
A Novel Yeast Surface Display Method for Large-Scale Screen Inhibitors of Sortase A


Abstract
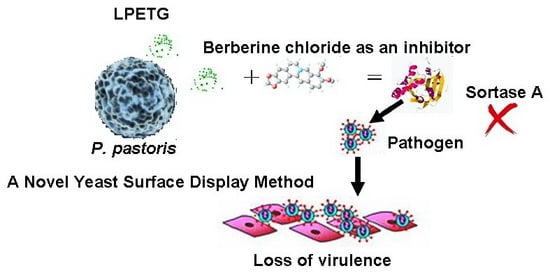
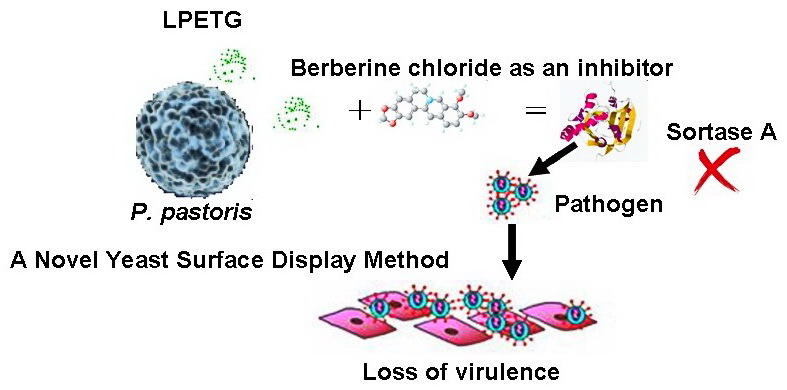
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Strains and Media
2.2. Optimization of Expression Condition of SrtA
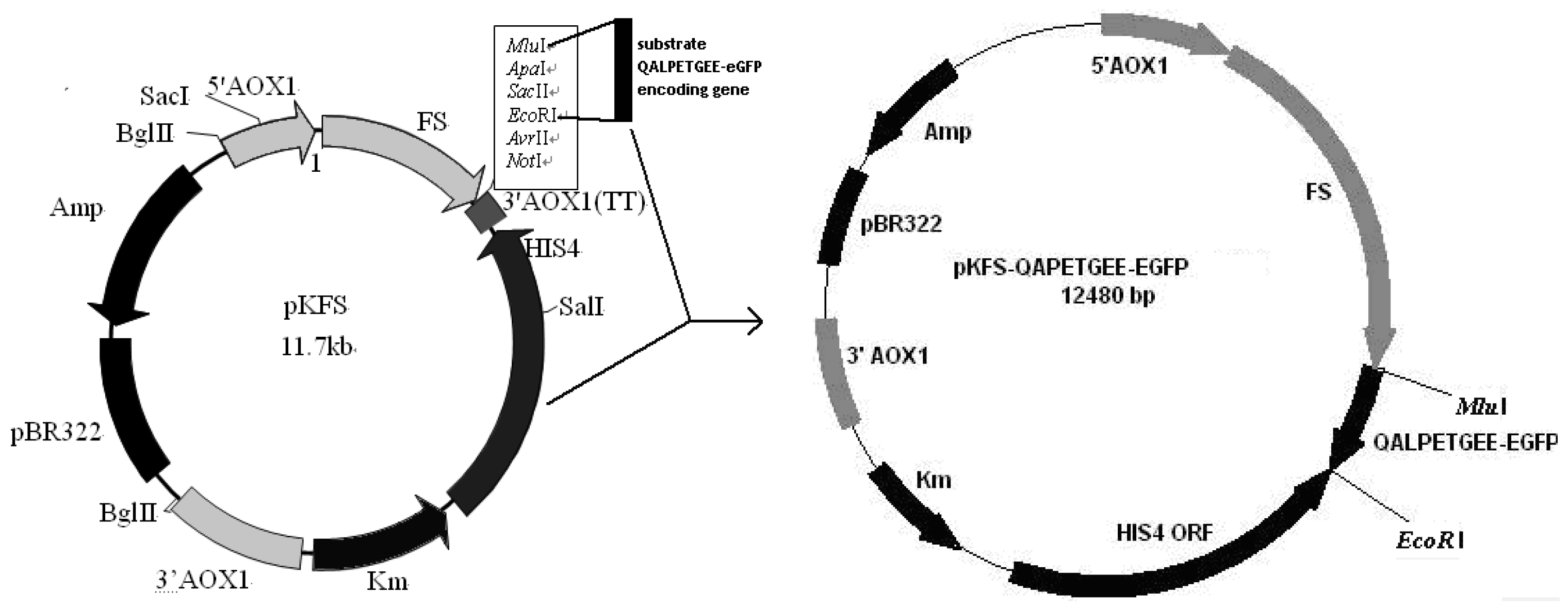
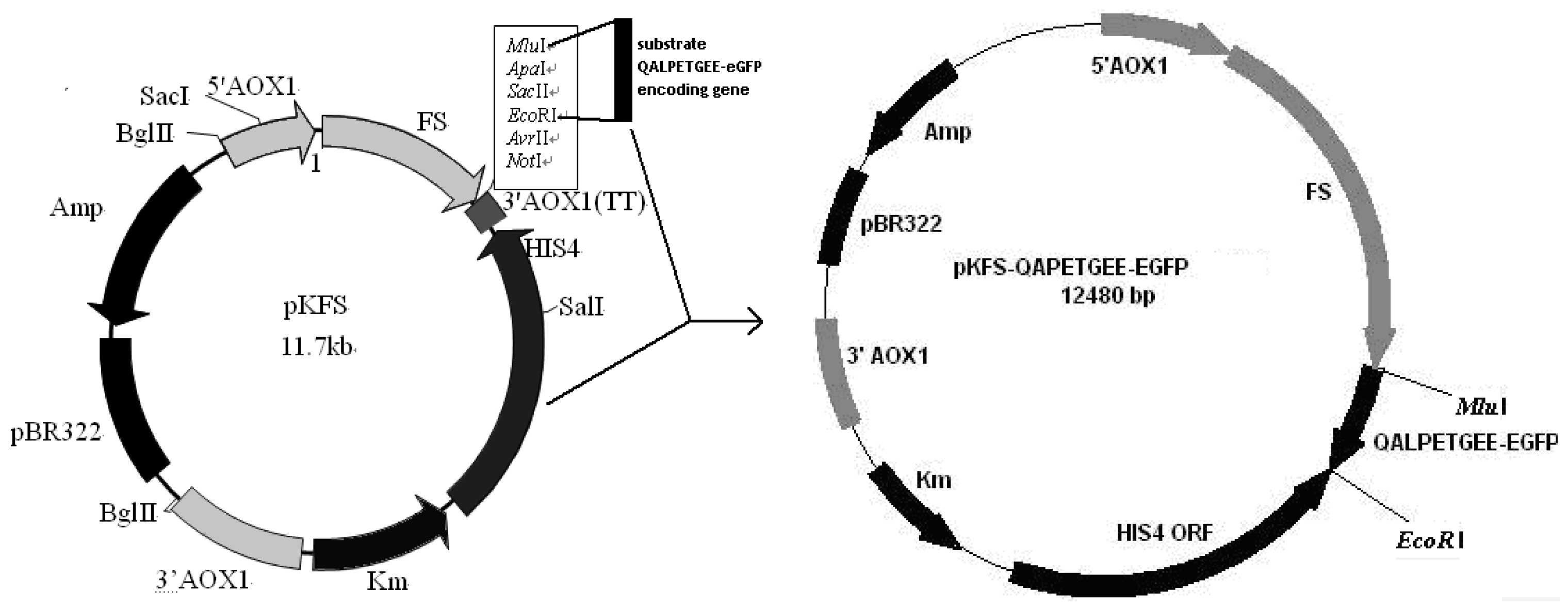
2.3. Construction of pKFS/LPETG Vector and Transformation
2.4. Yeast Culture
2.5. Fluorescence Assay
3. Results
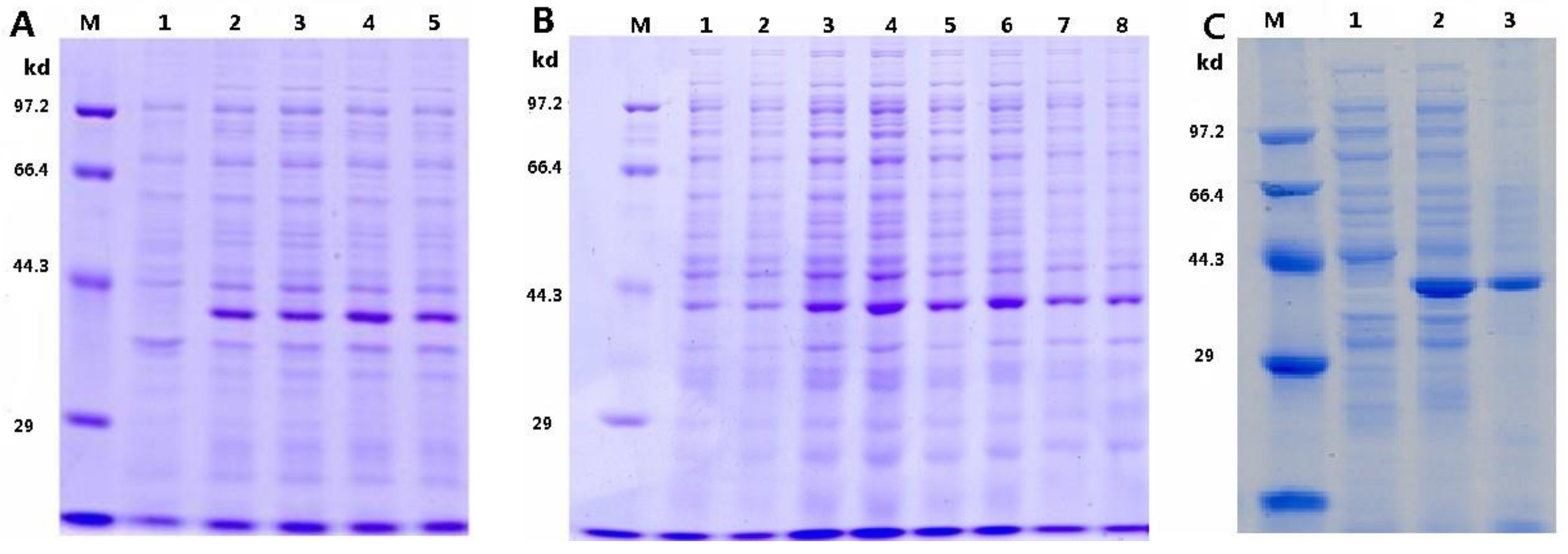
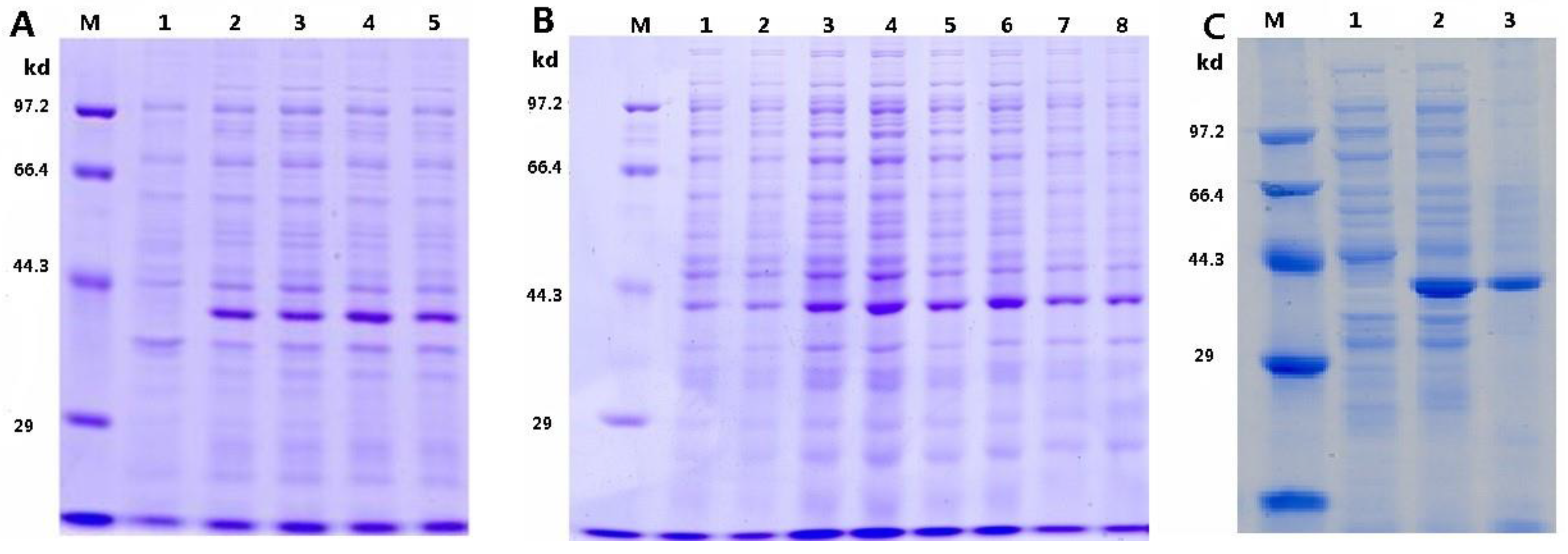
3.1. Optimization of pTRX-srtA Vector’s Expression
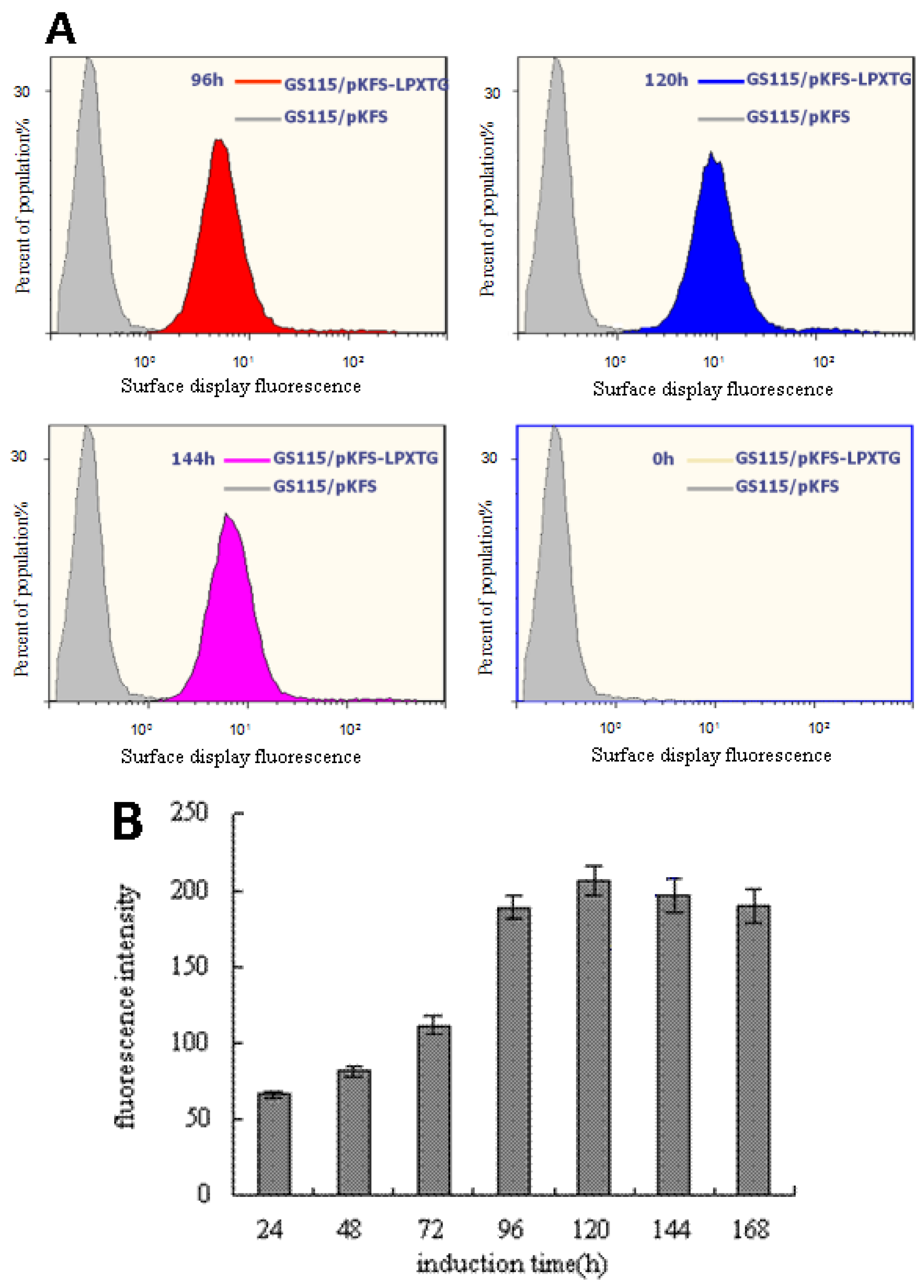
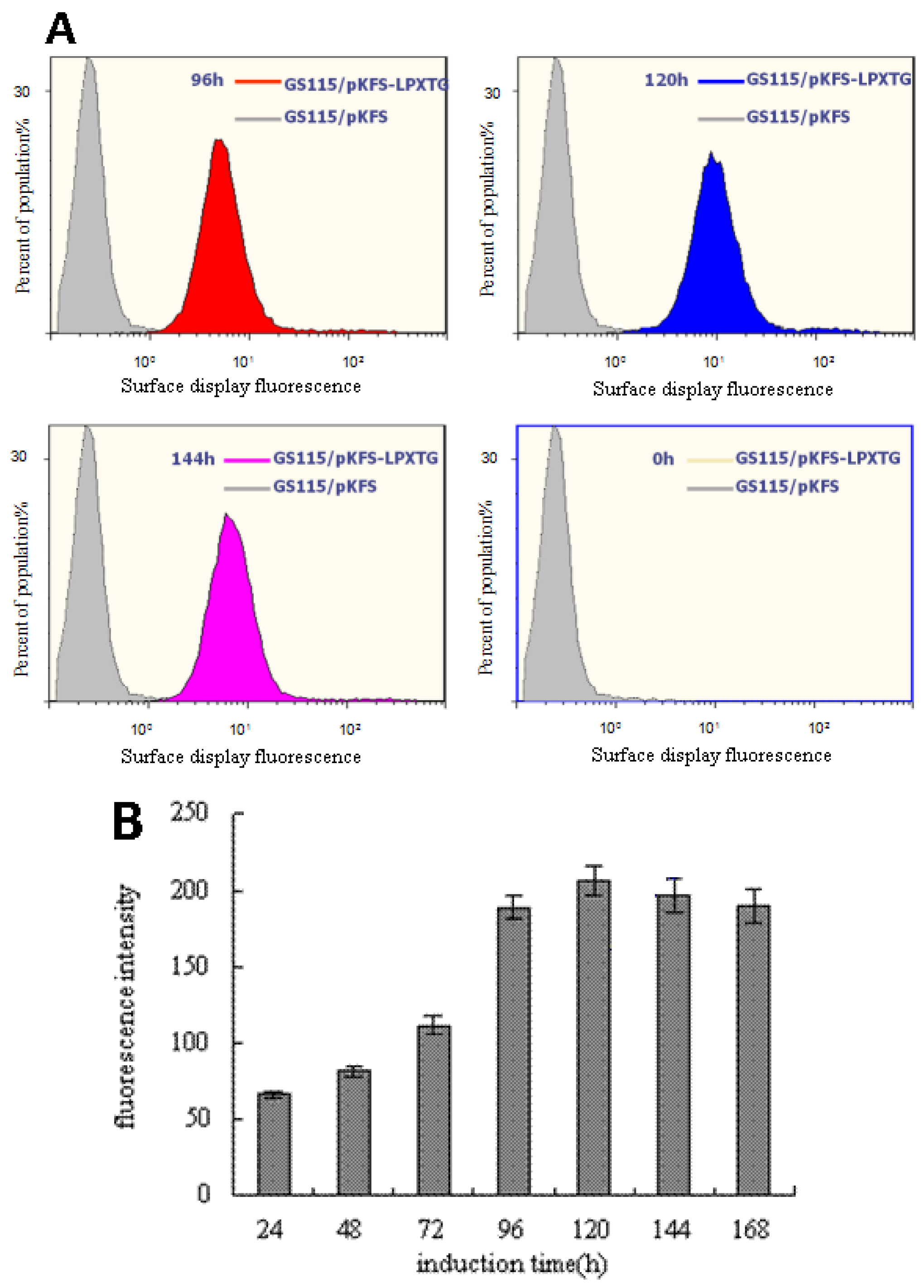
3.2. Analysis of the EGFP Protein Expressed on Cell Surface of P. pastoris
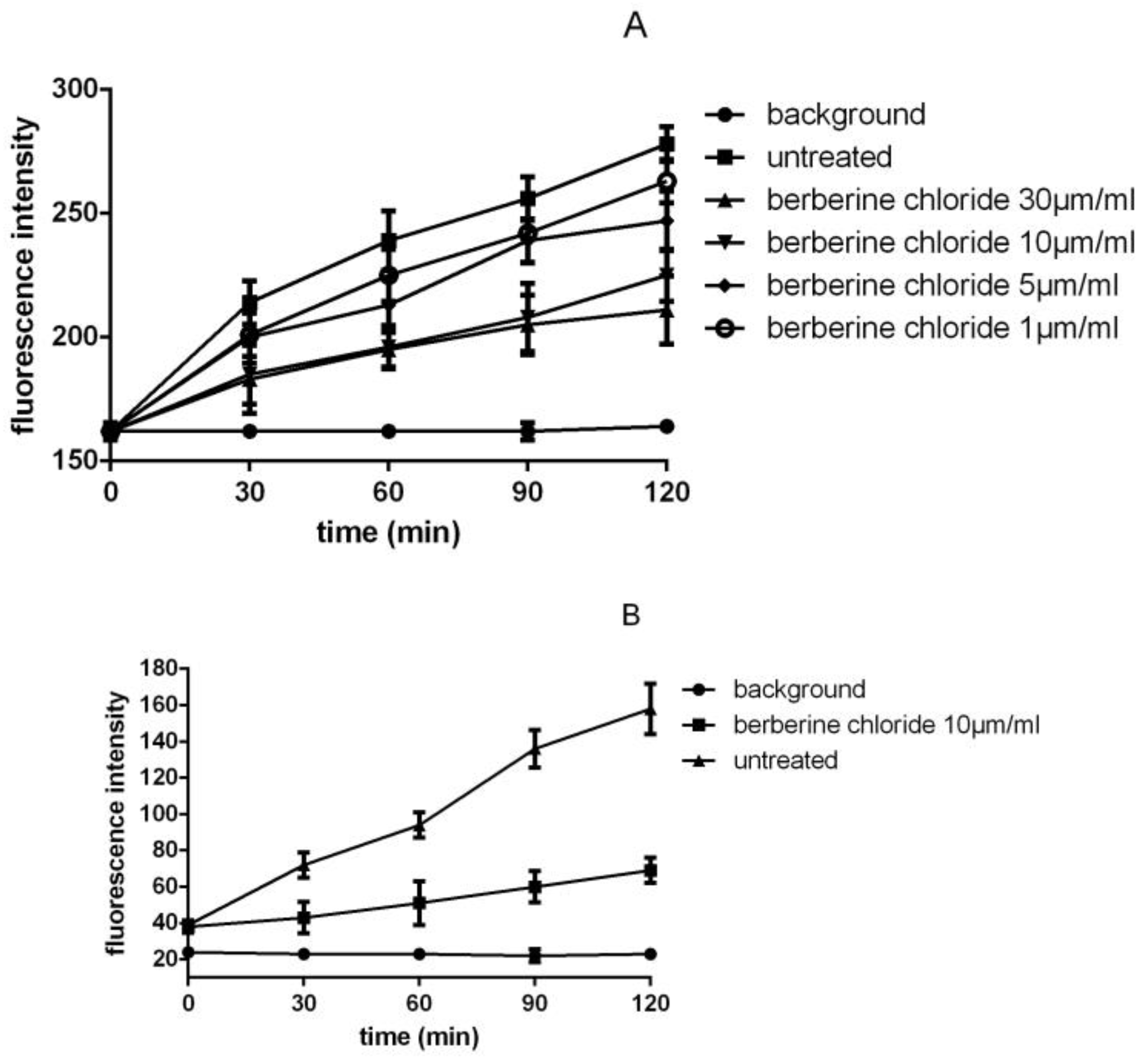
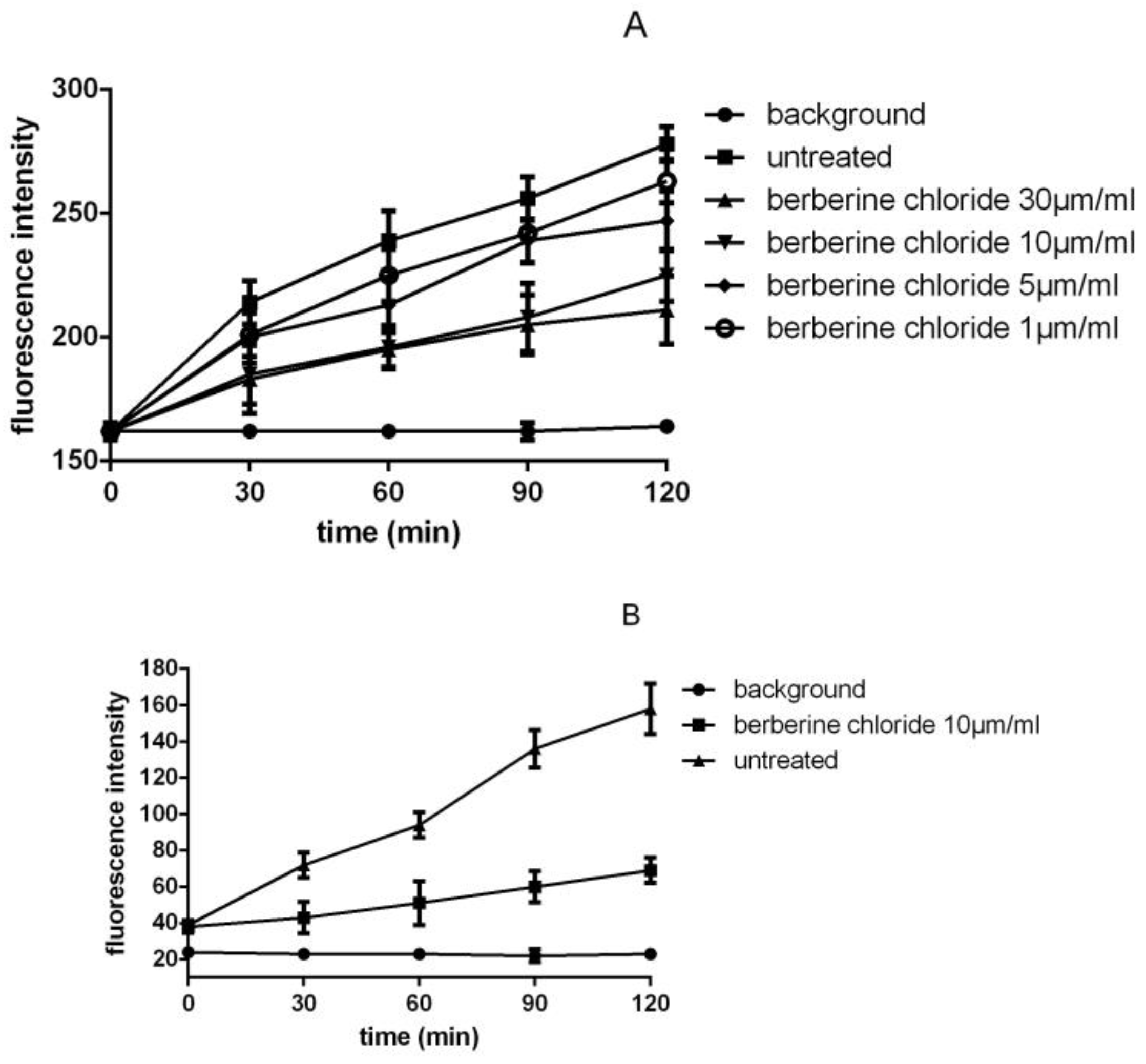
3.3. Detection of SrtA Activity
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. MMBR 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Totsika, M.; Schillaci, D. Sortase A: An ideal target for anti-virulence drug development. Microb. Pathog. 