
Development of Antibody–Drug Conjugates Using DDS and Molecular Imaging

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
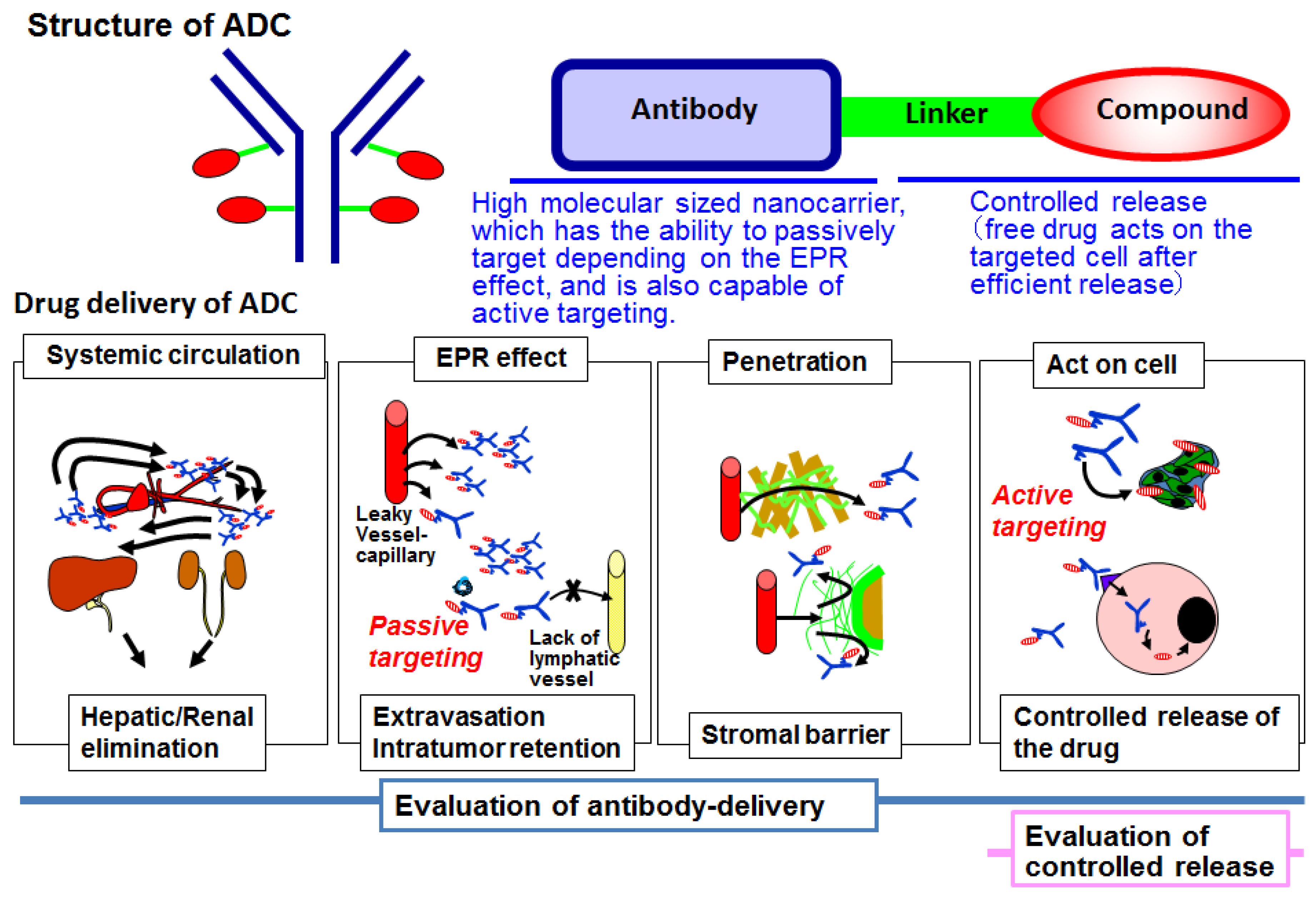
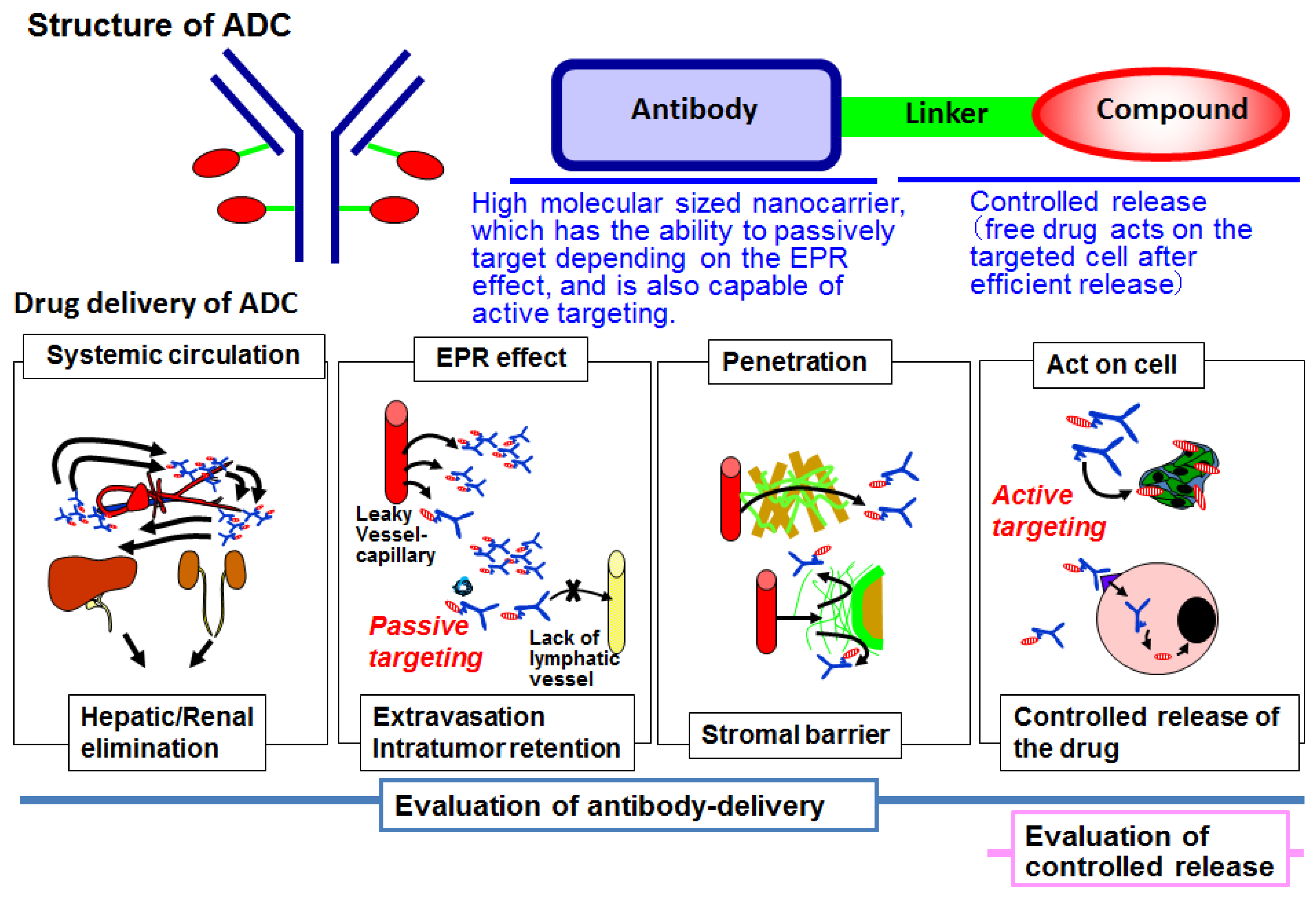
2. Antibody–Drug Conjugate
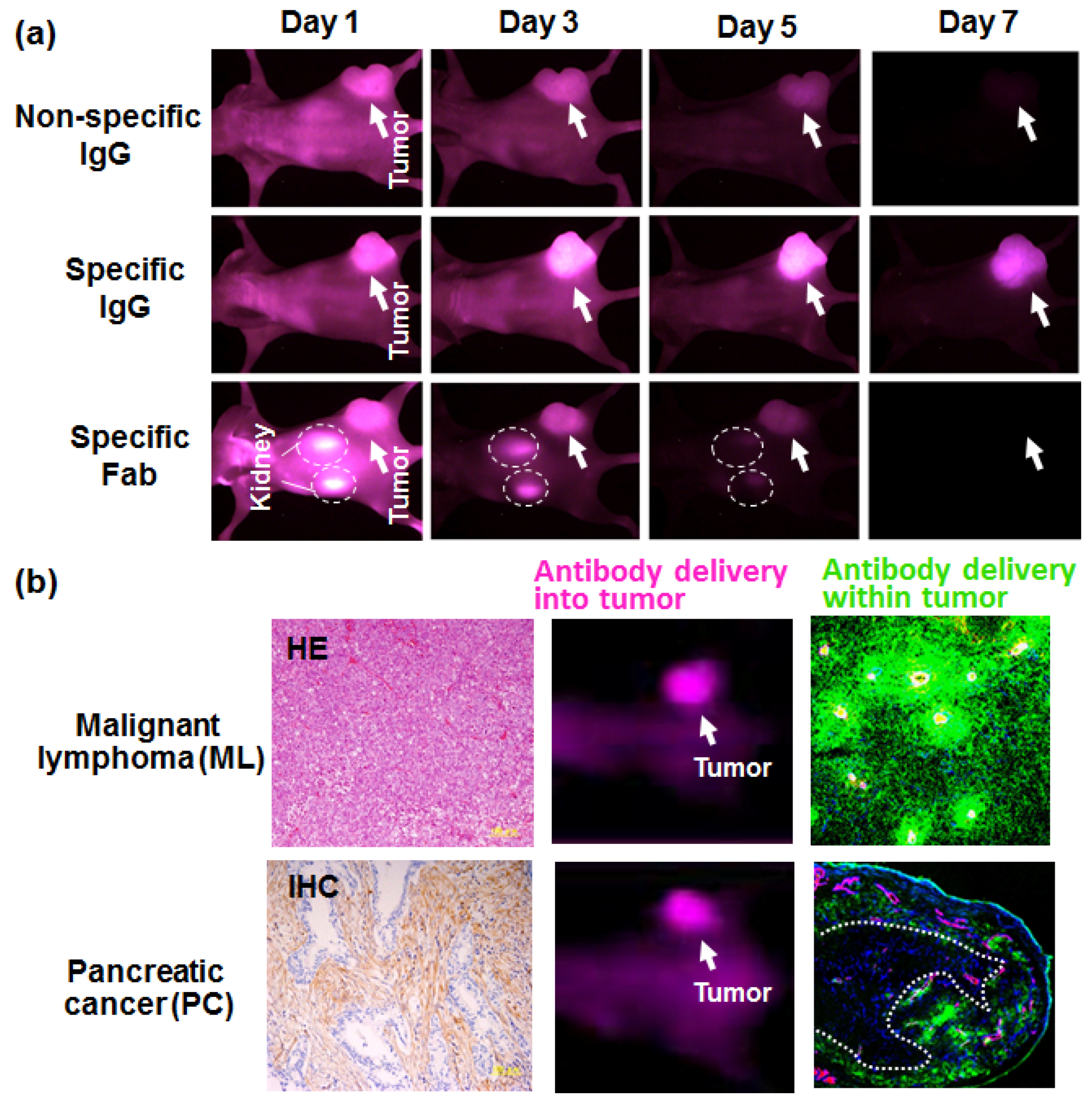
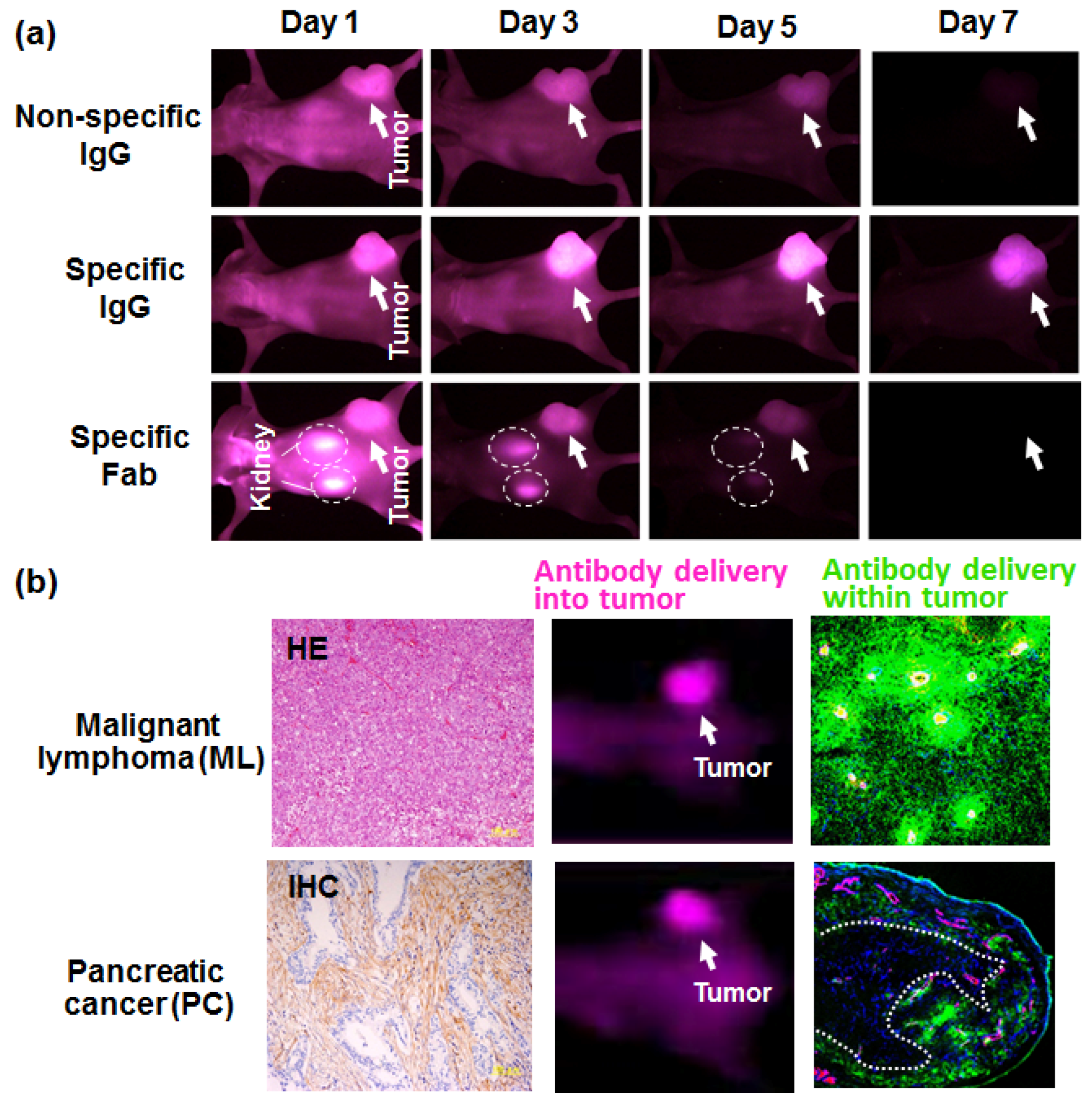
3. Antibody Delivery and the EPR Effect
4. Antibody Delivery and Tissue Penetration
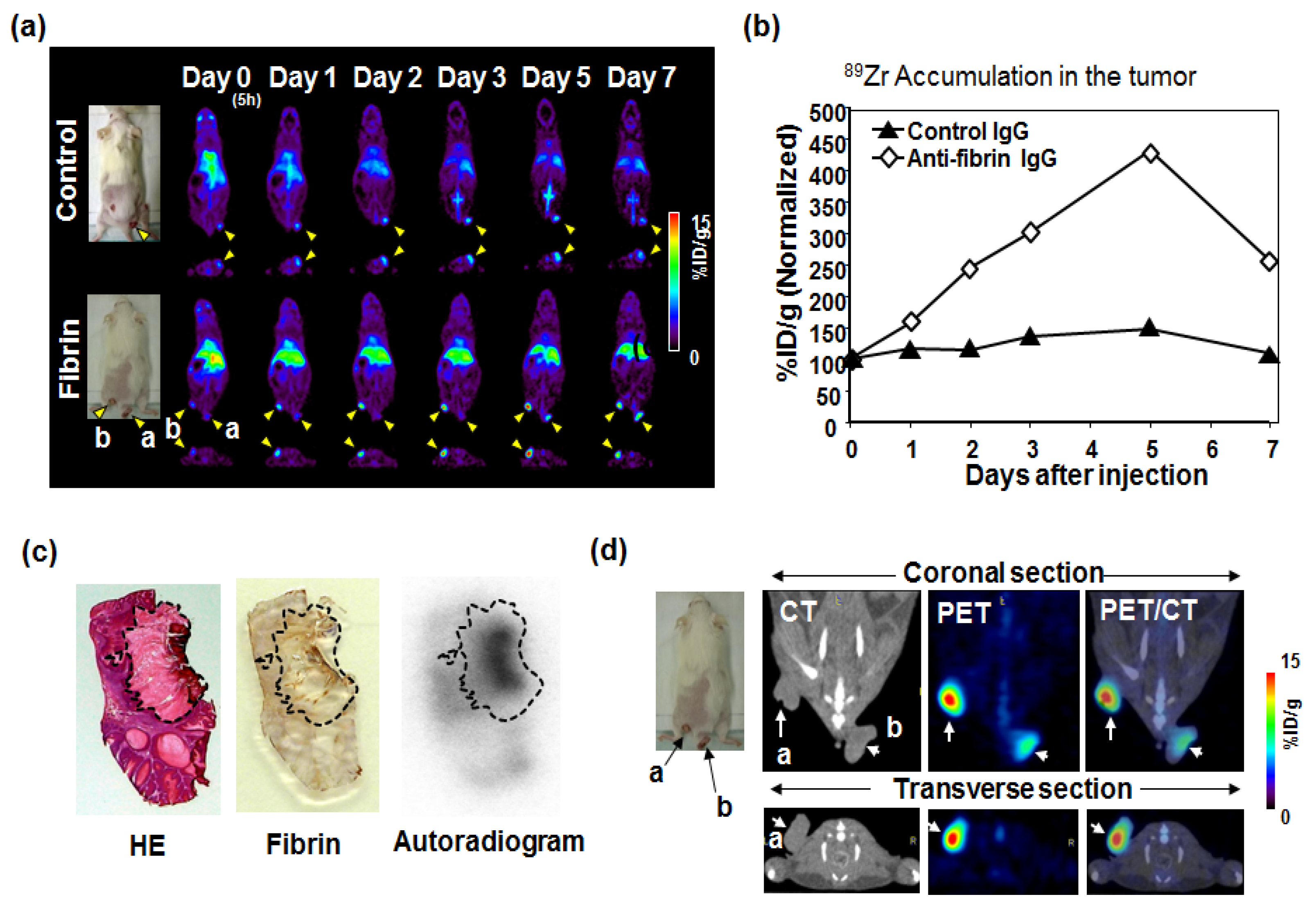
5. Immuno-PET Imaging
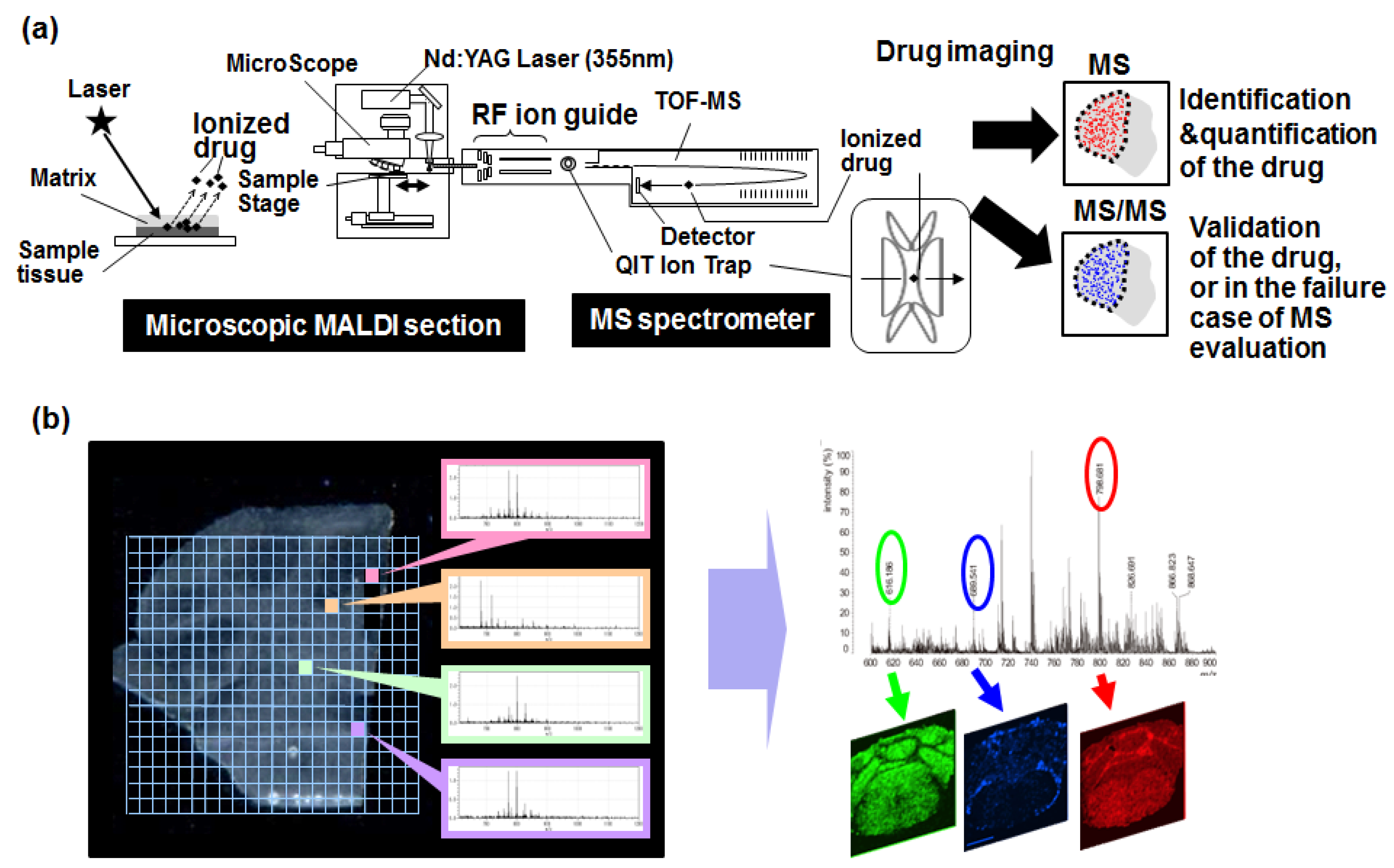
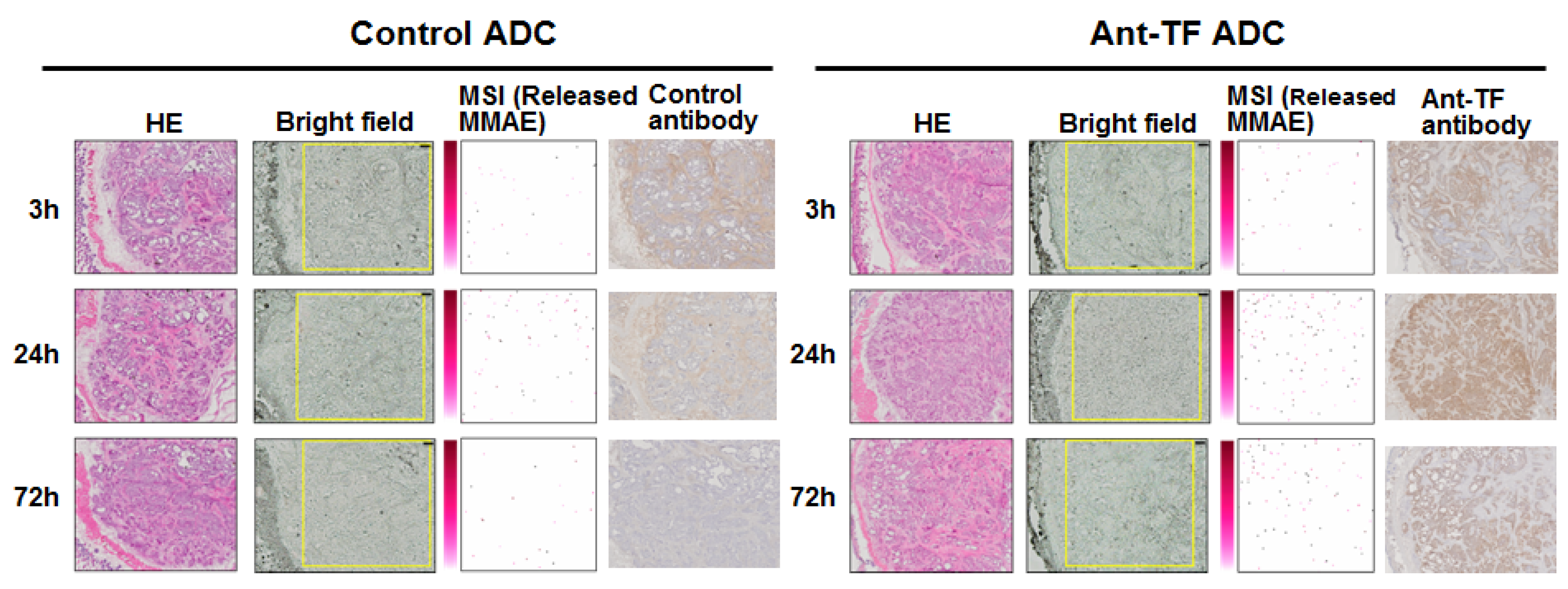
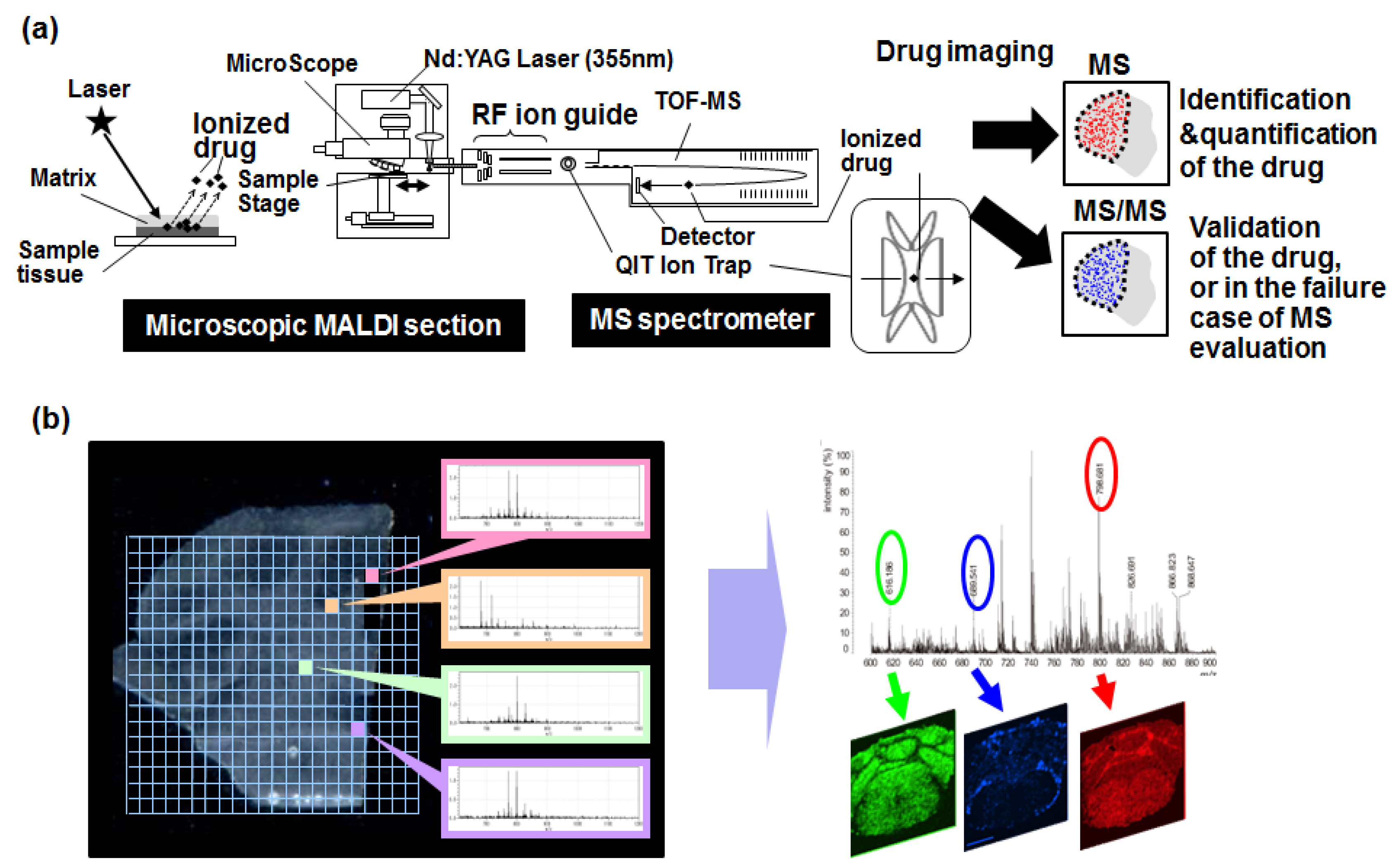
6. Mass Spectrometry Imaging
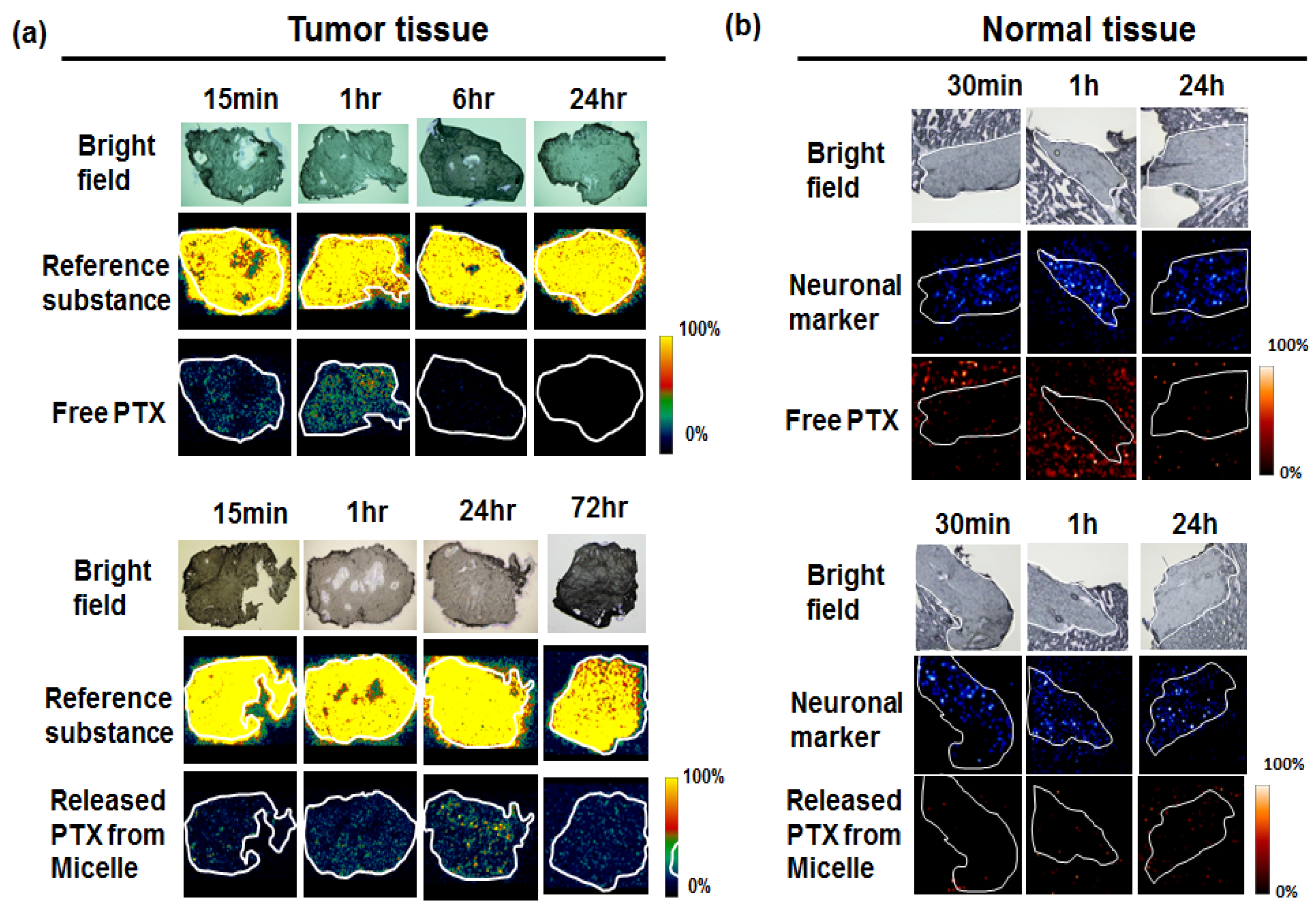
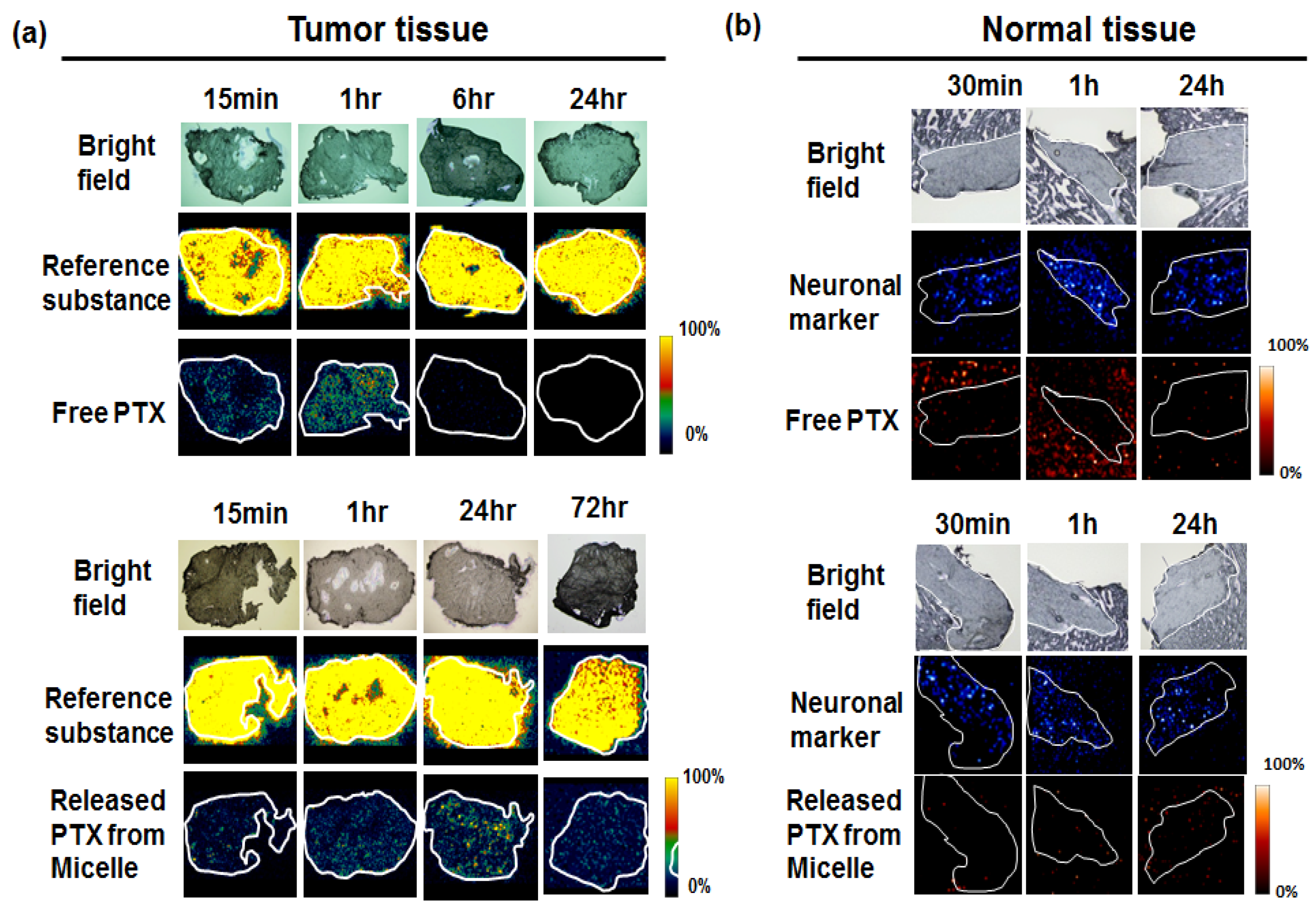
7. Visualization of the Controlled Release
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody-drug conjugates for cancer therapy. Lancet. Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—an emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Mack, F.; Ritchie, M.; Sapra, P. The next generation of antibody drug conjugates. Semin. Oncol. 2014, 41, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Damelin, M.; Zhong, W.; Myers, J.; Sapra, P. Evolving strategies for target selection for antibody-drug conjugates. Pharm. Res. 2015, 32, 3494–3507. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. Ds-8201a, a novel her2-targeting adc with a novel DNA topoisomerase i inhibitor, demonstrates a promising antitumor efficacy with differentiation from t-dm1. