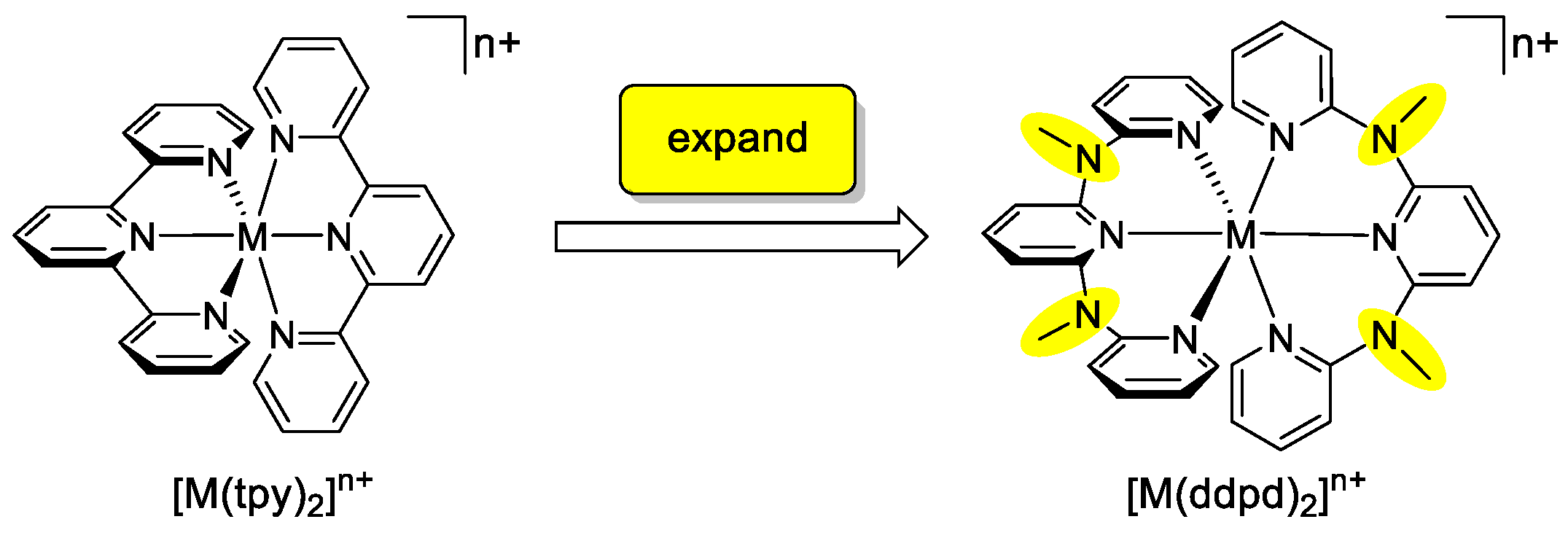
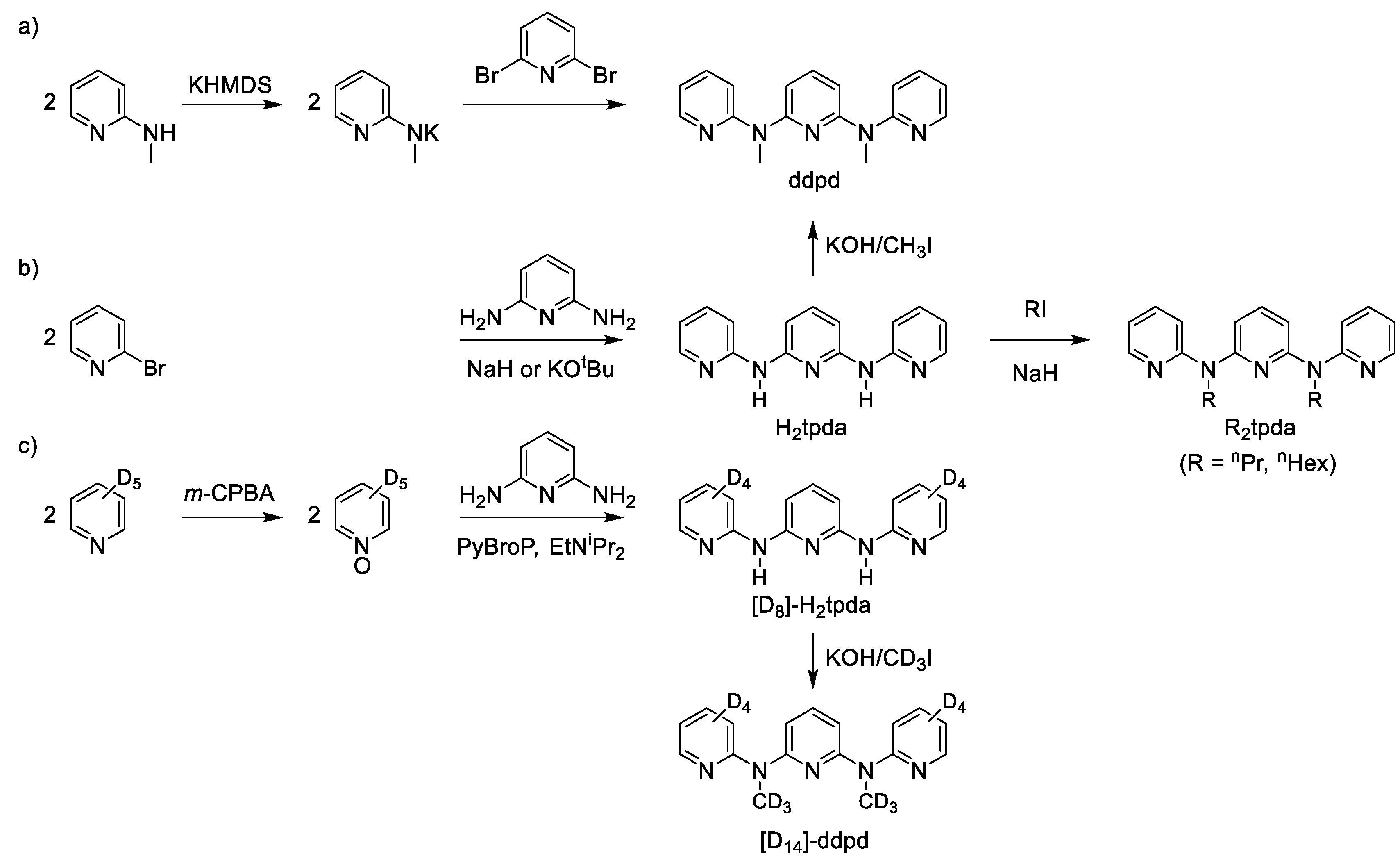
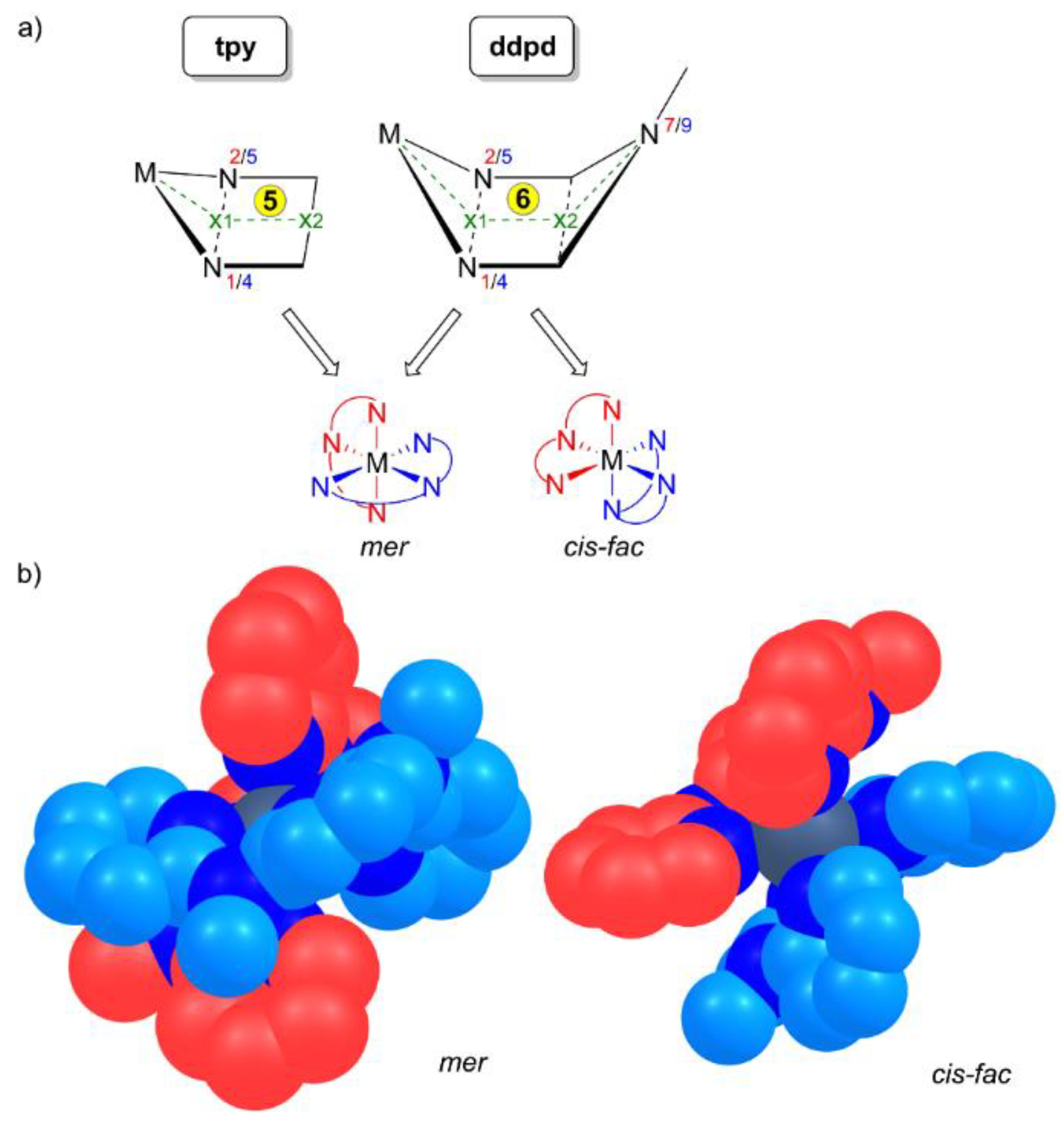
2. Properties of the Ligand ddpd
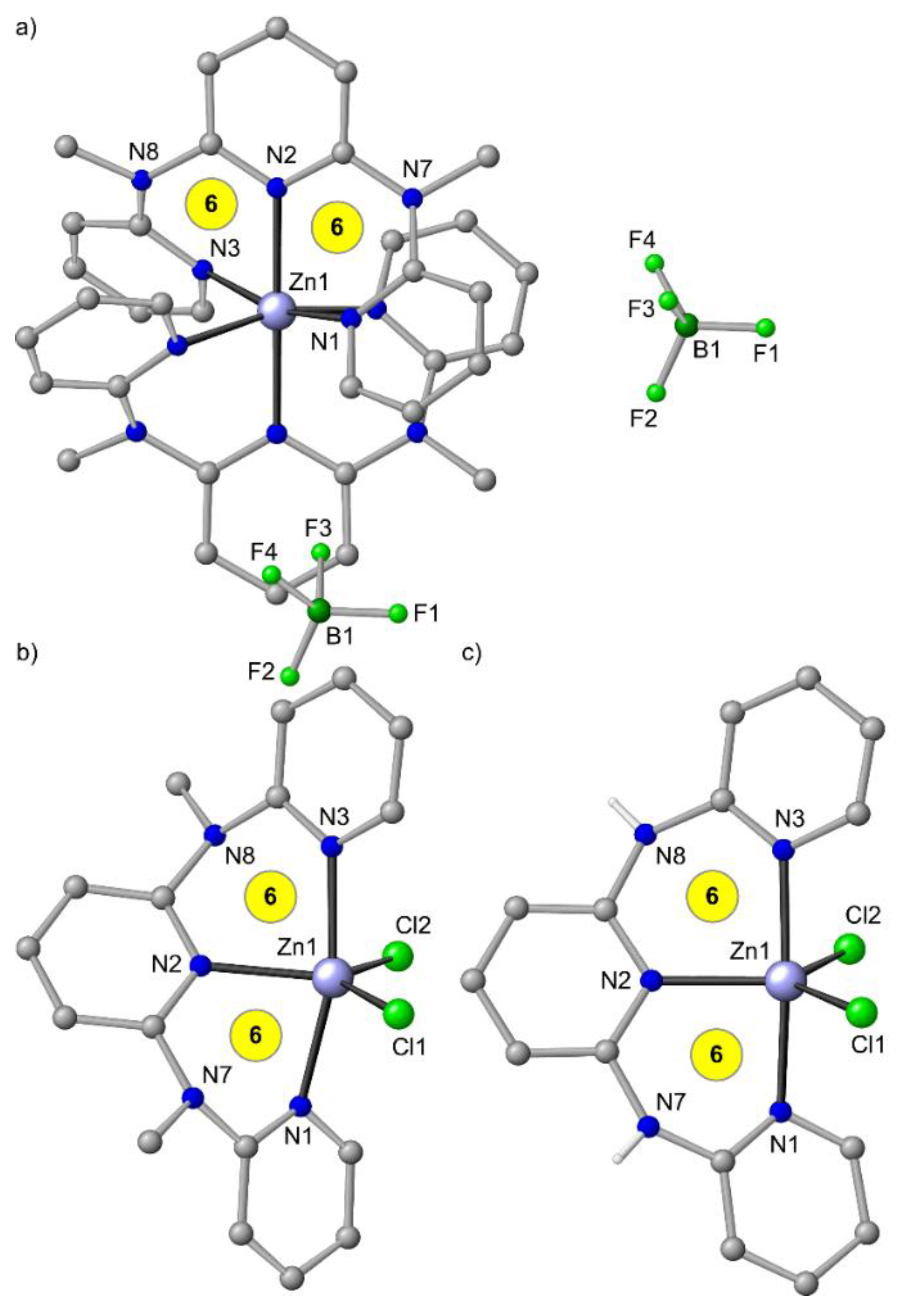
3. mer-[Zn(ddpd)2]2+ and ZnCl2(ddpd) [d10]
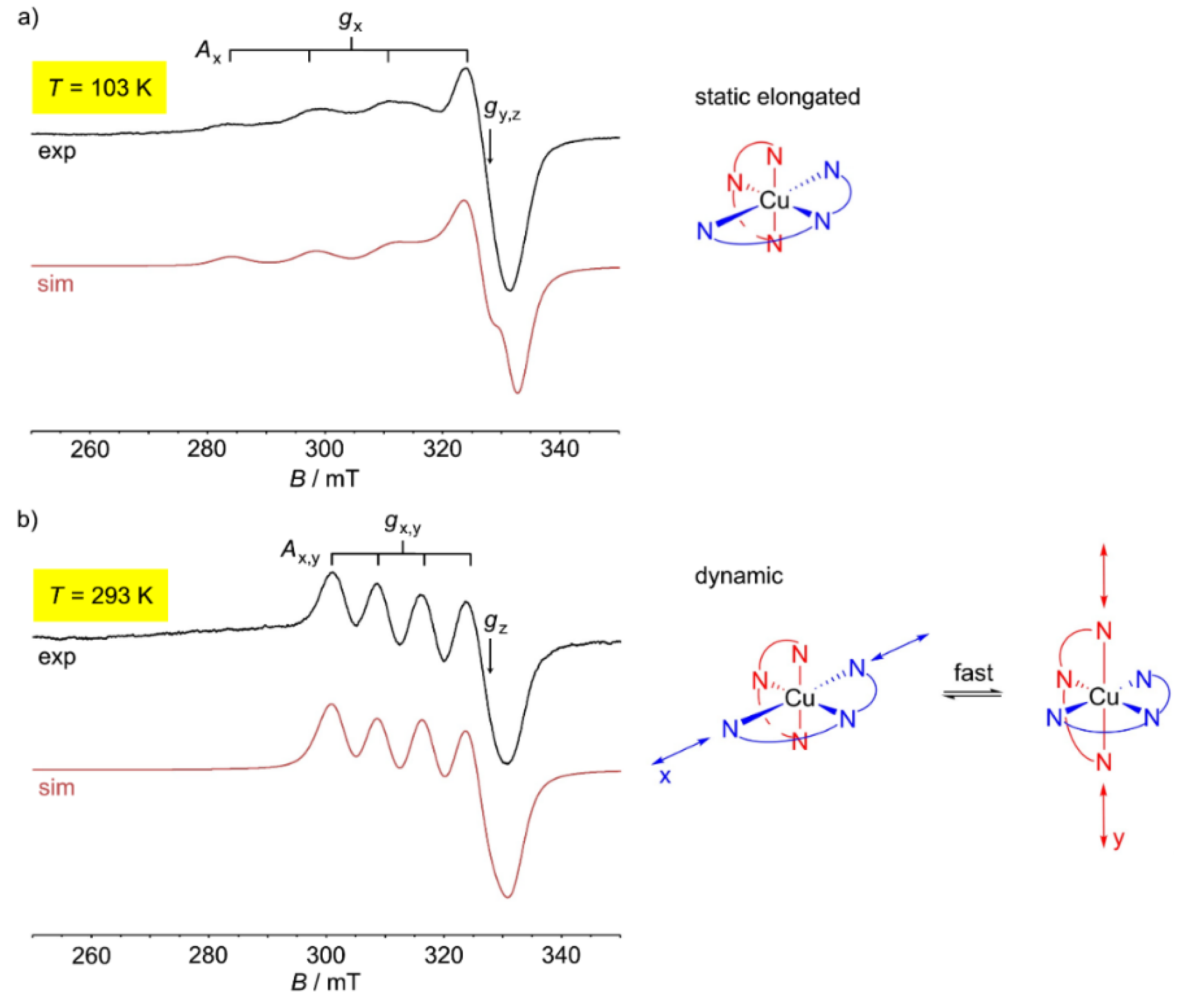
4. mer-[Cu(ddpd)2]2+ [d9]
5. mer-[Ni(ddpd)2]2+ [d8]
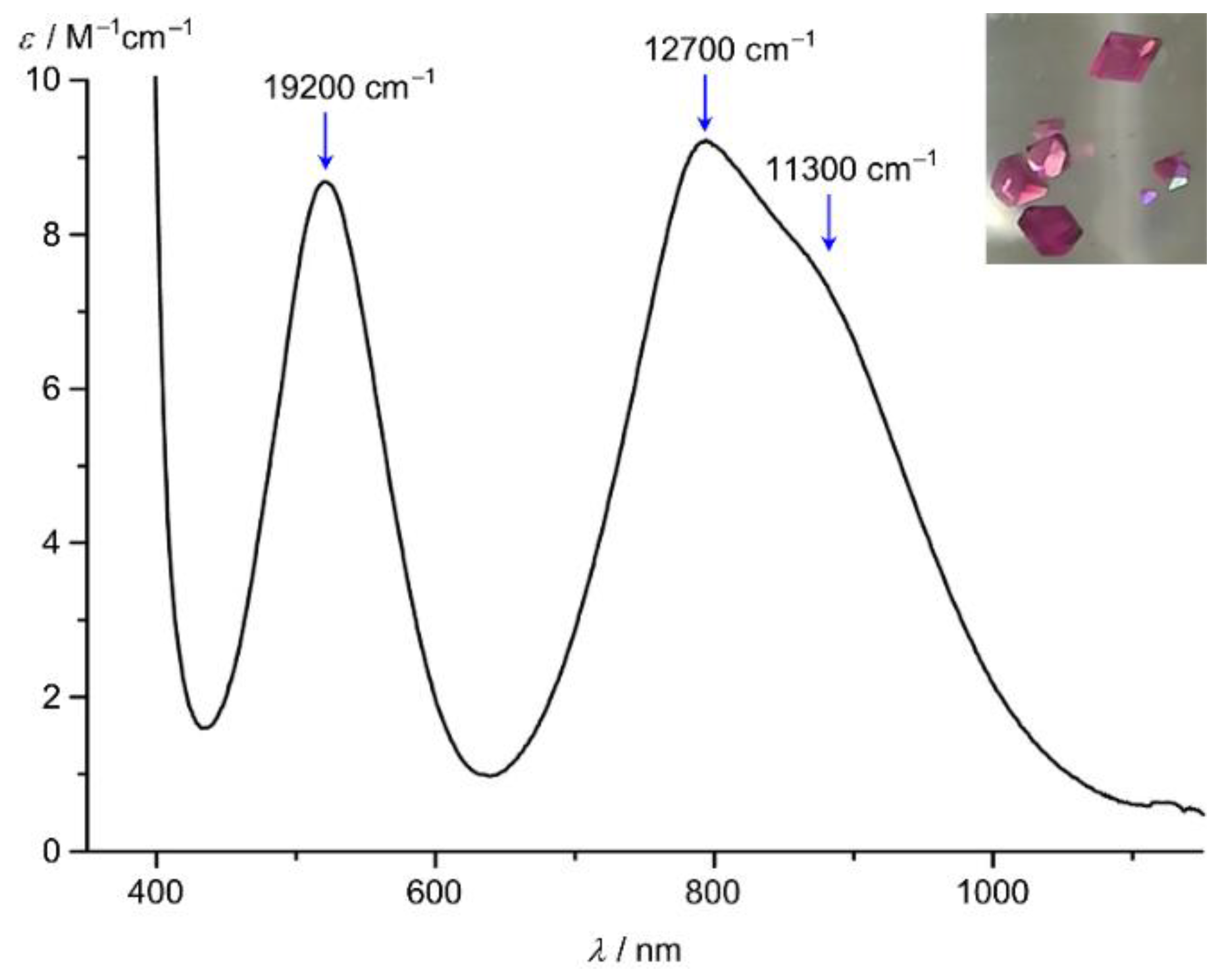
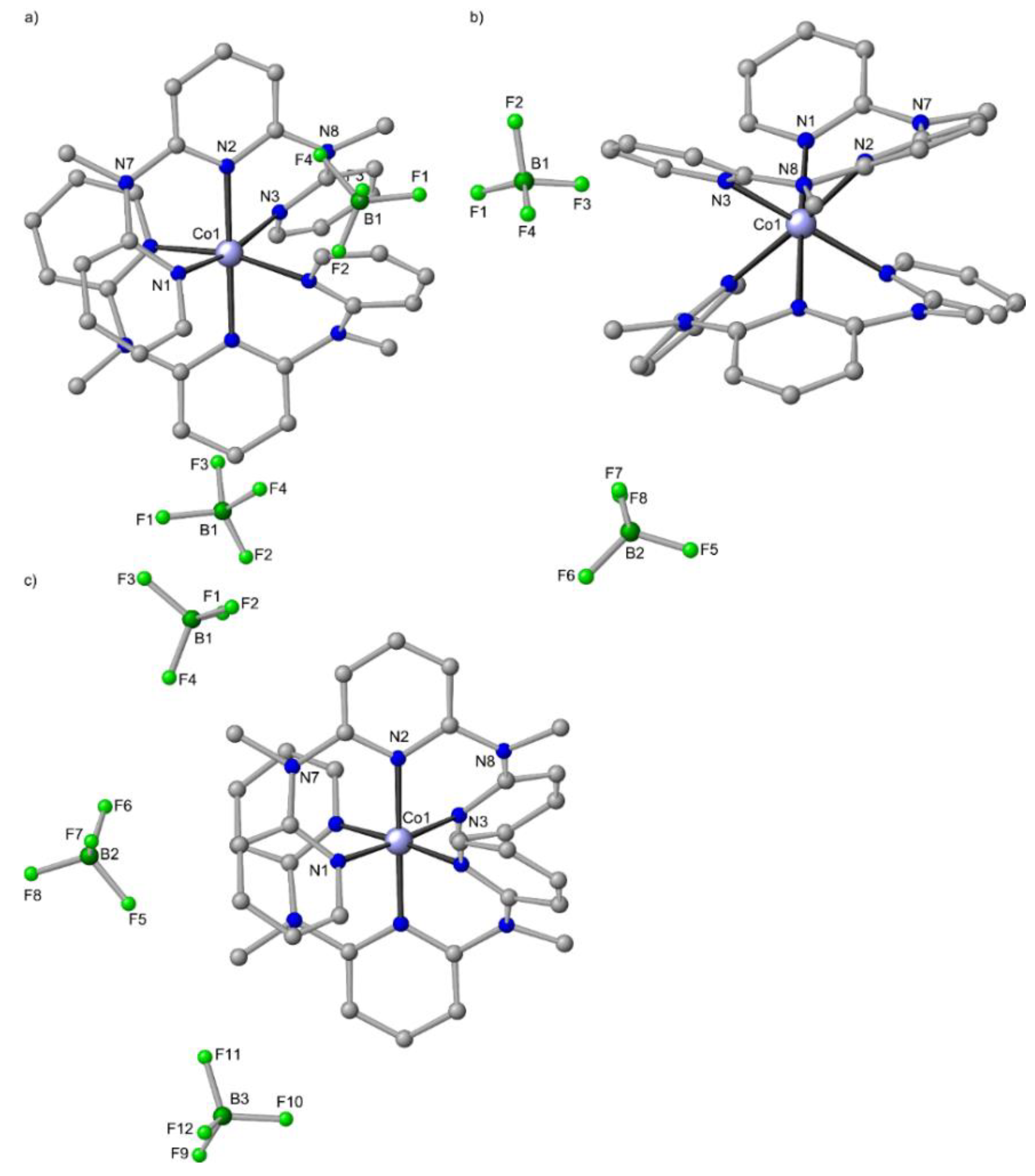
6. mer-[Co(ddpd)2]2+, cis-fac-[Co(ddpd)2]2+ [d7] and mer-[Co(ddpd)2]3+ [d6]
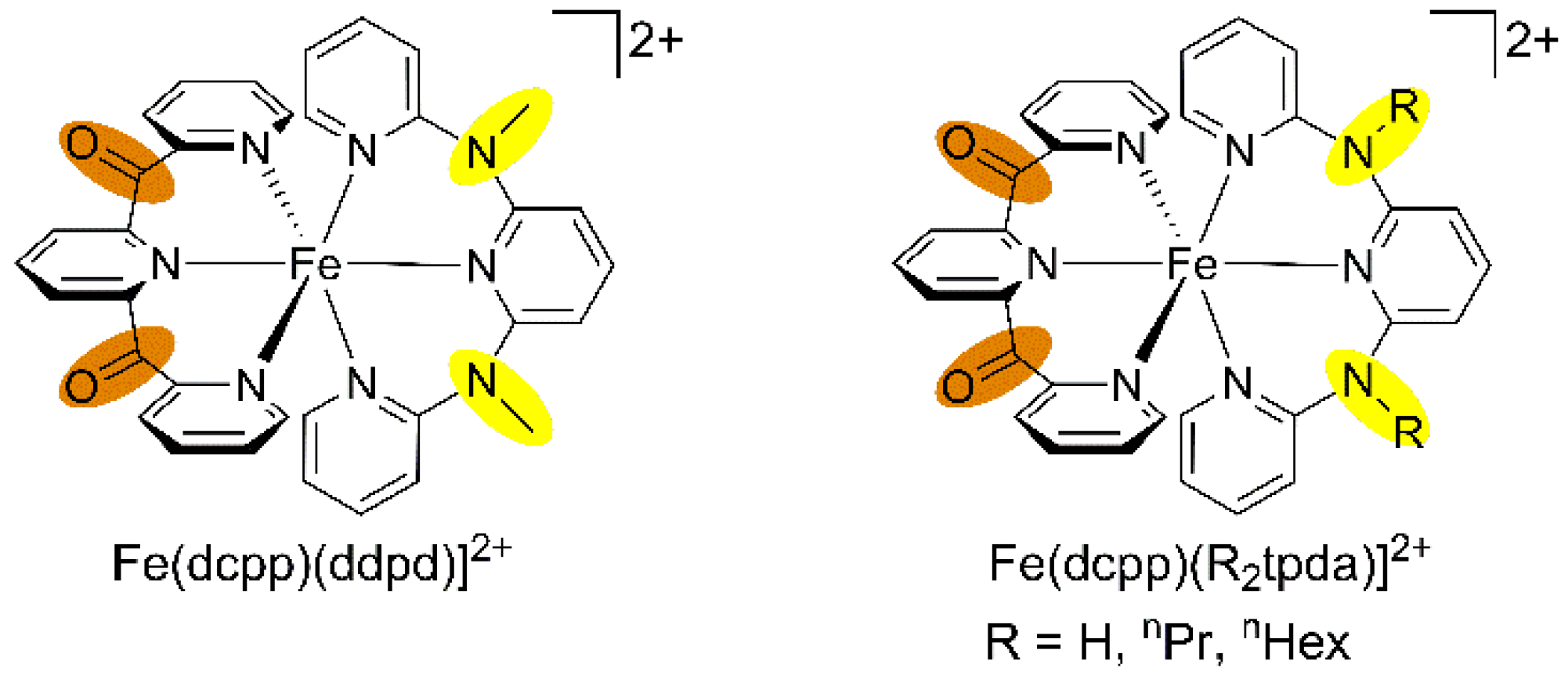
7. mer-[Fe(ddpd)2]2+ [d6] and mer-[Fe(ddpd)2]3+ [d5]
8. [Mn(ddpd)2]n+
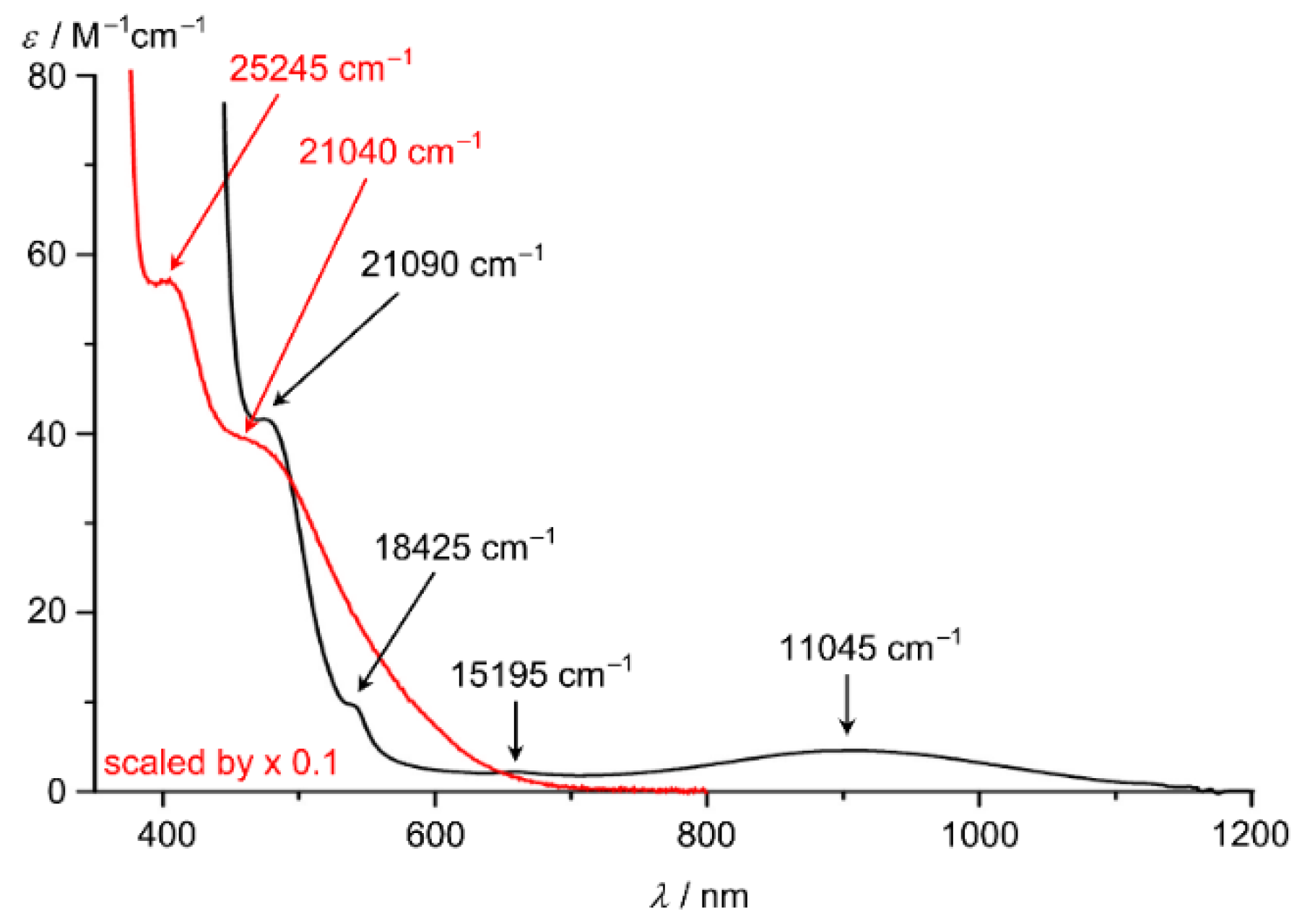
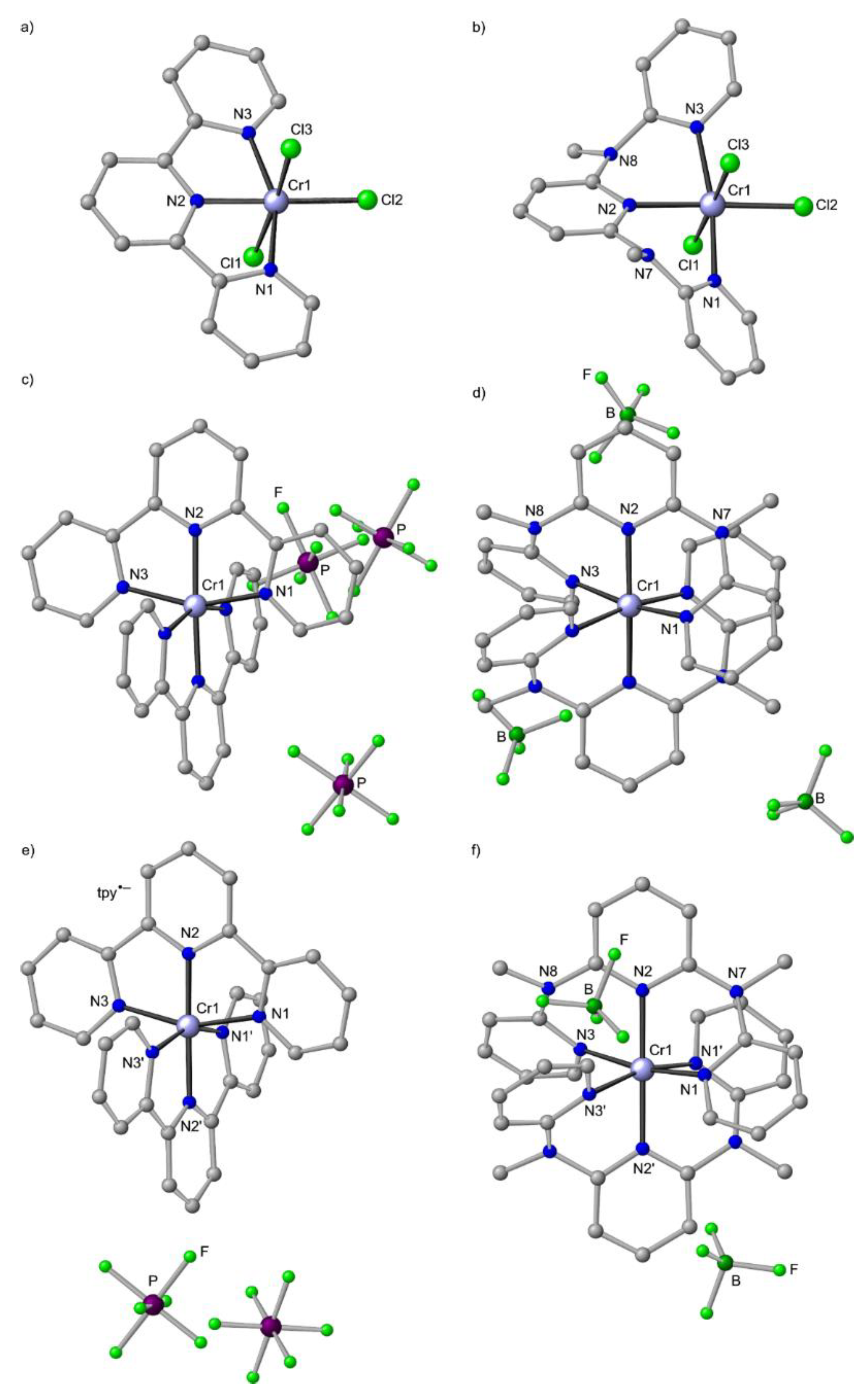
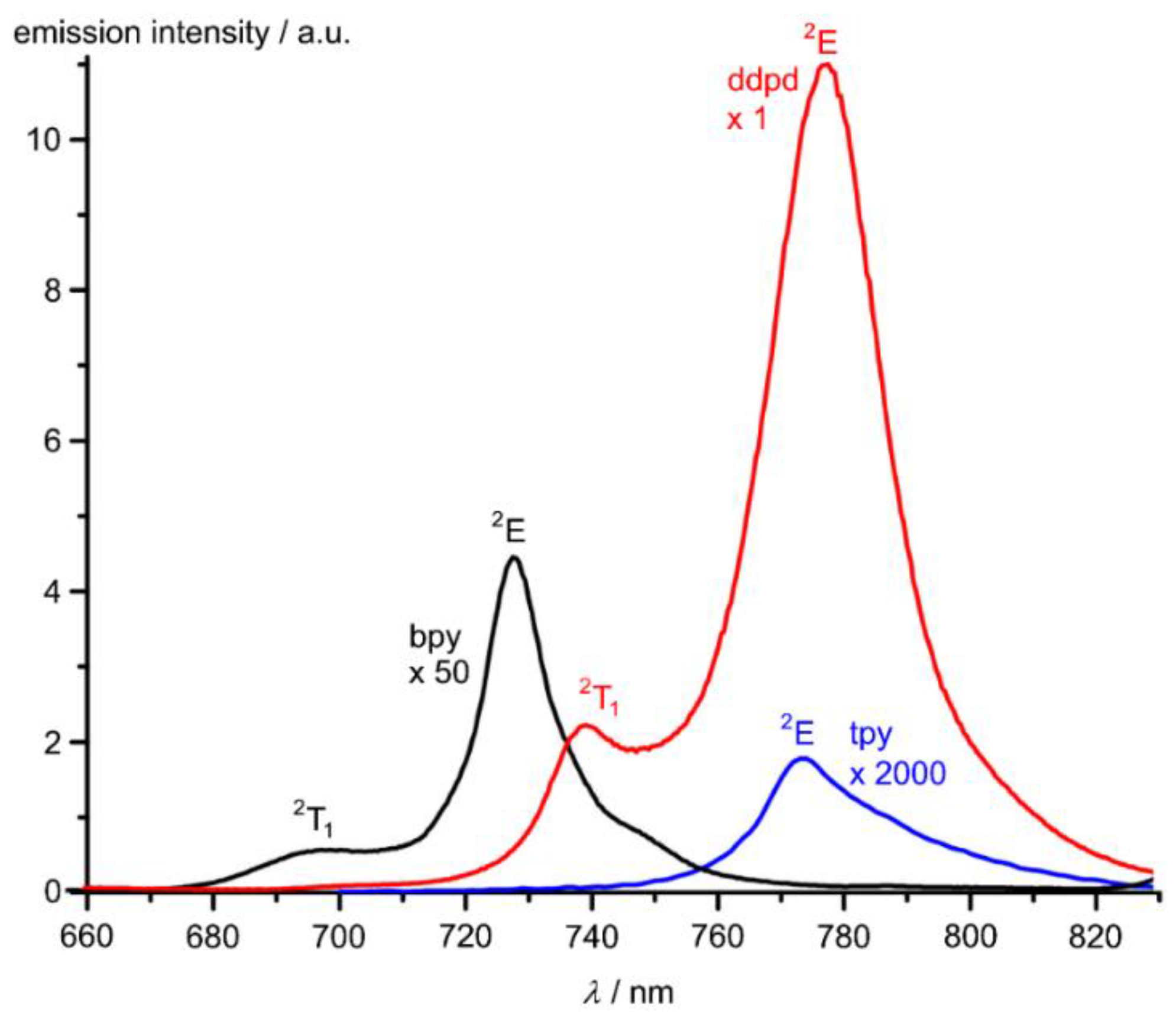
9. mer-[Cr(ddpd)2]3+, mer-CrCl3(ddpd) [d3] and mer-[Cr(ddpd)2]2+ [d4]
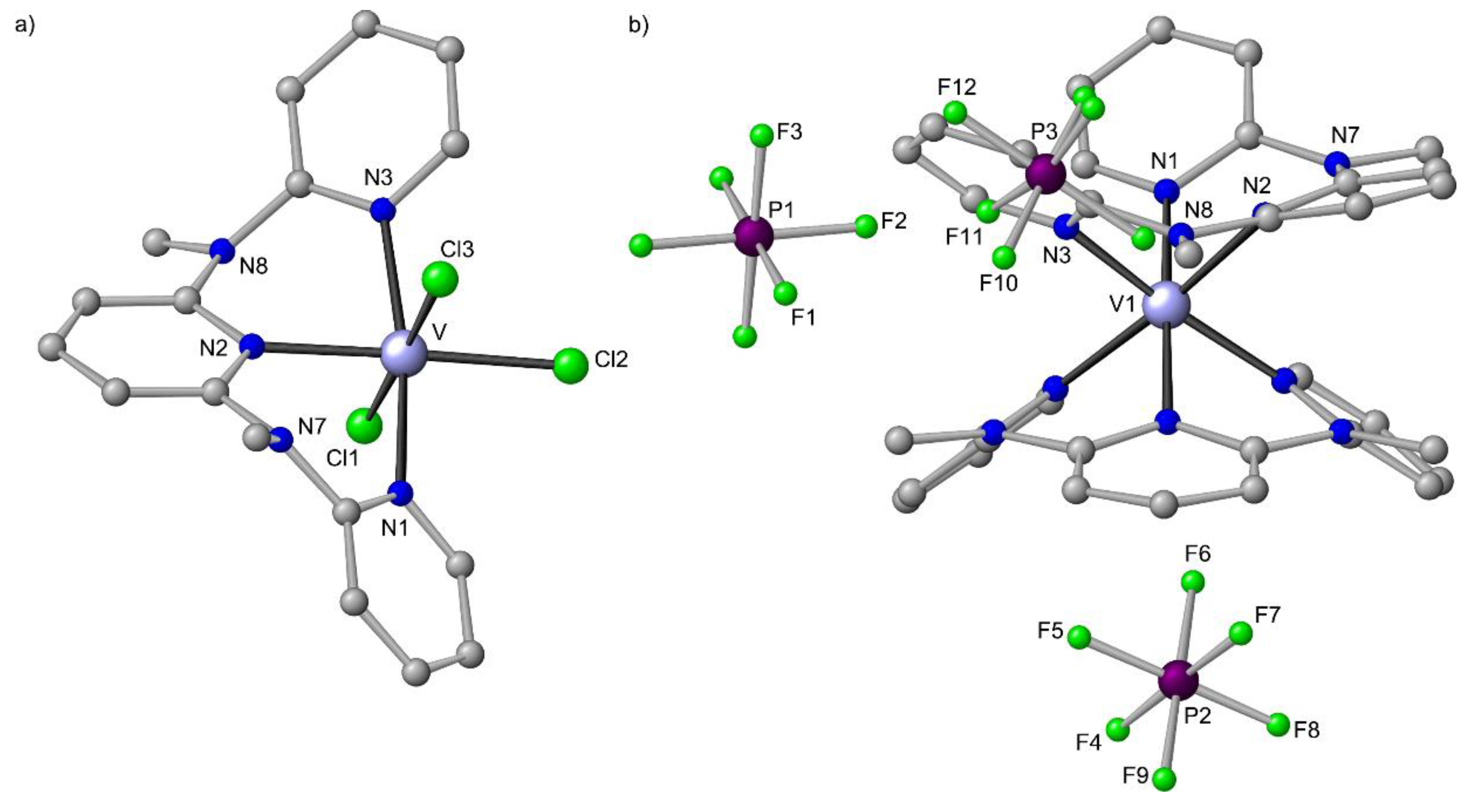
10. cis-fac-[V(ddpd)2]3+ and mer-VCl3(ddpd) [d2]
11. Experimental Section
12. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lawrence, M.A.W.; Green, K.-A.; Nelson, P.N.; Lorraine, S.C. Review: Pincer ligands—Tunable, versatile and applicable. Polyhedron 2018, 143, 11–27. [Google Scholar] [CrossRef]
- Peris, E.; Crabtree, R.H. Key factors in pincer ligand design. Chem. Soc. Rev. 2018, 47, 1959–1968. [Google Scholar] [CrossRef]
- Constable, E.C. The Coordination Chemistry of 2,2′:6′,2″-Terpyridine and Higher Oligopyridines. Adv. Inorg. Chem. 1986, 30, 69–121. [Google Scholar] [CrossRef]
- Constable, E.C. 2,2′:6′,2″-terpyridines: From chemical obscurity to common supramolecular motifs. Chem. Soc. Rev. 2007, 36, 246–253. [Google Scholar] [CrossRef]
- Hofmeier, H.; Schubert, U.S. Recent developments in the supramolecular chemistry of terpyridine–metal complexes. Chem. Soc. Rev. 2004, 33, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, J.P.; Collin, J.P.; Chambron, J.C.; Guillerez, S.; Coudret, C.; Balzani, V.; Barigelletti, F.; De Cola, L.; Flamigni, L. Ruthenium(II) and Osmium(II) Bis(terpyridine) Complexes in Covalently-Linked Multicomponent Systems: Synthesis, Electrochemical Behavior, Absorption Spectra, and Photochemical and Photophysical Properties. Chem. Rev. 1994, 94, 993–1019. [Google Scholar] [CrossRef]
- Flamigni, L.; Collin, J.P.; Sauvage, J.P. Iridium Terpyridine Complexes as Functional Assembling Units in Arrays for the Conversion of Light Energy. Acc. Chem. Res. 2008, 41, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.R.; Stang, P.J. Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.-Q.; Wang, H.; Wang, X.-Q.; Song, B.; Chen, L.-J.; Wang, L.; Hao, X.-Q.; Yang, H.-B.; Li, X. Self-assembly of emissive supramolecular rosettes with increasing complexity using multitopic terpyridine ligands. Nat. Commun. 2018, 9, 567. [Google Scholar] [CrossRef] [PubMed]
- Hasenknopf, B.; Lehn, J.-M.; Baum, G.; Fenske, D. Self-assembly of a heteroduplex helicate from two different ligand strands and Cu(II) cations. Proc. Natl. Acad. Sci. USA 1996, 93, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Bassani, D.M.; Lehn, J.-M.; Fromm, K.; Fenske, D. Toposelective and Chiroselective Self-Assembly of [2 × 2] Grid-Type Inorganic Arrays Containing Different Octahedral Metallic Centers. Angew. Chem. Int. Ed. 1998, 37, 2364–2367. [Google Scholar] [CrossRef]
- Cárdenas, D.J.; Livoreil, A.; Sauvage, J.-P. Redox Control of the Ring-Gliding Motion in a Cu-Complexed Catenane: A Process Involving Three Distinct Geometries. J. Am. Chem. Soc. 1996, 118, 11980–11981. [Google Scholar] [CrossRef]