2014, 77, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Dodson, K.W.; Pinkner, J.S.; Rose, T.; Magnusson, G.; Hultgren, S.J.; Waksman, G. Structural basis of the interaction of the pyelonephritic E. coli adhesin to its human kidney receptor. Cell 2001, 105, 733–743. [Google Scholar] [CrossRef]
- Allen, R.C.; Popat, R.; Diggle, S.P.; Brown, S.P. Targeting virulence: Can we make evolution-proof drugs? Nat. Rev. Microbiol. 2014, 12, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Schneewind, O.; Missiakas, D. Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochim. Biophys. Acta 2013, 1843, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Dhar, G.; Faull, K.F.; Schneewind, O. Anchor structure of cell wall surface proteins in Listeria monocytogenes. Biochemistry (Mosc.) 2000, 39, 3725–3733. [Google Scholar] [CrossRef]
- Oh, K.B.; Kim, S.H.; Lee, J.; Cho, W.J.; Lee, T.; Kim, S. Discovery of diarylacrylonitriles as a novel series of small molecule sortase A inhibitors. J. Med. Chem. 2004, 47, 2418–2421. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.-X.; Eriksson, L.A. Catalytic mechanism and roles of Arg197 and Thr183 in the Staphylococcus aureus sortase A enzyme. J. Phys. Chem. B 2011, 115, 13003–13011. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-Y.; Won, T.H.; Ahn, C.-H.; Lee, S.-H.; Yang, H.-C.; Shin, J.; Oh, K.-B. Streptococcus mutans sortase A inhibitory metabolites from the flowers of sophora japonica. Bioorg. Med. Chem. Lett. 2015, 25, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.B.; Mar, W.; Kim, S.; Kim, J.Y.; Oh, M.N.; Kim, J.G.; Shin, D.; Sim, C.J.; Shin, J. Bis(indole) alkaloids as sortase A inhibitors from the sponge Spongosorites sp. Bioorg. Med. Chem. Lett. 2005, 15, 4927–4931. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.; Yang, W.-Y.; Chung, S.-C.; Kim, T.-Y.; Oh, K.-B.; Shin, J. In vitro sortase A inhibitory and antimicrobial activity of flavonoids isolated from the roots of Sophora flavescens. Arch. Pharm. Res. 2011, 34, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Somoza, J.R.; Palmer, J.T.; Ho, J.D. The