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a pivotal phase ii study of brentuximab vedotin for patients with relapsed or refractory hodgkin's lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (sgn-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase ii study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [PubMed]
- Sau, S.; Alsaab, H.O.; Kashaw, S.K.; Tatiparti, K.; Iyer, A.K. Advances in antibody-drug conjugates: A new era of targeted cancer therapy. Drug Discov. Today 2017. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for her2-positive advanced breast cancer. New Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Matsumura, Y. The drug discovery by nanomedicine and its clinical experience. Jpn. J. Clin. Oncol. 2014, 44, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.P.; Weiner, L.M. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005, 23, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. Pd-1 and pd-l1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; Sapra, P.; Loganzo, F.; May, C. Combining antibody-drug conjugates and immune-mediated cancer therapy: What to expect? Biochem. Pharmacol. 2016, 102, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Kimura, M.; Yamamoto, T.; Maeda, H. Involvement of the kinin-generating cascade in enhanced vascular permeability in tumor tissue. Jpn. J. Cancer Res. Gann 1988, 79, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Steichen, S.D.; Caldorera-Moore, M.; Peppas, N.A. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2013, 48, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release Off. J. Control. Release Soc. 2011, 153, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Manchanda, P.; Vogelbaum, M.A.; Ohlfest, J.R.; Elmquist, W.F. Function of the blood-brain barrier and restriction of drug delivery to invasive glioma cells: Findings in an orthotopic rat xenograft model of glioma. Drug Metabol. Dispos. Biol. Fate Chem. 2013, 41, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A.; Yu, K.H.; O'Reilly, E.M. Pancreas adenocarcinoma: Novel therapeutics. Chin. Clin. Oncol. 2017, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, M.; Manabe, S.; Tarin, D.; Matsumura, Y. Cancer-stroma targeting therapy by cytotoxic immunoconjugate bound to the collagen 4 network in the tumor tissue. Bioconjugate Chem. 2011, 22, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, M.; Manabe, S.; Tarin, D.; Matsumura, Y. Tailored immunoconjugate therapy depending on a quantity of tumor stroma. Cancer Sci. 2013, 104, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. National Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, M.; Manabe, S.; Matsumura, Y. New concept of cytotoxic immunoconjugate therapy targeting cancer-induced fibrin clots. Cancer Sci. 2011, 102, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Manabe, S.; Aihara, Y.; Sato, R.; Tsumura, R.; Iwafuji, H.; Furuya, F.; Fuchigami, H.; Fujiwara, Y.; Hisada, Y.; et al. Antitumor effect of antitissue factor