The Nothoaspis amazoniensis Complete Mitogenome: A Comparative and Phylogenetic Analysis


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mitochondrial Genome Sequences of Argasidae Species
2.2. Sequencing of the Nothoaspis amazoniensis Mitochondrial Genome
2.2.1. DNA Extraction and Quantification
2.2.2. Library Preparation, Normalization, and Clustering
2.2.3. Library Sequencing
2.3. Assembling and Functional Annotation of the N. amazoniensis Mitochondrial Genome
2.4. Comparative Genomics
2.5. Phylogenetic Analysis
3. Results
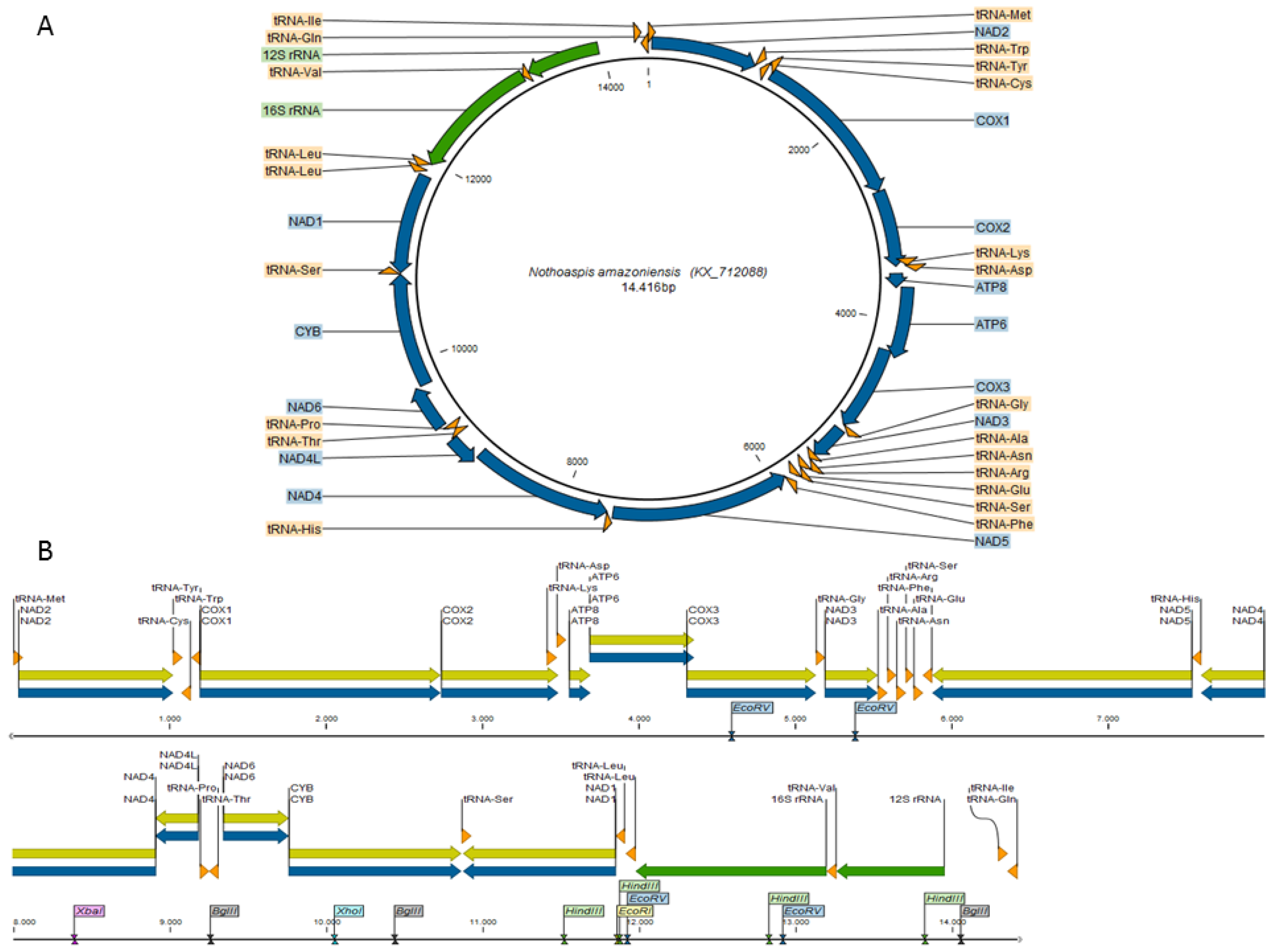
3.1. Characterization of the Nothoaspis amazoniensis Mitogenome
3.2. Comparative Genomics
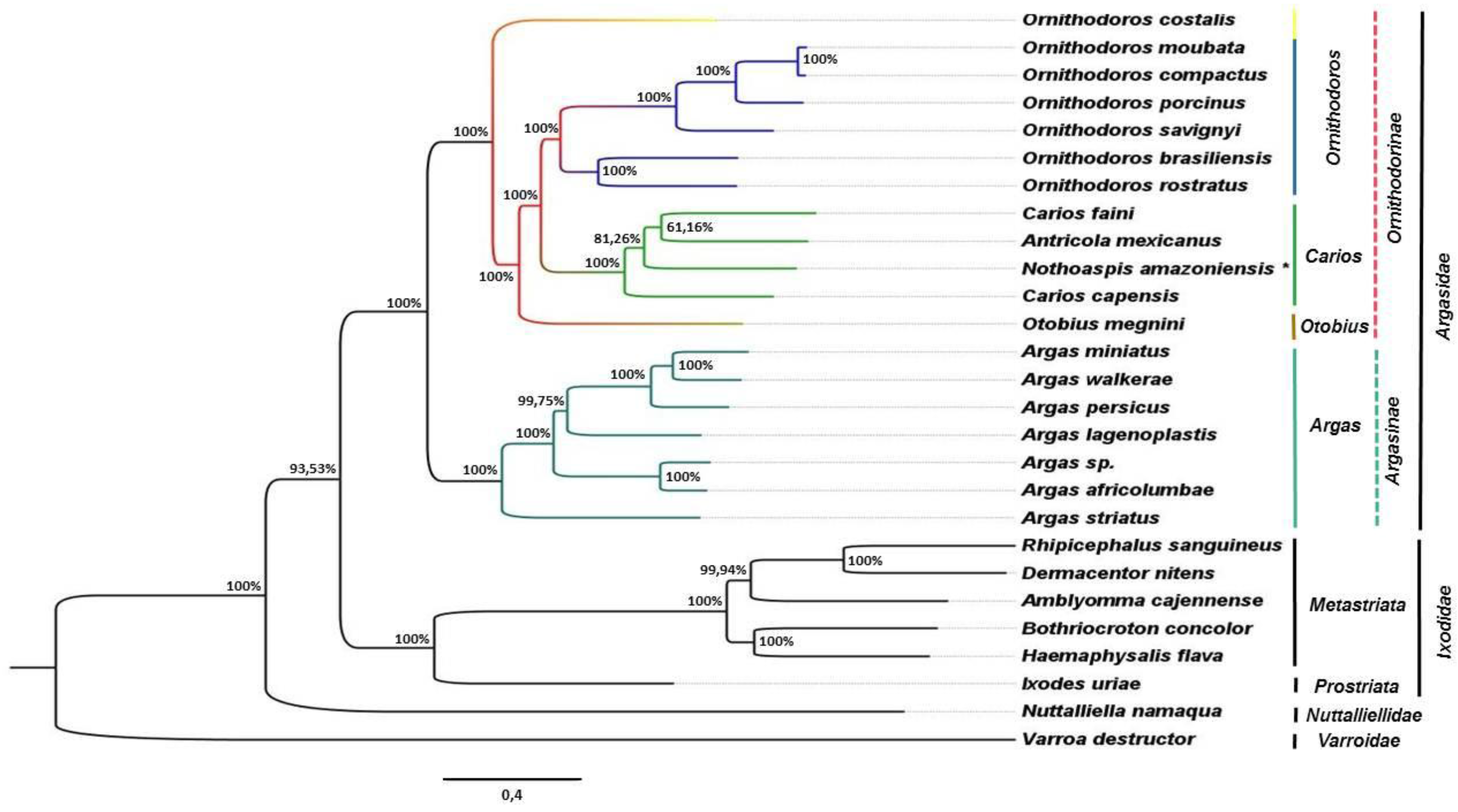
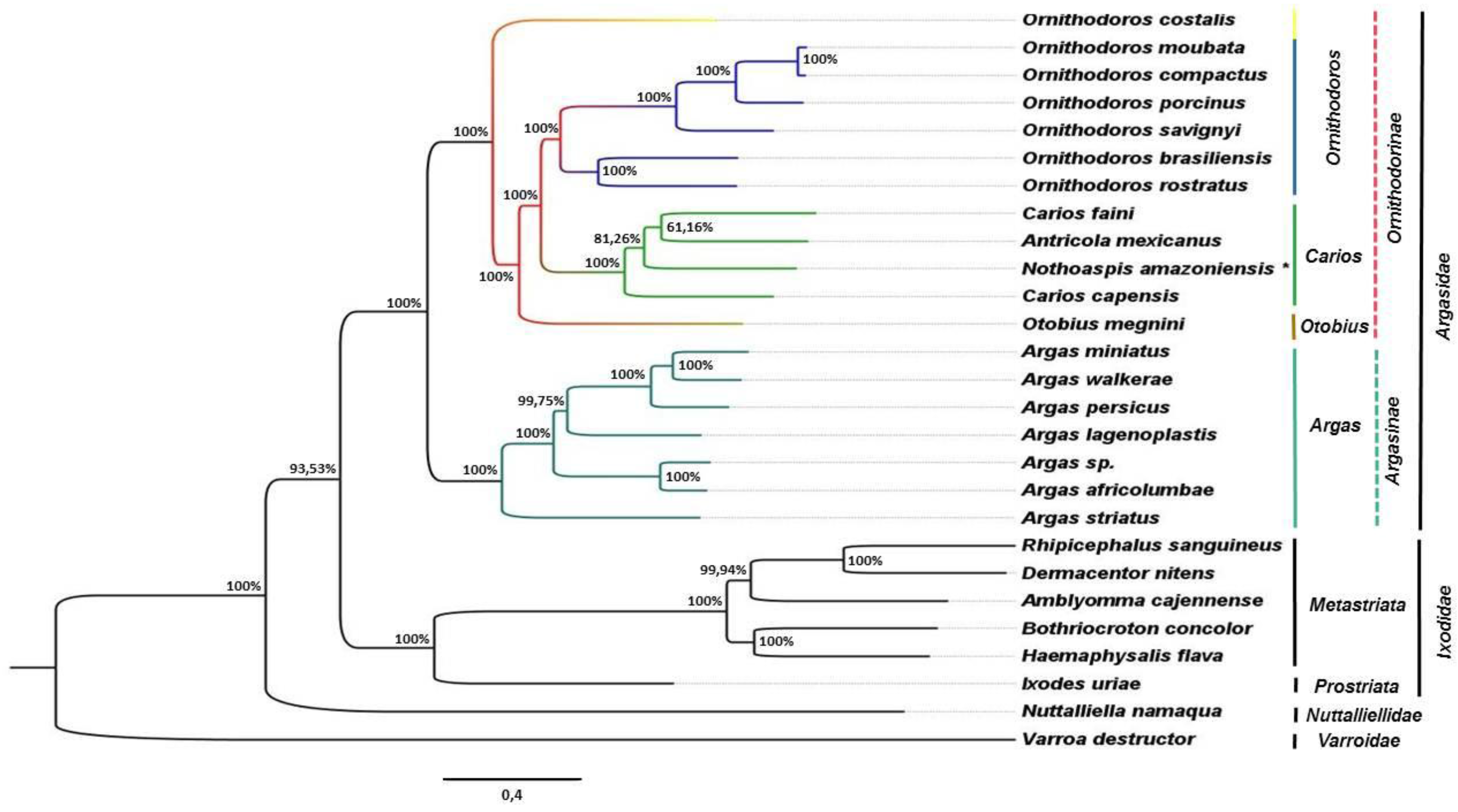
3.3. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guglielmone, A.A.; Robbins, R.G.; Apanaskevich, D.A.; Petney, T.N.; Estrada-Peña, A.; Horak, I.G.; Shao, R.; Barker, S.C. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida) of the world: A list of valid species names. Zootaxa 2010, 2528, 1–28. [Google Scholar]
- Hoogstraal, H. Argasid and nuttalliellid ticks as parasites and vectors. Adv. Parasitol. 1985, 24, 35–238. [Google Scholar]
- Estrada-Peña, A.; Mangold, A.J.; Nava, S.; Venzal, J.M.; Labruna, M.; Guglielmone, A.A. A review of the systematics of the tick family Argasidae (Ixodida). Acarologia 2010, 50, 317–333. [Google Scholar] [CrossRef]
- Nava, S.; Guglielmone, A.A.; Mangold, A.J. An overview of systematics and evolution of ticks. Front. Biosci. 2009, 14, 2857–2877. [Google Scholar] [CrossRef]
