
Workflow for Criticality Assessment Applied in Biopharmaceutical Process Validation Stage 1


Abstract
:
1. Introduction
- Risk assessment: to identify potential influential/critical parameters for each unit operation. This is usually performed using tools such as failure mode and effect analysis (FMEA) [4,5]. Ranking of potential criticality is performed using expert knowledge, historical process data, and interdependencies identified in development data.
- Scale down model establishment: Due to the costs related to large-scale experiments, in biopharmaceutical manufacturing it is necessary to develop appropriate scale down models (SDMs) that are appropriate to investigate the interdependency between process parameters and quality attributes.
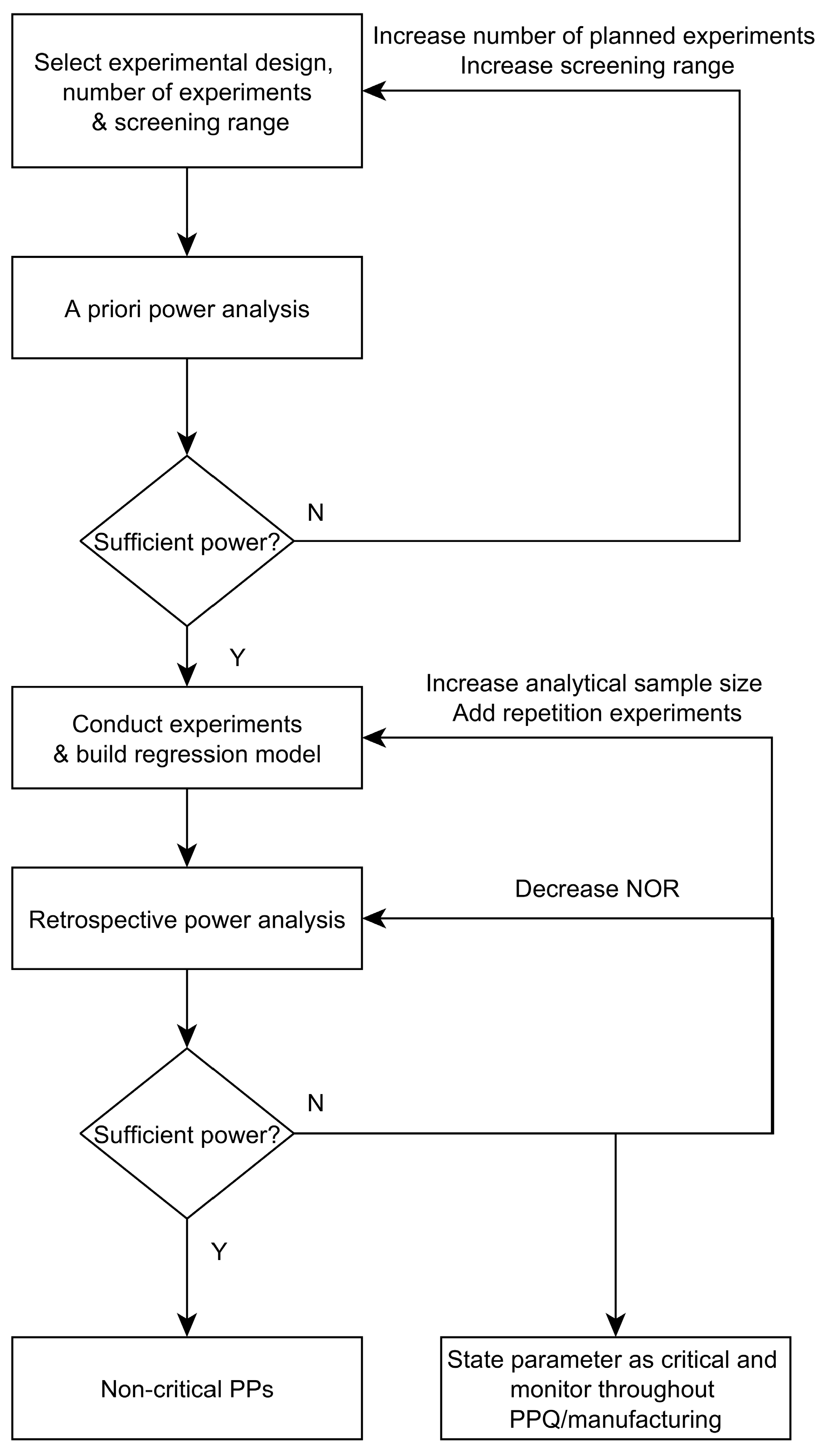
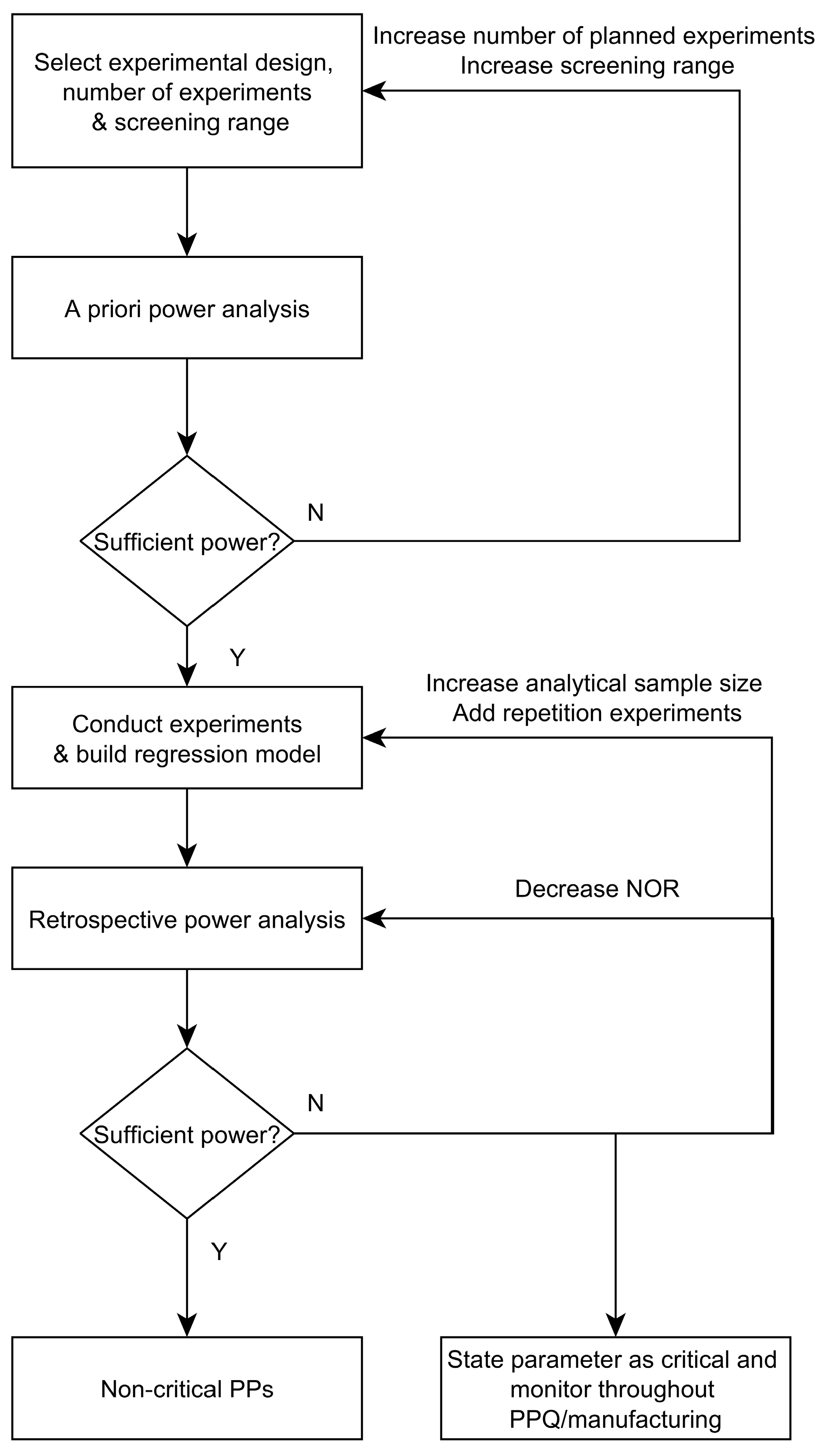
- Experimental designs: Design of Experiments are applied to quantify the impact of process parameters (PPs) on CQAs. Prior to conducting experiments, a priori power analysis is a good practice to evaluate if an effect that leads to a change in product quality—in the following defined as a critical effect—can be detected by the proposed design setting. Statistical power is defined as the probability that we are able to detect an effect if it is truly there [6]. This is done for a priori analysis by estimating the expected signal to noise ratio, which is thought to occur during the experiments [7]. As a result of this a priori power analysis, the number of required experiments, the intended screening range, or the design itself might be adjusted. After a sufficient power can be expected, potential influential/critical parameters are purposefully varied within experiments, which is done for each unit operation separately using the previously established SDMs.
