
Bioactive Compounds as Inhibitors of Inflammation, Oxidative Stress and Metabolic Dysfunctions via Regulation of Cellular Redox Balance and Histone Acetylation State

Abstract
:1. Introduction
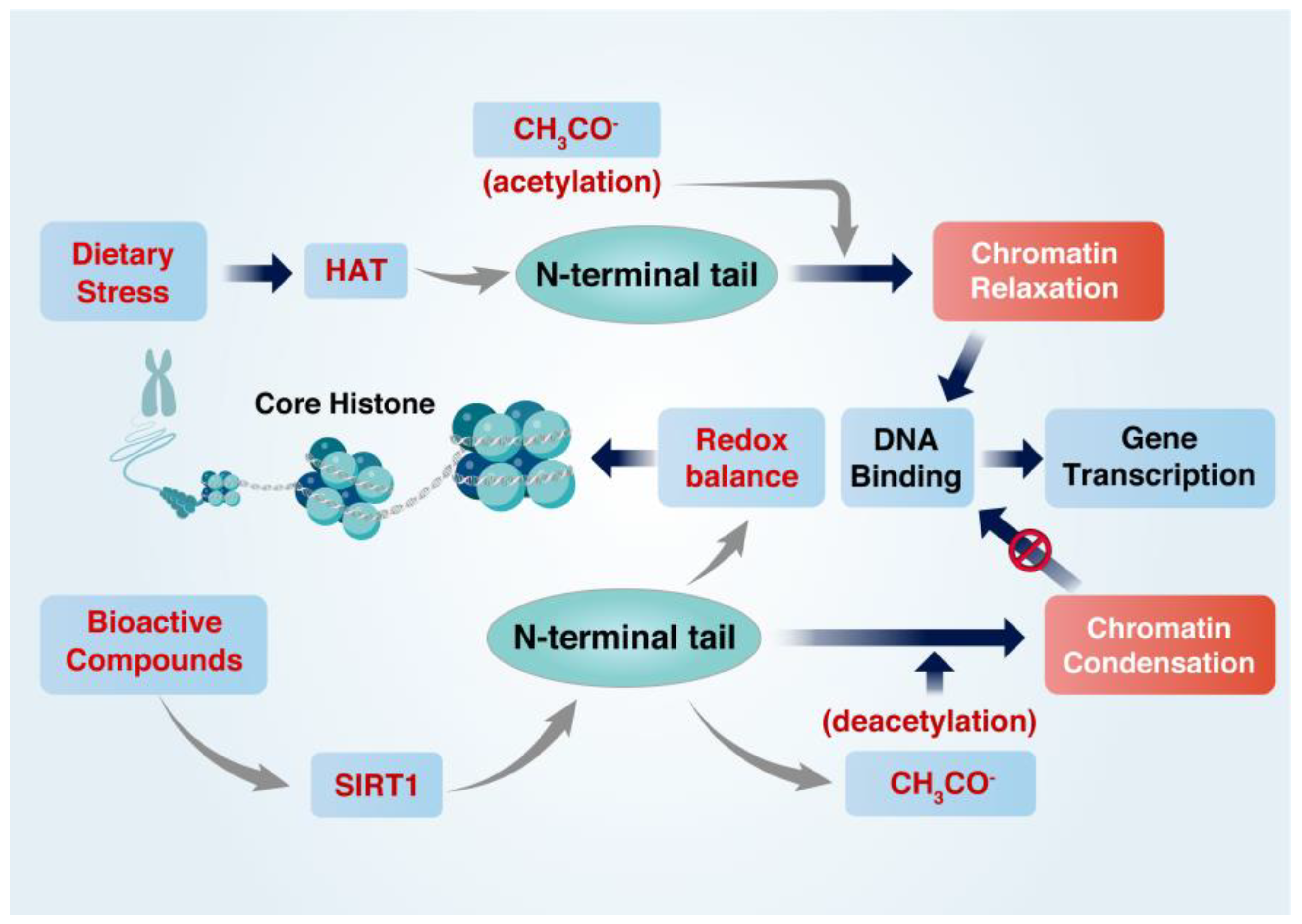
2. Regulation of the Cellular Redox Balance and Histone Acetylation State
2.1. Regulation of Cellular Redox Balance
2.2. Regulation of Histone Acetylation State
3. Diet-Induced Alterations of Redox Balance and Histone Acetylation State
3.1. Excessive Alcohol Use
3.2. High-Fat Diet
3.3. High-Glycemic Diet
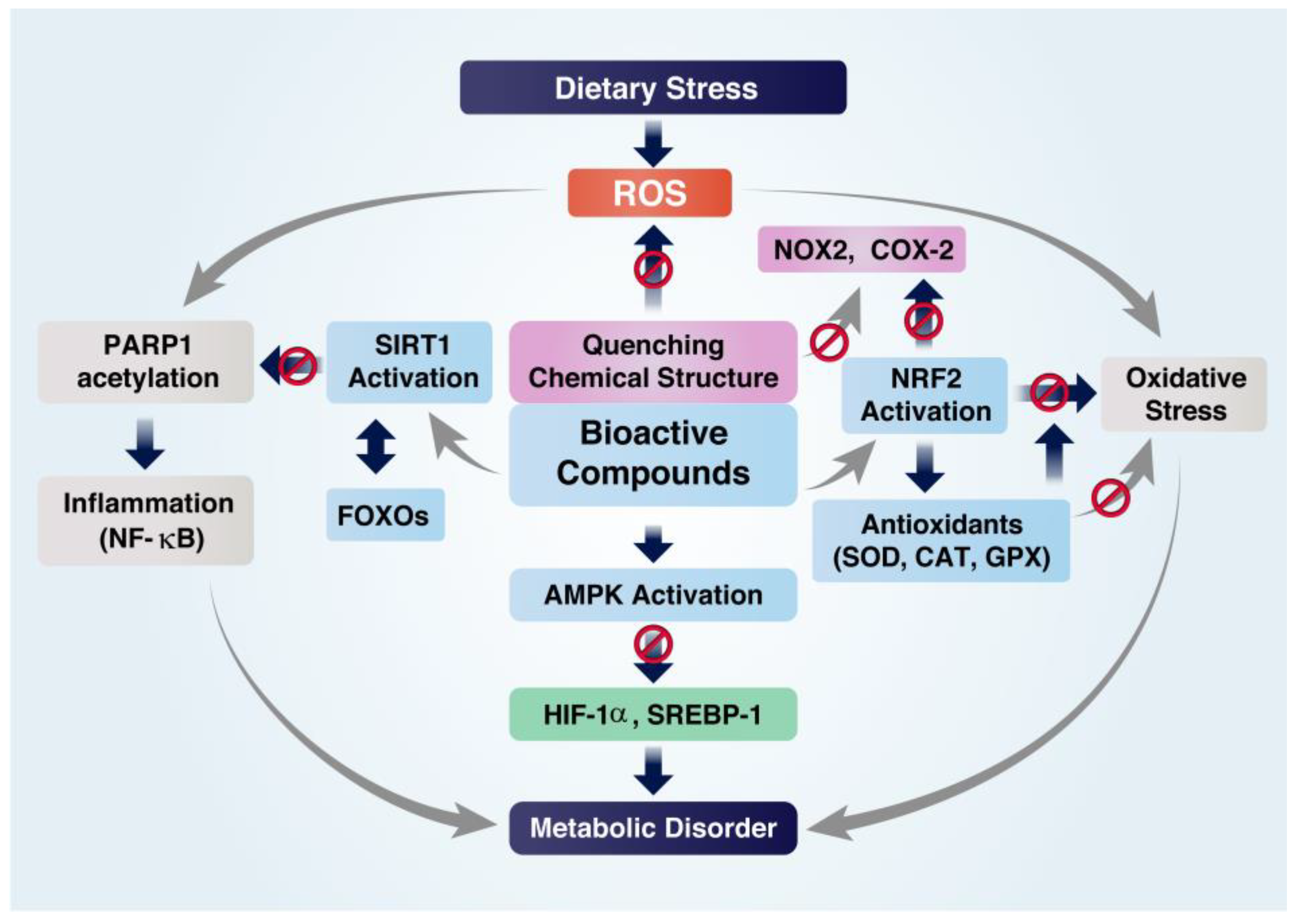
4. Protective Roles of Bioactive Compounds (BCs)
4.1. Inhibition of Inflammation
4.2. Inhibition of Oxidative Stress
4.3. Inhibition of Metabolic Disorders
5. Roles of BCs in Regulating Cellular Redox Balance and Histone Acetylation State
5.1. Astaxanthin
5.2. Butyrate
5.3. Polyphenols
5.3.1. Curcumin
5.3.2. Epigallocatechin-3-Gallate
5.3.3. Resveratrol
5.4. Nicotinamide Riboside
5.5. Sulforaphane
5.6. Ginsenoside
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pham, T.X.; Lee, J. Dietary regulation of histone acetylases and deacetylases for the prevention of metabolic diseases. Nutrients 2012, 4, 1868–1886. [Google Scholar] [CrossRef] [Green Version]
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.Y. ROS-Mediated Cancer Cell Killing through Dietary Phytochemicals. Oxid. Med. Cell. Longev. 2019, 2019, 9051542. [Google Scholar] [CrossRef]
- Sui, J.; Qiao, W.; Xiang, X.; Luo, Y. Epigenetic changes in Mycobacterium tuberculosis and its host provide potential targets or biomarkers for drug discovery and clinical diagnosis. Pharmacol. Res. 2022, 179, 106195. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Potempa, J. Epigenetic regulation in bacterial infections: Targeting histone deacetylases. Crit. Rev. Microbiol. 2018, 44, 336–350. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox Signal. 2010, 13, 1665–1678. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Dumollard, R.; Carroll, J.; Duchen, M.R.; Campbell, K.; Swann, K. Mitochondrial function and redox state in mammalian embryos. Semin. Cell Dev. Biol. 2009, 20, 346–353. [Google Scholar] [CrossRef]
- Yang, J.L.; Mukda, S.; Chen, S.D. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018, 16, 263–275. [Google Scholar] [CrossRef]
- Wang, H.; Yang, L.; Liu, M.; Luo, J. Protein post-translational modifications in the regulation of cancer hallmarks. Cancer Gene Ther. 2022. [Google Scholar] [CrossRef]
- Kaniskan, H.U.; Konze, K.D.; Jin, J. Selective inhibitors of protein methyltransferases. J. Med. Chem. 2015, 58, 1596–1629. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC inhibitors: Targets for tumor therapy, immune modulation and lung diseases. Transl. Oncol. 2022, 16, 101312. [Google Scholar] [CrossRef]
- Musselman, C.A.; Lalonde, M.E.; Cote, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaiswing, L.; Oberley, T.D. Extracellular/microenvironmental redox state. Antioxid. Redox Signal. 2010, 13, 449–465. [Google Scholar] [CrossRef]
- An, L.; Wang, X.; Cederbaum, A.I. Cytokines in alcoholic liver disease. Arch. Toxicol. 2012, 86, 1337–1348. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. 2012, 16, 180–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, N.; Reddy, M.A.; Guha, M.; Natarajan, R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 2003, 52, 1256–1264. [Google Scholar] [CrossRef] [Green Version]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef] [Green Version]
- Meli, R.; Mattace Raso, G.; Irace, C.; Simeoli, R.; Di Pascale, A.; Paciello, O.; Pagano, T.B.; Calignano, A.; Colonna, A.; Santamaria, R. High Fat Diet Induces Liver Steatosis and Early Dysregulation of Iron Metabolism in Rats. PLoS ONE 2013, 8, e66570. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Wu, J.; Jiang, Z.; Huang, W.; Jia, Y.; Sun, W.; Wu, H. P53/NRF2 mediates SIRT1’s protective effect on diabetic nephropathy. Biochim. Biophys. Acta. Mol. Cell Res. 2019, 1866, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, G.; Chen, L.; Ding, Y.; Lian, J.; Hong, G.; Lu, Z. Resveratrol protects mice from paraquat-induced lung injury: The important role of SIRT1 and NRF2 antioxidant pathways. Mol. Med. Rep. 2016, 13, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; Peterson, S.E.; Loring, J.F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014, 24, 143–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, P.S.; Boriek, A.M. SIRT1 Regulation in Ageing and Obesity. Mech. Ageing Dev. 2020, 188, 111249. [Google Scholar] [CrossRef]
- Kwon, H.S.; Ott, M. The ups and downs of SIRT1. Trends Biochem. Sci. 2008, 33, 517–525. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K. SIRT1 longevity factor suppresses NF-kappaB -driven immune responses: Regulation of aging via NF-kappaB acetylation? Bioessays 2008, 30, 939–942. [Google Scholar] [CrossRef]
- Ghosh, H.S.; Spencer, J.V.; Ng, B.; McBurney, M.W.; Robbins, P.D. Sirt1 interacts with transducin-like enhancer of split-1 to inhibit nuclear factor kappaB-mediated transcription. Biochem. J. 2007, 408, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.J.; Kho, J.H.; Kang, M.R.; Um, S.J. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol. Cell 2007, 28, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.M.; Huber, F.M.; Hoelz, A. Structural and functional analysis of human SIRT1. J. Mol. Biol. 2014, 426, 526–541. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Lee, Y.; Bae, M.; Park, Y.K.; Lee, J.Y. Astaxanthin inhibits alcohol-induced inflammation and oxidative stress in macrophages in a sirtuin 1-dependent manner. J. Nutr. Biochem. 2020, 85, 108477. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Park, Y.K.; Lee, J.Y. Nicotinamide riboside, an NAD(+) precursor, attenuates inflammation and oxidative stress by activating sirtuin 1 in alcohol-stimulated macrophages. Lab. Investig. 2021, 101, 1225–1237. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Lowenstein, C.J. MiR-34, SIRT1 and p53: The feedback loop. Cell Cycle. 2009, 8, 712–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Padhye, A.; Sharma, A.; Song, G.; Miao, J.; Mo, Y.Y.; Wang, L.; Kemper, J.K. A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J. Biol. Chem. 2010, 285, 12604–12611. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kemper, J.K. Controlling SIRT1 expression by microRNAs in health and metabolic disease. Aging 2010, 2, 527–534. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.Y.; Xu, X.Q.; Ji, C.B.; Shi, C.M.; Guo, X.R.; Fu, J.F. Aberrant hepatic microRNA expression in nonalcoholic fatty liver disease. Cell Physiol. Biochem. 2014, 34, 1983–1997. [Google Scholar] [CrossRef]