- Nava, S.; Venzal, J.M.; Terassini, F.A.; Mangold, A.J.; Camargo, L.M.A.; Labruna, M.B. Description of a new argasid tick (Acari: Ixodida) from bat caves in brazilian amazon. J. Parasitol. 2010, 96, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Keirans, J.E.; Cliffordand, C.M.; Redell, J.M. Description of immature stages of Nothoaspis reddelli (Ixodoidea: Argasidae) from bat caves in Mexico. Ann. Entomol. Soc. Am. 1977, 70, 591–595. [Google Scholar] [CrossRef]
- Burger, T.D.; Shao, R.; Labruna, M.B.; Barker, S.C. Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. TicksTick Borne Dis. 2014, 5, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R. The past, present and future of mitochondrial genomics: Have we sequenced mtDNA enough? Brief. Funct. Genom. 2015, 15, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R. Goodbye genome paper, hello genome report: The increasing popularity of genome announcements’ and their impact on science. Brief. Funct. Genom. 2016, 16, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. A decade’s perspective on DNA sequencing technology. Nature 2011, 470, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Rannala, B. Molecular phylogenetics: Principles and practice. Nat. Rev. Genet. 2012, 13, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Liolios, K.; Chen, I.M.A.; Mavromatis, K.; Tavernarakis, N.; Hugenholtz, P.; Markowitz, V.M.; Kyrpides, N.C. The Genomes on Line Database (GOLD) in 2009: Status of genomic and metagenomic projects and their associated metadata. Nucl. Acids Res. 2010, 38, D346–D354. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Topological variation in single-gene phylogenetic trees. Genome Biol. 2007, 8, 216. [Google Scholar] [PubMed]
- Pennisi, E. Building the tree of life, genome by genome. Science 2008, 320, 1716–1717. [Google Scholar] [CrossRef] [PubMed]