- Baumann, F.; Livoreil, A.; Kaim, W.; Sauvage, J.-P. Changeover in a multimodal copper(II) catenate as monitored by EPR spectroscopy. Chem. Commun. 1997, 35–36. [Google Scholar] [CrossRef]
- Livoreil, A.; Sauvage, J.-P.; Armaroli, N.; Balzani, V.; Flamigni, L.; Ventura, B. Electrochemically and Photochemically Driven Ring Motions in a Disymmetrical Copper [2]-Catenate. J. Am. Chem. Soc. 1997, 119, 12114–12124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayami, S.; Komatsu, Y.; Shimizu, T.; Kamihata, H.; Lee, Y.H. Spin-crossover in cobalt(II) compounds containing terpyridine and its derivatives. Coord. Chem. Rev. 2011, 255, 1981–1990. [Google Scholar] [CrossRef]
- Krivokapic, I.; Zerara, M.; Dakua, M.L.; Vargas, A.; Enachescu, C.; Ambrus, C.; Tregenna-Piggott, P.; Amstutz, N.; Krausz, E.; Hauser, A. Spin-crossover in cobalt(II) imine complexes. Coord. Chem. Rev. 2007, 251, 364–378. [Google Scholar] [CrossRef]
- Gütlich, P.; Garcia, Y.; Goodwin, H.A. Spin crossover phenomena in Fe(II) complexes. Chem. Soc. Rev. 2000, 29, 419–427. [Google Scholar] [CrossRef]
- Constable, E.C.; Baum, G.; Bill, E.; Dyson, R.; van Eldik, R.; Fenske, D.; Kaderli, S.; Morris, D.; Neubrand, A.; Neuburger, M.; et al. Control of Iron(II) Spin States in 2,2′:6′,2″-Terpyridine Complexes through Ligand Substitution. Chem. Eur. 1999, 5, 498–508. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Vicic, D.A.; Klein, A. Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review. Inorganics 2018, 6, 18. [Google Scholar] [CrossRef]
- Elgrishi, N.; Chambers, M.B.; Artero, V.; Fontecave, M. Terpyridine complexes of first row transition metals and electrochemical reduction of CO2 to CO. Phys. Chem. Chem. Phys. 2014, 16, 13635–13644. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.; Newkome, G.R.; Schubert, U.S. Catalytic Applications of Terpyridines and their Transition Metal Complexes. ChemCatChem 2011, 3, 1384–1406. [Google Scholar] [CrossRef]
- Winter, A.; Gottschaldt, M.; Newkome, G.R.; Schubert, U.S. Terpyridines and their complexes with first row transition metal ions: Cytotoxicity, nuclease activity and self-assembly of biomacromolecules. Curr. Top. Med. Chem. 2012, 12, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; England, J.; Weyhermüller, T.; Wieghardt, K. Electronic Structures of “Low-Valent” Neutral Complexes [NiL2]0 (S = 0; L = bpy, phen, tpy)—An Experimental and DFT Computational Study. Eur. J. Inorg. Chem. 2015, 1511–1523. [Google Scholar] [CrossRef]
- England, J.; Bill, E.; Weyhermüller, T.; Neese, F.; Atanasov, M.; Wieghardt, K. Molecular and Electronic Structures of Homoleptic Six-Coordinate Cobalt(I) Complexes of 2,2′:6′,2″-Terpyridine, 2,2′-Bipyridine, and 1,10-Phenanthroline. An Experimental and Computational Study. Inorg. Chem. 2015, 54, 12002–12018. [Google Scholar] [CrossRef] [PubMed]
- England, J.; Scarborough, C.C.; Weyhermüller, T.; Sproules, S.; Wieghardt, K. Electronic Structures of the Electron Transfer Series [M(bpy)3]n, [M(tpy)2]n, and [Fe(tbpy)3]n (M = Fe, Ru; n = 3+, 2+, 1+, 0, 1−): A Mössbauer Spectroscopic and DFT Study. Eur. J. Inorg. Chem. 2012, 4605–4621. [Google Scholar] [CrossRef]
