Strategy for Identification of Phosphorylation Levels of Low Abundance Proteins in Vivo for Which Antibodies Are not Available


Abstract
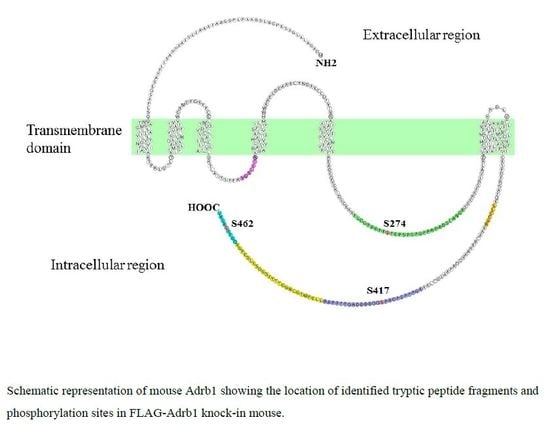
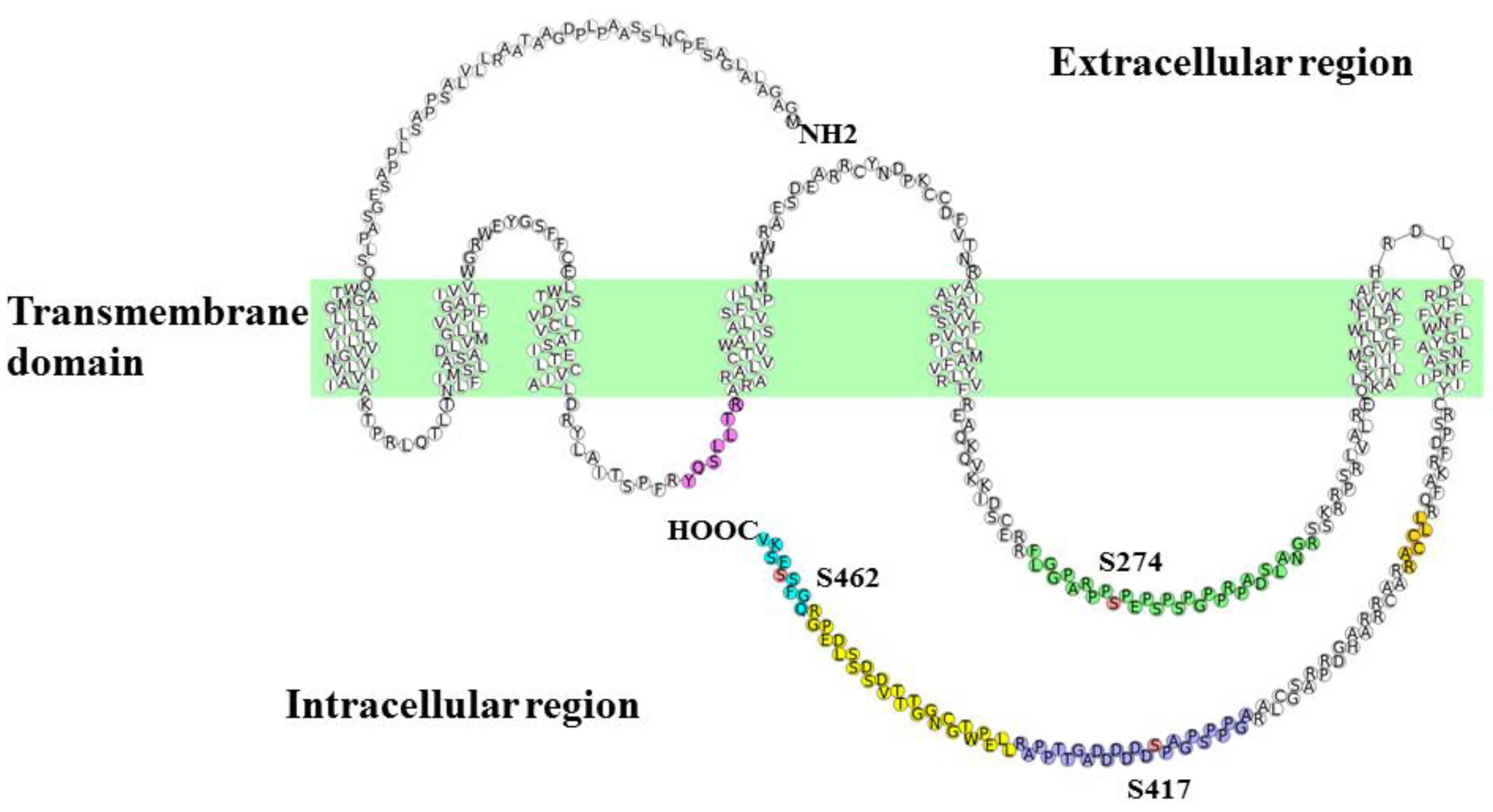