- Bapteste, E.; Brinkmann, H.; Lee, J.A.; Moore, D.V.; Sensen, C.W.; Gordon, P.; Durufle, L.; Gaasterland, T.; Lopez, P.; Müller, M.; et al. The analysis of 100 genes supports the grouping of three highly divergent amoebae: Dictyostelium, Entamoeba, and Mastigamoeba. Proc. Natl. Acad. Sci. USA 2002, 99, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Bapteste, E.; Susko, E.; Leigh, J.; Ruiz-Trillo, I.; Bucknam, J.; Doolittle, W.F. Alternative methods for concatenation of core genes indicate a lack of resolution in deep nodes of the prokaryotic phylogeny. Mol. Biol. Evol. 2008, 25, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Delsuc, F.; Brinkmann, H.; Philippe, H. Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 2005, 6, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.B.L.; Williams, N.; King, S.B.; Carroll, M.P.; Cummings, S.A.; Handley, D.S.; Myers, D.L.; Reed, K.; Winka, J.F. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Aoki, Y.; Mitani, H.; Tabuchi, N.; Barker, S.C.; Fukunaga, M. The mitochondrial genomes of soft ticks have an arrangement of genes that has remained unchanged for over 400 million years. Insect. Mol. Biol. 2004, 13, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Mitani, H.; Talbert, A.; Fukunaga, M. New World relapsing fever Borrelia found in Ornithodoros porcinus ticks in central Tanzania. Microbiol. Immunol. 2004, 48, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 April 2016).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. ClustalW and ClustalX version 2. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Bad, B.; Leg, N.T. Progressive MAUVE: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MrBayes: Bayesian inference of phylogenetic tree. