Development of Sunlight Driven Water Splitting Devices towards Future Artificial Photosynthetic Industry

Department of Chemical System Engineering, The University of Tokyo, Tokyo 113-8656, Japan
*
Author to whom correspondence should be addressed.
ChemEngineering 2018, 2(3), 36; https://doi.org/10.3390/chemengineering2030036
Submission received: 1 July 2018
/
Revised: 7 August 2018
/
Accepted: 9 August 2018
/
Published: 13 August 2018
(This article belongs to the Special Issue Process Design Issues for Hydrogen Production: From Catalyst Design to Reactor Modelling and Process Simulation)
Abstract
:The ongoing research and development of sunlight-driven water splitting in the “Japan Technological Research Association of Artificial Photosynthetic Chemical Process (ARPChem)” is overviewed. Water splitting photocatalysts, photoelectrochemical devices, large-scale reactor panels, product gas transportation, H2/O2 gas separation devices and safety measures against explosion are included as the research objectives. ARPChem was formed as a research union of Japan’s leading chemical firms, in which related elementary technologies have been cultivated. This article introduces our general scope for artificial photosynthesis and describes present research activities, mainly on solar driven water splitting photocatalysts/photoelectrodes and briefly on the processes and plans for plant construction for future industrial extension.
1. Introduction
The research and technological development of solar energy recovery is currently underway due to serious and urgent worldwide requests. Under global political, ecological, and economic trends, sustainable and renewable sources of energy are sought, desiring exodus from the heavy yoke of fossil energy, which is nonetheless still the main resource and the driving force of today’s civilization.
It is supposed that life was born 109 years ago on the earth, and that the photosynthesizing plants started converting the atmospheric CO2 into O2 4 × 108 years ago. Today’s accumulated fossil energy resources must have been deposited by photosynthesis since that era, under the irradiation of solar energy, which has slowly intensified and is now fluctuating around at a maximum of 1 kW·m−2 on the Earth’s surface.
The modern civilization based upon mass consumption of fossil energy began approximately 200 years ago, and will last until the recoverable fossil energy resources are all consumed. The speed of consumption is recognized to be much higher than the past speed of accumulation, when we see that fossil-fuel driven machines and vehicles are much more powerful than the growth of plants, in terms of spatial and temporal concentration. The condensed energy from fossil fuels is undoubtedly one of the base lines of the modern life style.
Direct industrial recovery of solar energy principally depends on a slow income of solar energy. Artificial photosynthesis, as a future category of mass-scale chemical industry, will be more similar to agricultural farming than to petroleum refining in terms of spatial and temporal energetic concentration. Although solar photovoltaic plants are now highly esteemed as a sustainable source of electricity and industrialized world-wide, aiming for free fuel from the sun, they are still costly. The photovoltaics as today’s industrial products are manufactured in energy-consuming processes. For this sake, for example in Japan, the renewable energy policy is partially supported by imposts over the monthly electricity fee, which is mostly generated from fossil fuels.
We consider that industrially successful application of solar energy depends on how we can reduce the cost of building the equipment and the sunlight-to-hydrogen plants to meet the essentially slow rate of solar energy production. In terms of H2 production, the industrialized steam reforming process of natural gas is far cheaper than the estimated cost of photovoltaic-driven water electrolysis. A recent announcement from the Japanese government states quantitative goals for hydrogen energy [1], in which a lower cost is aimed at for solar H2 than the natural gas H2. Photocatalysis is one of the approaches to reduce the cost under their consideration. The methods for saving energy and cost might be newly conceived as an unprecedented category of discipline but to some extent have already been put into practice in daily operation in the agricultural business. The slow energy production is essential as above, and moreover, the energy consumption within the solar plant itself should be reduced for stand-alone operation as much as possible. On the other hand, the risk for accidental uncontrollability, mainly of product H2 gas explosion, will be small due to slow gas production. Then there will be more freedom in designing the equipment involved in the plant.
2. Japan Technological Research Association of Artificial Photosynthetic Chemical Process (ARPChem)
ARPChem is our research project, originally founded as a union of Japanese leading firms Mitsubishi Chemical, Mitsui Chemical, Sumitomo Chemical, Fuji Films, Inpex and TOTO in 2012. As the members of this union, these companies deposited their capital funds and are dispatching highly experienced research staff to ARPChem from their regular employees. A number of university research groups and governmental research institutes are involved as individual taskforces of ARPChem by contract [2]. In particular, a central laboratory has been assembled at the University of Tokyo, gathering professors, postdoctoral researchers and the research staff from these companies. Approximately 100 research persons, involving engineers, currently serve for ARPChem. ARPChem is financially supported by the New Energy and Industrial Technology Development Organization (NEDO), an affiliated government agency of the Ministry of Economy, Trade and Industry (METI).
We, the University of Tokyo, participate in ARPChem to take on the photocatalytic and photoelectrochemical systems for water splitting to generate the stoichiometric ratio of 2H2 + O2. Our research scope involves fundamental studies and exploitation of visible-light-responding materials suitable for application to mass-scale solar H2 plants. The catalysts that are deposited on the photoactive materials to assist the evolution of H2 and O2, called “cocatalysts”, are inseparably included in our targets. In the same track, we also aim for the highest solar-to-hydrogen energetic efficiency (hereafter abbreviated as “STH”) by developing suitable photoactive materials for dual-photoelectrode water-splitting devices. The long-term stability and robustness of photocatalysts and photoelectrodes are another important and practical issue for us to pursue.
Mitsubishi Chemical (Tokyo, Japan) possesses the leading position among these companies in planning and conducting research and development in many of the categories. Mitsubishi makes basic plans and designs of the solar hydrogen plant, develops gas filtration devices for H2/O2 separation, and explores catalysts and catalytic reactors to synthesize light olefins from CO2 + H2. Fuji Film (Tokyo, Japan) has long-term experience in researching for photovoltaics and owning capabilities and facilities for photoactive thin film fabrication. They play a key role in our development for photoelectrochemical water splitting systems. Mitsui Chemical (Tokyo, Japan) participates in exploitation for novel photoactive materials. TOTO (Fukuoka, Japan), the world’s largest manufacturer for sanitary ceramics, copes with mass-scale production of photocatalysts for wide-area photoreactors. Inpex (Tokyo, Japan), Japan’s largest petroleum and natural gas supplier, is extending market and resource research for solar hydrogen plants, looking for a location in the sun-belt zone over the Earth. Sumitomo Chemical (Tokyo, Japan) took part in the development of catalysts and catalytic reactor systems for the CO2 + H2 reaction. Upon completion of their original aim for the catalytic systems, Sumitomo retired from this project.
ARPChem’s future artificial photosynthetic plant is schematized in Figure 1 [2], which is supposed to be socially implemented by the year 2030. The plants should be located in the sun-belt zone of the earth according to economical rationality for a large input of solar energy. Acres of land will be filled with sunlight harvesting photocatalyst panels arrayed similarly to solar photovoltaic plants. The photocatalyst panels are daisy-chained with pipes for water feed and product 2H2 + O2 exhalation.
The product transportation tubes are gathered and connected to the gas separation plant, which removes water vapor from the solar product gas and separates it into H2 and O2. A certain wattage of electric power is necessary for water feeding, gas transportation, gas desiccation, and gas separation. The power will be supplied by an additionally built solar photovoltaic plant. The areal overall efficiency for the H2 production rate will be normalized by the sum of the light acceptation areas for photocatalysts and photovoltaics. Streamlining of internal energy-consuming processes in the plant reduces the area of photovoltaics, and hence raises the overall STH. As the other mainstream product of this separation plant, O2 gas will also be a commercially profitable material and will help support the economic viability of this system.
Then the separated H2 gas is supplied to the synthetic plant, which is also fed with exhaust CO2 from a coal, oil or natural gas power plant. This arrangement is based on the primary resolution of ARPChem providing an essential solution for depletion of CO2 emissions and commercial delivery of olefins as the final products of this plant.
This whole mass-scale industrial system, composed of the solar H2 plant, the gas separation/purification plant, and the catalytic synthetic plant from H2 + CO2, is designed to be a self-standing industry to eat CO2 that would be emitted for energy production from carbon fuels and to deliver olefins that are the resources for various organic chemical products, without overlaid consumption of the fossil resources. This parasitic operation of artificial photosynthetic systems is ideally bifunctional, by reducing carbon emissions and generating profitable synthetic products with the aid of solar energy.
Hereafter, the solar energy harvesting part of ARPChem research will be overviewed.
3. Development of Photocatalysts and Photoelectrodes
The light-absorbing materials for photocatalysts or photoelectrodes should be able to absorb the photons from the sun, which is distributed centrally in the wavelength (or photon energy) range of the visible light. Figure 2 shows the solar spectra on the earth [3]. The extraterrestrial spectrum is recorded on the outer sphere atmosphere. The integrated spectral irradiance is 1.3661 kW·m−2. The ultraviolet end, the peak of intensity, and the infrared end of the spectrum are at 280 nm, 478 nm and over 3500 nm, respectively. The solar spectra on the ground level are reduced by absorption and scattering by the atmosphere. The maximum integrated intensity is approximately 1 kW·m−2 for the average latitude at the noon time (AM 1.5 G).
The photo-responding solid materials, on the other hand, are semiconductors that have an electronic band gap represented by a characteristic light absorption cutoff wavelength. The photons with wavelengths longer than the cutoff length are not absorbed and hence not utilized for photochemistry. Figure 2 shows the light absorption spectra of typical inorganic semiconductors applied for photocatalysis/photoelectrochemistry. Besides mass-produced commodity semiconductors such as Si or GaAs, visible-light absorbing materials are still under basic research. Nitrides and oxynitrides are in this category. Historically important oxides, such as TiO2 [13] and SrTiO3 [14], can absorb just the ultraviolet end of the solar spectrum, bearing a few % of the overall photon energy distribution from the sun.
According to those characteristics of materials, ARPChem’s current policy for photocatalytic/photoelectrochemical material research and development can be summarized as follows:
- (1)
- We do not pursue industrially established materials, such as Si and GaAs. Although they are successfully used for solar energy harvesting in photovoltaics, costly processes are involved in the conversion from ores to functional devices as today’s industrial culture. For this reason, we do not study water electrolyzer systems powered by conventional photovoltaics.
- (2)
- We survey nitrides, chalcogenides and oxide variations of those, as visible-light absorbers that can be assembled to the solar energy harvesting devices as powders or polycrystalline thin films. This is to open up a new category of semiconductive materials for sunlight absorption that can be produced in low-cost fabrication processes.
- (3)
- We utilize well-established photoactive oxides in order to study how to implement them in sunlight-driven water-splitting devices and plant-scale apparatuses from the viewpoint of chemical engineering. SrTiO3 and BiVO4 are in this category. Although the anticipated STH of these materials are not satisfactory for future application, technical knowledge so far learned from them is useful in planning and designing the devices in a solar hydrogen plant. This study covers microfabrication, such as deposition of cocatalysts, to ease the steps of H2 and O2 evolution, design for the solar reactors containing those materials, and towards all sorts of chemical engineering aimed to plan a mass-scale plant for solar H2 generation. Although the active wavelength range of those oxides are still limited, indeed they are currently available robust photocatalytic/photoelectrochemical materials that can be consumed in these various tests in a large amount.