- Zhong, W.; Weiss, H.L.; Jayswal, R.D.; Hensley, P.J.; Downes, L.M.; St Clair, D.K.; Chaiswing, L. Extracellular redox state shift: A novel approach to target prostate cancer invasion. Free Radic. Biol. Med. 2018, 117, 99–109. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Nanayakkara, G.K.; Wang, H.; Yang, X. Proton leak regulates mitochondrial reactive oxygen species generation in endothelial cell activation and inflammation—A novel concept. Arch. Biochem. Biophys. 2019, 662, 68–74. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Buelna-Chontal, M.; Zazueta, C. Redox activation of Nrf2 & NF-kappaB: A double end sword? Cell Signal. 2013, 25, 2548–2557. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Marwick, J.; Kirkham, P. Redox modulation of chromatin remodeling: Impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem. Pharmacol. 2004, 68, 1255–1267. [Google Scholar] [CrossRef]
- Bode, K.A.; Schroder, K.; Hume, D.A.; Ravasi, T.; Heeg, K.; Sweet, M.J.; Dalpke, A.H. Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology 2007, 122, 596–606. [Google Scholar] [CrossRef]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Sumida, Y.; Niki, E.; Naito, Y.; Yoshikawa, T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic. Res. 2013, 47, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.F.; Wan, T.; Ye, M.T.; Qiu, Y.; Pei, L.; Jiang, R.; Pang, N.Z.; Huang, Y.L.; Liang, B.X.; Ling, W.H.; et al. Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1 alpha/mitochondrial biosynthesis pathway. Redox Biol. 2018, 17, 89–98. [Google Scholar] [CrossRef]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 2004, 306, 2105–2108. [Google Scholar] [CrossRef]
- Xiong, S.; Salazar, G.; Patrushev, N.; Alexander, R.W. FoxO1 mediates an autofeedback loop regulating SIRT1 expression. J. Biol. Chem. 2011, 286, 5289–5299. [Google Scholar] [CrossRef] [Green Version]
- Noriega, L.G.; Feige, J.N.; Canto, C.; Yamamoto, H.; Yu, J.; Herman, M.A.; Mataki, C.; Kahn, B.B.; Auwerx, J. CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO Rep. 2011, 12, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1 alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Park, Y.K.; Lee, J.Y. Inhibition of alcohol-induced inflammation and oxidative stress by astaxanthin is mediated by its opposite actions in the regulation of sirtuin 1 and histone deacetylase 4 in macrophages. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2021, 1866, 158838. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver. Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Kraus, W.L. SIRT1-dependent regulation of chromatin and transcription: Linking NAD(+) metabolism and signaling to the control of cellular functions. BBA-Proteins Proteom. 2010, 1804, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios-Covian, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de Los Reyes-Gavilan, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [Green Version]
- Thakur, V.; McMullen, M.R.; Pritchard, M.T.; Nagy, L.E. Regulation of macrophage activation in alcoholic liver disease. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S53–S56. [Google Scholar] [CrossRef]
- Shen, Z.; Ajmo, J.M.; Rogers, C.Q.; Liang, X.; Le, L.; Murr, M.M.; Peng, Y.; You, M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-alpha production in cultured macrophage cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1047–G1053. [Google Scholar] [CrossRef]
- Park, P.H.; Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Nagy, L.E. Regulation of Kupffer cell activity during chronic ethanol exposure: Role of adiponectin. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. S3), S30–S33. [Google Scholar] [CrossRef]
- Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Wang, Q.; Nagy, L.E. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: Role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J. Leukoc. Biol. 2006, 79, 1348–1356. [Google Scholar] [CrossRef] [Green Version]
- Abdelmohsen, K.; Pullmann, R., Jr.; Lal, A.; Kim, H.H.; Galban, S.; Yang, X.; Blethrow, J.D.; Walker, M.; Shubert, J.; Gillespie, D.A.; et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 2007, 25, 543–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Oxidative stress modulates Sir2alpha in rat hippocampus and cerebral cortex. Eur. J. Neurosci. 2006, 23, 2573–2580. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sauve, A.A. NAD(+) content and its role in mitochondria. Methods Mol. Biol. 2015, 1241, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ye, J.P. Regulation of energy balance by inflammation: Common theme in physiology and pathology. Rev. Endocr. Metab. Dis. 2015, 16, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Kominsky, D.J.; Campbell, E.L.; Colgan, S.P. Metabolic Shifts in Immunity and Inflammation. J. Immunol. 2010, 184, 4062–4068. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sang, N. Hypoxia-Inducible Factor-1: A Critical Player in the Survival Strategy of Stressed Cells. J. Cell. Biochem. 2016, 117, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Hoyt, L.R.; Randall, M.J.; Ather, J.L.; DePuccio, D.P.; Landry, C.C.; Qian, X.; Janssen-Heininger, Y.M.; van der Vlieta, A.; Dixon, A.E.; Amiel, E.; et al. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. 2017, 12, 883–896. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, H.Z.; Liu, J.J.; Jia, Y.Y.; Zhang, Z.Q.; Yang, R.F.; Zhang, Y.; Xu, J.; Wei, Y.S.; Liu, D.P.; et al. SIRT1 Suppresses Activator Protein-1 Transcriptional Activity and Cyclooxygenase-2 Expression in Macrophages. J. Biol. Chem. 2010, 285, 7097–7110. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Fang, D. The Roles of SIRT1 in Cancer. Genes Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhao, J.; Zhang, S.J.; Weinman, S.A. FOXO3-dependent apoptosis limits alcohol-induced liver inflammation by promoting infiltrating macrophage differentiation. Cell Death Discov. 2018, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Li, M.; Zheng, D.; Chen, Q.; Liu, W.; Feng, L. Adipose tissue hypoxia and low-grade inflammation: A possible mechanism for ethanol-related glucose intolerance? Br. J. Nutr. 2015, 113, 1355–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, P.H.; Huang, H.; McMullen, M.R.; Mandal, P.; Sun, L.; Nagy, L.E. Suppression of lipopolysaccharide-stimulated tumor necrosis factor-alpha production by adiponectin is mediated by transcriptional and post-transcriptional mechanisms. J. Biol. Chem. 2008, 283, 26850–26858. [Google Scholar] [CrossRef] [Green Version]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Hong, F.; Sekhar, K.R.; Freeman, M.L.; Liebler, D.C. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 2005, 280, 31768–31775. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.Y.; Reddy, S.P.; Debiase, A.; Yamamoto, M.; Kleeberger, S.R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 2005, 38, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Lee, Y.; Kim, M.B.; Hu, S.; Jang, H.; Park, Y.K.; Lee, J.Y. The loss of histone deacetylase 4 in macrophages exacerbates hepatic and adipose tissue inflammation in male but not in female mice with diet-induced non-alcoholic steatohepatitis. J. Pathol. 2021, 255, 319–329. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Choi, M.J.; Kim, S.W.; Yun, J.W. Secreted protein acidic and rich in cysteine (SPARC) regulates thermogenesis in white and brown adipocytes. Mol. Cell. Endocrinol. 2020, 506, 110757. [Google Scholar] [CrossRef]