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Method 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Barker, S.C.; Mitani, H.; Aoki, Y.; Fukunaga, M. Evolution of duplicate control regions in the mitochondrial genomes of metazoan: A case study with australasian ixodes ticks. Mol. Biol. Evol. 2004, 22, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Castellana, S.; Vicario, S.; Saccone, C. Evolutionary patterns of the mitochondrial genome in metazoa: Exploring the role of mutation and selection in mitochondrial protein-coding genes. Gen. Biol. Evol. 2011, 3, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.F.; Barker, S.C.; Mitani, H.; Takahashi, M.; Fukunaga, M. Molecular mechanisms for the variation of mitochondrial gene content and gene arrangement among chigger mites of the genus Leptotrombidium (Acari: Acariformes). J. Mol. Evol. 2006, 63, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Filippova, N.A. Argasid Ticks (Argasidae); Zoologicheskogo Institut Akademii Nauk SSSR: Moscow-Leningrad, Russia, 1966. (In Russian) [Google Scholar]
- Pospelova-Shtrom, M.V. On the system of classification of ticks of the family Argasidae Can., 1890. Acarologia 1969, 11, 1–22. [Google Scholar] [PubMed]
- Clifford, C.M.; Kohls, G.M.; Sonenshine, D.E. The systematics of the subfamily Ornithodorinae (Acarina: Argasidae) I. The genera and subgenera. Ann. Entomol. Soc. Am. 1964, 57, 429–437. [Google Scholar] [CrossRef]
- Camicas, J.L.; Morel, P.C. Position systématique et classification des tiques (Acarida: Ixodida). Acarologia 1977, 18, 410–420. (In French) [Google Scholar]
- Camicas, J.L.; Hervy, J.P.; Adam, F.; Morel, P.C. Les Tiques du Monde. Nomenclature, Stades Décrits, Hôtes, Repártition (Acarina, Ixodida); Orstom: Paris, France, 1998; p. 300. (In French) [Google Scholar]