- Wang, M.; England, J.; Weyhermüller, T.; Wieghardt, K. Molecular and Electronic Structures of the Members of the Electron Transfer Series [Mn(bpy)3]n (n = 2+, 1+, 0, 1−) and [Mn(tpy)2]m (m = 4+, 3+, 2+, 1+, 0). An Experimental and Density Functional Theory Study. Inorg. Chem. 2014, 53, 2276–2287. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, C.C.; Lancaster, K.M.; DeBeer, S.; Weyhermüller, T.; Sproules, S.; Wieghardt, K. Experimental Fingerprints for Redox-Active Terpyridine in [Cr(tpy)2](PF6)n (n = 3–0), and the Remarkable Electronic Structure of [Cr(tpy)2]1–. Inorg. Chem. 2012, 51, 3718–3732. [Google Scholar] [CrossRef] [PubMed]
- Breivogel, A.; Kreitner, C.; Heinze, K. Redox and Photochemistry of Bis(terpyridine)ruthenium(II) Amino Acids and Their Amide Conjugates–from Understanding to Applications. Eur. J. Inorg. Chem. 2014, 5468–5490. [Google Scholar] [CrossRef]
- Parada, G.A.; Fredin, L.A.; Santoni, M.-P.; Jäger, M.; Lomoth, R.; Hammarström, L.; Johansson, O.; Persson, P.; Ott, S. Tuning the Electronics of Bis(tridentate)ruthenium(II) Complexes with Long-Lived Excited States: Modifications to the Ligand Skeleton beyond Classical Electron Donor or Electron Withdrawing Group Decorations. Inorg. Chem. 2013, 52, 5128–5137. [Google Scholar] [CrossRef] [PubMed]
- Laramée-Milette, B.; Hanan, G.S. Ruthenium bistridentate complexes with non-symmetrical hexahydro-pyrimidopyrimidine ligands: A structural and theoretical investigation of their optical and electrochemical properties. Dalton Trans. 2016, 45, 12507–12517. [Google Scholar] [CrossRef] [PubMed]
- Breivogel, A.; Wooh, S.; Dietrich, J.; Kim, T.Y.; Kang, Y.S.; Char, K.; Heinze, K. Anchor-Functionalized Push-Pull-Substituted Bis(tridentate) Ruthenium(II) Polypyridine Chromophores: Photostability and Evaluation as Photosensitizers. Eur. J. Inorg. Chem. 2014, 2720–2734. [Google Scholar] [CrossRef]
- Breivogel, A.; Park, M.; Lee, D.; Klassen, S.; Kühnle, A.; Lee, C.; Char, K.; Heinze, K. Push-Pull Design of Bis(tridentate) Ruthenium(II) Polypyridine Chromophores as Deep Red Light Emitters in Light-Emitting Electrochemical Cells. Eur. J. Inorg. Chem. 2014, 288–295. [Google Scholar] [CrossRef]
- Breivogel, A.; Meister, M.; Förster, C.; Laquai, F.; Heinze, K. Excited State Tuning of Bis(tridentate) Ruthenium(II) Polypyridine Chromophores by Push–Pull Effects and Bite Angle Optimization: A Comprehensive Experimental and Theoretical Study. Chem. Eur. J. 2013, 19, 13745–13760. [Google Scholar] [CrossRef] [PubMed]
- Friebe, C.; Görls, H.; Jäger, M.; Schubert, U.S. Linear Metallopolymers from Ruthenium(II)-2,6-di(quinolin-8-yl)pyridine Complexes by Electropolymerization–Formation of Redox-Stable and Emissive Films. Eur. J. Inorg. Chem. 2013, 4191–4202. [Google Scholar] [CrossRef]
- Breivogel, A.; Förster, C.; Heinze, K. A Heteroleptic Bis(tridentate)ruthenium(II) Polypyridine Complex with Improved Photophysical Properties and Integrated Functionalizability. Inorg. Chem. 2010, 49, 7052–7056. [Google Scholar] [CrossRef] [PubMed]