- Vegiopoulos, A.; Muller-Decker, K.; Strzoda, D.; Schmitt, I.; Chichelnitskiy, E.; Ostertag, A.; Berriel Diaz, M.; Rozman, J.; Hrabe de Angelis, M.; Nusing, R.M.; et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 2010, 328, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, S.; Uto, H.; Kumamoto, R.; Arima, S.; Hashimoto, S.; Nasu, Y.; Takami, Y.; Moriuchi, A.; Sakiyama, T.; Oketani, M.; et al. Liver regeneration after partial hepatectomy in rat is more impaired in a steatotic liver induced by dietary fructose compared to dietary fat. Biochem. Biophys. Res. Commun. 2011, 407, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Tian, D.; Jia, Y.; Huang, W.; Jiang, M.; Wang, J.; Sun, W.; Wu, H. MDM2 controls NRF2 antioxidant activity in prevention of diabetic kidney disease. Biochim. Biophys. Acta. Mol. Cell Res. 2018, 1865, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef] [Green Version]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Miller, R.A.; Patel, R.T.; Chen, J.; Dhir, R.; Wang, H.; Zhang, D.; Graham, M.J.; Unterman, T.G.; Shulman, G.I.; et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med. 2012, 18, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Jia, C.; Zhang, S.; Fan, G.; Li, Y.; Shan, P.; Sun, L.; Xiao, W.; Li, L.; Zheng, Y.; et al. The REGgamma proteasome regulates hepatic lipid metabolism through inhibition of autophagy. Cell Metab. 2013, 18, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Nanditha, A.; Ma, R.C.; Ramachandran, A.; Snehalatha, C.; Chan, J.C.; Chia, K.S.; Shaw, J.E.; Zimmet, P.Z. Diabetes in Asia and the Pacific: Implications for the Global Epidemic. Diabetes Care 2016, 39, 472–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Loomans, C.J.; van Haperen, R.; Duijs, J.M.; Verseyden, C.; de Crom, R.; Leenen, P.J.; Drexhage, H.A.; de Boer, H.C.; de Koning, E.J.; Rabelink, T.J.; et al. Differentiation of bone marrow-derived endothelial progenitor cells is shifted into a proinflammatory phenotype by hyperglycemia. Mol. Med. 2009, 15, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, Y.; Li, W.; Zhang, J.; Che, Y.; Cui, X.; Sun, B.; Zhao, G. Loss of histone deacetylase 2 inhibits oxidative stress induced by high glucose via the HO-1/SIRT1 pathway in endothelial progenitor cells. Gene 2018, 678, 1–7. [Google Scholar] [CrossRef]
- Mercado, N.; Thimmulappa, R.; Thomas, C.M.; Fenwick, P.S.; Chana, K.K.; Donnelly, L.E.; Biswal, S.; Ito, K.; Barnes, P.J. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem. Biophys. Res. Commun. 2011, 406, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Wang, Y.; Zhang, H.; Du, M.; Li, T. Acacetin attenuates mice endotoxin-induced acute lung injury via augmentation of heme oxygenase-1 activity. Inflammopharmacology 2018, 26, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [Green Version]
- Spaeth, J.M.; Walker, E.M.; Stein, R. Impact of Pdx1-associated chromatin modifiers on islet beta-cells. Diabetes Obes. Metab. 2016, 18 (Suppl. S1), 123–127. [Google Scholar] [CrossRef] [Green Version]
- Dewanjee, S.; Vallamkondu, J.; Kalra, R.S.; Chakraborty, P.; Gangopadhyay, M.; Sahu, R.; Medala, V.; John, A.; Reddy, P.H.; De Feo, V.; et al. The Emerging Role of HDACs: Pathology and Therapeutic Targets in Diabetes Mellitus. Cells 2021, 10, 1340. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Zhang, J.; Kheterpal, I.; Kennedy, N.; Davis, R.J.; Ye, J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J. Biol. Chem. 2011, 286, 22227–22234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.H. Dietary bioactive compounds and their health implications. J. Food Sci. 2013, 78 (Suppl. S1), A18–A25. [Google Scholar] [CrossRef] [PubMed]
- Camara, J.S.; Albuquerque, B.R.; Aguiar, J.; Correa, R.C.G.; Goncalves, J.L.; Granato, D.; Pereira, J.A.M.; Barros, L.; Ferreira, I. Food Bioactive Compounds and Emerging Techniques for Their Extraction: Polyphenols as a Case Study. Foods 2020, 10, 37. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Lagier, J.C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial culturomics: Paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 2012, 18, 1185–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erejuwa, O.O.; Sulaiman, S.A.; Ab Wahab, M.S. Modulation of gut microbiota in the management of metabolic disorders: The prospects and challenges. Int. J. Mol. Sci. 2014, 15, 4158–4188. [Google Scholar] [CrossRef] [Green Version]
- Lazar, V.; Ditu, L.M.; Pircalabioru, G.G.; Picu, A.; Petcu, L.; Cucu, N.; Chifiriuc, M.C. Gut Microbiota, Host Organism, and Diet Trialogue in Diabetes and Obesity. Front. Nutr. 2019, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarneshan, S.N.; Fakhri, S.; Farzaei, M.H.; Khan, H.; Saso, L. Astaxanthin targets PI3K/Akt signaling pathway toward potential therapeutic applications. Food Chem. Toxicol. 2020, 145, 111714. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Leonard, S.S.; Xia, C.; Jiang, B.H.; Stinefelt, B.; Klandorf, H.; Harris, G.K.; Shi, X. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochem. Biophys. Res. Commun. 2003, 309, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Das, D.K. Anti-inflammatory responses of resveratrol. Inflamm. Allergy Drug Targets 2007, 6, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Sohn, P.D.; Cho, S.H.; Swanson, R.A.; Gan, L. Sirtuins in neurodegenerative diseases: An update on potential mechanisms. Front. Aging Neurosci. 2013, 5, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassa, P.O.; Haenni, S.S.; Buerki, C.; Meier, N.I.; Lane, W.S.; Owen, H.; Gersbach, M.; Imhof, R.; Hottiger, M.O. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J. Biol. Chem. 2005, 280, 40450–40464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britton, G. Structure and properties of carotenoids in relation to function. FASEB J. 1995, 9, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Bailey-Downs, L.; Sosnowska, D.; Gautam, T.; Koncz, P.; Losonczy, G.; Ballabh, P.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Vascular oxidative stress in aging: A homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H363–H372. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, S.; Miyamoto, T.; Kobara, H.; Yamada, S.; Asaka, R.; Kikuchi, N.; Kashima, H.; Ohira, S.; Shiozawa, T. Trophoblast type-specific expression of senescence markers in the human placenta. Placenta 2019, 85, 56–62. [Google Scholar] [CrossRef]