antibody-mmae conjugate in human pancreatic tumor xenografts. Int. J. Cancer. J. Int. Du Cancer 2015, 137, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y. Cancer stromal targeting (cast) therapy. Adv. Drug Deliv. Rev. 2012, 64, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Yasunaga, M.; Hanaoka, S.; Saijou, S.; Sugino, T.; Tsuji, A.; Saga, T.; Tsumoto, K.; Manabe, S.; Kuroda, J.; et al. Discovery of an uncovered region in fibrin clots and its clinical significance. Sci. Rep. 2013, 3, 2604. [Google Scholar] [CrossRef] [PubMed]
- Bahce, I.; Yaqub, M.; Smit, E.F.; Lammertsma, A.A.; van Dongen, G.A.; Hendrikse, N.H. Personalizing nsclc therapy by characterizing tumors using tki-pet and immuno-pet. Lung Cancer Amst. Neth. 2017, 107, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Verel, I.; Visser, G.W.; Boellaard, R.; Stigter-van Walsum, M.; Snow, G.B.; van Dongen, G.A. 89zr immuno-pet: Comprehensive procedures for the production of 89zr-labeled monoclonal antibodies. J. Nucl. Med. Off. Publ. Society Nucl. Med. 2003, 44, 1271–1281. [Google Scholar]
- Wadas, T.J.; Wong, E.H.; Weisman, G.R.; Anderson, C.J. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for pet and spect imaging of disease. Chem. Rev. 2010, 110, 2858–2902. [Google Scholar] [CrossRef] [PubMed]