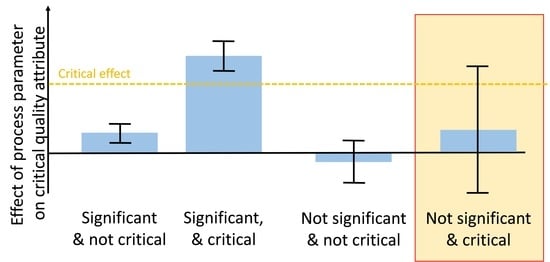
- Criticality assessment of process parameters by evaluating experimental designs: Identification of significant factors (rejection of the null hypothesis that the effect equals 0) at a desired significance level (typically α < 0.05) is performed using Pareto charts and analysis of significance of regression coefficients by means of ANOVA. Misleadingly, this does not imply that for non-significant factors the null hypothesis is true and their effect is zero [8]. Rather, it indicates that the uncertainty around these factors in the range examined—often indicated by large confidence intervals around the effect—is large and critical levels cannot be excluded. Commonly, only significant factors that have been observed to impact product quality or process performance are defined as critical or key, respectively. Those which cannot be stated as significantly impacting are stated as non-critical or non-key, respectively.
- Definition of control strategy: As a means to ensure all CQAs and quality specifications are met, a process control strategy for all critical and key process parameters must be put in place. Moreover, it has to be evaluated whether their mutual worst case setting would lead to acceptable product quality levels. Commonly for biopharmaceutical production, this is accomplished by setting normal operating ranges (NOR) and proven acceptable ranges (PAR).