- Deng, X.; Wang, M.; Hu, S.; Feng, Y.; Shao, Y.; Xie, Y.; Wu, M.; Chen, Y.; Shi, X. The Neuroprotective Effect of Astaxanthin on Pilocarpine-Induced Status Epilepticus in Rats. Front. Cell. Neurosci. 2019, 13, 123. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wu, H.; Zhuo, W.W.; Mao, Q.F.; Lan, H.; Zhang, Y.; Hua, S. Astaxanthin Normalizes Epigenetic Modifications of Bovine Somatic Cell Cloned Embryos and Decreases the Generation of Lipid Peroxidation. Reprod. Domest. Anim. 2015, 50, 793–799. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Horio, Y. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. PLoS ONE 2013, 8, e73875. [Google Scholar] [CrossRef] [PubMed]
- Kuscu, N.; Gungor-Ordueri, N.E.; Sozen, B.; Adiguzel, D.; Celik-Ozenci, C. FoxO transcription factors 1 regulate mouse preimplantation embryo development. J. Assist. Reprod. Genet. 2019, 36, 2121–2133. [Google Scholar] [CrossRef]
- Yin, L.; Xu, L.; Chen, B.; Zheng, X.; Chu, J.; Niu, Y.; Ma, T. SRT1720 plays a role in oxidative stress and the senescence of human trophoblast HTR8/SVneo cells induced by D-galactose through the SIRT1/FOXO3a/ROS signalling pathway. Reprod. Toxicol. 2022, 111, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [Green Version]
- Kawai, Y.; Garduno, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [Green Version]
- Shakespear, M.R.; Iyer, A.; Cheng, C.Y.; Das Gupta, K.; Singhal, A.; Fairlie, D.P.; Sweet, M.J. Lysine Deacetylases and Regulated Glycolysis in Macrophages. Trends Immunol. 2018, 39, 473–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, N.L.; Yeligar, S.M. Role of HIF-1alpha in Alcohol-Mediated Multiple Organ Dysfunction. Biomolecules 2018, 8, 170. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wang, S.; Li, Y.; Yu, S.; Zhao, Y. SIRT1/PGC-1alpha Signaling Promotes Mitochondrial Functional Recovery and Reduces Apoptosis after Intracerebral Hemorrhage in Rats. Front. Mol. Neurosci. 2017, 10, 443. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Anand, S.K.; Singh, N.; Dwarkanath, A.; Dwivedi, U.N.; Kakkar, P. Berbamine induced activation of the SIRT1/LKB1/AMPK signaling axis attenuates the development of hepatic steatosis in high-fat diet-induced NAFLD rats. Food Funct. 2021, 12, 892–909. [Google Scholar] [CrossRef]
- Daemen, S.; Kutmon, M.; Evelo, C.T. A pathway approach to investigate the function and regulation of SREBPs. Genes Nutr. 2013, 8, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Li, W.; Liu, Y.; Sun, Y.; Li, Y.; Yao, Q.; Li, J.; Zhang, Q.; Gao, Y.; Gao, L.; et al. Alpha-lipoic acid improves high-fat diet-induced hepatic steatosis by modulating the transcription factors SREBP-1, FoxO1 and Nrf2 via the SIRT1/LKB1/AMPK pathway. J. Nutr. Biochem. 2014, 25, 1207–1217. [Google Scholar] [CrossRef] [Green Version]
- Fakhri, S.; Aneva, I.Y.; Farzaei, M.H.; Sobarzo-Sanchez, E. The Neuroprotective Effects of Astaxanthin: Therapeutic Targets and Clinical Perspective. Molecules 2019, 24, 2640. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, T.; Kulkarni, A.; Terada, Y.; Maoka, T.; Etoh, H. Reaction of Astaxanthin with Peroxynitrite. Biosci. Biotechnol. Biochem. 2008, 72, 2716–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farruggia, C.; Kim, M.B.; Bae, M.; Lee, Y.; Pham, T.X.; Yang, Y.; Han, M.J.; Park, Y.K.; Lee, J.Y. Astaxanthin exerts anti-inflammatory and antioxidant effects in macrophages in NRF2-dependent and independent manners. J. Nutr. Biochem. 2018, 62, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bae, M.; Kim, B.; Park, Y.K.; Koo, S.I.; Lee, J.Y. Astaxanthin prevents and reverses the activation of mouse primary hepatic stellate cells. J. Nutr. Biochem. 2016, 29, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Min, J.; Nguyen, A.; Pham, T.X.; Park, H.J.; Park, Y.; Kim, B.; Bruno, R.S.; Lee, J. Astaxanthin-Rich Extract from the Green Alga Haematococcus pluvialis Lowers Plasma Lipid Concentrations and Enhances Antioxidant Defense in Apolipoprotein E Knockout Mice. J. Nutr. 2011, 141, 1611–1617. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kim, B.; Park, Y.K.; Koo, S.I.; Lee, J.Y. Astaxanthin prevents TGF beta 1-induced pro-fibrogenic gene expression by inhibiting Smad3 activation in hepatic stellate cells. BBA-Gen Subj. 2015, 1850, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Q.Z.; Zhao, S.H.; Ji, X.; Qiu, J.; Wang, J.; Zhou, Y.; Cai, Q.; Zhang, J.; Gao, H.Q. Astaxanthin attenuated pressure overload-induced cardiac dysfunction and myocardial fibrosis: Partially by activating SIRT1. BBA-Gen Subj. 2017, 1861, 1715–1728. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, W.J.; Li, Y.Y.; Hu, L.K.; Dai, Y.; Chen, J.Q.; Xu, S.Q.; Xu, X.F.; Jiang, H.Q. Astaxanthin ameliorates cerulein-induced acute pancreatitis in mice. Int. Immunopharmacol. 2018, 56, 18–28. [Google Scholar] [CrossRef]
- Rim, J.S.; Kozak, L.P. Regulatory motifs for CREB-binding protein and Nfe2l2 transcription factors in the upstream enhancer of the mitochondrial uncoupling protein 1 gene. J. Biol. Chem. 2002, 277, 34589–34600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, T.A.; Kalsoom, K.; Iqbal, A.; Asif, H.; Rahman, H.; Farooq, S.O.; Naveed, H.; Nasir, U.; Amin, M.U.; Hussain, M.; et al. A novel missense mutation in the NADPH binding domain of CYBB abolishes the NADPH oxidase activity in a male patient with increased susceptibility to infections. Microb. Pathogenes. 2016, 100, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yu, Y.; Sang, R.; Li, J.; Ge, B.; Zhang, X. Protective Effects of Taraxasterol against Ethanol-Induced Liver Injury by Regulating CYP2E1/Nrf2/HO-1 and NF-kappaB Signaling Pathways in Mice. Oxid. Med. Cell. Longev. 2018, 2018, 8284107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, K.L.; Lelis, D.D.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wu, C.; Kim, J.; Kim, B.; Lee, S.J. Astaxanthin reduces hepatic lipid accumulations in high-fat-fed C57BL/6J mice via activation of peroxisome proliferator-activated receptor (PPAR) alpha and inhibition of PPAR gamma and Akt. J. Nutr. Biochem. 2016, 28, 9–18. [Google Scholar] [CrossRef]
- Jourova, L.; Anzenbacherova, E.; Dostal, Z.; Anzenbacher, P.; Briolotti, P.; Rigal, E.; Daujat-Chavanieu, M.; Gerbal-Chaloin, S. Butyrate, a typical product of gut microbiome, affects function of the AhR gene, being a possible agent of crosstalk between gut microbiome, and hepatic drug metabolism. J. Nutr. Biochem. 2022, 107, 109042. [Google Scholar] [CrossRef]