- Klompen, J.S.H.; Oliver, J.H. Systematic relationships in the soft ticks (Acari: Ixodida: Argasidae). Syst. Entomol. 1993, 18, 313–331. [Google Scholar] [CrossRef]
- Rieppe, O. Monophyly, paraphyly, and natural kinds. Biol. Philos. 2005, 20, 465–487. [Google Scholar] [CrossRef]
- Labruna, M.B.; Venzal, J.M. Carios fonsecai sp. nov. (Acari Argasidae), a bat tick from the central-western region of Brazil. Acta Parasitol. 2009, 54, 355–363. [Google Scholar] [CrossRef]
- Estrada-Peña, A.; Venzal, J.M.; Barros-Battesti, D.M.; Onofrio, V.C.; Trajano, E.; Firmino, J.V.L. Three new species of Antricola (Acari: Argasidae) from Brazil, with a key for the known species in the genus. J. Parasitol. 2004, 90, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Venzal, J.M.; Estrada-Peña, A.; Mangold, A.J.; González-Acuña, D.; Guglielmone, A.A. The Ornithodoros (Alectorobius) talaje species group (Acari: Ixodida: Argasidae): Description of Ornithodoros (Alectorobius) rioplatensis n. sp. from southern South America. J. Med. Entomol. 2008, 45, 832–840. [Google Scholar] [PubMed]
- Barros-Battesti, D.M.; Ramirez, D.G.; Landulfo, G.A.; Faccini, J.H.; Dantas-Torres, F.; Labruna, M.B.; Venzal, J.M. Immature argasid ticks: Diagnosis and keys for Neotropical region. Rev. Bras. Parasitol. Vet. 2013, 22, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraal, H. Ornithodoros (Reticulinasus) faini sp. nov. (Ixodoidea, Argasidae) from Congo fruit bats, Rousettus leachii (Smith). Rev. Zottl. Bot. Afr. 1960, 62, 358–378. [Google Scholar]


{kind=link}
{kind=link}
| Species | ID GenBank (genome mt) | Reference |
|---|---|---|
| Antricola mexicanus | NC_023340.1 | Burger et al., 2014 [7] |
| Argas africolumbae | NC_019642.1 | - |
| Argas lagenoplastis | NC_023369.1 | Burger et al., 2014 [7] |