- Establishment of a methodology that prevents engineers, during process validation, from overlooking critical parameters;
- Setting a control strategy for critical and likely overlooked parameters that ensures a robust process design;
- A workflow that can be used during stage 1 process validation to assess PP criticality. Applying those guidelines, it will be possible to better understand potential process variability and provide an opportunity to reduce process variability, OOS events, and patient risk.
2. Methods
2.1. Description of Process and Design of Conducted Experiments
2.2. Calculation of Thresholds for Critical Effects
2.3. A Priori Power Analysis
- Estimate the mean () and variance () of the response variable from small-scale or pilot-scale experiments at set point conditions of manufacturing. We assume that residual error in the model is only due to process- and analytical variance. The latter estimate will be used to calculate the expected sum of squares of the residuals ():
- For each of the combinations (c) described above, we calculate critical effects for each parameter using its weight :In order to estimate the individual coefficient for the i-th parameters, from a risk-based approach, we divide by the longest distance from the set-point () to the nearest NOR border: where is the upper boundary of the NOR and is the lower boundary of the NOR of the parameter i. Note that this works for a symmetric as well as asymmetric NOR.
- Using the design matrix , obtained for a specific experimental design, we can simulate possible values at the screening range using:
- From that, the total sum of squares can be estimated:Together with the sum of squares of the residuals, the expected coefficient of variance can be calculated:
- Using Cohen’s effect size (), the non-centrality parameter λ and the critical F value (), the a priori power for the combination c of effects that no parameter has been overlooked can be calculated [7]:
- Confidence intervals for the a priori power for the combination c were calculated according towhere is the percentile from a χ2 distribution with degrees of freedom.
- where is the non-central F distribution with (number of DoE parameters) and , where n is the number of observations in the DoE.
- The mean power over all combinations of effects was estimated as the arithmetic mean of all :
2.4. Evaluation of DoEs
3. Results and Discussion
3.1. Permutation Test for Retrospective Power Analysis
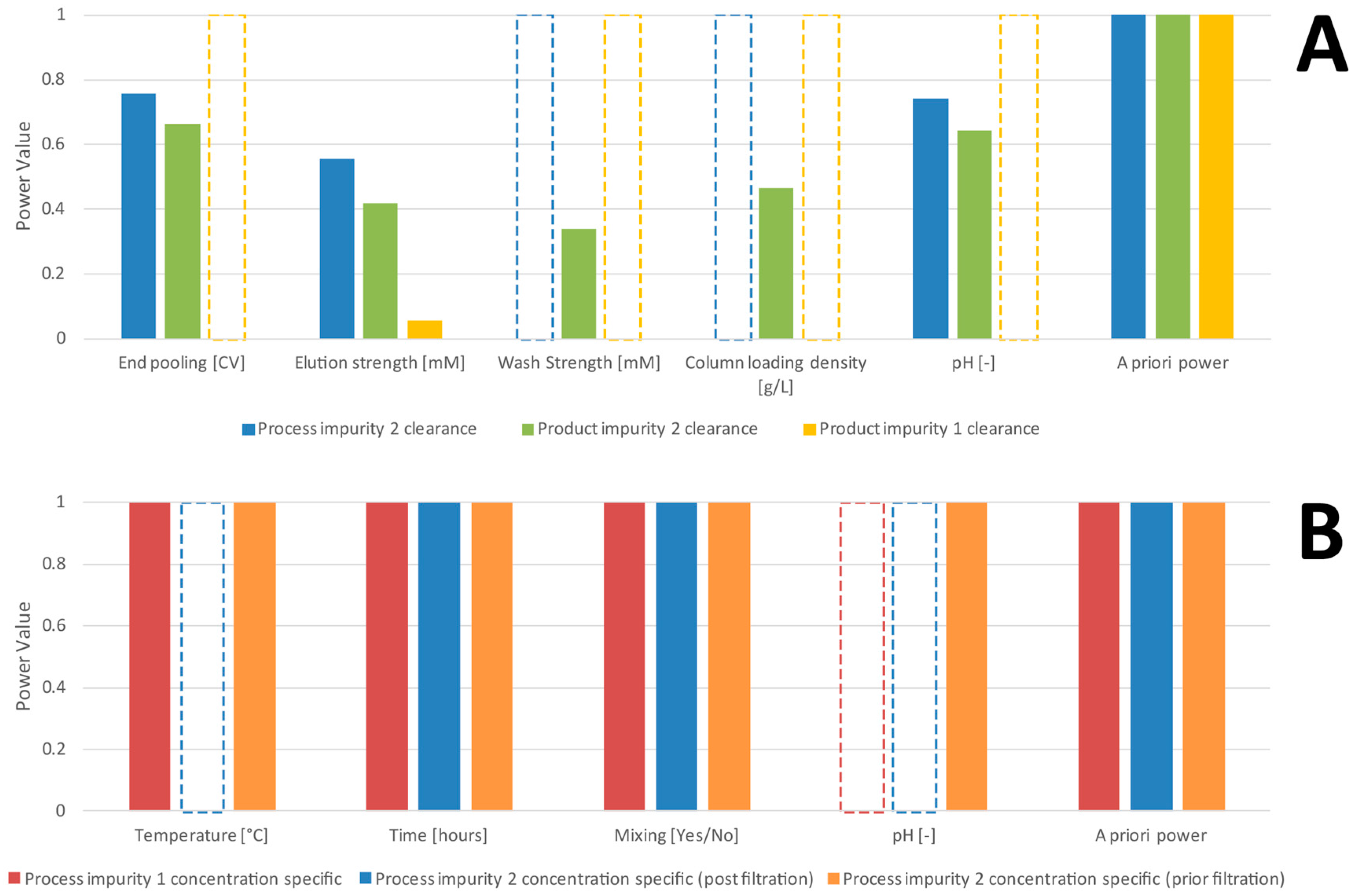
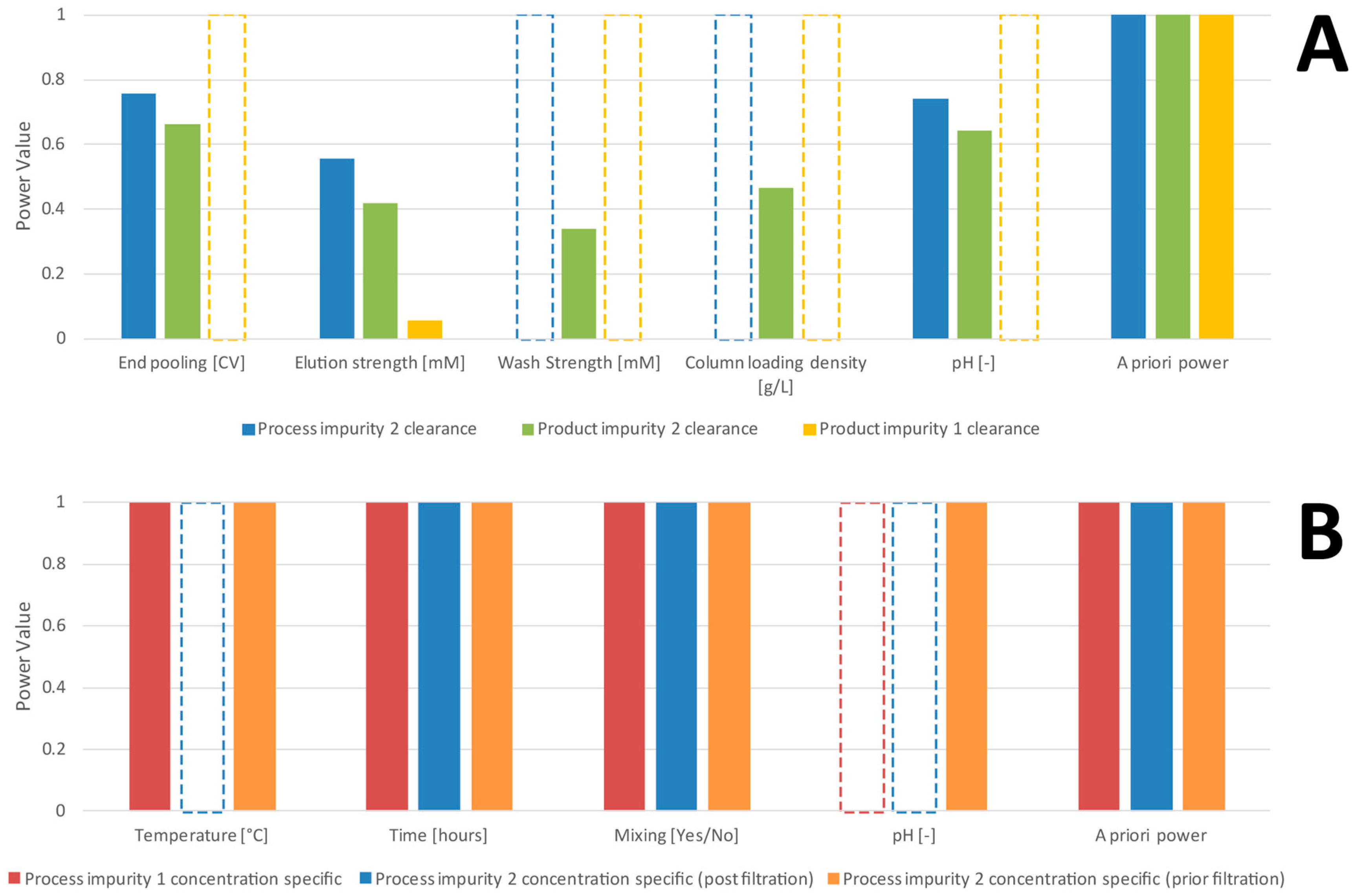
- Using variable selection procedures, we select a significant regression model (all included effects are not 0 to a certain significance level):where denotes the s significant parameters selected from a variable selection procedure (e.g., stepwise variable selection) and are the residuals of the obtained model. A list of those significantly selected parameters for the case studies of this work can be found in Table 1 and Table 2.