- Schramm, F.; Meded, V.; Fliegl, H.; Fink, K.; Fuhr, O.; Qu, Z.; Klopper, W.; Finn, S.; Keyes, T.E.; Ruben, M. Expanding the Coordination Cage: A Ruthenium(II)−Polypyridine Complex Exhibiting High Quantum Yields under Ambient Conditions. Inorg. Chem. 2009, 48, 5677–5684. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, M.; Jäger, M.; Österman, T.; Eriksson, L.; Persson, P.; Becker, H.-C.; Johansson, O.; Hammarström, L. A 3.0 μs Room Temperature Excited State Lifetime of a Bistridentate RuII−Polypyridine Complex for Rod-like Molecular Arrays. J. Am. Chem. Soc. 2006, 128, 12616–12617. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, M.; Wolpher, H.; Johansson, O.; Larsson, J.; Kritikos, M.; Eriksson, L.; Norrby, P.-O.; Bergquist, J.; Sun, L.; Åkermark, B.; et al. A New Strategy for the Improvement of Photophysical Properties in Ruthenium(II) Polypyridyl Complexes. Synthesis and Photophysical and Electrochemical Characterization of Six Mononuclear Ruthenium(II) Bisterpyridine-Type Complexes. Inorg. Chem. 2005, 44, 3215–3225. [Google Scholar] [CrossRef] [PubMed]
- Kröhnke, F. The Specific Synthesis of Pyridines and Oligopyridines. Synthesis 1976, 1–24. [Google Scholar] [CrossRef]
- Heller, M.; Schubert, U.S. Syntheses of Functionalized 2,2′:,2″-Terpyridines. Eur. J. Org. Chem. 2003, 947–961. [Google Scholar] [CrossRef]
- Ho, K.-Y.; Yu, W.-Y.; Cheung, K.-K.; Che, C.-M. Blue luminescent zinc(II) complexes with polypyridylamine ligands: Crystal structures and luminescence properties. J. Chem. Soc. Dalton Trans. 1999, 1581–1586. [Google Scholar] [CrossRef]
- Wang, C.; Otto, S.; Dorn, M.; Kreidt, E.; Lebon, J.; Sršan, L.; Di Martino-Fumo, P.; Gerhards, M.; Resch-Genger, U.; Seitz, M.; et al. Deuterated Molecular Ruby with Record Luminescence Quantum Yield. Angew. Chem. Int. Ed. 2018, 57, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Mengel, A.K.C.; Bissinger, C.; Dorn, M.; Back, O.; Förster, C.; Heinze, K. Boosting Vis/NIR Charge-Transfer Absorptions of Iron(II) Complexes by N-Alkylation and N-Deprotonation in the Ligand Backbone. Chem. Eur. J. 2017, 23, 7920–7931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessel, C.A.; See, R.F.; Jameson, D.L.; Churchill, M.R.; Takeuchi, K.J. Structural considerations of terdentate ligands: Crystal structures of 2,2′:6′,2″-terpyridine and 2,6-bis(pyrazol-1-yl)pyridine. J. Chem. Soc. Dalton Trans. 1992, 3223–3228. [Google Scholar] [CrossRef]
- Pearson, R.G.; Williams, F.V. Rates of Ionization of Pseudo Acids.1 V. Steric Effects in the Base-catalyzed Ionization of Nitroethane. J. Am. Chem. Soc. 1953, 75, 3073–3075. [Google Scholar] [CrossRef]
- Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I.A. Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales. J. Org. Chem. 2005, 70, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Staab, H.A.; Saupe, T. “Proton Sponges” and the Geometry of Hydrogen Bonds: Aromatic Nitrogen Bases with Exceptional Basicities. Angew. Chem. Int. Ed. Engl. 1988, 27, 865–879. [Google Scholar] [CrossRef]
- Hergold-Brundić, A.; Popović, Z.; Matković-Calogović, D. 2,2′:6′,2″-Terpyridinium Trifluoromethanesulfonate, [terpyH](CF3SO3). Acta Cryst. 1996, C52, 3154–3157. [Google Scholar] [CrossRef]
- Otto, S.; Moll, J.; Förster, C.; Geißler, D.; Wang, C.; Resch-Genger, U.; Heinze, K. Three-in-One Crystal: The Coordination Diversity of Zinc Polypyridine Complexes. Eur. J. Inorg. Chem. 2017, 5033–5040. [Google Scholar] [CrossRef]
- Otto, S.; Grabolle, M.; Förster, C.; Kreitner, C.; Resch-Genger, U.; Heinze, K. [Cr(ddpd)2]3+: A Molecular, Water-Soluble, Highly NIR-Emissive Ruby Analogue. Angew. Chem. Int. Ed. 2015, 54, 11572–11576. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, N.; Yamabe, S.; Kanehisa, N.; Takashima, H.; Tsukahara, K. A metal free blue emission by the protonated 2,2′:6′,2″-terpyridine hexafluorophosphate. J. Phys. Org. Chem. 2009, 22, 410–417. [Google Scholar] [CrossRef]