:
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
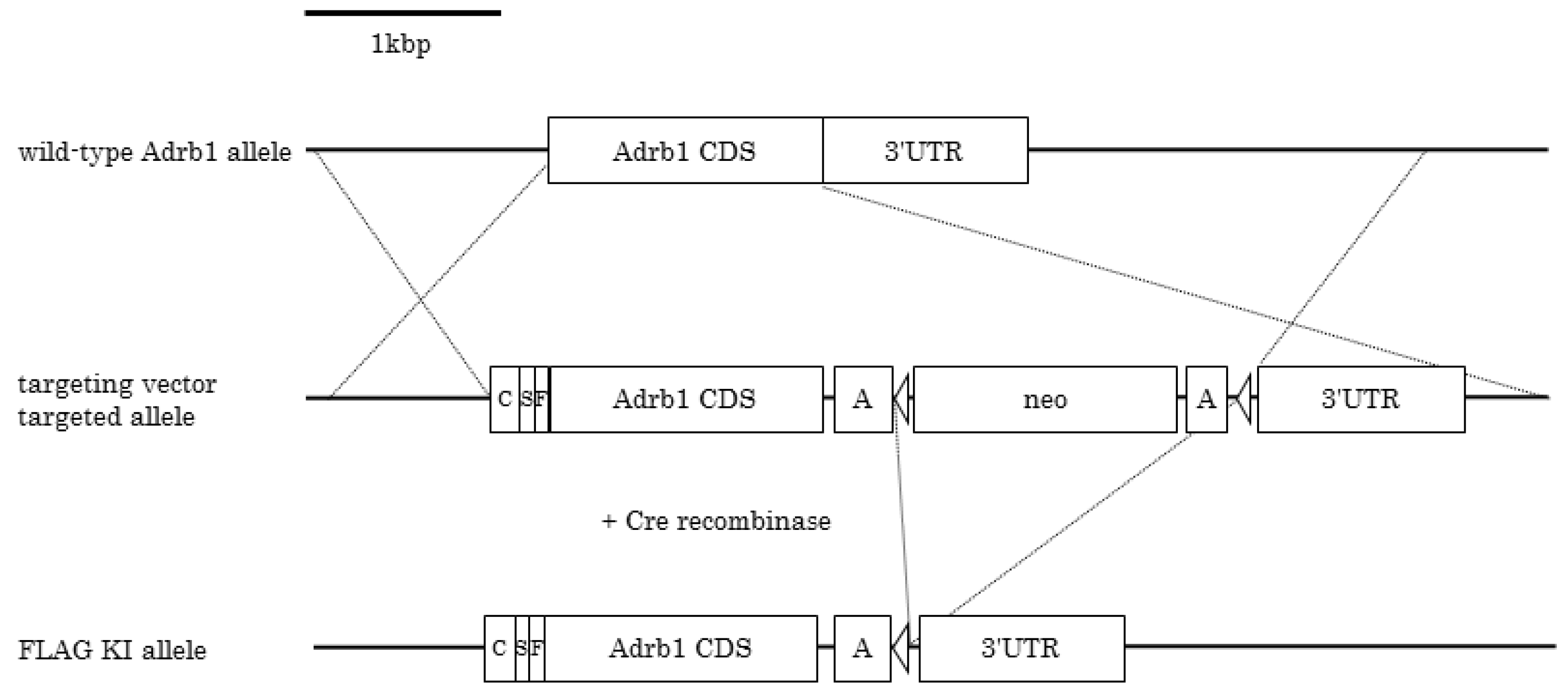