- Jauw, Y.W.; Menke-van der Houven van Oordt, C.W.; Hoekstra, O.S.; Hendrikse, N.H.; Vugts, D.J.; Zijlstra, J.M.; Huisman, M.C.; van Dongen, G.A. Immuno-positron emission tomography with zirconium-89-labeled monoclonal antibodies in oncology: What can we learn from initial clinical trials? Front. Pharmacol. 2016, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Dijkers, E.C.; Kosterink, J.G.; Rademaker, A.P.; Perk, L.R.; van Dongen, G.A.; Bart, J.; de Jong, J.R.; de Vries, E.G.; Lub-de Hooge, M.N. Development and characterization of clinical-grade 89zr-trastuzumab for her2/neu immunopet imaging. J. Nucl. Med. Off. Publ. Society Nucl. Med. 2009, 50, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Pichler, B.J.; Kolb, A.; Nagele, T.; Schlemmer, H.P. Pet/mri: Paving the way for the next generation of clinical multimodality imaging applications. J. Nuc. Med. Off. Publ. Society Nucl. Med. 2010, 51, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Clery, P.F.; Faivre-Chauvet, A.; Bourgeois, M.; Guerard, F.; Haddad, F.; Barbet, J.; Cherel, M.; Kraeber-Bodere, F.; Carlier, T.; et al. Immuno-pet for clinical theranostic approaches. Int. J. Mol. Sci. 2016, 18, 57. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, H.; Shimizu, C.; Miyakita, Y.; Yoshida, M.; Hamada, A.; Kanayama, Y.; Yonemori, K.; Hashimoto, J.; Tani, H.; Kodaira, M.; et al. Molecular imaging using pet for breast cancer. Breast Cancer Tokyo Jpn. 2016, 23, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Gebhart, G.; Lamberts, L.E.; Wimana, Z.; Garcia, C.; Emonts, P.; Ameye, L.; Stroobants, S.; Huizing, M.; Aftimos, P.; Tol, J.; et al. Molecular imaging as a tool to investigate heterogeneity of advanced her2-positive breast cancer and to predict patient outcome under trastuzumab emtansine (t-dm1): The zephir trial. Ann. Oncol. Off. J. Eur. Society Med. Oncol. 