- Hamacher, C.; Hurkes, N.; Kaiser, A.; Klein, A.; Schüren, A. Electrochemistry and Spectroscopy of Organometallic Terpyridine Nickel Complexes. Inorg. Chem. 2009, 48, 9947–9951. [Google Scholar] [CrossRef] [PubMed]
- Otto, S.; Dorn, M.; Förster, C.; Bauer, M.; Seitz, M.; Heinze, K. Understanding and exploiting long-lived near-infrared emission of a molecular ruby. Coord. Chem. Rev. 2018, 359, 102–111. [Google Scholar] [CrossRef]
- Alemany, P.; Casanova, D.; Alvarez, S.; Dryzun, C.; Avnir, D. Continuous Symmetry Measures: A New Tool in Quantum Chemistry. In Reviews in Computational Chemistry; Parrill, A.L., Lipkowitz, K.B., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2017; Volume 30, pp. 289–352. [Google Scholar]
- Alvarez, S. Distortion Pathways of Transition Metal Coordination Polyhedra Induced by Chelating Topology. Chem. Rev. 2015, 115, 13447–13483. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Alemany, P.; Casanova, D.; Cirera, J.; Llunell, M.; Avnir, D. Shape maps and polyhedral interconversion paths in transition metal chemistry. Coord. Chem. Rev. 2005, 249, 1693–1708. [Google Scholar] [CrossRef]
- Alvarez, S.; Avnir, D.; Llunell, M.; Pinsky, M. Continuous symmetry maps and shape classification. The case of six-coordinated metal compounds. New J. Chem. 2002, 26, 996–1009. [Google Scholar] [CrossRef]
- Zabrodsky, H.; Peleg, S.; Avnir, D. Continuous Symmetry Measures. J. Am. Chem. Soc. 1992, 114, 7843–7851. [Google Scholar] [CrossRef]
- Jahn, H.A.; Teller, E. Stability of polyatomic molecules in degenerate electronic states—I—Orbital degeneracy. Proc. R. Soc. Lond. A 1937, 161, 220–235. [Google Scholar] [CrossRef]
- Mack, K.; Wünsche von Leupoldt, A.; Förster, C.; Ezhevskaya, M.; Hinderberger, D.; Klinkhammer, K.W.; Heinze, K. Effect of chelate ring expansion on Jahn-Teller distortion and Jahn-Teller dynamics in copper(II) complexes. Inorg. Chem. 2012, 51, 7851–7858. [Google Scholar] [CrossRef] [PubMed]
- Dorn, M.; Mack, K.; Carrella, L.M.; Rentschler, E.; Förster, C.; Heinze, K. Structure and electronic properties of an expanded terpyridine complex of nickel(II) [Ni(ddpd)2](BF4)2. Z. Anorg. Allg. Chem. 2018, 644, 706–712. [Google Scholar] [CrossRef]
- Förster, C.; Mack, K.; Carrella, L.M.; Ksenofontov, V.; Rentschler, E.; Heinze, K. Coordination of expanded terpyridine ligands to cobalt. Polyhedron 2013, 52, 576–581. [Google Scholar] [CrossRef]
- Förster, C.; Gorelik, T.E.; Kolb, U.; Ksenofontov, V.; Heinze, K. Crystalline Non-Equilibrium Phase of a Cobalt(II) Complex with Tridentate Ligands. Eur. J. Inorg. Chem. 2015, 920–924. [Google Scholar] [CrossRef]
- Mengel, A.K.C.; Förster, C.; Breivogel, A.; Mack, K.; Ochsmann, J.R.; Laquai, F.; Ksenofontov, V.; Heinze, K. A Heteroleptic Push–Pull Substituted Iron(II) Bis(tridentate) Complex with Low-Energy Charge-Transfer States. Chem. Eur. J. 2015, 21, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Skelton, B.W.; Harrowfield, J.M. CCDC 1585922, Experimental Crystal Structure Determination (2017), doi:10.5517/ccdc.csd.cc1q78s0. Available online: https://www.ccdc.cam.ac.uk/structures/search?id=doi:10.5517/ccdc.csd.cc1q78s0&sid=DataCite (accessed on 26 July 2018).