crystal structure of human cathepsin F and its implications for the development of novel immunomodulators. J. Mol. Biol. 2002, 322, 559–568. [Google Scholar] [CrossRef]
- Kruger, R.G.; Barkallah, S.; Frankel, B.A.; McCafferty, D.G. Inhibition of the Staphylococcus aureus sortase transpeptidase SrtA by phosphinic peptidomimetics. Bioorg. Med. Chem. 2004, 12, 3723–3729. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Clemens, J.J.; Suree, N.; Liew, C.K.; Pilpa, R.; Campbell, D.O. Synthesis of (2R,3S) 3-amino-4-mercapto-2-butanol, a threonine analogue for covalent inhibition of sortases. Bioorg. Med. Chem. Lett. 2005, 15, 5076–5079. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Raffa, D.; Maggio, B.; Raimondi, M.V.; Schillaci, D.; Daidone, G. Sortase A Inhibitors: Recent advances and future perspectives. J. Med. Chem. 2015, 58, 9108–9123. [Google Scholar] [CrossRef] [PubMed]
- Aulabaugh, A.; Ding, W.; Kapoor, B.; Tabei, K.; Alksne, L. Development of an HPLC assay for Staphylococcus aureus sortase: Evidence for the formation of the kinetically competent acyl enzyme intermediate. Anal. Biochem. 2007, 360, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Kruger, R.G.; Dostal, P.; McCafferty, D.G. Development of a high-performance liquid chromatography assay and revision of kinetic parameters for the Staphylococcus aureus sortase transpeptidase SrtA. Anal. Biochem. 2004, 326, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Bice, T.W.; Ton-That, H.; Schneewind, O.; Narayana, S.V.L. Crystal strutures of Staphylococcus aureus sortase A and its substrate complex. J. Biol. Chem. 2004, 279, 31383–31389. [Google Scholar] [CrossRef] [PubMed]
- Chenna, B.C.; Shinkre, B.A.; King, J.R.; Lucius, A.L.; Narayana, S.V.L.; Velu, S.E. Identification of novel inhibitors of bacterial surface enzyme Staphylococcus aureus Sortase A. Bioorg. Med. Chem. Lett. 2008, 18, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Choi, J.H.; Xu, A. Microbial cell-surface display. Trends Biotechnol. 2003, 21, 45–52. [Google Scholar]