Under our resolutions (1) and (2), we have been searching for visible-light responding semiconductor compounds. The semiconductors are in principle divided into two categories: p-type and n-type. When those materials are in contact with aqueous electrolytic media, up- or down-slopes of chemical potentials are formed for electrons and holes in those solids, according to the chemical potentials of those in the electrolyte, which can be controlled by the solutes and also by a voltage-applied counter electrode. The appearance of potential slopes within the solids—namely “band bending”—determines the flow of the charge carriers. The minority carrier, that is, electrons for p-type and holes for n-type, conveys the electric current that is excited by absorption of light.
Henceforward, when the electrode potential is suitably controlled and light is irradiated, p-type semiconductors inject electrons into water that convert H+ into H2 (gas), and n-type semiconductors inject holes into water that convert OH− into O2 (gas). Those gas-evolving processes are mostly aided by cocatalysts attached on the light-absorbing substrate. Pt metal is a good catalyst for H2 evolution, and oxides of Ni, Co and Fe are good catalysts for O2 evolution.
Upon the above discussion of electron and hole chemical potentials, the natural trend of semiconductor electron and hole emissivity to water is determined by the electronic energetics within the solid without applying an external voltage. The semiconductors are electronically characterized as having a band gap as mentioned above. The band gap is composed in the electron energetic quantum-state distribution, as the valence band filled with electrons and the empty conduction band. The band gap is where no electronic state is allowed. The energetically highest end of the valence band (valence band maximum, VBM) and the lowest end of the conduction band (conduction band minimum, CBM) are specific to the semiconductor solid. Excitation by a photon creates an electron in the conduction band and a hole in the valence band. Those carriers can move along the potential towards the surface and manifests the chemical reactions.
Then it is needed for a semiconductor to have a higher natural CBM than the chemical potential of H+ to promote H2 production, that is, 0 V versus the reversible hydrogen electrode (RHE) reference potential. To promote O2 evolution, VBM of a n-type semiconductor should be lower by 1.23 V or more than 0 V, which corresponds to the free energy of H2O decomposition into H2 and O2. If a pair of p-type and n-type semiconductors meets this condition, and the band gaps of them overlap each other, then a system can be constructed for overall H2O splitting driven solely by solar energy.
Practically, the water splitting photodevice is built by electrically tying a p-type semiconductor (H2 evolving “photocathode”) and a n-type semiconductor (O2 evolving “photoanode”), both dressed with proper cocatalysts. If the two photoelectrodes are arranged separately to accept incident light, the device is called a “parallel cell”. If one of the photoelectrodes is transparent for a part of the incident spectrum and laid over the other, it is called a “tandem cell” (Figure 3). They are the benchmark test devices of ARPChem to investigate the intrinsic activity of the semiconductors without external power supply.
This scheme of the stand-alone overall water photo-splitting system is also limited by an electrochemical requirement for the photoanode and photocathode. The photoelectrodes are characterized individually by electrochemical current-versus-voltage (I-V) curves. The photocurrent is proportional to the input light intensity as a rough approximation. When no photon impinges, the I-V curve represents the “dark current” of the electrode. As shown in Figure 4A, when the input light is irradiated on a photoanode, the photocurrent flows positively (positive charges from the electrode to the solution) at a certain electrode potential or more positive region higher than that. In the more negative region than that potential, the photocurrent is zero. This characteristic potential for the photoanode is called the “onset potential”. Similarly, in Figure 4B, the onset potential for photocathodes separates the more negative region with a negative photocurrent and more positive region with zero photocurrent. Those curves are called photoelectrochemical characteristic (PEC) curves. The PEC curves are recorded by potentiostatic measurement with the absolute electrode potential defined by the reference electrode (such as RHE), mostly by sweeping the potential linearly to time (linear sweep voltammetry, LSV).
Figure 4C depicts the limiting relationship of the onset potentials of the photocathode and photoanode. If the photocathode onset potential is more negative than the photoanode onset potential, the photocurrent through the tying wire will be zero and no electrochemical evolution of H2 and O2 occurs. The stand-alone overall water photo-splitting proceeds if the PEC of photoanode and negated PEC of photocathode cross over each other at nonzero current region. The crossover point corresponds to the dual-electrode photocurrent directly proportional to the rate of 2H2 + O2 gas evolution and the absolute potential of operation.
This is a severe condition for constructing photoelectrodes. Especially for photoanodes, the onset potentials of presently available visible-light-active materials are mostly too positive to realize the crossover. The onset potential depends on the intrinsic electronic band structure of the material as well as on the charge transportation characteristics and electrochemistry of the electrode surfaces. The material-dependent issues for the photoelectrodes will be discussed later specifically.
The dual-photoelectrode device is essentially a water electrolyzer supplied with electromotive forces of the photoelectrodes. Actually, if the short circuit tie is replaced with a battery, a solar cell or a DC power supply fed with house electricity, the water splitting reaction is assisted by those energy sources. Some water splitting devices with photoelectrodes assembled with photovoltaics extrinsically have been frequently proposed with a high STH [15,16,17,18,19,20,21]. The external power application can overcome the above-mentioned onset potential problem instantly. We ARPChem on the other hand adhere to the simple dual photoelectrode systems to foster the development of photocathodic and photoanodic semiconductors and cocatalysts.
ARPChem’s last few years’ surveys on visible-light absorbing semiconductors have been conducted as fabrication of photoelectrodes and basic tests in photoelectrochemical characteristics, such as photocurrent density, electrochemical onset potentials for H2/O2 evolution, and durability tests, under simulated sunlight irradiation. These tests are designed to bring out the maximum photocurrent after careful choice of the cocatalyst. Most of the tests were performed on “particle-transferred” photoelectrodes [22], fabricated from powder of photoactive semiconductors bound on evaporated metal films and forged into planer sheets. The metal film facilitates the collection of carriers and electric conduction at the same time, conveniently, as electrodes in aqueous solutions. The particle-transferred photoelectrodes facilitate the observations in photoelectrochemical surveys, which usually reflect the activity of the composing powder grains.
Table 1 summarizes the ratings of ARPChem’s photoanodes (for O2 evolution) and photocathodes (for H2 evolution) so far published in the literature. For photocathodes, mixed copper chalcogenides have demonstrated high photocurrent densities [23,24,25]. Among these, CuIn1-xGaxSe2 (CIGS, cutoff wavelength 900~1100 nm) exhibits the most suitable performance in application for the tandem-type configuration. CIGS overcoated with CdS has been recognized as a low-cost material for photovoltaics [26] and the deposition of a cocatalyst (usually metallic Pt) alters CIGS into a highly efficient H2-evolving photocathode in neutral-pH electrolytes. The present best performance under solar simulator irradiation can cover the overall water photo-splitting up to 10% of STH.
As for the photoanodes, plenty of new materials have been synthesized and tested to realize breakthrough for high efficiency. We are still striving to find visible-light responding n-type materials that exhibit a high photocurrent, a negative onset potential, good durability, and adaptability to low-pH solutions for compatibility with the photocathode.
BiVO4 (cutoff wavelength 520 nm) has been so far widely investigated as a durable oxide photocatalyst [33] and many photoanodes with attractive properties have been fabricated across the world [34]. Semi-transparent BiVO4 photoanodes [6,35] are suitable for the tandem-type water splitting cells. The onset potentials of BiVO4 photoelectrodes are in general near 0.2 V vs. RHE and a good PEC crossover is realized with CIGS-based photocathodes. We achieved 3.7% of stand-alone STH by pairing with a CIGS photocathode [23].
The durability of the BiVO4 photoanode was fully demonstrated by the particle-transfer method. Particles of BiVO4 (0.3 atomic% Mo-doped, average diameter 500 nm) were evaporated with Ni and then Sn to form a conductive film as a photoanode, without adding cocatalyst materials. When applied in the potentiostatic amperometric test with simulated sunlight, the photocurrent gradually increased and reached a constant photocurrent density of 2.6 mA·cm−2 at 0.6 V vs. RHE of the fixed potential in potassium borate aqueous solution (pH 9). The cocatalytic activity for O2-evolution was conveyed by NiFeOx mixed oxide spontaneously deposited from the evaporated Ni and impurity Fe in water. The same photocurrent lasted for 1100 h [29]. This extraordinary stability was explained by regeneration of cocatalyst from the low-concentration solutes of Ni and Fe cations, replacing the cocatalyst nano-grains lost by dissolution or exfoliation.
Despite many interesting results on BiVO4, the photocurrent density is limited by the short cutoff wavelength of light absorption, and we need to explore new semiconductors with narrower bandgaps. A series of transition-metal nitrides and oxynitrides, that is, Ta3N5 [30], LaTiO2N [7], BaTaO2N [9] and BaNbO2N [10], were studied as n-type materials in ARPChem, as powders fabricated into photoanodes by the particle-transfer method. The maximum photocurrent densities at 1.23 V versus RHE (the potential of O2 evolution) mostly exceed 7 mA·cm−2, as listed in Table 1. Certainly, those materials with longer cutoff wavelength than 590 nm generate suitably high photocurrents.
However, the onset potentials for O2 evolution are mostly more positive than 0.6 V versus RHE and the crossover current with CIGS photocathodes cannot be high enough. Most of those N-containing materials have band alignment with the flat band potential near the H2 evolution potential and in principle the genuine onset potential should be near 0 V vs. RHE. Actually, small positive photocurrents have frequently been detected at 0 V vs. RHE on those materials. The deficiency of photocurrent at the negative potential region can be linked to the electronic properties originating from the crystalline imperfection of these synthesized solids, which are related to carrier trapping, rapid electron-hole recombination and high electric resistance within the boundaries between particles or particle versus the backing metal. Therefore, we are trying to improve the quality of those synthesized solid materials and interfaces.
One solution for this is to fabricate these N-containing materials into planer film on solid. This approach is so far less experienced and less formulated, as the thin-film deposition process of those materials often damage the underlying solid substrate. In this sense, thin film deposition is more sophisticated and limited in the choice of fabrication methods, than the powder synthesis. Widening the variety of fabrication methods is our main task in the experimental survey. Currently, we are using methods with organometallic reagents, such as recently progressing chemical vapor deposition (CVD) and atomic layer deposition (ALD) as well as conventional methods of molecular beam epitaxy (MBE) and plasma sputtering, under careful consideration of the background conditions.
Nonetheless, a Ta3N5 thin film (thickness ≅ 500 nm) was successfully deposited on a metal Ta sheet from amorphous tantalum semi-oxide film on Ta by treatment in NH3 flow at a high temperature. Such Ta3N5 thin films, decorated with cobalt phosphate cocatalyst, can generate approximately 7 mA·cm−2 of the photocurrent [30], higher than the particle-transferred one [36]. The Ta3N5 film is composed of densely packed crystallites with a good crystallographic orientation alignment [30].