- Docherty, R.; Tuna, F.; Kilner, C.A.; McInnes, E.J.L.; Halcrow, M.A. Suppression of the Jahn–Teller distortion in a six-coordinate copper(II) complex by doping it into a host lattice. Chem. Commun. 2012, 48, 4055–4057. [Google Scholar] [CrossRef] [PubMed]
- Anderer, C.; Näther, C.; Bensch, W. Bis(2,2′:6′,2″-terpyridine-κ3N,N′,N″)nickel(II) bis(perchlorate) hemihydrate. IUCrData 2016, 1, x161009. [Google Scholar] [CrossRef]
- Constable, E.C.; Harris, K.; Housecroft, C.E.; Neuburger, M.; Zampese, J.A. Turning {M(tpy)2}n+ embraces and CH…π interactions on and off in homoleptic cobalt(II) and cobalt(III) bis(2,2′:6′,2″-terpyridine) complexes. CrystEngComm 2010, 12, 2949–2961. [Google Scholar] [CrossRef]
- Oshio, H.; Spiering, H.; Ksenofontov, V.; Renz, F.; Gütlich, P. Electronic relaxation phenomena following 57Co(EC)57Fe nuclear decay in [Mn(II)(terpy)2](ClO4)2·1/2H2O and in the spin crossover complexes [Co(II)(terpy)2]X2·nH2O (X = Cl and ClO4): A Mössbauer emission spectroscopic study. Inorg. Chem. 2001, 40, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Romain, S.; Duboc, C.; Neese, F.; Rivière, E.; Hanton, L.R.; Blackman, A.G.; Philouze, C.; Leprêtre, J.-C.; Deronzier, A.; Collomb, M. An Unusual Stable Mononuclear MnIII Bis-terpyridine Complex Exhibiting Jahn–Teller Compression: Electrochemical Synthesis, Physical Characterisation and Theoretical Study. Chem. Eur. J. 2009, 15, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Schönle, J.; Zampese, J.A. The surprising lability of bis(2,2′:6′,2″-terpyridine)chromium(III) complexes. Dalton Trans. 2014, 43, 7227–7235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scudder, M.L.; Goodwin, H.A.; Dance, I.G. Crystal supramolecular motifs: Two-dimensional grids of terpy embraces in [ML2]z complexes (L = terpy or aromatic N3-tridentate ligand). New J. Chem. 1999, 23, 695–705. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, X. Homoleptic Zinc(II) Complexes Based on 4′-(2-Thienyl)-terpyridine: Preparation, Structures, and Properties. Z. Anorg. Allg. Chem. 2017, 643, 398–402. [Google Scholar] [CrossRef]
- Bozic-Weber, B.; Constable, E.C.; Hostettler, N.; Housecroft, C.E.; Schmitt, R.; Schönhofer, E. The d10 route to dye-sensitized solar cells: Step-wise assembly of zinc(II) photosensitizers on TiO2 surfaces. Chem. Commun. 2012, 48, 5727–5729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jameson, D.L.; Guise, L.E. An improved, two-step synthesis of 2,2′:6′,2″-terpyridine. Tetrahedron Lett. 1991, 32, 1999–2002. [Google Scholar] [CrossRef]
- Elsbernd, H.; Beattie, J.K. The NMR spectra of terpyridine and the bis-terpyridine complexes of cobalt(III) and iron(II). J. Inorg. Nucl. Chem. 1972, 34, 771–774. [Google Scholar] [CrossRef]
- Machan, C.W.; Adelhardt, M.; Sarjeant, A.A.; Stern, C.L.; Sutter, J.; Meyer, K.; Mirkin, C.A. One-Pot Synthesis of an Fe(II) Bis-Terpyridine Complex with Allosterically Regulated Electronic Properties. J. Am. Chem. Soc. 2012, 134, 16921–16924. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Scaife, D.B. Electro-oxidation and electro-reduction of some iron(II), cobalt(II) and nickel(II) polypyridyl complexes in acetonitrile. J. Electroanal. Chem. 1977, 84, 373–386. [Google Scholar] [CrossRef]
- Rao, J.M.; Hughes, M.C.; Macero, D.J. Voltammetry of terpyridine and terosine complexes of cobalt(II) and iron(II). Inorg. Chim. Acta 1976, 16, 231–236. [Google Scholar] [CrossRef]
- Dobson, J.C.; Taube, H. Coordination chemistry and redox properties of polypyridyl complexes of vanadium(II). Inorg. Chem. 1989, 28, 1310–1315. [Google Scholar] [CrossRef]
- Albano, G.; Balzani, V.; Constable, E.C.; Maestri, M.; Smith, D.R. Photoinduced processes in 4′-(9-anthryl)-2,2′:6′,2″-terpyridine, its protonated forms and Zn(II), Ru(II) and Os(II) complexes. Inorg. Chim. Acta 1998, 277, 225–231. [Google Scholar] [CrossRef]
- Morgan, G.; Burstall, F.H. Researches on residual affinity and co-ordination. Part XXXVII. Complex metallic salts containing 2:6-di-2′-pyridylpyridine (2:2′:2″-tripyridyl). J. Chem. Soc. 1937, 1649–1655. [Google Scholar] [CrossRef]
- Kahn, O. Molecular Magnetism; Wiley-VCH: New York, NY, USA, 1993; pp. 9–10. [Google Scholar]
- Saravani, H.; Rezvani, A.R.; Hadadzadeh, H.; Safari, N. An Investigation of Z-in Distortion in Mononuclear Cu(II) Complex with Terpyridine Ligands, [Cu(terpy)2](PF6)2. Iran. J. Chem. Chem. Eng. 2007, 26, 103–110. [Google Scholar]
- Folgado, J.-V.; Henke, W.; Allmann, R.; Stratemeier, H.; Beltrán-Porter, D.; Roja, T.; Reinen, D. Fluxionality in hexacoordinated copper(II) complexes with 2,2′:6′,2″-terpyridine (terpy) and related ligands: Structural and spectroscopic investigations. Inorg. Chem. 1990, 29, 2035–2042. [Google Scholar] [CrossRef]
- Hogg, R.; Wilkins, R.G. Exchange studies of certain chelate compounds of the transitional metals. Part VIII. 2,2′,2″-terpyridine complexes. J. Chem. Soc. 1962, 341–350. [Google Scholar] [CrossRef]
- Rao, J.M.; Hughes, M.C.; Macero, D.J. Redox behavior of aromatic tridentate imine ligand complexes of manganese and chromium. Inorg. Chim. Acta 1976, 18, 127–131. [Google Scholar] [CrossRef]
- Scarborough, C.C.; Sproules, S.; Weyhermüller, T.; DeBeer, S.; Wieghardt, K. Electronic and Molecular Structures of the Members of the Electron Transfer Series [Cr(tbpy)3]n (n = 3+, 2+, 1+, 0): An X-ray Absorption Spectroscopic and Density Functional Theoretical Study. Inorg. Chem. 2011, 50, 12446–12462. [Google Scholar] [CrossRef] [PubMed]
- Casellato, U.; Graziani, R.; Bonomo, R.P.; Di Bilio, A.J. X-ray crystal structures and electron spin resonance spectroscopic characterization of mixed-ligand chromium(III) complexes with L-aspartate or pyridine-2,6-dicarboxylate and 1,10-phenanthroline or 2,2′:6′,2″-terpyridyl. J. Chem. Soc. Dalton Trans. 1991, 23–31. [Google Scholar] [CrossRef]
- Herzog, S.; Aul, H. Über elektronenreiche Komplexe des Vanadins mit 2,2′,2″-Tripyridyl. Zeitschr. Chem. 1966, 6, 343–344. [Google Scholar] [CrossRef]
- Henke, W.; Reinen, D. Spektroskopische Untersuchungen zum Jahn-Teller-Effekt des Cu2+-Ions in Terpyridin-Komplexen Cu(terpy)2X2·nH2O [X = NO3–, ClO4–, Br–]. Z. Anorg. Allg. Chem. 1977, 436, 187–200. [Google Scholar] [CrossRef]
- Waldmann, O.; Hassmann, J.; Müller, P.; Volkmer, D.; Schubert, U.S.; Lehn, J.-M. Magnetism of self-assembled mono- and tetranuclear supramolecular Ni2+ complexes. Phys. Rev. B 1998, 58, 3277–3285. [Google Scholar] [CrossRef]
- González, E.; Rodrigue-Witchel, A.; Reber, C. Absorption spectroscopy of octahedral nickel(II) complexes: A case study of interactions between multiple electronic excited states. Coord. Chem. Rev. 2007, 251, 351–363. [Google Scholar] [CrossRef]
- Kuehnel, M.F.; Orchard, K.L.; Dalle, K.E.; Reisner, E. Selective Photocatalytic CO2 Reduction in Water through Anchoring of a Molecular Ni Catalyst on CdS Nanocrystals. J. Am. Chem. Soc. 2017, 139, 7217–7223. [Google Scholar] [CrossRef] [PubMed]
- Judge, J.S.; Baker, W.A., Jr. On the spin equilibrium in bis(2,2′,2″-terpyridine) cobalt(II) salts. Inorg. Chim. Acta 1967, 1, 68–72. [Google Scholar] [CrossRef]
- Kremer, S.; Henke, W.; Reinen, D. High-spin-low-spin equilibria of cobalt(2+) in the terpyridine complexes Co(terpy)2X2.nH2O. Inorg. Chem. 1982, 21, 3013–3022. [Google Scholar] [CrossRef]
- Komatsu, Y.; Kato, K.; Yamamoto, Y.; Kamihata, H.; Lee, Y.H.; Fuyuhiro, A.; Kawata, S.; Hayami, S. Spin-Crossover Behaviors Based on Intermolecular Interactions for Cobalt(II) Complexes with Long Alkyl Chains. Eur. J. Inorg. Chem. 2012, 2769–2775. [Google Scholar] [CrossRef]
- Hayami, S.; Shigeyoshi, Y.; Akita, M.; Inoue, K.; Kato, K.; Osaka, K.; Takata, M.; Kawajiri, R.; Mitani, T.; Maeda, Y. Reverse Spin Transition Triggered by a Structural Phase Transition. Angew. Chem. Int. Ed. 2005, 44, 4899–4903. [Google Scholar] [CrossRef] [PubMed]
- Kilner, C.A.; Halcrow, M.A. An unusual discontinuity in the thermal spin transition in [Co(terpy)2][BF4]2. Dalton Trans. 2010, 39, 9008–9012. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.S.; Constable, E.C.; Housecroft, C.E.; Kulicke, K.J.; Tao, Y. When electron exchange is chemical exchange–assignment of 1H NMR spectra of paramagnetic cobalt(II)-2,2′:6′,2″-terpyridine complexes. Dalton Trans. 2005, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Beattie, J.K.; Binstead, R.A.; Kelso, M.T.; Del Favero, P.; Dewey, T.G.; Turner, D.H. Dynamics of cobalt(II) spin-equilibrium complexes. Inorg. Chim. Acta 1995, 235, 245–251. [Google Scholar] [CrossRef]
- Mengel, A.K.C.; Cho, W.; Breivogel, A.; Char, K.; Kang, Y.S.; Heinze, K. A Bis(tridentate)cobalt Polypyridine Complex as Mediator in Dye-Sensitized Solar Cells. Eur. J. Inorg. Chem. 2015, 3299–3306. [Google Scholar] [CrossRef]
- Kreitner, C.; Mengel, A.K.C.; Lee, T.K.; Cho, W.; Char, K.; Kang, Y.S.; Heinze, K. Strongly Coupled Cyclometalated Ruthenium Triarylamine Chromophores as Sensitizers for DSSCs. Chem. Eur. J. 2016, 22, 8915–8928. [Google Scholar] [CrossRef] [PubMed]
- Wentworth, R.A.D.; Piper, T.S. A Crystal Field Model for the Spectral Relationships in Monoacidopentaammine and Diacidotetraammine Complexes of Cobalt(III). Inorg. Chem. 1965, 4, 709–714. [Google Scholar] [CrossRef]
- Sharma, R.P.; Singh, A.; Brandão, P.; Felix, V.; Venugopalan, P. Second sphere coordination in binding of fluoroanions: Synthesis, spectroscopic characterization and single crystal X-ray structure determination of [Co(phen)3](BF4)3·H2O and [Co(phen)3](PF6)3·CH3COCH3. J. Mol. Struct. 2009, 920, 119–127. [Google Scholar] [CrossRef]
- Sutin, N.; Brunschwig, B.S.; Creutz, C.; Winkler, J.R. Nuclear reorganization barriers to electron transfer. Pure Appl. Chem. 1988, 60, 1817–1830. [Google Scholar] [CrossRef]
- Cummins, D.; Gray, H.B. Electron-transfer protein reactivities. Kinetic studies of the oxidation of horse heart cytochrome c, Chromatium vinosum high potential iron-sulfur protein, Pseudomonas aeruginosa azurin, bean plastocyanin, and Rhus vernicifera stellacyanin by pentaammminepyridineruthenium(III). J. Am. Chem. Soc. 1977, 99, 5158–5167. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.; Creutz, C.; Sutin, N. Rate constants and activation parameters for outer-sphere electron-transfer reactions and comparisons with the predictions of Marcus theory. J. Am. Chem. Soc. 1977, 99, 5615–5623. [Google Scholar] [CrossRef]