- Sengupta, S.; Muir, J.G.; Gibson, P.R. Does butyrate protect from colorectal cancer? J. Gastroenterol. Hepatol. 2006, 21, 209–218. [Google Scholar] [CrossRef]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, J.; He, T.; Becker, S.; Zhang, G.; Li, D.; Ma, X. Butyrate: A Double-Edged Sword for Health? Adv. Nutr. 2018, 9, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoni, L.; Nuding, S.; Wehkamp, J.; Stange, E.F. Intestinal barrier in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 1165–1179. [Google Scholar] [CrossRef]
- Li, H.; Gao, Z.; Zhang, J.; Ye, X.; Xu, A.; Ye, J.; Jia, W. Sodium butyrate stimulates expression of fibroblast growth factor 21 in liver by inhibition of histone deacetylase 3. Diabetes 2012, 61, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Boffa, L.C.; Vidali, G.; Mann, R.S.; Allfrey, V.G. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J. Biol. Chem. 1978, 253, 3364–3366. [Google Scholar] [CrossRef]
- Yaku, K.; Enami, Y.; Kurajyo, C.; Matsui-Yuasa, I.; Konishi, Y.; Kojima-Yuasa, A. The enhancement of phase 2 enzyme activities by sodium butyrate in normal intestinal epithelial cells is associated with Nrf2 and p53. Mol. Cell. Biochem. 2012, 370, 7–14. [Google Scholar] [CrossRef]
- Kibbie, J.J.; Dillon, S.M.; Thompson, T.A.; Purba, C.M.; McCarter, M.D.; Wilson, C.C. Butyrate directly decreases human gut lamina propria CD4 T cell function through histone deacetylase (HDAC) inhibition and GPR43 signaling. Immunobiology 2021, 226, 152126. [Google Scholar] [CrossRef] [PubMed]
- Salkowska, A.; Karas, K.; Walczak-Drzewiecka, A.; Dastych, J.; Ratajewski, M. Differentiation stage-specific effect of histone deacetylase inhibitors on the expression of RORgammaT in human lymphocytes. J. Leukoc. Biol. 2017, 102, 1487–1495. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kim, M.; Kang, S.G.; Jannasch, A.H.; Cooper, B.; Patterson, J.; Kim, C.H. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015, 8, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Cao, T.; Zhang, X.; Chen, D.; Zhang, P.; Li, Q.; Muhammad, A. The epigenetic modification during the induction of Foxp3 with sodium butyrate. Immunopharmacol. Immunotoxicol. 2018, 40, 309–318. [Google Scholar] [CrossRef]
- Sharma, R.A.; Gescher, A.J.; Steward, W.P. Curcumin: The story so far. Eur. J. Cancer 2005, 41, 1955–1968. [Google Scholar] [CrossRef]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Zhan, P.; Wang, Q.; Wang, C.; Liu, Y.; Yu, Z.; Zhang, S. Curcumin upregulates the Nrf2 system by repressing inflammatory signaling-mediated Keap1 expression in insulin-resistant conditions. Biochem. Biophys. Res. Commun. 2019, 514, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [Green Version]
- Maradana, M.R.; Thomas, R.; O’Sullivan, B.J. Targeted delivery of curcumin for treating type 2 diabetes. Mol. Nutr. Food Res. 2013, 57, 1550–1556. [Google Scholar] [CrossRef]
- Huang, M.T.; Smart, R.C.; Wong, C.Q.; Conney, A.H. Inhibitory effect of curcumin, chlorogenic acid, caffeic acid, and ferulic acid on tumor promotion in mouse skin by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1988, 48, 5941–5946. [Google Scholar] [PubMed]
- Chun, K.S.; Keum, Y.S.; Han, S.S.; Song, Y.S.; Kim, S.H.; Surh, Y.J. Curcumin inhibits phorbol ester-induced expression of cyclooxygenase-2 in mouse skin through suppression of extracellular signal-regulated kinase activity and NF-kappaB activation. Carcinogenesis 2003, 24, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.K.; Cha, S.H.; Jeon, H.G. Curcumin-induced histone hypoacetylation enhances caspase-3-dependent glioma cell death and neurogenesis of neural progenitor cells. Stem. Cells Dev. 2006, 15, 165–174. [Google Scholar] [CrossRef]
- Shishodia, S.; Amin, H.M.; Lai, R.; Aggarwal, B.B. Curcumin (diferuloylmethane) inhibits constitutive NF-kappaB activation, induces G1/S arrest, suppresses proliferation, and induces apoptosis in mantle cell lymphoma. Biochem. Pharmacol. 2005, 70, 700–713. [Google Scholar] [CrossRef]
- El-Moselhy, M.A.; Taye, A.; Sharkawi, S.S.; El-Sisi, S.F.; Ahmed, A.F. The antihyperglycemic effect of curcumin in high fat diet fed rats. Role of TNF-alpha and free fatty acids. Food Chem. Toxicol. 2011, 49, 1129–1140. [Google Scholar] [CrossRef]
- Yun, J.M.; Jialal, I.; Devaraj, S. Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J. Nutr. Biochem. 2011, 22, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.; Khan, Z.A.; Farhangkhoee, H.; Chakrabarti, S. Curcumin prevents diabetes-associated abnormalities in the kidneys by inhibiting p300 and nuclear factor-kappaB. Nutrition 2009, 25, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Chun, K.S.; Kim, D.H.; Kim, S.J.; Kim, S.H.; Cho, N.C.; Na, H.K.; Surh, Y.J. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem. Pharmacol. 2020, 173, 113820. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Chiba, T.; Suzuki, T.; Iwai, K.; Yamanaka, K.; Minato, N.; Suzuki, H.; Shimbara, N.; Hidaka, Y.; Osaka, F.; et al. NEDD8 recruits E2-ubiquitin to SCF E3 ligase. EMBO J. 2001, 20, 4003–4012. [Google Scholar] [CrossRef] [Green Version]
- Chuengsamarn, S.; Rattanamongkolgul, S.; Luechapudiporn, R.; Phisalaphong, C.; Jirawatnotai, S. Curcumin extract for prevention of type 2 diabetes. Diabetes Care 2012, 35, 2121–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.D.; Yang, C.S. Mechanisms of cancer prevention by tea constituents. J. Nutr. 2003, 133, 3262S–3267S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, M.W.; Cho, C.H. Pharmacological effects of green tea on the gastrointestinal system. Eur. J. Pharmacol. 2004, 500, 177–185. [Google Scholar] [CrossRef]
- Cabrera, C.; Artacho, R.; Gimenez, R. Beneficial effects of green tea—A review. J. Am. Coll. Nutr. 2006, 25, 79–99. [Google Scholar] [CrossRef]
- Thakur, V.S.; Gupta, K.; Gupta, S. Green tea polyphenols causes cell cycle arrest and apoptosis in prostate cancer cells by suppressing class I histone deacetylases. Carcinogenesis 2012, 33, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.C.; Jung, M.G.; Lee, Y.H.; Yoon, J.C.; Kwon, S.H.; Kang, H.B.; Kim, M.J.; Cha, J.H.; Kim, Y.J.; Jun, W.J.; et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009, 69, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.K.; Cheung, C.; Reuhl, K.R.; Liu, A.B.; Lee, M.J.; Lu, Y.P.; Yang, C.S. Effects of green tea polyphenol (−)-epigallocatechin-3-gallate on newly developed high-fat/Western-style diet-induced obesity and metabolic syndrome in mice. J. Agric. Food Chem. 2011, 59, 11862–11871. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Zhu, W.; Shen, C.L.; Gao, W. Green tea polyphenols reduce body weight in rats by modulating obesity-related genes. PLoS ONE 2012, 7, e38332. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.D.; Kennett, M.J.; Sang, S.; Reuhl, K.R.; Ju, J.; Yang, C.S. Hepatotoxicity of high oral dose (−)-epigallocatechin-3-gallate in mice. Food Chem. Toxicol. 2010, 48, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domazetovic, V.; Bonanomi, A.G.; Stio, M.; Vincenzini, M.T.; Iantomasi, T. Resveratrol decreases TNFalpha-induced ICAM-1 expression and release by Sirt-1-independent mechanism in intestinal myofibroblasts. Exp. Cell Res. 2019, 382, 111479. [Google Scholar] [CrossRef]