One of the drawbacks of these N-containing materials is their insufficient stability as operated in electrolytic solutions. The photoanodes decorated with cocatalysts lose the photocurrent within a few minutes or a few hours. In most of the cases, surface O2 evolution induces rapid oxidation of the substrate and extraction of N; therefore, attempts have been made to protect the light absorber substrate surface with a hole-conductive, visible-light-transparent thin overlayer, the surface of which is modified with the cocatalyst.
On Ta3N5, coating with GaN has been attempted. GaN is a stable solid that is transparent in the visible region, and its electric conductivity can be controlled by doping. Various methods can be used to make GaN a thin film, and we took a simple approach of Ga metal or GaOx evaporation and successive nitridation in NH3 flow [30]. This NH3 treatment at high temperatures (<1000 °C) prevents decomposition of the underlying Ta3N5 layer, and a sharp boundary between GaN and Ta3N5 is formed. The thickness of GaN layer is 50~100 nm over the 500 nm Ta3N5 layer. The active period of O2 evolution under solar-simulator irradiation was multiplied by an order of magnitude compared to the bare one [30]. Although our fabrication methods of the GaN thin layer at this point in time have not gained an extensive freedom of changing morphology and doping, we will be able to pursue a breakthrough by adjusting the electronic properties of GaN, by which a preferable GaN/Ta3N5 junction for Ta3N5 protection, smooth charge transportation and stable electrochemical properties. By using this approach as a prototype, we can cover new visible-light-absorbing semiconductors suitable for photocatalysis.
To realize effective control of polycrystalline grain size, defects in grains, crystallographic orientation, and epitaxy with respect to the substrate, the conditions for deposition should be finely controlled. The crystallinity of Ta3N5 film is changeable by the choice of substrate material and crystallinity. Moreover, we pay attention to the sputtering condition of the precursor TaOδ film and post-nitridation in NH3 gas flow, so as to gain good reproducibility of the film morphology and the photoelectrochemical performance.
4. Benchmark Tests for Water-Splitting Photocatalytic Plants
The overall agenda of the ARPChem project involves research and planning for future mass-scale water photo-splitting systems from the photocatalytic/photoelectrochemical elements towards the catalytic reactors for commodity chemical products from CO2 and photosynthesized H2. In this series of technology, the water photo-splitting plant is the only one component that has not been studied intensively for applications in the real world. The research on water photo-splitting is mostly still in the stage of basic surveys for photoactive materials and mechanistic studies of catalytic water splitting elementary reactions. Little has been experienced in magnifying the reactor to a scale compatible with mega-solar photovoltaic power plants. Therefore, it is necessary for us to perform benchmark tests for building large-area photoreactors using primitive photocatalytic systems.
In starting from scratch, we chose a simple photocatalytic system composed of a single-component photocatalyst. Strontium titanate (SrTiO3) has been studied as the ultraviolet-absorbing substrate semiconductor since the beginning of the history of water photo-splitting [14]. By irradiation with highly energetic ultraviolet rays, H2 and O2 were at the ratio of 2:1 in pure water at a small but definite rate [14]. The band gap energy of SrTiO3 is 3.2 eV by far exceeding the free energy of water splitting (1.23 eV). The band gap fortunately straddles the potentials of both H2 evolution and O2 evolution, potentially enabling simultaneous stoichiometric evolution of H2 and O2, that is, the overall stoichiometric stand-alone water splitting.
The photocatalytic activity of SrTiO3 as a flat powder layer was recently improved dramatically by doping with Al and decoration with Rh/CrOx H2 evolution cocatalyst up to STH = 0.6% excited by simulated sunlight. The quantum efficiency for photons in the ultraviolet region (<350 nm) reaches 30% [4,27] (Figure 5). The quantum efficiency was recently further improved up to 69% by addition of MoOy [37].
This series of Al:SrTiO3 photocatalysts, although its STH is far behind comparison with today’s practical photovoltaics (>10%), possesses a preferable simplicity for designing the reactor system. The reactor just needs an ultraviolet-transparent cassette (pressurizable by the evolved gas and supplied water, with inlet and outlet tubes) housing a mechanically robust thin layer of photocatalyst painted on a planer substrate.
A square-meter scale photoreactor panel was recently assembled and tested under natural sunlight [38]. The panel consists of UV-transparent Plexiglas plate (5 mm in thickness) for sunlight inlet and the same Plexiglas backing plate. The photocatalytic sheet was prepared on a 33 cm × 33 cm glass plate. The Al:SrTiO3 was in advance loaded with RhCrOx cocatalyst and painted with an aqueous mixture with colloidal silica. Then the glass plate was calcined at 623 K in air to gain the best photo-activity and mechanical robustness. Nine pieces of 33 cm × 33 cm photocatalyst sheets were arranged between the Plexiglas plates with spacers to adjust the thickness of water layer (Figure 6). Finally, pure water was confined in the inner space, and the evolution of the product gas mixture from a gate on the upper edge was connected to a water-filled gas burette for quantification.
Upon exposure to sunlight on a sunny day in Tokyo (0.65–0.75 kW·cm−2), bubbles with submillimeter diameters were seen sliding up the slope of photocatalyst sheets. The quantified volume of H2 + O2 gas mixture in a unit time corresponded to 0.4% STH, reflecting the activity of the RhCrOx/Al:SrTiO3 photocatalyst. Minor technical improvements have been made for the optimal thickness of the water layer, the hydrophilicity/hydrophobicity of the window surface, catalyst surface and so on. They are related to the size of gas bubbles, which is associated with the smooth gathering and exhalation of product gas at the outlet tube connection. More importantly, the intrinsic control of water level, the rate of water feeding and gas output around the panel should be coped with.
Photocatalyst with STH less than a few % will not be adaptable for practical product gas delivery for future social implementation. At present, we are studying the flow of product gas and water within the photoreactor in detail. The applicability of the reactor panels for photocatalysts with STH = 10% can be examined by using an UV light source that is intense enough for STH = 0.6% photocatalyst to generate the product gas at the same rate as that of STH = 10% photocatalyst irradiated with a normal 1-kW·m−2 solar simulator. This sort of acceleration tests has also been conducted to obtain preliminary knowledge on the overall operation of the plant, in regard with the systematic control of gas and water flows, which vary according to the daily positions of the sun and weather.
Using such a type of photoreactors, construction of an outdoor pilot plant is now under our consideration as schematized in Figure 7. The pilot plant will consist of an array of photocatalytic reactors containing STH = 0.6% RhCrOx/Al:SrTiO3 photocatalyst sheets, water delivery and product gas tubing, and a gas desiccation / separation apparatus to put out H2 and O2 separately.
A special issue in gathering this type of single-powder photoreactors is that the product is 2:1 mixture of H2 + O2. The mixture is moreover moistened with water vapor at high relative humidity. To utilize H2 in the following industrial process, H2 must be isolated from the mixture. The isolation process is definitely necessary for safe operation. This is a drawback of a single-powder system, compared to water electrolyzers or photoelectrochemical dual-electrode cells, in which a conventional gas separation membrane can be assembled to set apart H2 and O2 from the beginning. This choice, whether single-powder/mixed product gas or dual electrodes/separated gases, is a point of careful discussions in our project. This is partly an economic issue depending on the balance of the costs for construction and the accumulated profit from the daily operation. The member companies are pursuing one of these two choices according to their background. The single-powder benchmark plant has been temporality chosen on the basis of the current scale of this project.
To realize the steady and safe operation of the mixed H2 + O2 evolution type, an efficient device to filter H2 out of the mixture is necessary, and today, the suitable filtering material for mass production can be inorganic gas-selective permeation membranes. The filter membranes should be highly permeable for H2 and not transmitting O2. The filter membranes should also be robust against H2O. The materials for filtering membranes to meet these requirements are not readily available, and ARPChem’s Separation Membrane Team is challenging for a breakthrough in the development of filter materials.
To operate a separation membrane, a certain pressure difference is needed between the input gas mixture and the filtrate gas. The pressure difference can be generated by compressing the 2H2 + O2 product by a motor-driven pump, or by evacuating the filtrate H2 gas by a motor-driven vacuum pump.
Another idea to generate pressure is to choke the gas exhalation tube by a variable-conductance valve or an orifice to confine the reactor. The photocatalytic 2H2 + O2 generation reaction also generates the product pressure in principle. Static hydraulic pressure from the reactant water also helps this method. According to thermodynamics, the overall STH is reduced by a part of the received solar energy that is redirected into the pressure of the gas. In some cases, the reverse reaction of H2O production is favored in high pressure 2H2 + O2 under a dynamic condition [32]. The wide-area reactor containers must be inner-pressure proof against the outer atmosphere, which is a drawback of this method.
The ideal thermodynamic energy consumption for gas separation is much less than 1% of the solar hydrogen energy in the product in the average operation condition. However, the energetic efficiency of the pumps or the photocatalytic reactors is a technical parameter that cannot be better than the theoretical thermodynamic efficiency and can be beefed up by smart mechanical design for the pumps or by improvement of hydro/gaseous-dynamics in the reactor.
Since the single-powder photoreactor generates explosive 2:1 H2 + O2 mixture (oxyhydrogen detonative mixture, or “Brown Gas”) as the essential function, we definitely need the means to avoid all hazards that can be brought out in the array of photocatalytic reactors, gas transportation tubes, the filtering devices and pumps. The handling of oxyhydrogen detonative mixture has been considered as an untouchable issue on the grounds of the petrochemical industry, in which all flammable gases are strictly isolated from O2, air, and other gases supporting combustion. Apart from elaborate studies on combustion and detonation of 2H2 + O2 in astronautic engines and related fields, not much knowledge is shared as guiding principles in practical engineering.
However, some good news nonetheless consequential in the solar energy industry, is that the degree of energetic concentration in a solar plant will be apparently lower than that in a petrochemical plant in general. The distribution of 1 kW·m−2 on a sunny day lunch time does not hurt people and environment in the daily life. The maximum rate of solar 2H2 + O2 generation at STH = 10% is 15.5 mL·s−1 per 1 m2 at the atmospheric pressure. As long as the product oxyhydrogen mixture is confined in a small volume per the unit area, the damage of an explosion will be also sparsely distributed and the damage by explosion will be minute. This is a much lower level than that for the hazard prevention activities in petrochemical plants, in which pressurized H2 gas and other flammable materials are always transported.
Based on such a philosophy, we plan to perform two kinds of preliminary tests for oxyhydrogen explosion. They are for convincing the administration and the fire department that are in charge of the area in which our pilot plant will be sited. One is to estimate the natural frequency of explosion without ignition. Tubes, panel reactors and gas filter cartridges are filled with oxyhydrogen fed slowly and continuously for a long period of time (weeks or months) and we just keep watching, aided by video recording. In most past experiences of hazardous explosion and fire, the cause of firing has been attributed to electrostatic discharging, which is not always truly verified. The probability of catching fire must depend on the equipment materials as well as the environmental condition, and we have to find the realistic upper limit of firing under the conditions the plants will be operated in.