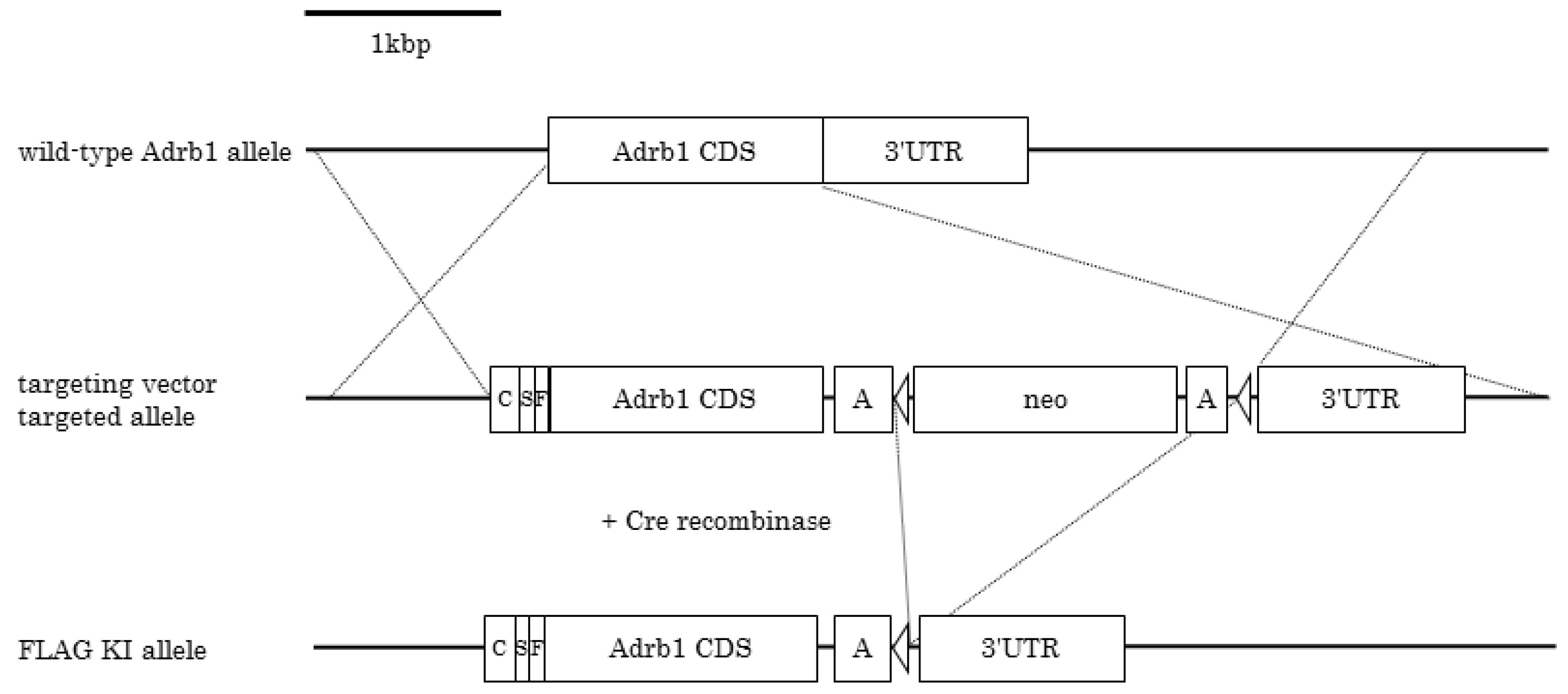
2.2. Generation of FLAG-β1AR KI Mice
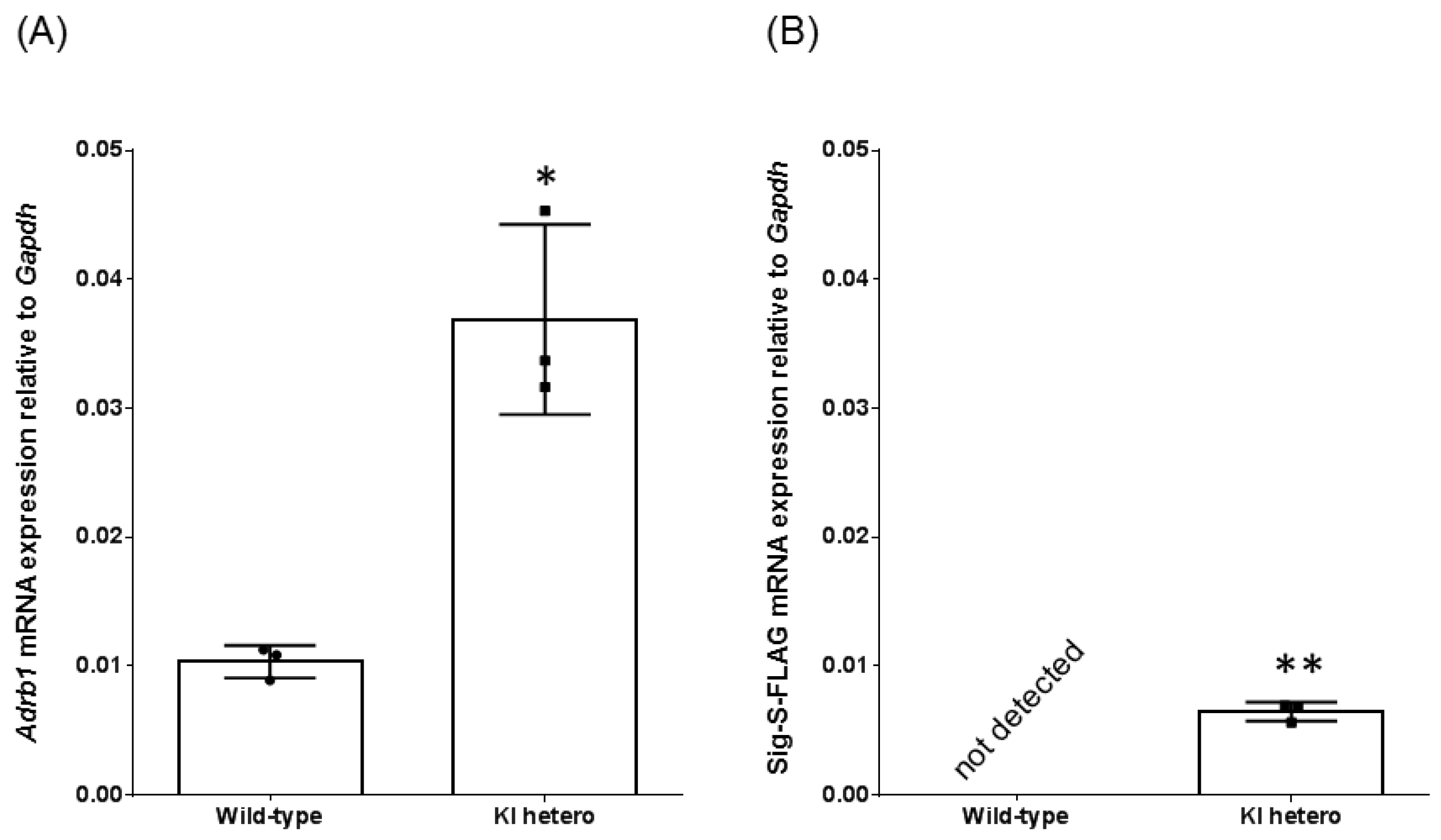