- Wernérus, H.; Lehtiö, J.; Samuelson, P.; Ståhl, S. Engineering of staphylococcal surfaces for biotechnological applications. J. Biotechnol. 2002, 96, 67–78. [Google Scholar] [CrossRef]
- Oh, K.B.; Oh, M.N.; Kim, J.G.; Shin, D.S.; Shin, J. Inhibition of sortase-mediated Staphylococcus aureus adhesion to fibronectin via fibronectin-binding protein by sortase inhibitors. Appl. Microbiol. Biotechnol. 2006, 70, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Shin, D.S.; Oh, M.N.; Chung, S.C.; Lee, J.S.; Oh, K.B. Inhibition of the bacterial surface protein anchoring transpeptidase sortase by isoquinoline alkaloids. Biosci. Biotechnol. Biochem. 2004, 68, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Tanino, T.; Fukuda, H.; Kondo, A. Construction of a Pichia pastoris cell-surface display system using Flo1p anchor system. Biotechnol. Prog. 2006, 22, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Gai, S.A.; Wittrup, K.D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struct. Biol. 2007, 17, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Jiang, B.; Chen, M. Cloning and expression of the novelm antimicrobial target enzyme Sortase gene in two prokaryotic vectors. Acta Microbial. Sin. 2009, 49, 186–190. (In Chinese) [Google Scholar]