The other is to investigate the damage of an explosion by filling those containers with 2H2 + O2 and observing the process of combustion/detonation ignited by a spark gap intentionally. Previous studies showed that an explosion of oxyhydrogen would not shatter or tear soft polymer materials enveloping the gas in a small volume at the atmospheric pressure [39]. Thanks to the low degree of flammable accumulation, the damage was mostly negligible. The scope of this test involves novel devices to avoid or weaken the hazard of explosion, such as emergency pressure-releasing devices (water traps, rupture disks, flame-arresting fillers, etc.) applicable to slightly higher pressure of oxyhydrogen than the atmosphere.
Those two kinds of tests should be repeated for all equipment that will be involved in the pilot plant, to guarantee safe operation.
5. Conclusions
This article has reviewed ARPChem’s current research and development activities over water photo-splitting catalytic and photoelectrochemical materials, macro-scale water photo-splitting devices, product gases (H2 and O2) separation by filtering membranes, and safety issues regarding with the explosive product gas mixture. The light-responding semiconductors for photcatalysts and photoelectrodes have been developed to cover the visible portion of the solar spectrum, and approaching STH of the same level as the practical photovoltaics. Reactor systems for massive-scale solar hydrogen plants have been planned and preliminary tests have been conducted. The test catalyst was a single-powder Al:SrTiO3-based photocatalyst with STH = 0.6%, in order to find and familiarize the practical maneuvers of product gas transportation, H2/O2 separation, and safety means in advance. The technology for avoiding the product 2H2 + O2 explosion, and for nullifying the damage of combustion has been anticipated for consideration.
ARPChem conducts research on the individual important themes in parallel simultaneously, aiming at the best performance for each. Photocatalytic reactors are designed and tested for the photocatalysts that are supposed to be as efficient as today’s practical photovoltaics, although the development of photocatalysts has not attained that level. The gas filtration membranes are also under development and have not yet been completed as an attachable assembly. A rather extended period is allowed for the whole set of ARPChem research, and this parallel conduction in the multiple inter-conversing disciplines will complete a novel embodied variation of solar energy utilization in the long run. So far, we have done many trials, however there are still plenty of materials, fabrication processes, and chemical engineering devices left untouched in this field.
Author Contributions
Conceptualization, K.D.; Writing-Original Draft Preparation, T.Y.; Writing-Review & Editing, T.Y. and K.D.
Funding
The research and development described in this report have been funded by Japan Technological Research Association of Artificial Photosynthetic Chemical Process (ARPChem), New Energy and Industrial Technology Development Organization (NEDO), a subside agency of Ministry of Economy, Trade and Industry of Japan (METI).
Acknowledgments
The authors are thankful to Taisei Nishimi (ARPChem), Director Hiroyuki Sato (ARPChem) and Project Leader Tohru Setoyama (Mitsubishi Chemical) for their encouragement in writing this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Basic Hydrogen Strategy. Available online: www.meti.go.jp/english/press/2017/pdf/1226_003b.pdf (accessed on 10 August 2018).
- Development of Basic Chemical Processes for Carbon Dioxide as Raw Material. Available online: www.nedo.go.jp/activities/EV_00296.html (accessed on 10 August 2018).
- Solar Spectra. Available online: rredc.nrel.gov/solar/spectra/am1.5/ (accessed on 10 August 2018).
- Ham, Y.-L.; Minegishi, T.; Histomi, T.; Domen, K. A SrTiO3 photoanode prepared by the particle transfer method for oxygen evolution from water with high quantum efficiencies. Chem. Commun. 2016, 52, 5011–5014. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Xu, X.-X.; Fang, J.-Z.; Zhou, G.-G.; Liu, Z.; Wu, S.-X.; Xu, W.-C.; Chu, J.-H.; Zhu, X.-M. Synthesis of BiOI-TiO2 composite nanoparticles by microemulsion method and study on their photocatalytic activities. Sci. World J. 2014, 2014, 1–8. [Google Scholar]
- Kuang, Y.-B.; Jia, Q.-X.; Nishiyama, H.; Yamada, T.; Kudo, A.; Domen, K. A Front-Illuminated Nanostructured transparent BiVO4 photoanode for >2% efficient water splitting. Adv. Energ. Mater. 2016, 6, 1501645. [Google Scholar] [CrossRef]
- Akiyama, S.; Nakabayashi, M.; Shibata, N.; Minegishi, T.; Asakura, Y.; Abdulla-Al-Mamun, M.; Hisatomi, T.; Nishiyama, H.; Katayama, M.; Yamada, T.; et al. Highly efficient water oxidation photoanode made of surface modified LaTiO2N particles. Small 2016, 12, 5468–5476. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Magesh, G.; Youn, D.-H.; Jang, J.-W.; Kubota, J.; Domen, K.; Lee, J.-S. Single-crystalline, wormlike hematite photoanodes for efficient solar water splitting. Sci. Rep. 2013, 3, 2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, K.; Minegishi, T.; Clune, J.; Nakabayashi, M.; Hisatomi, T.; Nishiyama, H.; Katayama, M.; Shibata, N.; Kubota, J.; Yamada, T.; et al. Photoelectrochemical oxidation of water using BaTaO2N photoanodes prepared by particle transfer method. J. Am. Chem. Soc. 2015, 137, 2227–2230. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Hisatomi, T.; Nakabayashi, M.; Shibata, N.; Minegishi, T.; Katayama, M.; Domen, K. Efficient solar-driven water oxidation over perovskite-type BaNbO2N photoanodes absorbing visible light up to 740 nm. Adv. Energy Mater. 2018. [Google Scholar] [CrossRef]
- Green, M.A.; Keevers, M. Optical properties of intrinsic silicon at 300 K. Prog. Photovolt. 1995, 3, 189–192. [Google Scholar] [CrossRef]
- Palik, E.D. Handbook of Optical Constants of Solids; Academic Press: Cambridge, MA, USA, 1985; pp. 429–443. [Google Scholar]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Domen, K.; Naito, S.; Soma, M.; Onishi, T.; Tamaru, K. Photocatalytic decomposition of water-vapor on an NiO-SrTiO3 catalyst. J. Chem. Soc. Chem. Commun. 1980, 12, 543–544. [Google Scholar] [CrossRef]
- Luo, J.-S.; Im, J.-H.; Mayer, M.T.; Schreier, M.; Nazeeruddin, M.D.K.; Park, N.-G.; Tilley, S.D.; Fan, H.J.; Grätzel, M. Water photolysis at 12.3% efficiency via perovskite photovoltaics and Earth-abundant catalysts. Science 2014, 345, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- McKone, J.R.; Lewis, N.S.; Gray, H.B. Will solar-driven water-splitting devices see the light of day? Chem. Mater. 2014, 26, 407–414. [Google Scholar] [CrossRef]
- Reece, S.Y.; Hamel, J.A.; Sung, K.; Jarvi, T.D.; Esswein, A.J.; Pijpers, J.J.H.; Nocera, D.G. Wireless solar water splitting using silicon-based semiconductors and earth-abundant catalysts. Science 2011, 334, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, S.W.; Warren, E.L.; Putnam, M.C.; Santori, E.A.; Turner-Evans, D.; Kelzenberg, M.D.; Walter, M.G.; McKone, J.R.; Brunschwig, B.S.; Atwater, H.A.; et al. Photoelectrochemical hydrogen evolution using Si microwire arrays. J. Am. Chem. Soc. 2011, 133, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Warren, E.L.; McKone, J.R.; Atwater, H.A.; Graya, H.B.; Lewis, N.S. Hydrogen-evolution characteristics of Ni-Mo-coated, radial junction, n(+)p-silicon microwire array photocathodes. Energy Environ. Sci. 2012, 5, 9653. [Google Scholar]