- Manna, S.K.; Mukhopadhyay, A.; Aggarwal, B.B. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: Potential role of reactive oxygen intermediates and lipid peroxidation. J. Immunol. 2000, 164, 6509–6519. [Google Scholar] [CrossRef] [Green Version]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Gardner, P.J.; Joshi, L.; Lee, R.W.; Dick, A.D.; Adamson, P.; Calder, V.L. SIRT1 activation protects against autoimmune T cell-driven retinal disease in mice via inhibition of IL-2/Stat5 signaling. J. Autoimmun. 2013, 42, 117–129. [Google Scholar] [CrossRef]
- Yang, H.; Hu, J.; Chen, Y.J.; Ge, B. Role of Sirt1 in innate immune mechanisms against Mycobacterium tuberculosis via the inhibition of TAK1 activation. Arch. Biochem. Biophys. 2019, 667, 49–58. [Google Scholar] [CrossRef]
- Huang, K.; Huang, J.; Xie, X.; Wang, S.; Chen, C.; Shen, X.; Liu, P.; Huang, H. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-beta1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med. 2013, 65, 528–540. [Google Scholar] [CrossRef]
- Huang, K.; Chen, C.; Hao, J.; Huang, J.; Wang, S.; Liu, P.; Huang, H. Polydatin promotes Nrf2-ARE anti-oxidative pathway through activating Sirt1 to resist AGEs-induced upregulation of fibronetin and transforming growth factor-beta1 in rat glomerular messangial cells. Mol. Cell. Endocrinol. 2015, 399, 178–189. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD(+) in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trammell, S.A.J.; Yu, L.P.; Redpath, P.; Migaud, M.E.; Brenner, C. Nicotinamide Riboside Is a Major NAD(+) Precursor Vitamin in Cow Milk. J. Nutr. 2016, 146, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1 alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Cassavaugh, J.; Lounsbury, K.M. Hypoxia-Mediated Biological Control. J. Cell. Biochem. 2011, 112, 735–744. [Google Scholar] [CrossRef]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.H.; Kang, Y.K.; Lee, K.M.; Kang, M.H.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A Lactate-Induced Response to Hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.X.; Han, J.Y.; Epstein, P.N.; Liu, Y.Q. Regulation of PDK mRNA by high fatty acid and glucose in pancreatic islets. Biochem. Biophys. Res. Commun. 2006, 344, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Hrelia, P. Sulforaphane as a promising molecule for fighting cancer. Mutat. Res. 2007, 635, 90–104. [Google Scholar] [CrossRef]
- Ho, E.; Clarke, J.D.; Dashwood, R.H. Dietary sulforaphane, a histone deacetylase inhibitor for cancer prevention. J. Nutr. 2009, 139, 2393–2396. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tian, Z.; Yang, Q.; Li, H.; Guan, H.; Shi, B.; Hou, P.; Ji, M. Sulforaphane inhibits thyroid cancer cell growth and invasiveness through the reactive oxygen species-dependent pathway. Oncotarget 2015, 6, 25917–25931. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.Y.; Kee, H.J.; Jin, L.; Ryu, Y.; Sun, S.; Kim, G.R.; Jeong, M.H. Inhibition of class IIa histone deacetylase activity by gallic acid, sulforaphane, TMP269, and panobinostat. Biomed. Pharmacother. 2018, 101, 145–154. [Google Scholar] [CrossRef]
- Rajendran, P.; Delage, B.; Dashwood, W.M.; Yu, T.W.; Wuth, B.; Williams, D.E.; Ho, E.; Dashwood, R.H. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: Competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol. Cancer 2011, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Myzak, M.C.; Dashwood, W.M.; Orner, G.A.; Ho, E.; Dashwood, R.H. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J. 2006, 20, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.M.; Lee, Y.S.; Sin, D.M.; Lee, S.; Lee, M.K.; Lee, Y.M.; Hong, J.T.; Yun, Y.P.; Yoo, H.S. Sulforaphane inhibits mitotic clonal expansion during adipogenesis through cell cycle arrest. Obesity 2012, 20, 1365–1371. [Google Scholar] [CrossRef]
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhauser, C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J. Biol. Chem. 2001, 276, 32008–32015. [Google Scholar] [CrossRef] [Green Version]
- Bose, C.; Alves, I.; Singh, P.; Palade, P.T.; Carvalho, E.; Borsheim, E.; Jun, S.R.; Cheema, A.; Boerma, M.; Awasthi, S.; et al. Sulforaphane prevents age-associated cardiac and muscular dysfunction through Nrf2 signaling. Aging Cell 2020, 19, e13261. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Kozumbo, W.J. The phytoprotective agent sulforaphane prevents inflammatory degenerative diseases and age-related pathologies via Nrf2-mediated hormesis. Pharmacol. Res. 2021, 163, 105283. [Google Scholar] [CrossRef]
- Zhang, C.; Su, Z.Y.; Khor, T.O.; Shu, L.; Kong, A.N. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.N.; Choi, H.Y.; Kim, Y.K. Regulation of adipocyte differentiation by histone deacetylase inhibitors. Arch. Pharm. Res. 2009, 32, 535–541. [Google Scholar] [CrossRef]
- Yoo, E.J.; Chung, J.J.; Choe, S.S.; Kim, K.H.; Kim, J.B. Down-regulation of histone deacetylases stimulates adipocyte differentiation. J. Biol. Chem. 2006, 281, 6608–6615. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, T.K.; Idelman, G.; Blanco, V.; Blomkalns, A.L.; Piegore, M.G., Jr.; Weintraub, D.S.; Kumar, S.; Rajsheker, S.; Manka, D.; Rudich, S.M.; et al. Histone deacetylase 9 is a negative regulator of adipogenic differentiation. J. Biol. Chem. 2011, 286, 27836–27847. [Google Scholar] [CrossRef] [Green Version]
- Catalioto, R.M.; Maggi, C.A.; Giuliani, S. Chemically distinct HDAC inhibitors prevent adipose conversion of subcutaneous human white preadipocytes at an early stage of the differentiation program. Exp. Cell Res. 2009, 315, 3267–3280. [Google Scholar] [CrossRef]
- Lagace, D.C.; Nachtigal, M.W. Inhibition of histone deacetylase activity by valproic acid blocks adipogenesis. J. Biol. Chem. 2004, 279, 18851–18860. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.G.; Zhang, H.; Dai, X.; Zhu, R.Y.; Chen, B.B.; Xia, B.K.; Ye, Z.M.W.; Zhao, D.D.; Gao, S.H.; Orekhov, A.N.; et al. A comprehensive review on the phytochemistry, pharmacokinetics, and antidiabetic effect of Ginseng. Phytomedicine 2021, 92, ARTN 153717. [Google Scholar] [CrossRef]