- We define a critical gap (CG) that we must not surpass as the difference of the threshold and the worst case model prediction within the NOR (, which is the parameter setting where the model prediction () is closest to the :
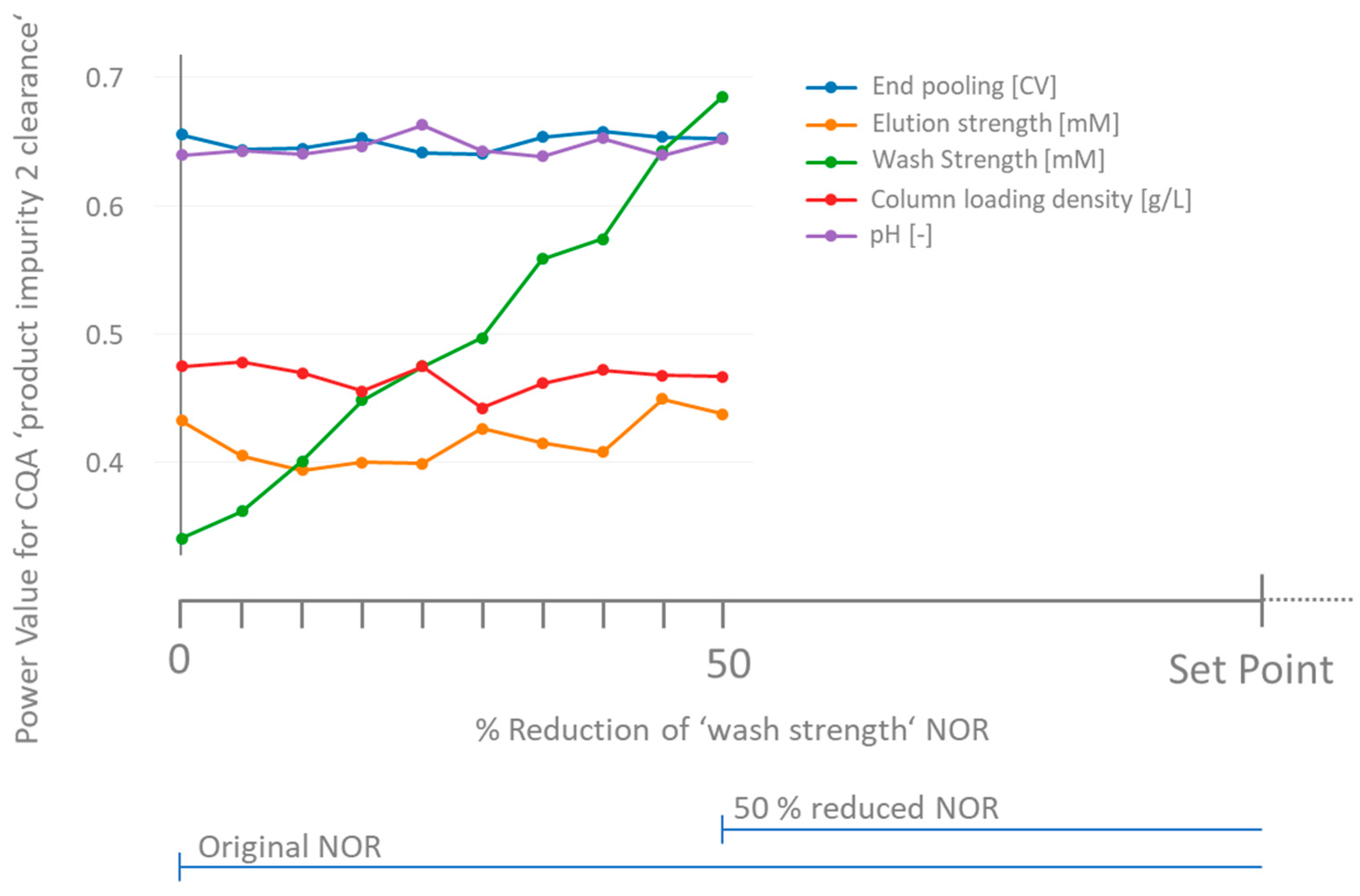
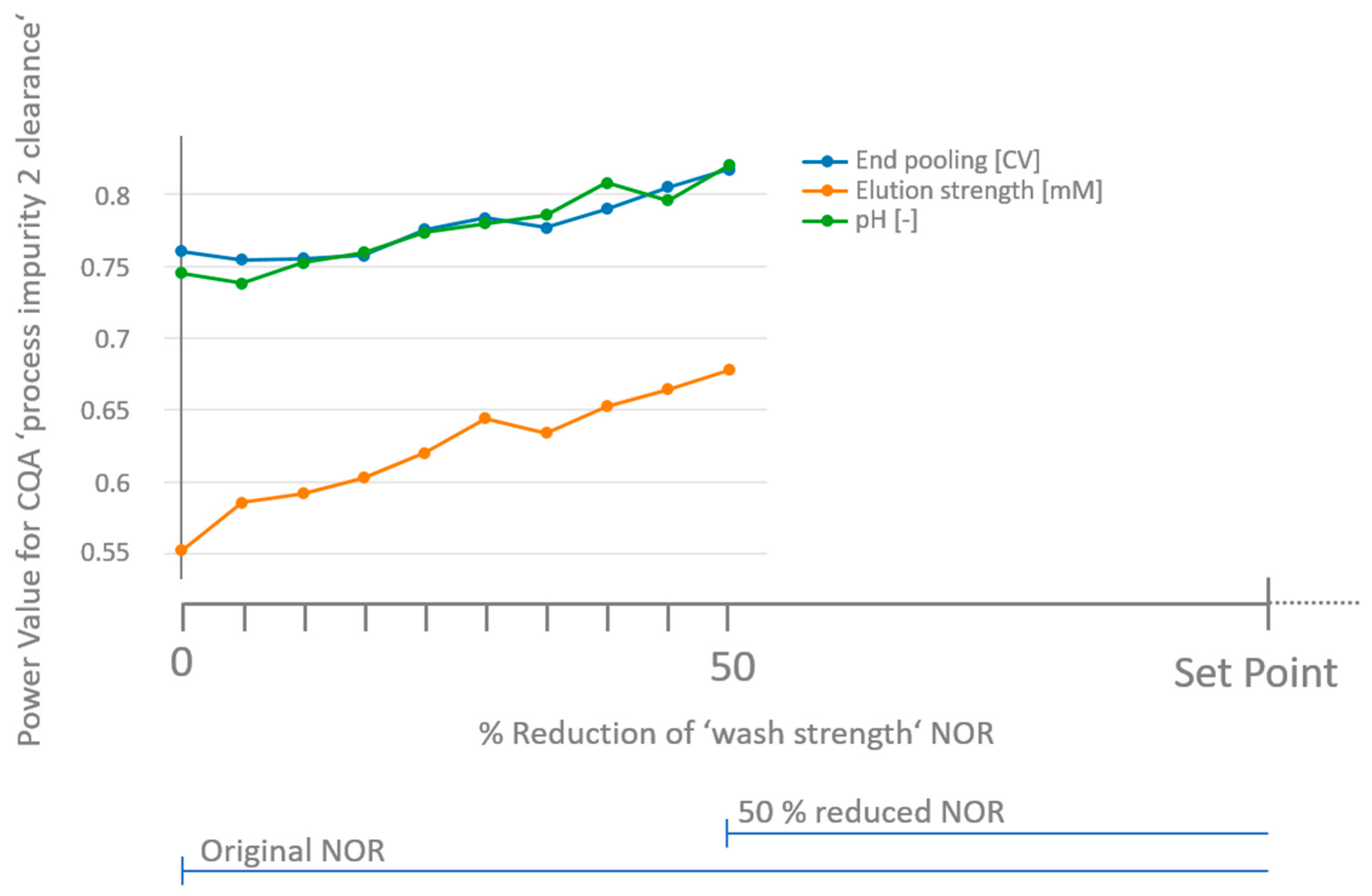
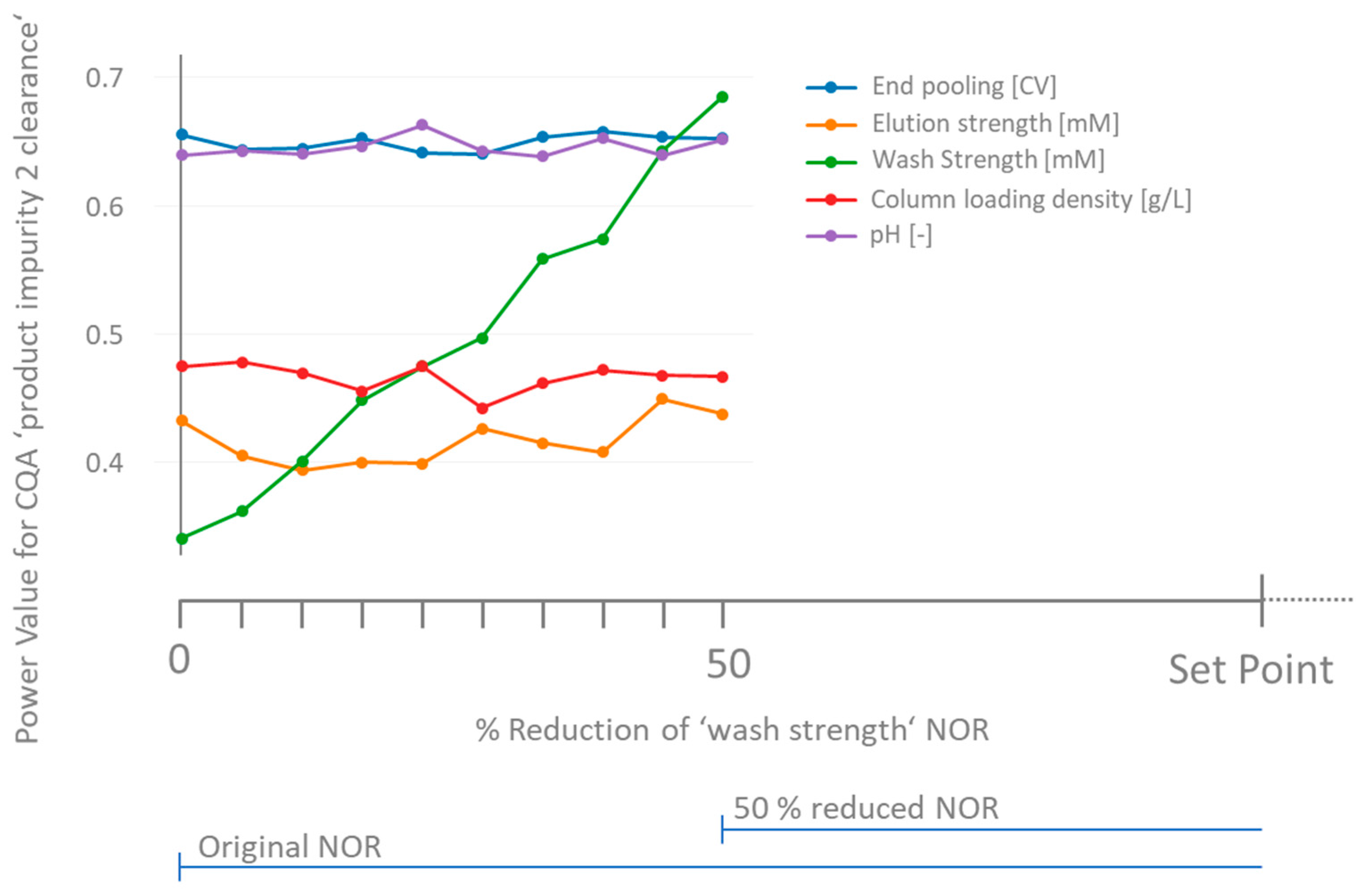
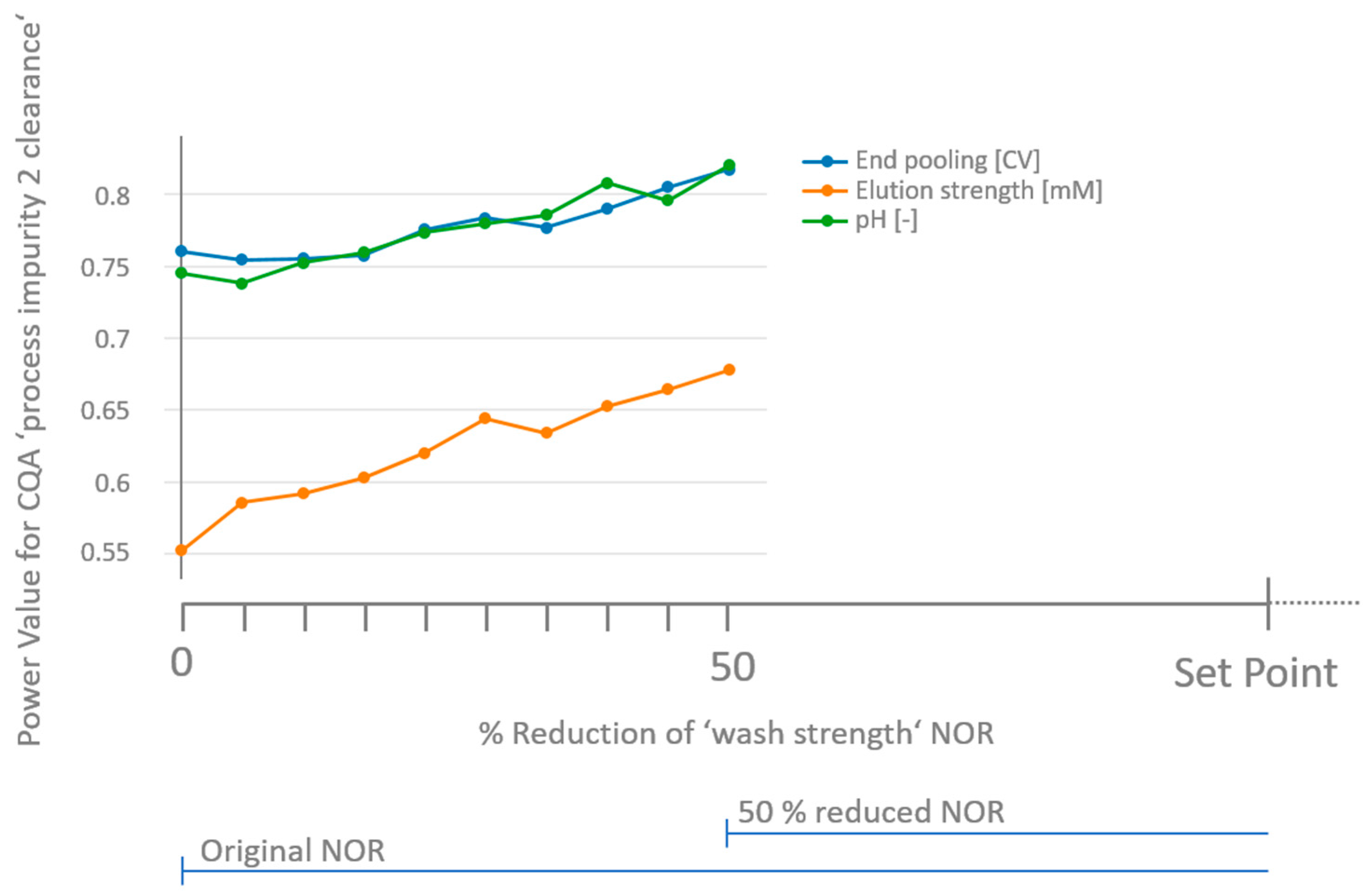
- Similar to the approach discussed in Section 2.3 for the a priori power analysis, for non-significant parameters, a variety of combinations (in total C) of effects for those parameters exist that lead to surpassing a critical threshold. In order to estimate the mean likelihood of not overlooking a specific parameter, we vary the relative impact on the threshold of each parameter gradually between 0 and 1 in 100 steps. The fraction of the CG which is attributed to the non-significant parameter is expressed as the weight for the combination c. Equation (5) can be used to calculate the critical effect of the parameter .
- The residuals are permuted randomly, producing .
- New response values are calculated from the permuted residuals assuming that the critical effect is present under the alternative hypothesis ():where is a vector of regression coefficients for the non-significant parameters and is the design matrix for all non-significant parameters.
- Make a model for based on X and Z and record significance of at a certain significance level (here α = 0.05)
- Repeat steps 4, 5 and 6 a large number of times (here 1000) and count the number of significant outcomes for each at a certain significance level (here α = 0.05). The fraction of significant outcomes of all iteration cycles equals the retrospective power of parameter .