- Warren, E.L.; Atwater, H.A.; Lewis, N.S. Silicon microwire arrays for solar energy-conversion applications. J. Phys. Chem. C 2014, 118, 747–759. [Google Scholar] [CrossRef]
- Abdi, F.F.; Han, L.-H.; Smets, A.H.M.; Zeman, M.; Dam, B.; van de Krol, R. Efficient solar water splitting by enhanced charge separation in a bismuth vanadate-silicon tandem photoelectrode. Nat. Commun. 2013, 4, 2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minegishi, T.; Nishimura, N.; Kubota, J.; Domen, K. Photoelectrochemical properties of LaTiO2N electrodes prepared by particle transfer for sunlight-driven water splitting. Chem. Sci. 2013, 4, 1120–1124. [Google Scholar] [CrossRef]
- Kobayashi, H.; Sato, N.; Orita, M.; Kuang, Y.; Kaneko, H.; Minegishi, T.; Yamada, T.; Domen, K. Development of highly efficient CuIn0.5Ga0.5Se2-based photocathode and application to overall solar driven water splitting. Energy Environ. Sci. 2018. [Google Scholar] [CrossRef]
- Zhao, J.; Minegishi, T.; Zhang, L.; Zhong, M.; Gunawan; Nakabayashi, M.; Ma, G.-J.; Hisatomi, T.; Katayama, M.; Ikeda, S.; et al. Enhancement of solar hydrogen evolution from water by surface modification with CdS and TiO2 on porous CuInS2 photocathodes prepared by an electrodeposition-sulfurization method. Angew. Chem. Int. Ed. 2014, 53, 11808–11812. [Google Scholar]
- Hayashi, T.; Niishiro, R.; Ishihara, H.; Yamaguchi, M.; Jia, Q.; Kuang, Y.; Higashi, T.; Iwase, A.; Minegishi, T.; Yamada, T.K.; et al. Powder-based (CuGa1−yIny)1−xZn2xS2 solid solution photocathodes with a largely positive onset potential for solar water splitting. Sustain. Energy Fuels 2018. [Google Scholar] [CrossRef]
- Peter, L.M. Towards sustainable photovoltaics: the search for new materials. Philos. Trans. R. Soc. A 2011, 369, 1840–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ham, Y.-L.; Hisatomi, T.; Goto, Y.; Moriya, Y.; Sakata, Y.; Yamakata, A.; Kubota, J.; Domen, K. Flux-mediated doping of SrTiO3 photocatalysts for efficient overall water splitting. J. Mater. Chem. A 2016, 4, 3027–3033. [Google Scholar] [CrossRef]
- Niishiro, R.; Takano, Y.; Jia, Q.; Yamaguchi, M.; Iwase, A.; Kuang, Y.-B.; Minegishi, T.; Yamada, T.; Domen, K.; Kudo, A. A CoOx-modified SnNb2O6 photoelectrode for highly efficient oxygen evolution from water. Chem. Commun. 2017, 53, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.-B.; Jia, Q.-X.; Ma, G.-J.; Hisatomi, T.; Minegishi, T.; Nishiyama, H.; Nakabayashi, M.; Shibata, N.; Yamada, T.; Kudo, A.; et al. Ultrastable low-bias water splitting photoanodes via photocorrosion inhibition and in situ catalyst regeneration. Nat. Energy 2016, 2, 16191. [Google Scholar] [CrossRef]
- Zhong, M.; Hisatomi, T.; Sasaki, Y.; Suzuki, S.; Teshima, K.; Nakabayashi, M.; Shibata, N.; Nishiyama, H.; Katayama, M.; Yamada, T.; et al. Highly active GaN-stabilized Ta3N5 thin-film photoanode for solar water oxidation. Angew. Chem. Int. Ed. 2017, 56, 4739–4743. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Minegishi, T.; Kageshima, Y.; Kobayashi, H.; Hisatomi, T.; Higashi, T.; Katayama, M.; Domen, K. CdTe-Based photoanode for oxygen evolution from water under simulated sunlight. J. Phys. Chem. Lett. 2017, 8, 5712–5717. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hisatomi, T.; Jia, Q.; Tokudome, H.; Zhong, M.; Wang, C.; Pan, Z.-H.; Takata, T.; Nakabayashi, M.; Shibata, N.; et al. Scalable water splitting on particulate photocatalyst sheets with a solar-to-hydrogen energy conversion efficiency exceeding 1%. Nat. Mater. 2016, 15, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Kudo, A.; Omori, K.; Kato, H. A novel aqueous process for preparation of crystal form-controlled and highly crystalline BiVO4 powder from layered vanadates at room temperature and its photocatalytic and photophysical properties. J. Am. Chem. Soc. 1999, 121, 11459–11467. [Google Scholar] [CrossRef]
- Moniz, S.J.A.; Shevlin, S.A.; Martin, D.J.; Guo, Z.-X.; Tang, J.-W. Visible-light driven heterojunction photocatalysts for water splitting—A critical review. Energy Environ. Sci. 2015, 8, 731–759. [Google Scholar] [CrossRef]
- Kim, T.-W.; Choi, K.-S. Nanoporous BiVO4 photoanodes with dual-layer oxygen evolution catalysts for solar water splitting. Science 2014, 343, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Asakura, Y.; Higashi, T.; Nishiyama, H.; Kobayashi, H.; Nakabayashi, M.; Shibata, N.; Mineghishi, T.; Hisatomi, T.; Katayama, M.; Yamada, T.; et al. Activation of particulate Ta3N5 water-oxidation photoanode with GaN hole-blocking layer. Sustain. Energy Fuels 2018, 2, 73–78. [Google Scholar] [CrossRef]
- Chiang, T.-H.; Hao, L.; Hisatomi, T.; Goto, Y.; Takata, T.; Katayama, M.; Minegishi, T.; Domen, K. Efficient Photocatalytic water splitting using Al-doped SrTiO3 coloaded with molybdenum oxide and rhodium−chromiusm oxide. ACS Catal. 2018, 8, 2782–2788. [Google Scholar] [CrossRef]
- Goto, Y.; Hisatomi, T.; Wang, Q.; Higashi, T.; Ishikiriyama, K.; Maeda, T.; Sakata, Y.; Okunaka, S.; Tokudome, H.; Katayama, M.; et al. A particulate photocatalyst water-splitting panel for large-scale solar hydrogen generation. Joule 2018, 2, 509–520. [Google Scholar] [CrossRef]
- Shinohara, K. Investigation of safe and effective condition on detonating gas performable experiment by vinyl plastic tube. Kagaku-to-Kyoiku (Chem. Educ.) 2004, 52, 471–474. (In Japanese) [Google Scholar]
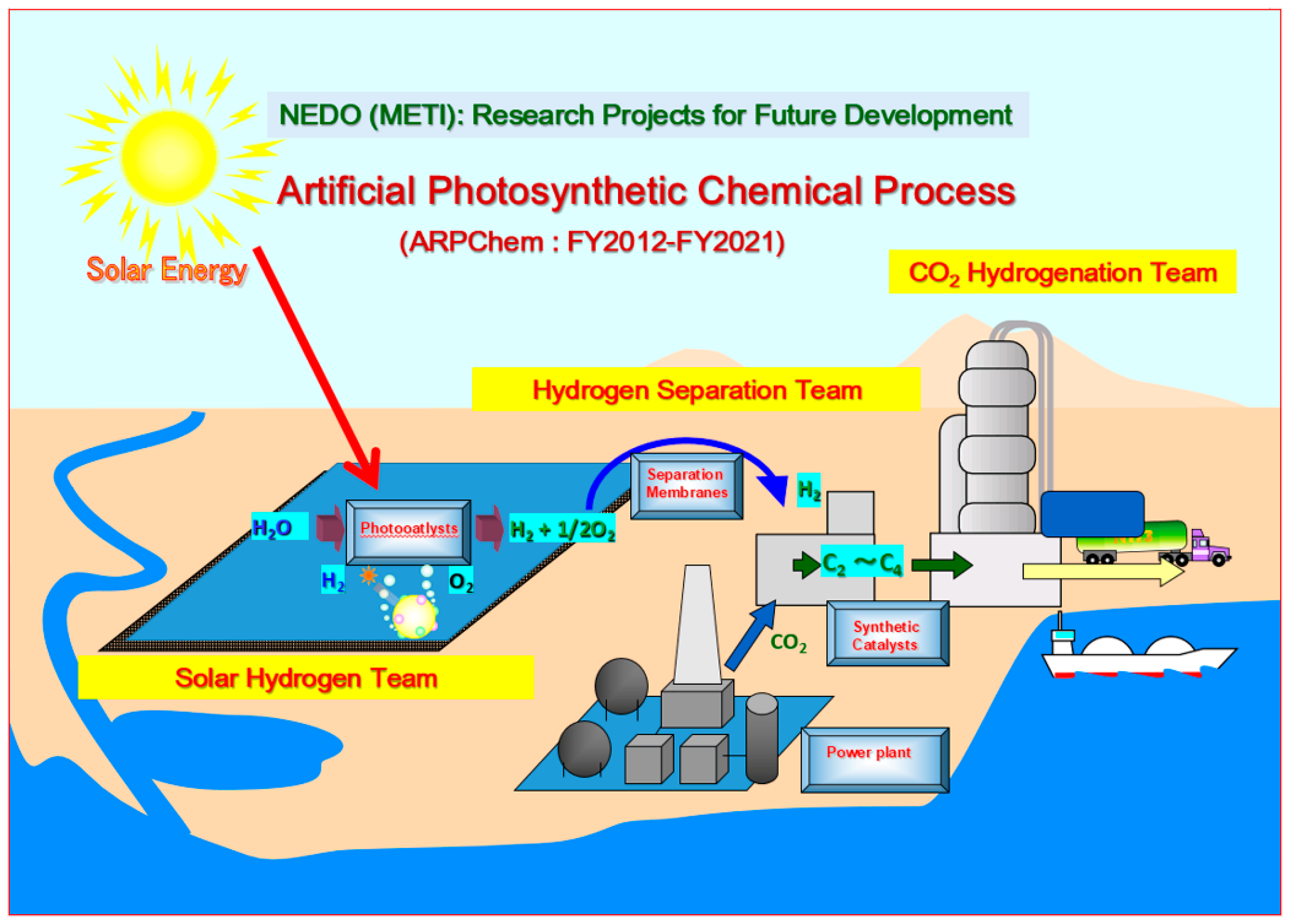
Figure 1.
Schematic diagram of ARPChem. ARPChem is a research and development union of leading chemical firms of Japan, funded by New Energy and Industrial Technology Development Organization (NEDO), a subside agency of Ministry of Economy, Trade and Industry (METI) [2]. There are three research teams: Solar Hydrogen Team for water photo-splitting catalysts, Hydrogen Separation Team for gas separation membranes, and Synthetic Catalyst Team for CO2 hydrogenation catalysts, all concentrating in materials research and development. These teams also have their missions for future planning and designing industrial photocalatlytic plants, gas separation apparatuses, and catalytic synthesis plants, respectively.
Figure 1.
Schematic diagram of ARPChem. ARPChem is a research and development union of leading chemical firms of Japan, funded by New Energy and Industrial Technology Development Organization (NEDO), a subside agency of Ministry of Economy, Trade and Industry (METI) [2]. There are three research teams: Solar Hydrogen Team for water photo-splitting catalysts, Hydrogen Separation Team for gas separation membranes, and Synthetic Catalyst Team for CO2 hydrogenation catalysts, all concentrating in materials research and development. These teams also have their missions for future planning and designing industrial photocalatlytic plants, gas separation apparatuses, and catalytic synthesis plants, respectively.

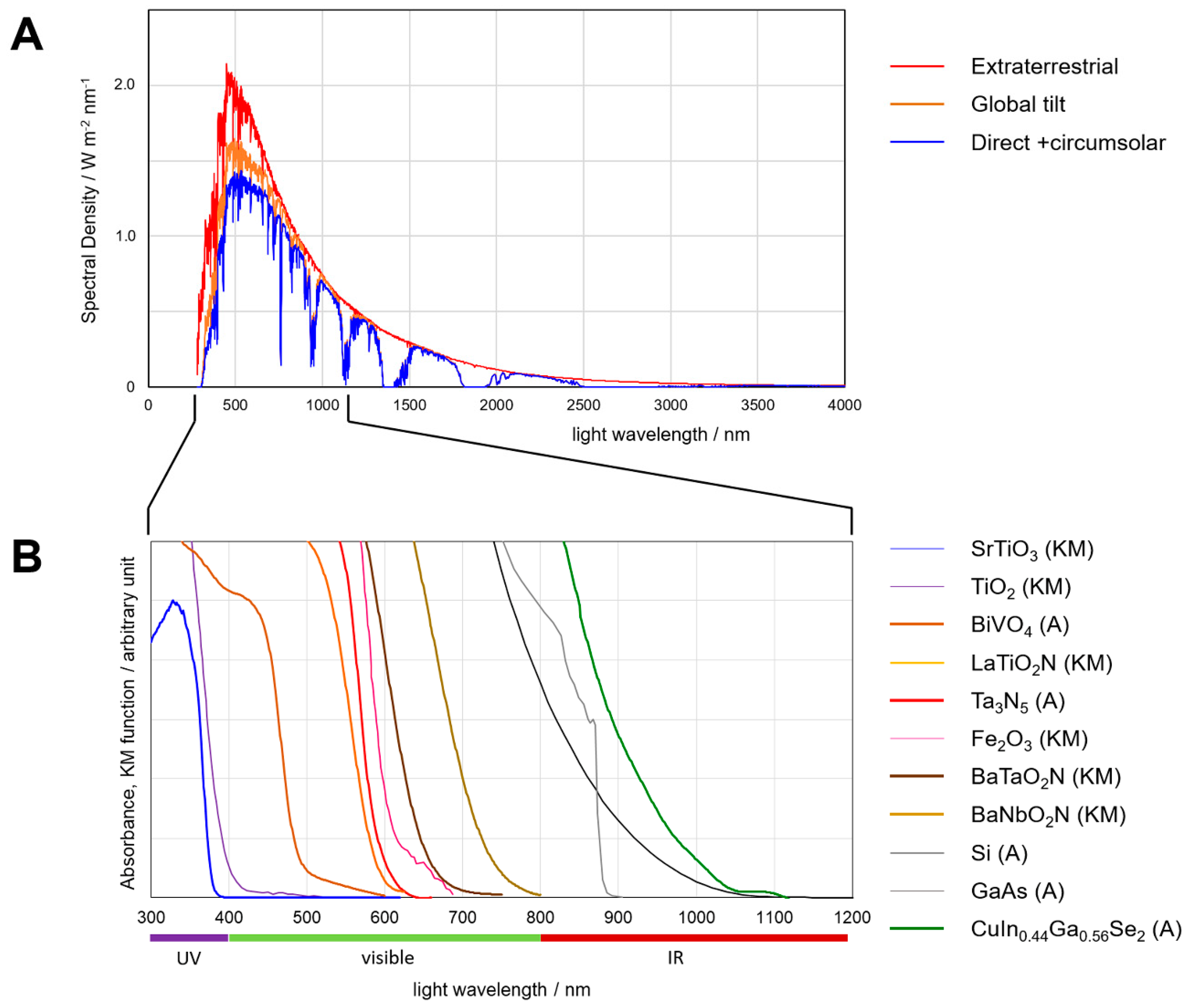
Figure 2.