- Liu, H.; Lu, X.; Hu, Y.; Fan, X. Chemical constituents of Panax ginseng and Panax notoginseng explain why they differ in therapeutic efficacy. Pharmacol. Res. 2020, 161, 105263. [Google Scholar] [CrossRef]
- Wu, T.; Kwaku, O.R.; Li, H.Z.; Yang, C.R.; Ge, L.J.; Xu, M. Sense Ginsenosides From Ginsengs: Structure-Activity Relationship in Autophagy. Nat. Prod. Commun. 2019, 14, 1934578x19858223. [Google Scholar] [CrossRef]
- Kang, H.; Lim, J.W.; Kim, H. Inhibitory effect of Korean Red Ginseng extract on DNA damage response and apoptosis in Helicobacter pylori-infected gastric epithelial cells. J. Ginseng Res. 2020, 44, 79–85. [Google Scholar] [CrossRef]
- Huang, Y.; Kwan, K.K.L.; Leung, K.W.; Yao, P.; Wang, H.; Dong, T.T.; Tsim, K.W.K. Ginseng extracts modulate mitochondrial bioenergetics of live cardiomyoblasts: A functional comparison of different extraction solvents. J. Ginseng Res. 2019, 43, 517–526. [Google Scholar] [CrossRef]
- Huang, Q.; Gao, S.; Zhao, D.; Li, X. Review of ginsenosides targeting mitochondrial function to treat multiple disorders: Current status and perspectives. J. Ginseng Res. 2021, 45, 371–379. [Google Scholar] [CrossRef]
- Huang, Q.; Lou, T.; Lu, J.; Wang, M.; Chen, X.; Xue, L.; Tang, X.; Qi, W.; Zhang, Z.; Su, H.; et al. Major ginsenosides from Panax ginseng promote aerobic cellular respiration and SIRT1-mediated mitochondrial biosynthesis in cardiomyocytes and neurons. J. Ginseng Res. 2022, 46, 759–770. [Google Scholar] [CrossRef]
- Irrcher, I.; Adhihetty, P.J.; Sheehan, T.; Joseph, A.M.; Hood, D.A. PPARgamma coactivator-1alpha expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am. J. Physiol. Cell Physiol. 2003, 284, C1669–C1677. [Google Scholar] [CrossRef] [Green Version]
- Chu, S.F.; Zhang, Z.; Zhou, X.; He, W.B.; Chen, C.; Luo, P.; Liu, D.D.; Ai, Q.D.; Gong, H.F.; Wang, Z.Z.; et al. Ginsenoside Rg1 protects against ischemic/reperfusion-induced neuronal injury through miR-144/Nrf2/ARE pathway. Acta. Pharmacol. Sin. 2019, 40, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Zhang, M.; Ling, C.; Zhu, Y.; Ren, H.; Hong, C.; Qin, J.; Liu, T.; Wang, J. Neuroprotective Effects of Ginsenosides against Cerebral Ischemia. Molecules 2019, 24, 1102. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Yi, Y.S.; Kim, M.Y.; Cho, J.Y. Role of ginsenosides, the main active components of Panax ginseng, in inflammatory responses and diseases. J. Ginseng Res. 2017, 41, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M. Anti-inflammatory effects of ginsenosides Rg5, Rz1, and Rk1: Inhibition of TNF-alpha-induced NF-kappaB, COX-2, and iNOS transcriptional expression. Phytother. Res. 2014, 28, 1893–1896. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.S.; Shin, J.A.; Park, E.M.; Lee, J.E.; Kang, Y.S.; Min, S.W.; Kim, D.H.; Hyun, J.W.; Shin, C.Y.; Kim, H.S. Anti-inflammatory mechanism of ginsenoside Rh1 in lipopolysaccharide-stimulated microglia: Critical role of the protein kinase A pathway and hemeoxygenase-1 expression. J. Neurochem. 2010, 115, 1668–1680. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, S.; Liu, J.; Cai, E.; Zhu, H.; He, Z.; Gao, Y.; Li, P.; Zhao, Y. Sesquiterpenoids from the root of Panax Ginseng protect CCl(4)-induced acute liver injury by anti-inflammatory and anti-oxidative capabilities in mice. Biomed. Pharmacother. 2018, 102, 412–419. [Google Scholar] [CrossRef]
- Su, G.Y.; Li, Z.Y.; Wang, R.; Lu, Y.Z.; Nan, J.X.; Wu, Y.L.; Zhao, Y.Q. Signaling pathways involved in p38-ERK and inflammatory factors mediated the anti-fibrosis effect of AD-2 on thioacetamide-induced liver injury in mice. Food Funct. 2019, 10, 3992–4000. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Li, H.; Zhao, Y.; Cai, E.; Zhu, H.; Li, P.; Liu, J. Protective Effects of Sesquiterpenoids from the Root of Panax ginseng on Fulminant Liver Injury Induced by Lipopolysaccharide/d-Galactosamine. J. Agric. Food Chem. 2018, 66, 7758–7763. [Google Scholar] [CrossRef] [PubMed]
- Ning, C.; Gao, X.; Wang, C.; Huo, X.; Liu, Z.; Sun, H.; Yang, X.; Sun, P.; Ma, X.; Meng, Q.; et al. Protective effects of ginsenoside Rg1 against lipopolysaccharide/d-galactosamine-induced acute liver injury in mice through inhibiting toll-like receptor 4 signaling pathway. Int. Immunopharmacol. 2018, 61, 266–276. [Google Scholar] [CrossRef]
- Majeed, F.; Malik, F.Z.; Ahmed, Z.; Afreen, A.; Afzal, M.N.; Khalid, N. Ginseng phytochemicals as therapeutics in oncology: Recent perspectives. Biomed. Pharmacother. 2018, 100, 52–63. [Google Scholar] [CrossRef]
- Yuan, Z.; Jiang, H.; Zhu, X.; Liu, X.; Li, J. Ginsenoside Rg3 promotes cytotoxicity of Paclitaxel through inhibiting NF-kappaB signaling and regulating Bax/Bcl-2 expression on triple-negative breast cancer. Biomed. Pharmacother. 2017, 89, 227–232. [Google Scholar] [CrossRef]
- Bae, J.K.; Kim, Y.J.; Chae, H.S.; Kim, D.Y.; Choi, H.S.; Chin, Y.W.; Choi, Y.H. Korean red ginseng extract enhances paclitaxel distribution to mammary tumors and its oral bioavailability by P-glycoprotein inhibition. Xenobiotica 2017, 47, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; He, R.; Xu, Q.; Yang, Y.; Hu, Q.; Hou, H.; Liu, X.; Li, J. Ginsenoside 20(S)-protopanaxadiol inhibits triple-negative breast cancer metastasis in vivo by targeting EGFR-mediated MAPK pathway. Pharmacol. Res. 2019, 142, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Zhang, Y.; Zhou, J.; Zhi, Q.; Wu, M.Y.; Gong, F.R.; Shen, M.; Liu, L.; Tao, M.; Shen, B.; et al. Ginsenoside Rg3 targets cancer stem cells and tumor angiogenesis to inhibit colorectal cancer progression in vivo. Int. J. Oncol. 2018, 52, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Xue, H.; Zhao, Z.; Lin, Z.; Geng, J.; Guan, Y.; Song, C.; Zhou, Y.; Tai, G. Selective effects of ginseng pectins on galectin-3-mediated T cell activation and apoptosis. Carbohydr. Polym. 2019, 219, 121–129. [Google Scholar] [CrossRef]
- Li, X.; Tsauo, J.; Geng, C.; Zhao, H.; Lei, X.; Li, X. Ginsenoside Rg3 Decreases NHE1 Expression via Inhibiting EGF-EGFR-ERK1/2-HIF-1 alpha Pathway in Hepatocellular Carcinoma: A Novel Antitumor Mechanism. Am. J. Chin. Med. 2018, 46, 1915–1931. [Google Scholar] [CrossRef]
- Meng, L.; Ji, R.; Dong, X.; Xu, X.; Xin, Y.; Jiang, X. Antitumor activity of ginsenoside Rg3 in melanoma through downregulation of the ERK and Akt pathways. Int. J. Oncol. 2019, 54, 2069–2079. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Gao, J.; Wei, F.; Zhao, J.; Wang, D.; Wei, J. Therapeutic Potential of Ginsenosides as an Adjuvant Treatment for Diabetes. Front. Pharmacol. 2018, 9, 423. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, W.; Li, X.; Zhang, M.; Chen, L.; Zheng, Y.N.; Sun, G.Z.; Ruan, C.C. Antidiabetic effects of malonyl ginsenosides from Panax ginseng on type 2 diabetic rats induced by high-fat diet and streptozotocin. J. Ethnopharmacol. 2013, 145, 233–240. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| BCs | Effects | Model | Mechanisms | Ref |