- Ondersma, J.W.; Hamann, T.W. Recombination and redox couples in dye-sensitized solar cells. Coord. Chem. Rev. 2103, 257, 1533–1543. [Google Scholar] [CrossRef]
- Yum, J.-H.; Baranoff, E.; Kessler, F.; Moehl, T.; Ahmad, S.; Bessho, T.; Marchioro, A.; Ghadiri, E.; Moser, J.-E.; Yi, C.; et al. A cobalt complex redox shuttle for dye-sensitized solar cells with high open-circuit potentials. Nat. Commun. 2012, 3, 631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harzmann, G.D.; Neuburger, M.; Mayor, M. 4,4’’-Disubstituted Terpyridines and Their Homoleptic FeII Complexes. Eur. J. Inorg. Chem. 2013, 3334–3347. [Google Scholar] [CrossRef]
- Pazderski, L.; Pawlak, T.; Sitkowski, J.; Kozerski, L.; Szlyk, E. 1H, 13C, 15N NMR coordination shifts in Fe(II), Ru(II) and Os(II) cationic complexes with 2,2′:6′,2″-terpyridine. Magn. Reson. Chem. 2011, 49, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Mack, K. Neue Übergangsmetall-Komplexe des N,N′-dimethyl-N,N′-dipyridin-2-ylpyridin-2,6-diamins-Strukturelle, magnetische und optische Eigenschaften. Diploma Thesis, Johannes Gutenberg University, Mainz, Germany, 2012. [Google Scholar]
- Shepard, S.G.; Fatur, S.M.; Rappé, A.K.; Damrauer, N.H. Highly Strained Iron(II) Polypyridines: Exploiting the Quintet Manifold To Extend the Lifetime of MLCT Excited States. J. Am. Chem. Soc. 2016, 138, 2949–2952. [Google Scholar] [CrossRef] [PubMed]
- Fatur, S.M.; Shepard, S.G.; Higgins, R.F.; Shores, M.P.; Damrauer, N.H. A Synthetically Tunable System To Control MLCT Excited-State Lifetimes and Spin States in Iron(II) Polypyridines. J. Am. Chem. Soc. 2017, 139, 4493–4505. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lin, T.-W.; Chou, C.-C.; Lee, H.-C.; Chang, H.-C.; Lee, G.-H.; Leung, M.; Peng, S.-P. New oligo-α-pyridylamino ligands and their metal complexes. Chem. Commun. 1997, 2279–2280. [Google Scholar] [CrossRef]
- Jamula, L.L.; Brown, A.M.; Guo, D.; McCusker, J.K. Synthesis and Characterization of a High-Symmetry Ferrous Polypyridyl Complex: Approaching the 5T2/3T1 Crossing Point for FeII. Inorg. Chem. 2014, 53, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Harlang, T.; Canton, S.E.; Chábera, P.; Suárez-Alcántara, K.; Fleckhaus, A.; Vithanage, D.A.; Göransson, E.; Corani, A.; Lomoth, R.; et al. Towards longer-lived metal-to-ligand charge transfer states of iron(II) complexes: An N-heterocyclic carbene approach. Chem. Commun. 2013, 49, 6412–6414. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kjær, K.S.; Fredin, L.A.; Chábera, P.; Harlang, T.; Canton, S.E.; Lidin, S.; Zhang, J.; Lomoth, R.; Bergquist, K.-E.; et al. A Heteroleptic Ferrous Complex with Mesoionic Bis(1,2,3-triazol-5-ylidene) Ligands: Taming the MLCT Excited State of Iron(II). Chem. Eur. J. 2015, 21, 3628–3639. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Duchanois, T.; Etienne, T.; Monari, A.; Beley, M.; Assfeld, X.; Haacke, S.; Gros, P.C. A new record excited state 3MLCT lifetime for metalorganic iron(II) complexes. Phys. Chem. Chem. Phys. 2016, 18, 12550–12556. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, P.; Burkhardt, L.; Friedrich, A.; Steube, J.; Neuba, A.; Schepper, R.; Müller, P.; Flörke, U.; Huber, M.; Lochbrunner, S.; et al. The Connection between NHC Ligand Count and Photophysical Properties in Fe(II) Photosensitizers: An Experimental Study. Inorg. Chem. 2018, 57, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Chábera, P.; Kjaer, K.S.; Prakash, O.; Honarfar, A.; Liu, Y.; Fredin, L.A.; Harlang, T.C.B.; Lidin, S.; Uhlig, J.; Sundström, V.; et al. FeII Hexa N-Heterocyclic Carbene Complex with a 528 ps Metal-to-Ligand Charge-Transfer Excited-State Lifetime. J. Phys. Chem. Lett. 2018, 9, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Romain, S.; Baffert, C.; Duboc, C.; Leprêtre, J.-C.; Deronzier, A.; Collomb, M.-N. Mononuclear MnIII and MnIV Bis-terpyridine Complexes: Electrochemical Formation and Spectroscopic Characterizations. Inorg. Chem. 2009, 48, 3125–3131. [Google Scholar] [CrossRef] [PubMed]
- Limburg, J.; Vrettos, J.S.; Liable-Sands, L.M.; Rheingold, A.L.; Crabtree, R.H.; Brudvig, G.W. A Functional Model for O-O Bond Formation by the O2-Evolving Complex in Photosystem II. Science 1999, 283, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Young, K.J.; Brennan, B.J.; Tagore, R.; Brudvig, G.W. Photosynthetic Water Oxidation: Insights from Manganese Model Chemistry. Acc. Chem. Res. 2015, 48, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Zare, D.; Doistau, B.; Nozary, H.; Besnard, C.; Guénée, L.; Suffren, Y.; Pelé, A.-L.; Hauser, A.; Piguet, C. CrIII as an alternative to RuII in metallo-supramolecular chemistry. Dalton Trans. 2017, 46, 8992–9009. [Google Scholar] [CrossRef] [PubMed]
- Cloete, N.; Visser, H.G.; Roodt, A. mer-Trichloro(2,2′,2″-terpyridine)chromium(III) dimethyl sulfoxide solvate. Acta Crystallogr. Sect. E Struct. Rep. Online 2007, 63, m45–m47. [Google Scholar] [CrossRef]
- Henriques, R.T.; Herdtweck, E.; Kühn, F.E.; Lopes, A.D.; Mink, J.; Romão, C.C. Synthesis, characterization, and reactions of tetrakis(nitrile)chromium(II) tetrafluoroborate complexes. J. Chem. Soc. Dalton Trans. 1998, 1293–1297. [Google Scholar] [CrossRef]
- Åkesson, R.; Pettersson, L.G.M.; Sandström, M.; Wahlgren, U. Theoretical calculations of the Jahn-Teller effect in the hexahydrated Copper(II), chromium(II), and manganese(III) ions, hexaaquacopper(2+), hexaaquachromium(2+) and hexaaquamanganese(3+), and comparisons with the hexahydrated copper(I), chromium(III), and manganese(II) clusters. J. Phys. Chem. 1992, 96, 150–156. [Google Scholar] [CrossRef]
- Thangavel, A.; Wieliczko, M.; Scarborough, C.; Dittrich, B.; Bacsa, J. An investigation of the electron density of a Jahn–Teller-distorted CrII cation: The crystal structure and charge density of hexakis(acetonitrile-κN)chromium(II) bis(tetraphenylborate) acetonitrile disolvate. Acta Crystallogr. Sect. C 2015, C71, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Hieber, W.; Mühlbauer, F. Über Metallcarbonyle. XII. Reaktionen und Derivate der Hexacarbonyle des Chroms und Molybdäns. Z. Anorg. Allg. Chem. 1935, 221, 337–348. [Google Scholar] [CrossRef]
- Hieber, W.; Abeck, W.; Platzer, H.K. Über Metallcarbonyle. 69. Über Tricarbonyl-triammin-Chrom und seine Derivate. Z. Anorg. Allg. Chem. 1955, 280, 252–263. [Google Scholar] [CrossRef]
- Kuo, C.-Y.; Fuh, Y.-S.; Shiue, J.-Y.; Yu, S.J.; Lee, G.-H.; Peng, S.-M. Syntheses and chemistry of Tris(2-pyridyl)phosphine complexes of Group VI transition metals. X-ray structural studies of the molybdenum complexes. J. Organomet. Chem. 1999, 588, 260–267. [Google Scholar] [CrossRef]
- Howie, R.A.; McQuillan, G.P. Trisubstituted Group 6 metal carbonyl complexes with di-2-pyridylamine ligands. Crystal structures of (2,2′-bipyridyl-N,N′) tricarbonyl(di-2-pyridylamine-N′)-molybdenum(0) and -tungsten(0). J. Chem. Soc. Dalton Trans. 1986, 759–764. [Google Scholar] [CrossRef]
- Otto, S. Synthese, experimentelle und theoretische Charakterisierung neuartiger Polypyridyl-Komplexe des Chrom, Mangan und Zink. Diploma Thesis, Johannes Gutenberg University, Mainz, Germany, 2015. [Google Scholar]
- Xiang, H.; Cheng, J.; Ma, X.; Zhou, X.; Chruma, J.J. Near-infrared phosphorescence: Materials and applications. Chem. Soc. Rev. 2013, 42, 6128–6185. [Google Scholar] [CrossRef] [PubMed]
- Barbour, J.C.; Kim, A.J.I.; de Vries, E.; Shaner, S.E.; Lovaasen, B.M. Chromium (III) Bis-Arylterpyridyl Complexes with Enhanced Visible Absorption via Incorporation of Intraligand Charge-Transfer Transitions. Inorg. Chem. 2017, 56, 8212–8222. [Google Scholar] [CrossRef] [PubMed]
- Otto, S.; Förster, C.; Wang, C.; Resch-Genger, U.; Heinze, K. A strongly luminescent chromium(III) complex acid. Chem. Eur. J. 2018, 24. in press. [Google Scholar] [CrossRef]
- Otto, S.; Nauth, A.M.; Ermilov, E.; Scholz, N.; Friedrich, A.; Resch-Genger, U.; Lochbrunner, S.; Opatz, T.; Heinze, K. Photo-Chromium: Sensitizer for Visible-Light-Induced Oxidative C−H Bond Functionalization—Electron or Energy Transfer? ChemPhotoChem 2017, 1, 344–349. [Google Scholar] [CrossRef]
- Vaidyanathan, V.G.; Nair, B.U. Nucleobase Oxidation of DNA by (Terpyridyl)chromium(III) Derivatives. Eur. J. Inorg. Chem. 2004, 1840–1846. [Google Scholar] [CrossRef]
- Stevenson, S.M.; Shores, M.P.; Ferreira, E.M. Photooxidizing Chromium Catalysts for Promoting Radical Cation Cycloadditions. Angew. Chem. Int. Ed. 2015, 54, 6506–6510. [Google Scholar] [CrossRef] [PubMed]
- Higgins, R.F.; Fatur, S.M.; Shepard, S.G.; Stevenson, S.M.; Boston, D.J.; Ferreira, E.M.; Damrauer, N.H.; Rappé, A.K.; Shores, M.P. Uncovering the Roles of Oxygen in Cr(III) Photoredox Catalysis. J. Am. Chem. Soc. 2016, 138, 5451–5464. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, S.M.; Higgins, R.F.; Shores, M.P.; Ferreira, E.M. Chromium photocatalysis: Accessing structural complements to Diels–Alder adducts with electron-deficient dienophiles. Chem. Sci. 2017, 8, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Otto, S.; Scholz, N.; Behnke, T.; Resch-Genger, U.; Heinze, K. Thermo-Chromium: A Contactless Optical Molecular Thermometer. Chem. Eur. J. 2017, 23, 12131–12135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, S.; Harris, J.P.; Heinze, K.; Reber, C. Molecular Ruby under Pressure. Angew. Chem. Int. Ed. 2018, 57, 11069–11073. [Google Scholar] [CrossRef] [PubMed]
- Forman, R.A.; Piermarini, G.J.; Barnett, J.D.; Block, S. Pressure measurement made by the utilization of ruby sharp-line luminescence. Science 1972, 176, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Pajdowski, L.; Karwecka, Z.; Fried, K.; Adamczak, H. Complex compounds of vanadium(III) in pyridine solutions. J. Inorg. Nucl. Chem. 1974, 36, 585–589. [Google Scholar] [CrossRef]
- Casey, A.T.; Clark, R.J.H. Preparations and properties of vanadium(III) complexes of bidentate and terdentate nitrogen-donor ligands. Transit. Met. Chem. 1977, 2, 76–80. [Google Scholar] [CrossRef]
- Silverman, L.D.; Dewan, J.C.; Giandomenico, C.M.; Lippard, S.J. Molecular structure and ligand-exchange reactions of trichlorotris(tert-butyl isocyanide)vanadium(III). Synthesis of the hexakis(tert-butyl isocyanide)vanadium(II) cation. Inorg. Chem. 1980, 19, 3379–3383. [Google Scholar] [CrossRef]
- Manzer, L.E.; Deaton, J.; Sharp, P.; Schrock, R.R. Tetrahydrofuran Complexes of Selected Early Transition Metals. Inorg. Synth. 1982, 21, 135–140. [Google Scholar] [CrossRef]