(A) Natural solar spectra. “Extraterrestrial” = average radiation at the top of earth atmosphere facing the sun, “Global Tilt” = radiation on a plane at the ground facing the south and tilted by 37° from horizontal, involving the sun light, atmosphere-scattered light and reflection from the ground, “Direct + Circumsolar” = tracked radiation from the sun on the ground, and scattered light within ±2.5° from the center of sun. Global Tilt represents the air-mass 1.5 G (AM 1.5 G) spectrum. The coordinate is given in the absolute areal energetic spectral density in W·m−2·nm−1. These data are obtained from National Renewable Energy Laboratory, USA (rredc.nrel.gov/solar/spectra/am1.5/ [3]). (B) Absorption spectra of inorganic semiconductors, indicated as either the absorption coefficient converted from the transmission spectra (A) or Kubelka-Munk diffuse-absorption function (KM), both arbitrarily normalized to magnify the edge of absorption by each material. The spectral curves were reproduced for SrTiO3 from [4], TiO2 [5], BiVO4 [6], LaTiO2N [7], Ta3N5 by our own dedicated measurement, Fe2O3 [8], BaTaO2N [9], BaNbO2N [10], Si [11], GaAs [12] and CuIn0.44Ga0.56Se2 by our own dedicated measurement.
Figure 2.
(A) Natural solar spectra. “Extraterrestrial” = average radiation at the top of earth atmosphere facing the sun, “Global Tilt” = radiation on a plane at the ground facing the south and tilted by 37° from horizontal, involving the sun light, atmosphere-scattered light and reflection from the ground, “Direct + Circumsolar” = tracked radiation from the sun on the ground, and scattered light within ±2.5° from the center of sun. Global Tilt represents the air-mass 1.5 G (AM 1.5 G) spectrum. The coordinate is given in the absolute areal energetic spectral density in W·m−2·nm−1. These data are obtained from National Renewable Energy Laboratory, USA (rredc.nrel.gov/solar/spectra/am1.5/ [3]). (B) Absorption spectra of inorganic semiconductors, indicated as either the absorption coefficient converted from the transmission spectra (A) or Kubelka-Munk diffuse-absorption function (KM), both arbitrarily normalized to magnify the edge of absorption by each material. The spectral curves were reproduced for SrTiO3 from [4], TiO2 [5], BiVO4 [6], LaTiO2N [7], Ta3N5 by our own dedicated measurement, Fe2O3 [8], BaTaO2N [9], BaNbO2N [10], Si [11], GaAs [12] and CuIn0.44Ga0.56Se2 by our own dedicated measurement.

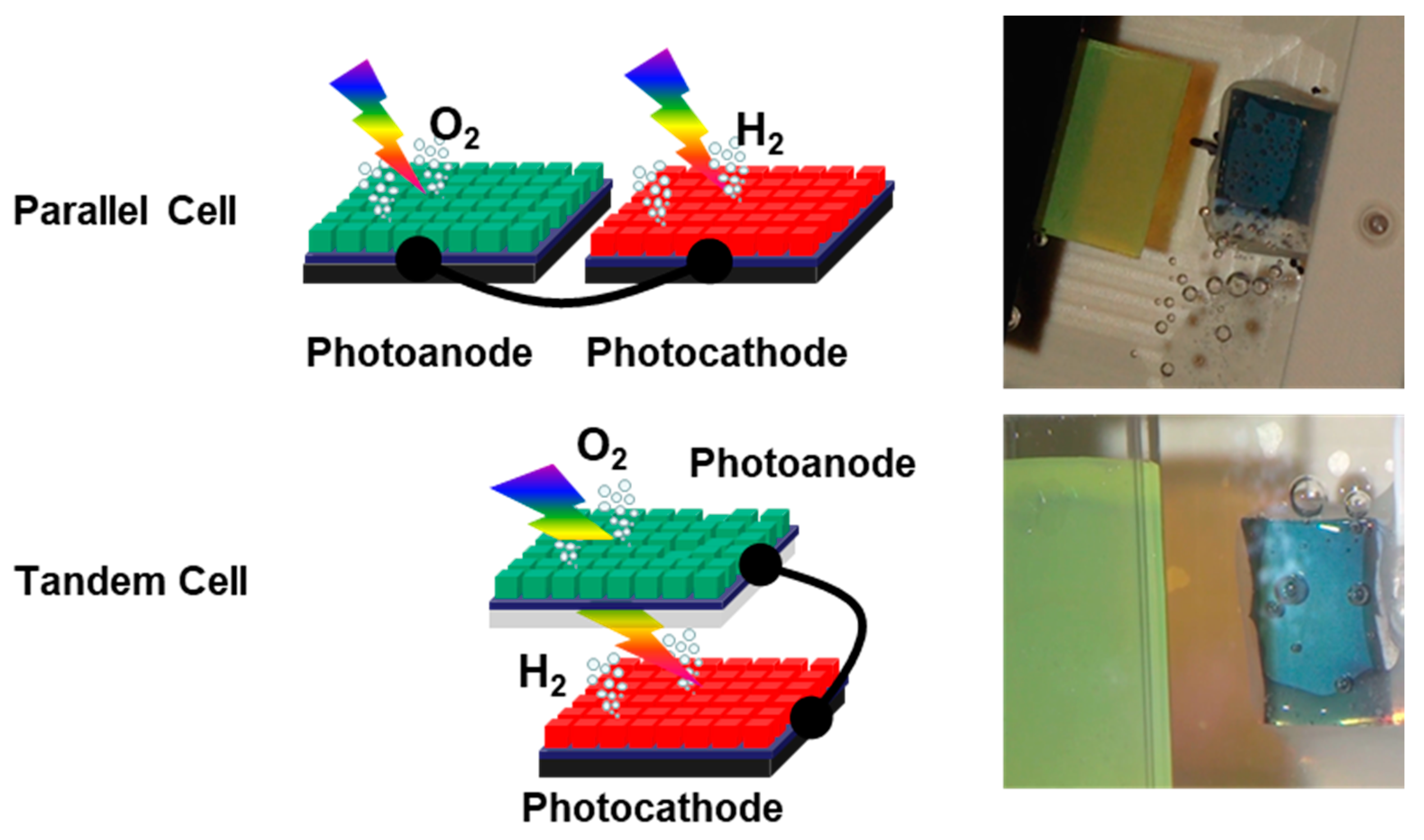
Figure 3.
Schematic diagrams of dual-photoelectrode stand-alone overall water photo-splitting devices. In the parallel cell, a pair of photoanode and photocathode is electrically shunted and immersed in electrolyte. The nominal light-acceptation area is the sum for those two electrodes. In the tandem cell, a semi-transparent photoanode/photocathode placed in front of photocathode/photoanode. The semi-transparent one should have a shorter cutoff wavelength than that of the other, so as not to block all of the utilizable wavelength range. The nominal light-acceptation area is that of the larger one. The ratio of photoanode/photocathode light acceptation areas can be optimized to maximize the solar-to-H2 energetic efficiency (STH) both in the parallel cell and the tandem cell. The photographs show these two types of cells in operation under solar-simulator irradiation.
Figure 3.
Schematic diagrams of dual-photoelectrode stand-alone overall water photo-splitting devices. In the parallel cell, a pair of photoanode and photocathode is electrically shunted and immersed in electrolyte. The nominal light-acceptation area is the sum for those two electrodes. In the tandem cell, a semi-transparent photoanode/photocathode placed in front of photocathode/photoanode. The semi-transparent one should have a shorter cutoff wavelength than that of the other, so as not to block all of the utilizable wavelength range. The nominal light-acceptation area is that of the larger one. The ratio of photoanode/photocathode light acceptation areas can be optimized to maximize the solar-to-H2 energetic efficiency (STH) both in the parallel cell and the tandem cell. The photographs show these two types of cells in operation under solar-simulator irradiation.

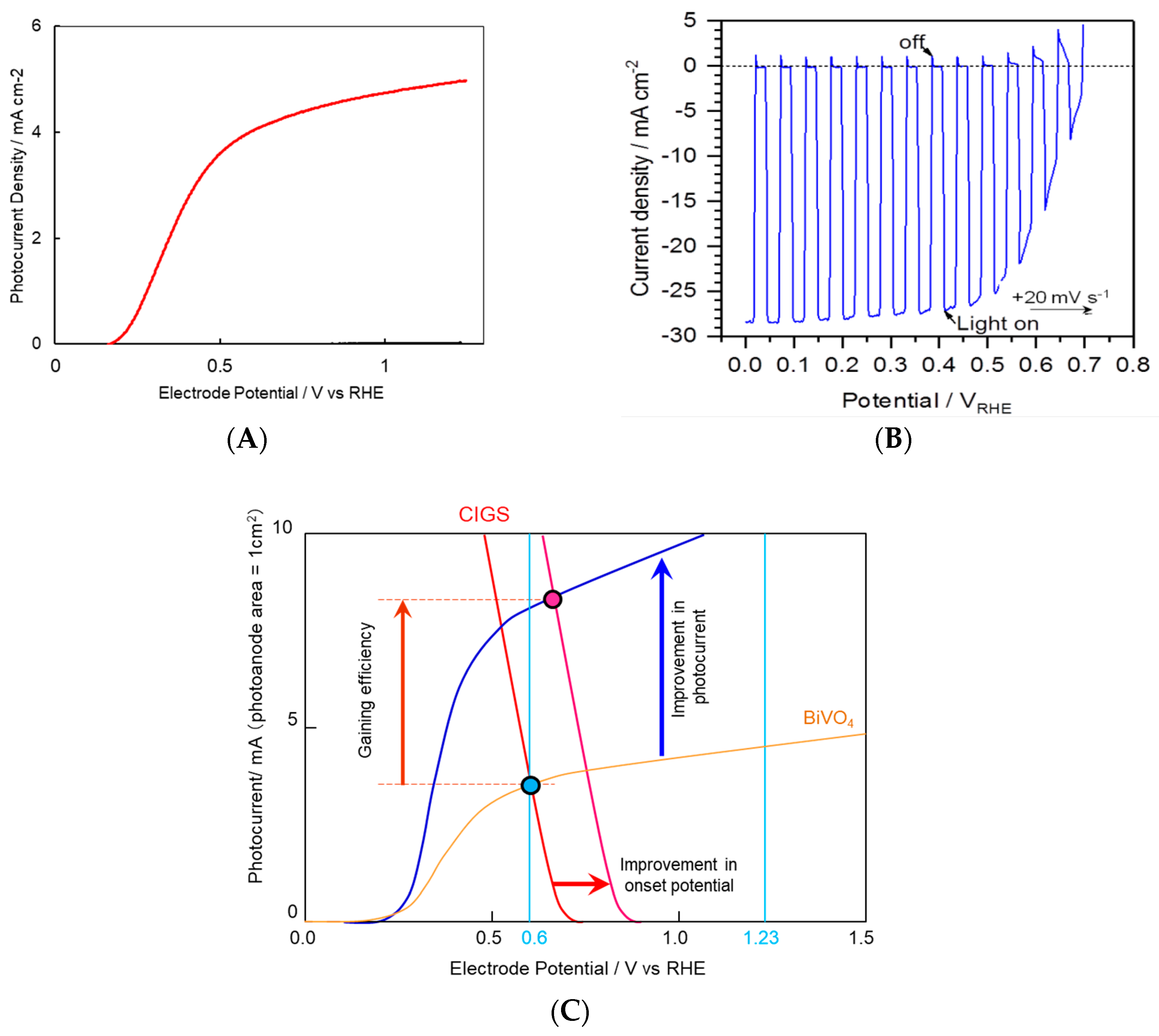
Figure 4.