2016, 27, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, H.; Hamada, A.; Yoshida, M.; Shimma, S.; Hashimoto, J.; Yonemori, K.; Tani, H.; Miyakita, Y.; Kanayama, Y.; Wada, Y.; et al. (64)cu-dota-trastuzumab pet imaging and her2 specificity of brain metastases in her2-positive breast cancer patients. EJNMMI Res. 2015, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Cornett, D.S.; Reyzer, M.L.; Chaurand, P.; Caprioli, R.M. Maldi imaging mass spectrometry: Molecular snapshots of biochemical systems. Nat. Methods 2007, 4, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Rompp, A.; Spengler, B. Mass spectrometry imaging with high resolution in mass and space. Histochem. Cell Biol. 2013, 139, 759–783. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Dill, A.L.; Eberlin, L.S.; Cooks, R.G.; Ifa, D.R. Mass spectrometry imaging under ambient conditions. Mass Spectrom. Rev. 2013, 32, 218–243. [Google Scholar] [CrossRef] [PubMed]
- Levenson, R.M.; Mansfield, J.R. Multispectral imaging in biology and medicine: Slices of life. Cytom. Part A J. Int. Society Anal. Cytol. 2006, 69, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, D.; Feldman, D.R.; Norton, I.; Brastianos, P.K.; Dunn, I.F.; Santagata, S.; Agar, N.Y. Molecular typing of meningiomas by desorption electrospray ionization mass spectrometry imaging for surgical decision-making. Interna. J. Mass Spectrom. 