3.2. Comparison of a Priori and Retrospective Power
3.3. How to Deal with Low-Powered Parameters?
3.4. Workflow for Criticality Assessment
4. Conclusions
- reduce the chance of overlooking potential CPPs
- develop a control strategy for potentially overlooked CPPs in order to increase process robustness
- lower OOS events and finally contribute to increased patient safety.
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Ahir, K.B.; Singh, K.D.; Yadav, S.P.; Patel, H.S.; Poyahari, C.B. Overview of Validation and Basic Concepts of Process Validation. Sch. Acad. J. Pharm. 2014, 3, 178–190. [Google Scholar]
- FDA Guidance for Industry: Process Validation: General Principles and Practices. 2011. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm070336.pdf (accessed on 10 October 2017).
- Katz, P.; Campbell, C. FDA 2011 process validation guidance: Process validation revisited. J. GXP Compliance 2012, 16, 18–29. [Google Scholar]
- Mollah, A.H. Application of failure mode and effect analysis (FMEA) for process risk assessment. BioProcess Int. 2005, 3, 12–20. [Google Scholar]
- ICH Harmonised Tripartite Guideline. Pharmaceutical Development Q8 (R2). Current Step 4 Version. Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf (accessed on 10 October 2017).
- Peres-Neto, P.R.; Olden, J.D. Assessing the robustness of randomization tests: Examples from behavioural studies. Anim. Behav. 2001, 61, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences; Revised Edition; Academic Press: New York, NY, USA, 1977; ISBN 978-0-12-179060-8. [Google Scholar]
- Nickerson, R.S. Null hypothesis significance testing: A review of an old and continuing controversy. Psychol. Methods 2000, 5, 241–301. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L. Retrospective Power Analysis. Conserv. Biol. 1997, 11, 276–280. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.; Krebs, C.J. A review of statistical power analysis software. Bull. Ecol. Soc. Am. 1997, 78, 126–138. [Google Scholar]
- Jones, B.; Nachtsheim, C.J. A class of three-level designs for definitive screening in the presence of second-order effects. J. Qual. Technol. 2011, 43, 1–15. [Google Scholar]
- Tai, M.; Ly, A.; Leung, I.; Nayar, G. Efficient high-throughput biological process characterization: Definitive screening design with the Ambr250 bioreactor system. Biotechnol. Prog. 2015, 31, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Shari, K.; Pat, W.; Mark, A. Handbook for Experimenters; Stat-Ease, Inc.: Minneapolis, MN, USA, 2005. [Google Scholar]
- Freedman, D.; Lane, D. A Nonstochastic Interpretation of Reported Significance Levels. J. Bus. Econ. Stat. 1983, 1, 292–298. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| End Pooling [CV] | Elution Strength [mM] | Wash Strength [mM] | Column Loading Density [g/L] | pH [–] | |||
|---|---|---|---|---|---|---|---|
| CQA | NOR 1 | −1.1–0 | −1.1–0.65 | −1.1–1.1 | −0.51–1.1 | −0.55–0.55 | |
| Threshold | |||||||
| Process impurity 2 clearance | 0.85 | - | - | 0.059 | 0.099 | - | 7.79 |
| Product impurity 1 clearance | 1.08 | 0.028 | - | 0.098 | 0.089 | 0.027 | 18.12 |
| Product impurity 2 clearance | 0.1 | - | - | - | - | - | 256.06 |
| Temperature [°C] | Time [Hours] | Mixing [Yes/No] | pH [–] | |||
|---|---|---|---|---|---|---|
| CQA | NOR 1 | −1.71–0.41 | 0.33–0.41 | −0.95–0.95 | −0.61–0.61 | |
| Threshold | ||||||
| Process impurity 1 concentration specific | 9 × 105 | 9 × 10−5 * | - | - | 0.07 | 64.89 |
| Process impurity 2 concentration specific (prior filtration) | 9 × 104 | - | - | - | - | 2.68 |
| Process impurity 2 concentration specific (post filtration) | 784.7 | - | - | - | 0.021 | 0.55 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahel, T.; Marschall, L.; Abad, S.; Vasilieva, E.; Maurer, D.; Mueller, E.M.; Murphy, P.; Natschläger, T.; Brocard, C.; Reinisch, D.; et al. Workflow for Criticality Assessment Applied in Biopharmaceutical Process Validation Stage 1. Bioengineering 2017, 4, 85. https://doi.org/10.3390/bioengineering4040085
Zahel T, Marschall L, Abad S, Vasilieva E, Maurer D, Mueller EM, Murphy P, Natschläger T, Brocard C, Reinisch D, et al. Workflow for Criticality Assessment Applied in Biopharmaceutical Process Validation Stage 1. Bioengineering. 2017; 4(4):85. https://doi.org/10.3390/bioengineering4040085
Chicago/Turabian StyleZahel, Thomas, Lukas Marschall, Sandra Abad, Elena Vasilieva, Daniel Maurer, Eric M. Mueller, Patrick Murphy, Thomas Natschläger, Cécile Brocard, Daniela Reinisch, and et al. 2017. "Workflow for Criticality Assessment Applied in Biopharmaceutical Process Validation Stage 1" Bioengineering 4, no. 4: 85. https://doi.org/10.3390/bioengineering4040085