- Han, S.; Han, Z.; Lin, Y.; Zheng, S. Construction of high efficiency pichia pastoris surface display system based on Flo1 protein. Prog. Biochem. Biophys. 2010, 37, 200–207. [Google Scholar] [CrossRef]
- Luo, L.; Wu, L.; Lin, Y. Sortase analysis by displaying its substrates on yeast surface. Acta Microbial. Sin. 2009, 49, 1534–1539. (In Chinese) [Google Scholar]
- Budzik, J.M.; Oh, S.Y.; Schneewind, O. Sortase D forms the covalent bond that links BcpB to the tip of Bacillus cereus pili. J. Biol. Chem. 2009, 284, 12989–12997. [Google Scholar] [CrossRef] [PubMed]
- Lowy, F.D. Antimicrobial resistance: The example of Staphylococcus aureus. J. Clin. Investig. 2003, 111, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Aulabaugh, A.; Ding, W.; Kapoor, B.; Alksne, L.; Tabei, K.; Ellestad, G. Kinetic mechanism of Staphylococcus aureus Sortase Srt A. Biochemistry 2003, 42, 11307–11315. [Google Scholar] [CrossRef]
- Oh, K.B.; Nam, K.W.; Ahn, H.; Shin, J.; Kim, S.; Mar, W. Therapeutic effect of (Z)-3-(2,5-dimethoxyphenyl)-2-(4-methoxyphenyl) acrylonitrile (DMMA) against Staphylococcus aureus infection in a murine model. Biochem. Biophys. Res. Commun. 2010, 396, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.M.; Ton-That, H.; Mazmanian, S.K.; Schneewind, O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. J. Biol. Chem. 2002, 277, 16241–16248. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.; Li, H.; Tang, T. A Novel Yeast Surface Display Method for Large-Scale Screen Inhibitors of Sortase A. Bioengineering 2017, 4, 6. https://doi.org/10.3390/bioengineering4010006
Wu L, Li H, Tang T. A Novel Yeast Surface Display Method for Large-Scale Screen Inhibitors of Sortase A. Bioengineering. 2017; 4(1):6. https://doi.org/10.3390/bioengineering4010006
Chicago/Turabian StyleWu, Lin, Huijun Li, and Tianle Tang. 2017. "A Novel Yeast Surface Display Method for Large-Scale Screen Inhibitors of Sortase A" Bioengineering 4, no. 1: 6. https://doi.org/10.3390/bioengineering4010006