| Argas miniatus | NC_023371.1 | Burger et al., 2014 [7] |
| Argas persicus | NC_029174.1 | - |
| Argas sp. SpringbokSA-QMS95171 | KC_769588.1 | Burger et al., 2014 [7] |
| Argas striatus | NC_029175.1 | - |
| Argas walkerae | NC_029176.1 | - |
| Carios capensis | NC_005291.1 | - |
| Carios faini | NC_029177.1 | - |
| Ornithodoros brasiliensis | NC_023373.1 | Burger et al., 2014 [7] |
| Ornithodoros compactus | NC_029178.1 | - |
| Ornithodoros costalis | NC_029179.1 | - |
| Ornithodoros moubata | NC_004357.1 | Shao et al., 2004 [20] |
| Ornithodoros porcinus | NC_005820.1 | Mitani et al.,2004 [21] |
| Ornithodoros rostratus | NC_023372.1 | Burger et al., 2014 [7] |
| Ornithodoros savignyi | NC_029180.1 | - |
| Otobius megnini | NC_023370.1 | Burger et al., 2014 [7] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, P.H.C.; Vidigal, P.M.P.; Barcelos, R.M.; Klein, R.C.; Montandon, C.E.; Fabres-Klein, M.H.; Dergam, J.A.; Venzal, J.M.; Mafra, C. The Nothoaspis amazoniensis Complete Mitogenome: A Comparative and Phylogenetic Analysis. Vet. Sci. 2018, 5, 37. https://doi.org/10.3390/vetsci5020037
Lima PHC, Vidigal PMP, Barcelos RM, Klein RC, Montandon CE, Fabres-Klein MH, Dergam JA, Venzal JM, Mafra C. The Nothoaspis amazoniensis Complete Mitogenome: A Comparative and Phylogenetic Analysis. Veterinary Sciences. 2018; 5(2):37. https://doi.org/10.3390/vetsci5020037
Chicago/Turabian StyleLima, Paulo H. C., Pedro M. P. Vidigal, Rafael M. Barcelos, Raphael C. Klein, Carlos E. Montandon, Mary H. Fabres-Klein, Jorge A. Dergam, José M. Venzal, and Cláudio Mafra. 2018. "The Nothoaspis amazoniensis Complete Mitogenome: A Comparative and Phylogenetic Analysis" Veterinary Sciences 5, no. 2: 37. https://doi.org/10.3390/vetsci5020037