Examples of photoelectrochemical curves (current density-versus-potential curves in linear-sweep voltammetry) for (A) an O2-evolving semi-transparent NiFeOx/BiVO4/ITO photoanode (reproduced from [6], in 1 M potassium borate (pH 9.3) solution containing 0.2 M Na2SO3 with (red curve) or without (black curve, “dark current”) AM 1.5 G solar simulator irradiation at room temperature, with a sweep rate of 10 mV·s−1), (B) a H2-evolving Pt/CdS/CuIn0.44Ga0.56Se2/Mo/soda lime glass photoanode (our dedicated experiment in a pH 9.8 phosphate buffer with chopped AM 1.5 G solar simulator, showing the dark current and lighted current on one curve, swept at 20 mV·s−1). (C) shows a schematic viewgraph for the stand-alone dual photoelectrode operation for overall water photo-splitting. The curves of a real BiVO4 semi-transparent photoanode (blue curve) and a CIGS photocathode (red curve) (The sign of photocurrent is inverted.) under AM 1.5 G solar simulator irradiation are reproduced. The light acceptation areas were 1 cm2 for both. The cross point of these curves corresponds to the steady-state operational photocurrent evolving 2H2 and O2, which is proportional to STH. It is need for those two curves to cross over each other at a positive photocurrent, to gain the product gases. If the onset potential of photocathode is more negative than that of photoanode, the photocurrent will be zero and no product gas is obtained. We focus our development to obtain photo-responsive materials that provide higher photocurrents and suitable onset potential arrangements, to realize a high STH.
Figure 4.
Examples of photoelectrochemical curves (current density-versus-potential curves in linear-sweep voltammetry) for (A) an O2-evolving semi-transparent NiFeOx/BiVO4/ITO photoanode (reproduced from [6], in 1 M potassium borate (pH 9.3) solution containing 0.2 M Na2SO3 with (red curve) or without (black curve, “dark current”) AM 1.5 G solar simulator irradiation at room temperature, with a sweep rate of 10 mV·s−1), (B) a H2-evolving Pt/CdS/CuIn0.44Ga0.56Se2/Mo/soda lime glass photoanode (our dedicated experiment in a pH 9.8 phosphate buffer with chopped AM 1.5 G solar simulator, showing the dark current and lighted current on one curve, swept at 20 mV·s−1). (C) shows a schematic viewgraph for the stand-alone dual photoelectrode operation for overall water photo-splitting. The curves of a real BiVO4 semi-transparent photoanode (blue curve) and a CIGS photocathode (red curve) (The sign of photocurrent is inverted.) under AM 1.5 G solar simulator irradiation are reproduced. The light acceptation areas were 1 cm2 for both. The cross point of these curves corresponds to the steady-state operational photocurrent evolving 2H2 and O2, which is proportional to STH. It is need for those two curves to cross over each other at a positive photocurrent, to gain the product gases. If the onset potential of photocathode is more negative than that of photoanode, the photocurrent will be zero and no product gas is obtained. We focus our development to obtain photo-responsive materials that provide higher photocurrents and suitable onset potential arrangements, to realize a high STH.

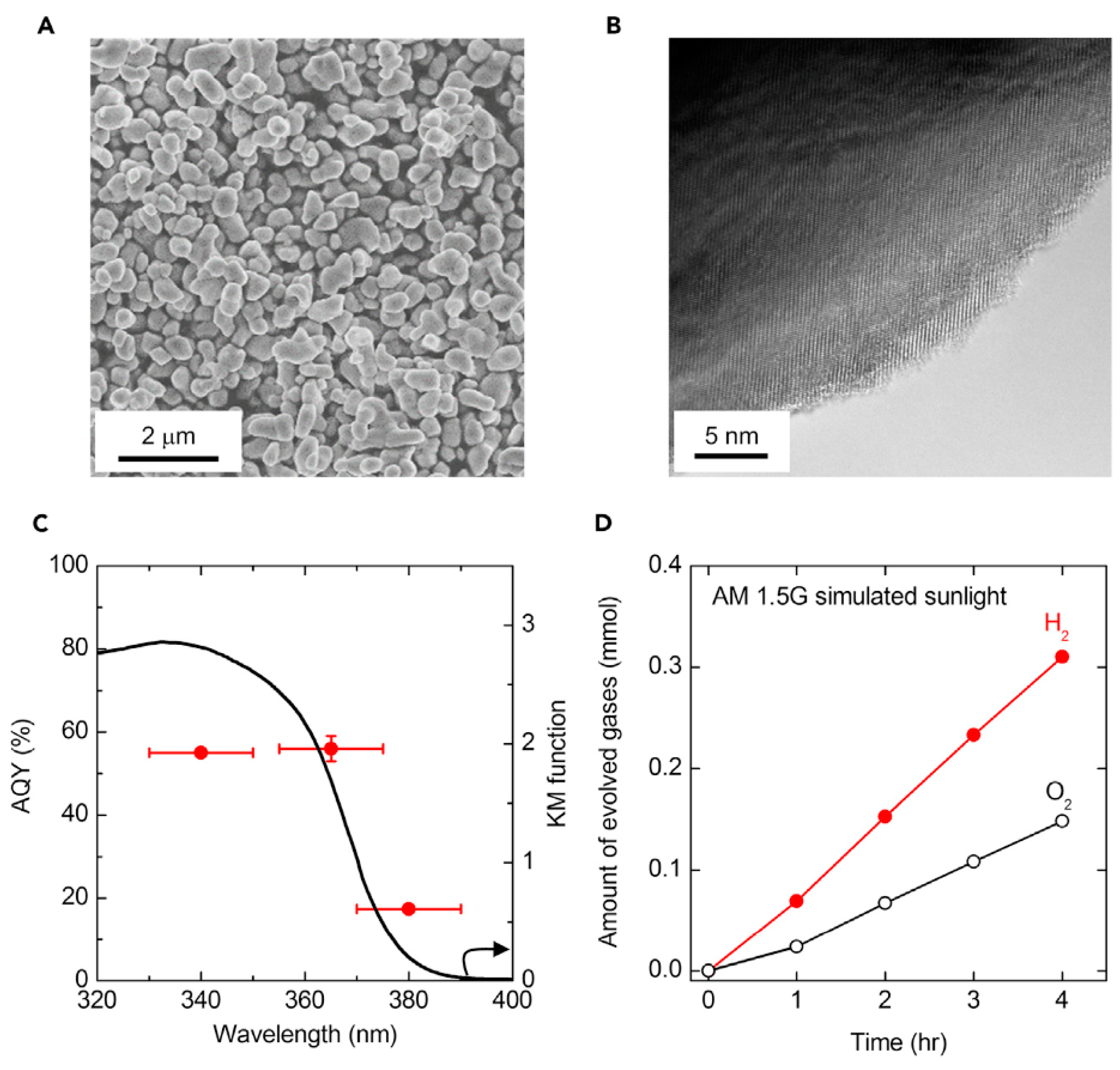
Figure 5.
Microscopic views and photocatalytic water-splitting reactitvity of Al-doped SrTiO3 single-powder photocatalyst, loaded with RhCrOx cocatalyst. (A) scanning electron microscopic image of Al:SrTiO3 powder. (B) high-resolution transmission microscopic image of a RhCrOx/Al:SrTiO3 grain. (C) apparent quantum yield (AQY, red plots) for overall water photo-splitting in pure water as a function of the incident wavelength. The light was delivered by a Xe lamp and the wavelength was controlled by corresponding bandpass filters with the window widths indicated by the error bars. The evolved H2 and O2 were quantified by gas chromatography. The coinciding diffuse absorption spectrum of this catalyst is also indicated (black curve as Kubelka-Munk function). The active wavelength range is in the ultraviolet region. (D) The time course of overall water photo-splitting by RhCrOx/Al:SrTiO3 under simulated sunlight (AM 1.5 G). Reaction conditions: catalyst weight 100 mg in 100 mL of pure H2O, at 288 K and 10 kPa. Stoichiometric decomposition of water is demonstrated for 4 h. (Reproduced from [38]).
Figure 5.
Microscopic views and photocatalytic water-splitting reactitvity of Al-doped SrTiO3 single-powder photocatalyst, loaded with RhCrOx cocatalyst. (A) scanning electron microscopic image of Al:SrTiO3 powder. (B) high-resolution transmission microscopic image of a RhCrOx/Al:SrTiO3 grain. (C) apparent quantum yield (AQY, red plots) for overall water photo-splitting in pure water as a function of the incident wavelength. The light was delivered by a Xe lamp and the wavelength was controlled by corresponding bandpass filters with the window widths indicated by the error bars. The evolved H2 and O2 were quantified by gas chromatography. The coinciding diffuse absorption spectrum of this catalyst is also indicated (black curve as Kubelka-Munk function). The active wavelength range is in the ultraviolet region. (D) The time course of overall water photo-splitting by RhCrOx/Al:SrTiO3 under simulated sunlight (AM 1.5 G). Reaction conditions: catalyst weight 100 mg in 100 mL of pure H2O, at 288 K and 10 kPa. Stoichiometric decomposition of water is demonstrated for 4 h. (Reproduced from [38]).