2015, 377, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, D.; Caragacianu, D.; Liu, X.; Norton, I.; Thompson, C.J.; Richardson, A.L.; Golshan, M.; Easterling, M.L.; Santagata, S.; Dillon, D.A.; et al. Application of desorption electrospray ionization mass spectrometry imaging in breast cancer margin analysis. Proc. National Acad. Sci. U.S.A. 2014, 111, 15184–15189. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, M.; Furuta, M.; Ogata, K.; Koga, Y.; Yamamoto, Y.; Takigahira, M.; Matsumura, Y. The significance of microscopic mass spectrometry with high resolution in the visualisation of drug distribution. Sci. Rep. 2013, 3, 3050. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Furuta, M.; Manabe, S.; Koga, Y.; Yasunaga, M.; Matsumura, Y. Imaging mass spectrometry for the precise design of antibody-drug conjugates. Sci. Rep. 2016, 6, 24954. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Waki, M.; Hameed, S.; Hayasaka, T.; Setou, M. Development of imaging mass spectrometry. Biol. Pharm. Bull. 2012, 35, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, Y.; Honda, K.; Suematsu, M. Development of an imaging mass spectrometry technique for visualizing localized cellular signaling mediators in tissues. Mass Spectrom. Tokyo Jpn. 2015, 4, A0040. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Yuba-Kubo, A.; Sugiura, Y.; Zaima, N.; Hayasaka, T.; Goto-Inoue, N.; Wakui, M.; Suematsu, M.; Takeshita, K.; Ogawa, K.; et al. Visualization of volatile substances in different organelles with an atmospheric-pressure mass microscope. Anal. Chem. 2009, 81, 9153–9157. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Matsumura, Y.; Suzuki, M.; Shimizu, K.; Goda, R.; Nakamura, I.; Nakatomi, I.; Yokoyama, M.; Kataoka, K.; Kakizoe, T. Nk105, a paclitaxel-incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br. J. Cancer 2005, 92, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, M.; Manabe, S.; Matsumura, Y. Immunoregulation by il-7r-targeting antibody-drug conjugates: Overcoming steroid-resistance in cancer and autoimmune disease. Sci. Rep. 2017, 7, 10735. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasunaga, M.; Manabe, S.; Tsuji, A.; Furuta, M.; Ogata, K.; Koga, Y.; Saga, T.; Matsumura, Y. Development of Antibody–Drug Conjugates Using DDS and Molecular Imaging. Bioengineering 2017, 4, 78. https://doi.org/10.3390/bioengineering4030078
Yasunaga M, Manabe S, Tsuji A, Furuta M, Ogata K, Koga Y, Saga T, Matsumura Y. Development of Antibody–Drug Conjugates Using DDS and Molecular Imaging. Bioengineering. 2017; 4(3):78. https://doi.org/10.3390/bioengineering4030078
Chicago/Turabian StyleYasunaga, Masahiro, Shino Manabe, Atsushi Tsuji, Masaru Furuta, Koretsugu Ogata, Yoshikatsu Koga, Tsuneo Saga, and Yasuhiro Matsumura. 2017. "Development of Antibody–Drug Conjugates Using DDS and Molecular Imaging" Bioengineering 4, no. 3: 78. https://doi.org/10.3390/bioengineering4030078