|---|---|---|---|---|
| Astaxanthin | ↓ Oxidative stress ↓ Inflammation | In vitro | ↓ ROS, ↓ NOX2 and COX-2 | [119,145] |
| Macrophage stimulated with LPS and H2O2 | ↑ SIRT1, ↑ NRF2, ↑ antioxidant enzymes ↓ NF-κB nuclear translocation | [146,147] | ||
| apolipoprotein E-knockout mice | ↑ NRF2, ↑ antioxidant enzymes | [148] | ||
| Hepatic stellate cells | ↑ NRF2, ↑ antioxidant enzymes | [149] | ||
| Macrophages stimulated with ethanol | ↑ SIRT1, ↑ NRF2, ↑ NAD+ levels | [30] | ||
| ↓ hepatic lipid | High-fat diet fed mice | ↑ PPARα, ↓ PPARγ and Akt ↓ SREBP1c nuclear translocation | [109,156] | |
| ↑ hepatic autophagy | Cerulein-induced acute pancreatitis in mice | ↑ PPARα, ↓ PPARγ and Akt | [151] | |
| Butyrate | ↓ Insulin resistance | High-fat diet fed mice | ↓ Fasting blood glucose and insulin ↑ PPARα, ↑ AMPK and p38 | [160] |
| ↓ Inflammation | Colorectal cancer | ↓ HDACs | [105] | |
| ↑ Hepatic fatty acid oxidation | High-fat diet fed mice HepG2 cells | ↓ HDAC3 activity ↑ FGF21 and PGC1α expression | [164] | |
| ↑ Hepatic drug metabolizing ability | HepG2-C3 cells Primary human hepatocytes | ↑ Aryl hydrocarbon receptor ↑ Cytochrome p450 | [157] | |
| ↑ Thermogenesis in brown fat | High-fat diet fed mice | ↑ PGC1α and UCP1, | [156] | |
| ↓ Glucose intolerance Insulin resistance | High-fat diet fed mice | ↓ HDAC activity | [160] | |
| ↑ Antioxidant effects in intestine | IEC-6 epithelial cells HT-29 colorectal cancer cells | ↑ NRF2, ↓ HDAC activity | [166] | |
| ↓ Inflammation | Human gut lamina propria CD4 T cell Murine splenic cell Human peripheral blood CD4 T cells | ↓ HDAC activity ↓ Inflammatory cytokine production ↑ PPARγ signaling pathways | [167,168,169] | |
| Curcumin | ↑ Antioxidant | Oxidative or insulin-resistant conditions | ↑ Keap1 expression and NRF2 | [173] |
| Obesity and diabetes | ↑ Antioxidant function of NRF2 | [174,175] | ||
| Mouse skin Murine epidermal cells | ↑ HO-1 expression and NRF2 | [177,183] | ||
| ↓ Skin tumorigenesis | Mouse skin | ↓ COX-2 expression, ↓ NF-κB activity | [176,177] | |
| ↑ Apoptosis | Glioma cell | ↓ Histone acetylation, ↓ p300/CBP | [172,178] | |
| ↓ Inflammation | Mantle cell lymphoma High-fat diet-fed rats | ↓ Acetylation state ↓ NF-κB activation and inflammation | [179,180] | |
| High glucose-induced monocyte | ↓ NF-κB activity, ↓ IL-6 and TNFα | [181] | ||
| ↓ Hyperglycemia and Diabetes | High-fat diet-fed rats | ↑ HAT levels, ↑ insulin sensitivity | [180] | |
| STZ-induced diabetic rats | ↓ iNOS and TGF-β1 ↓ p300/CBP and NF-κB | [182] | ||
| EGCG | ↑ Apoptosis in prostate cancer cells | LNCaP and PC3 cells | ↓ HAT activity, ↑ cell cycle arrest ↑ p21/waf1 and Bax | [189] |
| ↓ Inflammation | High-fat/western-style diet-induced obese mice | ↓ HAT activity, ↓ body weight ↓ Hepatic lipids and inflammation | [191] | |
| High-fat-fed rat (female) | ↓ IL-1β and IL-6, ↑ SOD and COMT | [192] | ||
| Resveratrol | ↓ Inflammation | RAW 264.7 cells JB6 cells | ↑ Scavenge free radicals ↓ Lipid peroxidation and DNA damage | [111] |
| Paraquat-induced lung injured mice | ↑ SIRT1 expression and NRF2 activation | [21] | ||
| Human colon-derived myofibroblast cells | ↓ TNFα-induced ROS and ICAM-1 ↑ SIRT1 activation | [196] | ||
| Jurkat lymphoid HeLa and H4 epithelial cells | ↓ TNFα-induced NF-κB and MAPK ↓ cytotoxicity and caspase activation | [197] | ||
| Mycobacterium tuberculosis-infected mice | ↓ TAK1, NF-κB and MAPK ↑ SIRT1 activation | [200] | ||
| Diabetic rats Mesangial cells | ↓ Fibronectin and TGF-β1 | [201,202] | ||
| NR | ↑ Antioxidant ↓ Inflammation | Ethanol-exposed macrophages | ↑ SIRT1 activation | [31,205] |
| ↓ Lipid accumulation | Lieber-DeCarli ethanol liquid diet-fed mice | ↑ SIRT1 and PGC1α activation ↑ Mitochondrial function | [48] | |
| Energy metabolism | Ethanol-exposed macrophages | ↑ SIRT1 activation, HIF-1α activation ↓ GLUT1, PDK1, LDHα | [31] | |
| SFN | ↑ Antioxidant | Cancer cells | ↑NRF2 activation ↑ Detoxifying/antioxidant enzymes | [212] |
| ↑ Cancer prevention | HCT116 colon cells Human embryonic kidney 293 cells | ↓ HDAC2 and HDAC3 | [213,215] | |
| Cancer cell lines | ↑ ROS for initiation of apoptosis ↓ ROS during apoptosis ↑ Caspase 3 and 8, ↑ p21 and BAX ↑ NF-κB activation, ↓ HDAC6 activity ↓ HDAC1, 4, 5, and 7 ↑ Histone 3 acetylation, ↑ NRF2 | [213,214] [220,223] | ||
| Adipocyte differentiation | 3T3-L1 adipocytes | ↓ HDAC2, HDAC4, 5, 7, and 9 ↓ PPARγ, FABP4, FAS, ↓ p300 and C/EBPα | [215] [79,218] [225,226] | |
| TGS | ↑ Energy metabolism | Cardiomyocytes Skeletal myoblasts Vascular endothelial cells Epithelial cells | ↑ Mitochondrial oxygen consumption, ↑ Cellular respiration ↑ ATP production | [233,234] |
| Cardiomyocytes and neurons | ↑ SIRT1/PGC1α axis, ↑ NRF2 ↑ Mitochondrial biosynthesis | [235,236] | ||
| ↓ Inflammation | Inflammatory condition | ↓ NF-κB and proinflammatory cytokines | [239,240] | |
| LPS-stimulated microglia | ↓ MAPK phosphorylation, ↑ CREB | 241 | ||
| LPS-induced RAW 264.7 macrophages | ↓ JAK2/STAT3 activation | [230] | ||
| CCl(4)-induced acute liver injured mice | ↓ NF-κB subunit p65 and COX-2 ↓ ERK, JNK, and MAPK, p38 | [242] | ||
| Thioacetamide-induced liver injured mice D-GalN and LPS-induced liver injured mice | ↓ TNFα, IL-1β, IL-6, and caspase-1 ↓ liver fibrosis, ↓ NF-κB, p38/ERK ↑ SIRT1 and NRF2 | [243,244] | ||
| D-GalN and LPS-induced acute injured mice | ↓ TLR4, ↓ NF-κB and MAPK | [245] | ||
| Cancer cell lines | ↓ EGF and EGFR phosphorylation ↓ ERK1/2, p38, and c-JNK activation ↓ ROS/ERK pathway, ↓ ERK-Akt pathway | [249] [250,251] [252,253] | ||
| ↑ Anti-diabetes | High-fat diet and STZ-induced diabetic rats | ↓ Gluconeogenesis, ↑ glucose transport ↑ insulin production | [254,255] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.; Kim, B. Bioactive Compounds as Inhibitors of Inflammation, Oxidative Stress and Metabolic Dysfunctions via Regulation of Cellular Redox Balance and Histone Acetylation State. Foods 2023, 12, 925. https://doi.org/10.3390/foods12050925
Kang H, Kim B. Bioactive Compounds as Inhibitors of Inflammation, Oxidative Stress and Metabolic Dysfunctions via Regulation of Cellular Redox Balance and Histone Acetylation State. Foods. 2023; 12(5):925. https://doi.org/10.3390/foods12050925
Chicago/Turabian StyleKang, Hyunju, and Bohkyung Kim. 2023. "Bioactive Compounds as Inhibitors of Inflammation, Oxidative Stress and Metabolic Dysfunctions via Regulation of Cellular Redox Balance and Histone Acetylation State" Foods 12, no. 5: 925. https://doi.org/10.3390/foods12050925