- Abbo, H.S.; Titinchi, S.J.J. A New Vanadium (III) Complex of 2,6-Bis(3,5-diphenylpyrazol-1-ylmethyl)pyridine as a Catalyst for Ethylene Polymerization. Molecules 2013, 18, 4728–4738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, M.R.; Allan, L.E.N.; Decken, A.; Shaver, M.P. Organometallic mediated radical polymerization of vinyl acetate using bis(imino)pyridine vanadium trichloride complexes. Dalton Trans. 2013, 42, 9157–9165. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.E.; Taube, H. An Investigation of the Vanadium(II)-Vanadium(III) Couple with Polypyridine Ligands. Inorg. Chem. 1968, 7, 254–261. [Google Scholar] [CrossRef]
- Behrens, H.; Brandl, H.; Lutz, K. Über Bis-[tripyridyl(2.2′.2″)]-vanadin(0). Z. Naturforsch. 1967, 22b, 99–100. [Google Scholar] [CrossRef]
- Wang, M.; Weyhermüller, T.; England, J.; Wieghardt, K. Molecular and Electronic Structures of Six-Coordinate “Low-Valent” [M(Mebpy)3]0 (M = Ti, V, Cr, Mo) and [M(tpy)2]0 (M = Ti, V, Cr), and Seven-Coordinate [MoF(Mebpy)3](PF6) and [MX(tpy)2](PF6) (M = Mo, X = Cl and M = W, X = F). Inorg. Chem. 2013, 52, 12763–12776. [Google Scholar] [CrossRef] [PubMed]
- STOE & Cie. X-Red; STOE & Cie: Darmstadt, Germany, 2002. [Google Scholar]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Crystallogr. A 1995, 51, 33–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXL-2014/7; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Sinnecker, S.; Rajendran, A.; Klamt, A.; Diedenhofen, M.; Neese, F. Calculation of Solvent Shifts on Electronic g-Tensors with the Conductor-Like Screening Model (COSMO) and Its Self-Consistent Generalization to Real Solvents (Direct COSMO-RS). J. Phys. Chem. A 2006, 110, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.F. The Determination of the Paramagnetic Susceptibility of Substances in Solution by Nuclear Magnetic Resonance. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Evans, D.F.; Fazakerley, G.V.; Phillips, R.F. Organometallic Compounds of Bivalent Europium, Ytterbium, and Samarium. J. Chem. Soc. A 1971, 1931–1934. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L = ddpd(A) | L = ddpd(B) | ||||||
|---|---|---|---|---|---|---|---|
| M | M–N1/M–N2/M–N3 | PL N7/PL N8 | N1–M–N2/N1–M–N3/N2–M–N3 | M–N4/M–N5/M–N6 | PL N9/PL N10 | N4–M–N5/N4–M–N6/N5–M–N6 | S(OC-6) [g] |
| ZnII [49] | 2.1427(16)/2.127(2)/2.1427(16) | 88/88 | 82.40(4)/164.79(8)/82.40(4) | 2.1427(16)/2.127(2)/2.1427(16) | 88/88 | 82.40(4)/164.79(8)/82.40(4) | 1.18 |
| CuII [a] [60] | 2.149(5)/2.064(5)/2.149(5) | 93/93 | 83.25(12)/166.5(2)/83.25(12) | 2.191(4)/1.978(6)/2.191(4) | 81/81 | 83.51(12)/167.0(2)/83.51(12) | 1.11 |
| CuII [b] [60] | 2.0256(15)/1.989(3)/2.0256(15) | 91/91 | 86.06(5)/172.12(9)/86.06(5) | 2.3318(16)/2.060(3)/2.3318(16) | 90/90 | 80.03(4)/160.06(8)/80.03(4) | 1.60 |
| NiII [61] | 2.0922(15)/2.058(2)/2.0922(15) | 88/88 | 84.64(4)/169.28(8)/84.64(4) | 2.0922(15)/2.058(2)/2.0922(15) | 88/88 | 84.64(4)/169.28(8)/84.64(4) | 0.60 |
| CoII [62] | 2.1199(19)/2.090(2)/2.1199(19) | 88/88 | 83.19(5)/166.38(10)/83.19(5) | 2.1199(19)/2.090(2)/2.1199(19) | 88/88 | 83.19(5)/166.38(10)/83.19(5) | 0.95 |
| CoII [c] [63] | 2.20(2)/2.19(2)/2.14(2) | 97/96 | 83.6(8)/94.4(7)/83.0(8) | 2.25(2)/2.11(2)/2.19(2) | 99/92 | 81.3(8)/94.3(8)/81.7(8) | 0.81 |
| CoIII [d] | 1.954(3)/1.942(3)/1.952(3) | 89/88 | 87.66(14)/176.18(15)/88.54(14) | 1.947(3)/1.945(3)/1.941(3) | 87/84 | 87.92(15)/175.85(16)/87.93(14) | 0.10 |
| FeII [64] | 1.9938(17)/1.963(2)/1.9938(17) | 89/89 | 87.73(5)/175.46(9)/87.73(5) | 1.9938(17)/1.963(2)/1.9938(17) | 89/89 | 87.73(5)/175.46(9)/87.73(5) | 0.12 |
| FeII [f] [64] | 1.9939(18)/1.9593(18)/1.9922(18) | 89/88 | 88.39(7)/177.01(8)/88.63(7) | 1.9921(19)/1.9661(18)/1.9855(19) | 89/84 | 88.89(8)/176.48(7)/88.23(8) | 0.08 |
| FeII [d,e] | 1.983(3)/1.964(5)/1.983(3) | 87/87 | 87.90(10)/175.8(2)/87.90(10) | 1.983(3)/1.964(5)/1.983(3) | 87/87 | 87.90(10)/175.8(2)/87.90(10) | 0.10 |
| CrII [d] | 2.117(7)/2.070(11)/2.117(7) | 87/87 | 83.4(2)/166.7(4)/83.4(2) | 2.089(6)/2.064(9)/2.089(6) | 88/88 | 84.8(2)/169.7(4)/84.8(2) | 0.74 |
| CrIII [50] | 2.0485(18)/2.0393(18)/2.0394(19) | 82/88 | 85.13(8)/170.86(8)/85.74(7) | 2.0446(17)/2.0444(18)/2.0485(18) | 87/88 | 85.89(7)/170.88(7)/84.99(7) | 0.43 |
| CrIII [f] mol.A [50] | 2.041(6)/2.054(7)/2.028(6) | 81/87 | 85.7(2)/172.3(3)/86.7(3) | 2.040(7)/2.054(7)/2.048(7) | 85/90 | 85.4(3)/171.0(3)/85.6(3) | 0.37 |
| CrIII [f] mol. B [50] | 2.039(7)/2.030(7)/2.033(7) | 88/84 | 86.8(3)/172.6(3)/86.0(3) | 2.040(6)/2.046(7)/2.030(6) | 84/80 | 85.5(3)/173.0(3)/87.5(3) | 0.29 |
| VIII [c,d,f] | 2.104(8)/2.054(8)/2.121(9) | 88/94 | 81.3(3)/92.3(3)/84.6(4) | 2.101(7)/2.036(8)/2.106(8) | 86/90 | 82.7(3)/95.0(3)/84.0(3) | 0.64 |
| L = tpy(A) | L = tpy(B) | ||||
|---|---|---|---|---|---|
| M | M–N1/M–N2/M–N3 | N1–M–N2/N1–M–N3/N2–M–N3 | M–N4/M–N5/M–N6 | N4–M–N5/N4–M–N6/N5–M–N6 | S(OC-6) [e] |
| ZnII [a] [65] | 2.1952(15)/2.089(2)/2.1952(15) | 75.16(4)/150.31(8)/75.16(4) | 2.1882(15)/2.089(2)/2.1882(15) | 75.36(4)/150.71(9)/75.36(4) | 4.46 |
| CuII [66] | 2.225(6)/2.061(5)/2.232(6) | 77.8(2)/154.4(2)/76.7(2) | 2.207(5)/1.971(9)/2.224(5) | 76.9(2)/153.6(2)/76.8(2) | 3.70 |
| NiII [c] [67] | 2.110(2)/1.999(2)/2.119(2) | 78.00(8)/155.42(8)/77.50(8) | 2.120(2)/2.000(2)/2.117(2) | 77.40(8)/155.37(8)/78.06(8) | 3.18 |
| CoII [68] | 2.151(3)/2.013(5)/2.147(3) | 77.11(14)/154.30(13)/77.21(15) | 2.141(3)/2.003(4)/2.153(3) | 76.46(12)/153.34(12)/77.01(12) | 3.62 |
| CoIII [b] mol A [68] | 1.9545(16)/1.8590(16)/1.9408(16) | 82.60(7)/164.93(7)/82.46(7) | 1.9595(16)/1.8596(16)/1.9369(16) | 82.51(7)/165.08(7)/82.66(7) | 1.22 |
| CoIII [b] mol B [68] | 1.9348(15)/1.8550(15)/1.9440(15) | 83.06(7)/165.54(7)/82.50(7) | 1.9457(15)/1.8555(15)/1.9474(16) | 82.87(7)/165.55(7)/82.74(7) | 1.15 |
| FeII [c] [69] | 1.990(3)/1.888(3)/1.990(3) | 80.60(13)/161.45(12)/80.94(12) | 1.988(3)/1.887(3)/1.981(3) | 80.88(12)/161.64(12)/80.87(12) | 1.81 |
| MnII [c] [70] | 2.269(2)/2.196(2)/2.257(2) | 72.09(7)/144.45(8)/72.36(7) | 2.246(2)/2.178(2)/2.249(2) | 72.68(7)/144.70(8)/72.47(7) | 6.70 |
| MnIII [b,d] [70] | 2.1238(13)/1.9749(12)/2.1087(13) | 77.31(5)/155.03(5)/77.93(5) | 2.1238(13)/1.9749(12)/2.1087(13) | 77.31(5)/155.03(5)/77.93(5) | 3.42 |
| “CrII” [b] mol A[27] | 2.049(4)/1.916(4)/2.033(4) | 79.28(15)/158.19(15)/78.94(15) | 2.060(4)/1.997(4)/2.069(4) | 78.07(14)/156.50(15)/78.43(14) | 2.68 |
| “CrII” [b] mol B[27] | 2.036(4)/1.919(4)/2.053(4) | 78.83(15)/157.59(16)/78.80(15) | 2.073(4)/2.006(4)/2.062(4) | 78.01(15)/156.12(16)/78.18(14) | 2.80 |
| CrIII [b] mol A[71] | 2.054(3)/2.042(3)/2.058(4) | 78.78(14)/157.06(14)/78.34(14) | 2.042(3)/1.987(3)/2.062(4) | 79.00(14)/157.17(14)/78.22(15) | 2.73 |
| CrIII [b] mol B[71] | 2.055(3)/1.976(3)/2.057(3) | 79.01(13)/158.24(13)/79.29(14) | 2.062(3)/1.989(3)/2.042(3) | 79.12(13)/157.98(13)/78.97(14) | 2.57 |
| CrIII [b] mol C[71] | 2.099(4)/2.003(4)/2.035(4) | 77.39(15)/156.93(16)/79.59(17) | 2.063(4)/1.983(3)/2.049(3) | 78.37(14)/157.19(14)/78.92(14) | 2.74 |
| CrIII [b] mol D[71] | 2.039(4)/1.981(4)/2.049(4) | 78.80(15)/157.98(16)/79.41(15) | 2.053(4)/1.978(4)/2.057(4) | 78.43(15)/157.76(15)/79.38(15) | 2.63 |
| L = ddpd | L = tpy | |||
|---|---|---|---|---|
| M | ∆δ(Hα)/ppm [a] | M…Hα from XRD/Å | ∆δ(Hα)/ppm [b] | M…Hα from XRD/Å |
| ZnII | −0.56 [49] | 3.06 [49] | −0.95 [74] | 3.18–3.28 [d] |
| CoIII | −1.39 [62] | 2.92–2.94 [c] | −1.46 [68] | 3.07 [e] |
| FeII | −1.21 [64] | 2.99 [64] | −1.33 [f] | 3.01–3.12 [g] |
| L = ddpd | L = tpy | |||
|---|---|---|---|---|
| M | Oxidation Processes | Reduction Processes | Oxidation Processes | Reduction Processes |
| L only | +0.55 (irrev.), +1.06 (irrev.) [a] | −3.27 (irrev.) [a] | not reported | −2.55 (rev.) [b] |
| ZnII | +1.17, (irrev.), +1.34, (irrev.), +1.67 (irrev.) [a] | −2.38 (irrev.) [a] | not reported | −1.68, −1.81 (rev.) [c] |
| CuII | +0.95 (irrev.), +1.53 (irrev.), +1.75 (irrev.), +2.26 (irrev.) [d] | −0.91 (qrev. CuII/CuI), −3.10 (irrev.) [d] | not reported | −0.79 (irrev. CuII/CuI) [e] |
| NiII | +1.22 (qrev.), +1.71(irrev.) [f] | −2.03 (irrev.) [f] | +1.29 (qrev.) [g] | −1.62 (qrev.) [g] −1.62 (qrev.) −1.88 (qrev.) [c] |
| CoII | −0.17 (rev.) [h] | −1.80 (irrev.) [h] | –0.11 (rev.) [i] | −1.16 (rev.) [i] −1.17 (rev.), −2.03 (qrev.) [c] |
| FeII | +0.33 (rev.), +1.63 (irrev.) [j] | −2.33 (irrev.) [j] | +0.71 (rev.) [k] | −1.64 (rev.),−1.80 (rev.) [k] |
| MnII | - | - | +0.86 (rev.) [l] | −1.52 (rev.),−1.86 (rev.),−2.37 (rev.) [l] |
| CrIII | +1.71 (irrev.) [m] | −1.11 (rev. CrIII/CrII), −1.94 (irrev.) [m] | not reported | −0.53 (rev.),−0.95 (rev.) [n] |
| VII | - | - | –0.09 (qrev.) [o] | −1.25 (rev.),−1.40 (rev.),−1.93 (rev.) [o] |
| L = ddpd | L = tpy | Spin-Only Value [a] | |||
|---|---|---|---|---|---|
| M | χT/cm3 K mol−1 | giso | χT/cm3 K mol–1 | giso | χT/cm3 K mol–1 |
| CuII | 0.443 [b] | 2.122 (EPR) [b] | 0.475 [c] | 2.135 (EPR) [d] | 0.375 |
| NiII | 1.11 [e] | 2.12 (from χT) [e] | 1.20 [f] | 2.19 (from χT) [f] | 1.000 |
| CoII | 2.53 [g] | 2.32 (from χT) [g] | 2.31–3.00 [h] | 2.22–2.58 (from χT) [h] | 1.875 |
| MnII | - | - | 4.62 [i] | - | 4.375 |
| MnIII | - | - | 2.68 [j] | 1.89 (from χT) [j] | 3.000 |
| CrII | 2.67 | 1.89 (from χT) | 0.938 [k] | 1.94 (from χT) [k] | 3.000 |
| CrIII | 1.838 [l] | 1.990 (EPR, 77 K) [l] | 1.921 [k] | 1.95 (EPR, 150 K) [m] | 1.875 |
| VII | - | - | 1.721 [n] | 1.92 (from χT) [n] | 1.875 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Förster, C.; Dorn, M.; Reuter, T.; Otto, S.; Davarci, G.; Reich, T.; Carrella, L.; Rentschler, E.; Heinze, K. Ddpd as Expanded Terpyridine: Dramatic Effects of Symmetry and Electronic Properties in First Row Transition Metal Complexes. Inorganics 2018, 6, 86. https://doi.org/10.3390/inorganics6030086
Förster C, Dorn M, Reuter T, Otto S, Davarci G, Reich T, Carrella L, Rentschler E, Heinze K. Ddpd as Expanded Terpyridine: Dramatic Effects of Symmetry and Electronic Properties in First Row Transition Metal Complexes. Inorganics. 2018; 6(3):86. https://doi.org/10.3390/inorganics6030086
Chicago/Turabian StyleFörster, Christoph, Matthias Dorn, Thomas Reuter, Sven Otto, Güllü Davarci, Tobias Reich, Luca Carrella, Eva Rentschler, and Katja Heinze. 2018. "Ddpd as Expanded Terpyridine: Dramatic Effects of Symmetry and Electronic Properties in First Row Transition Metal Complexes" Inorganics 6, no. 3: 86. https://doi.org/10.3390/inorganics6030086