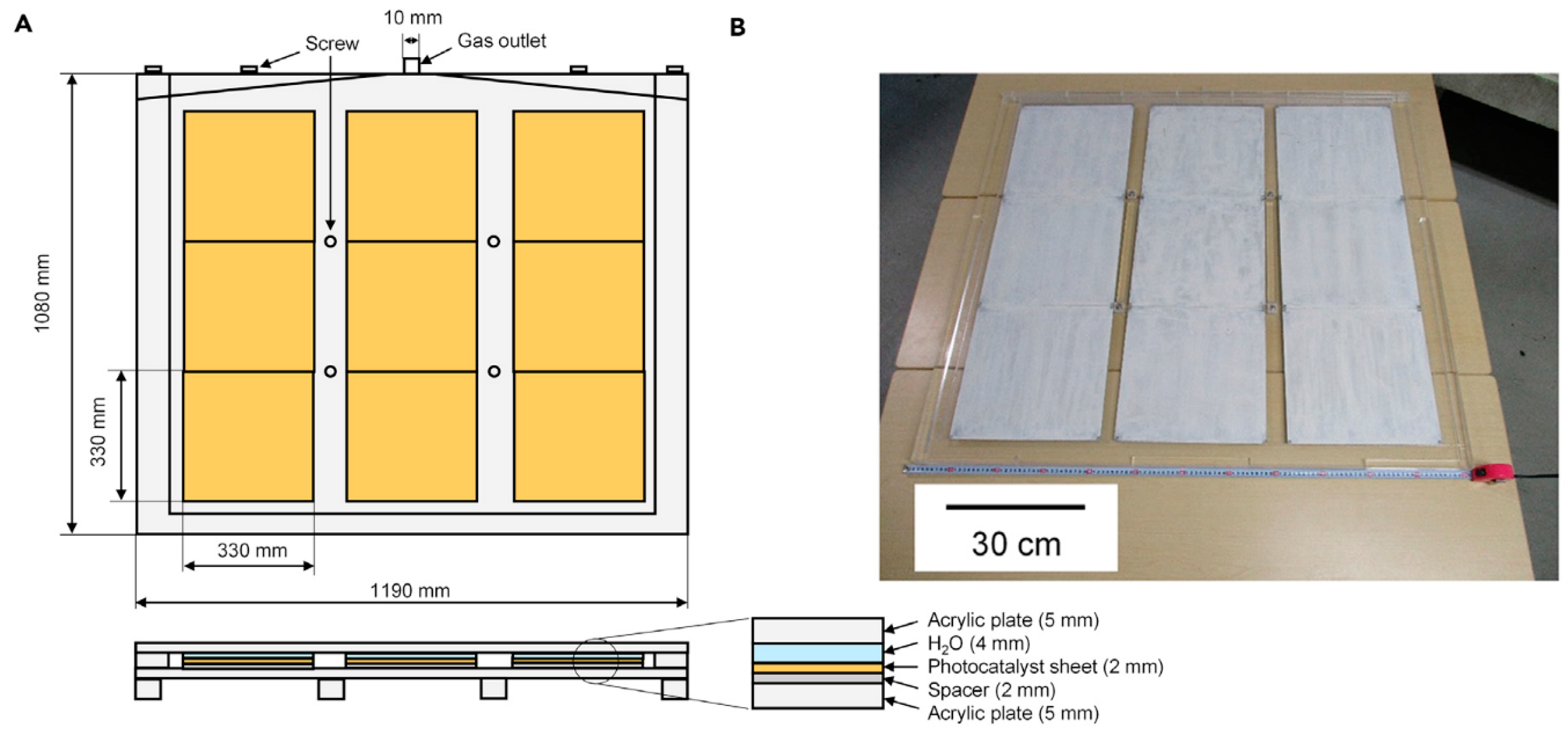
Figure 6.
(A) Schematics of the 1 m square water photo-splitting panel. Nine 0.6%-STH RhCrOx/Al:SrTiO3 photocatalyst sheets (33 cm × 33 cm for each) were arrayed in the reactor enveloped with transparent acrylic Plexiglas boards. A hydrophilized acrylic plate was used as the window. This panel was placed outdoor, with 10° tilt angle from the ground. The total weight of this panel, involving the reactant pure water, was approximately 4 kg. (B) A photograph of a 1 m square Al:SrTiO3 panel. (Reproduced from [38].)
Figure 6.
(A) Schematics of the 1 m square water photo-splitting panel. Nine 0.6%-STH RhCrOx/Al:SrTiO3 photocatalyst sheets (33 cm × 33 cm for each) were arrayed in the reactor enveloped with transparent acrylic Plexiglas boards. A hydrophilized acrylic plate was used as the window. This panel was placed outdoor, with 10° tilt angle from the ground. The total weight of this panel, involving the reactant pure water, was approximately 4 kg. (B) A photograph of a 1 m square Al:SrTiO3 panel. (Reproduced from [38].)

Figure 7.
A schematic drawing of our overall water photo-splitting pilot plant. A number of unit water splitting photocatalytic reactors, charged with 0.6%-STH RhCrOx/Al:SrTiO3 photocatalyst sheets, are arrayed filling the plant area. The reactors are daisy-chained with tubes for transporting the product 2H2 + O2 gas, as well as with pure water supplying tubes. The product transportation tubes are gathered and connected to a gas separator unit to put H2 and O2 into two output tubes. The gas separator unit consists of inorganic filter membranes and motor-driven gas pumps for filtration as well as for desiccation.
Figure 7.
A schematic drawing of our overall water photo-splitting pilot plant. A number of unit water splitting photocatalytic reactors, charged with 0.6%-STH RhCrOx/Al:SrTiO3 photocatalyst sheets, are arrayed filling the plant area. The reactors are daisy-chained with tubes for transporting the product 2H2 + O2 gas, as well as with pure water supplying tubes. The product transportation tubes are gathered and connected to a gas separator unit to put H2 and O2 into two output tubes. The gas separator unit consists of inorganic filter membranes and motor-driven gas pumps for filtration as well as for desiccation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
ARPChem score board for photoanode and photocathode materials. The performing parameters (photocurrent density, onset potential and durable period) were all experimentally obtained within ARPChem and published in literature previously. The tests were conducted with solar simulator 1 kW·m−2 AM 1.5 G irradiation (Those marked with * were tested by 300 W Xe lamp and a λ > 420 nm filter, which is approximately 3 times more intense than solar simulator, with a similar spectrum.). The durable period is defined as the time for the photocurrent density to reach 80% of the initial value. The photocurrent density, the onset potential, and the durable period were measured at an electrode potential of 0 V vs. RHE for all photocathodes (that is, the potential of H2 evolution), and 1.23 V vs. RHE for all photocathodes (that is, the potential of O2 evolution). For photocathode durability tests, the data marked with # were recorded at 0.6 V vs. RHE, which is the median potential of 0 V and 1.23 V vs. RHE, representing the practical operation crossover potential of the dual-electrode cell.
Table 1.
ARPChem score board for photoanode and photocathode materials. The performing parameters (photocurrent density, onset potential and durable period) were all experimentally obtained within ARPChem and published in literature previously. The tests were conducted with solar simulator 1 kW·m−2 AM 1.5 G irradiation (Those marked with * were tested by 300 W Xe lamp and a λ > 420 nm filter, which is approximately 3 times more intense than solar simulator, with a similar spectrum.). The durable period is defined as the time for the photocurrent density to reach 80% of the initial value. The photocurrent density, the onset potential, and the durable period were measured at an electrode potential of 0 V vs. RHE for all photocathodes (that is, the potential of H2 evolution), and 1.23 V vs. RHE for all photocathodes (that is, the potential of O2 evolution). For photocathode durability tests, the data marked with # were recorded at 0.6 V vs. RHE, which is the median potential of 0 V and 1.23 V vs. RHE, representing the practical operation crossover potential of the dual-electrode cell.
| Photoanodes | ||||||||||
| Light Absorber Material | Fabrication Technology | Substrate | Modifier Material | Cocatalyst | Absorption Band Edge/nm | Onset Potential/V vs. RHE | Photocurrent Density/mA·cm−2 | Test Solution | Durable Period | Ref. |
| Al:SrTiO3 | particle transfer | Ti | none | 390 | −0.15 * | 7.0 * | pH 13 Na2SO4 | >1000 s * | [27] | |
| SnNb2O6 | particle transfer | TI | CoOx | 517 | 0.20 | 2.0 | pH 9.4 K2B4O7 | a few minutes | [28] | |
| Mo:BiVO4 | particle transfer | Ni | FeNiOx | 517 | 0.29 | 4.6 | pH 9 K3BO3 | >1100 h | [29] | |
| Ta3N5 | NH3 nitridation | Ta | GaN | CoPi | 590 | 0.65 | 8.7 | pH 13 K3PO4 | 12 h | [30] |
| LaTiO2N | particle transfer | Ti | CoOx | 600 | 0.60 | 8.9 | pH 13.5 NaOH | a few minutes | [7] | |
| BaTaO2N | particle transfer | Ti | CoOx | 660 | 0.65 | 4.2 | pH 13 K3PO4 | 6 h | [9] | |
| BaNbO2N | particle transfer | Ti | CoOx | 740 | 0.65 | 5.2 | pH 9 K3BO3 | a few minutes | [10] | |
| CdTe | vacuum evaporation | CdS/FTO | MoOx, TiOx, CdCl2 | Ni | 830 | 0.20 | 5.1 | pH 8 K3PO4 | 60 min | [31] |
| Photocathodes | ||||||||||
| Light Absorber Material | Fabrication Technology | Substrate | Modifier Material | Cocatalyst | Absorption Band Edge/nm | Onset Potential/V vs. RHE | Photocurrent Density/mA·cm−2 | Test Solution | Durable Period | Ref. |
| CuInS2 (CIS) | electrodeposition and H2S suflurization | Mo/SLG | TiO2 CdS | Pt | 900 | 0.70 | 14.0 | pH 10 K3PO4 | >60 min | [24] |
| Cu(In1−x,Gax)Se2 x = 0.5 (CIGS) | MBE | Mo/SLG | CdS | Pt | 940 | 0.75 | 28.5 | pH 6.8 K3PO4 | 10 min # | [23] |
| (CuGa1−yIny)1−xZn2xS2 x = 0.2, y = 0.5 | particle transfer | Au | TiO2 CdS | Pt | 700 | 1.00 | 6.0 | pH 6.4 K3PO4 | 30 min # | [25] |
| La,Rh:SrTiO3 | particle transfer | Au | none | none | 480 | 1.2 * | 0.5 * | pH 6.8 K3PO4 | not tested | [32] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yamada, T.; Domen, K. Development of Sunlight Driven Water Splitting Devices towards Future Artificial Photosynthetic Industry. ChemEngineering 2018, 2, 36. https://doi.org/10.3390/chemengineering2030036
AMA Style
Yamada T, Domen K. Development of Sunlight Driven Water Splitting Devices towards Future Artificial Photosynthetic Industry. ChemEngineering. 2018; 2(3):36. https://doi.org/10.3390/chemengineering2030036
Chicago/Turabian StyleYamada, Taro, and Kazunari Domen. 2018. "Development of Sunlight Driven Water Splitting Devices towards Future Artificial Photosynthetic Industry" ChemEngineering 2, no. 3: 36. https://doi.org/10.3390/chemengineering2030036