2.3. Quantitative Reverse Transcription PCR
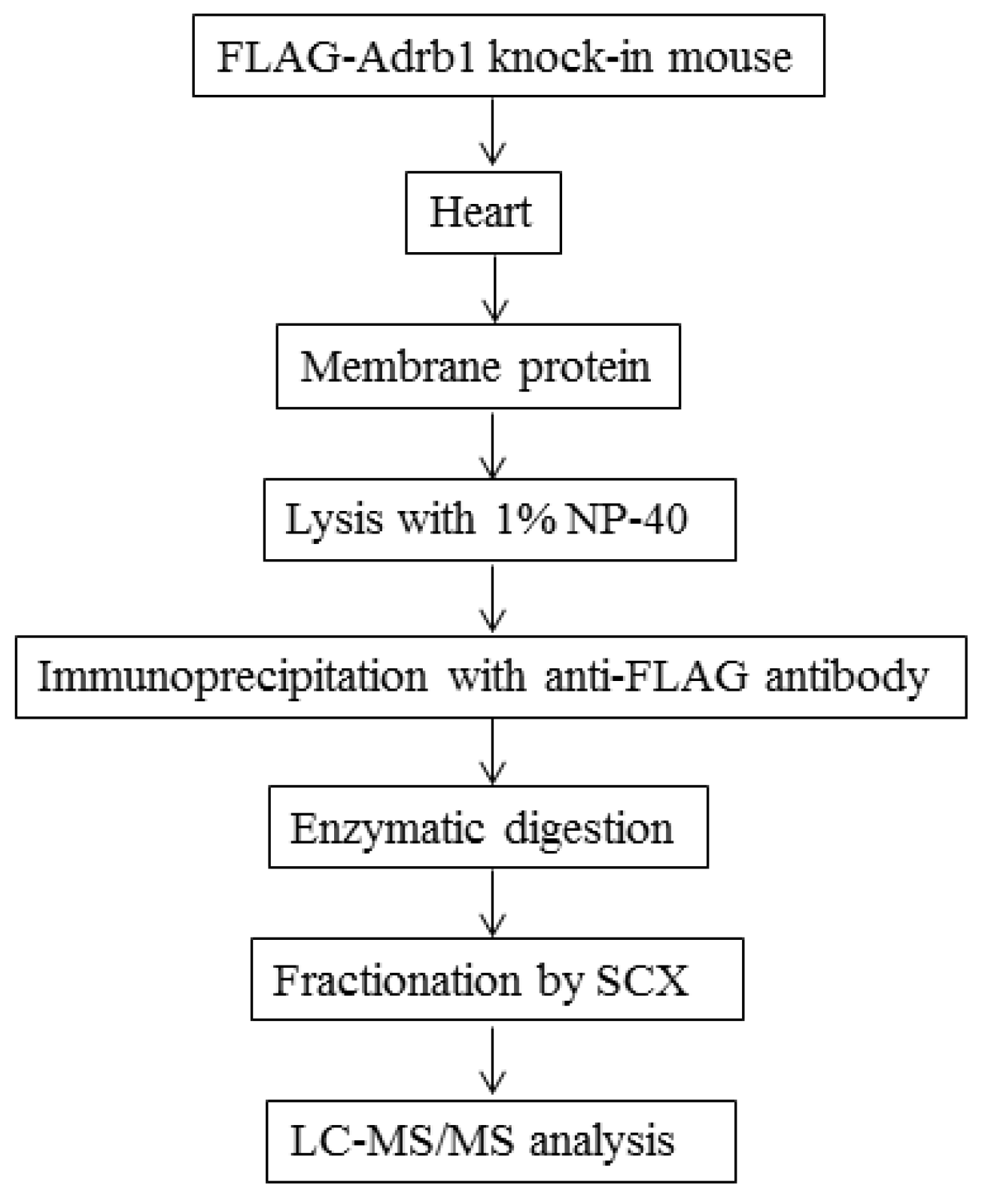
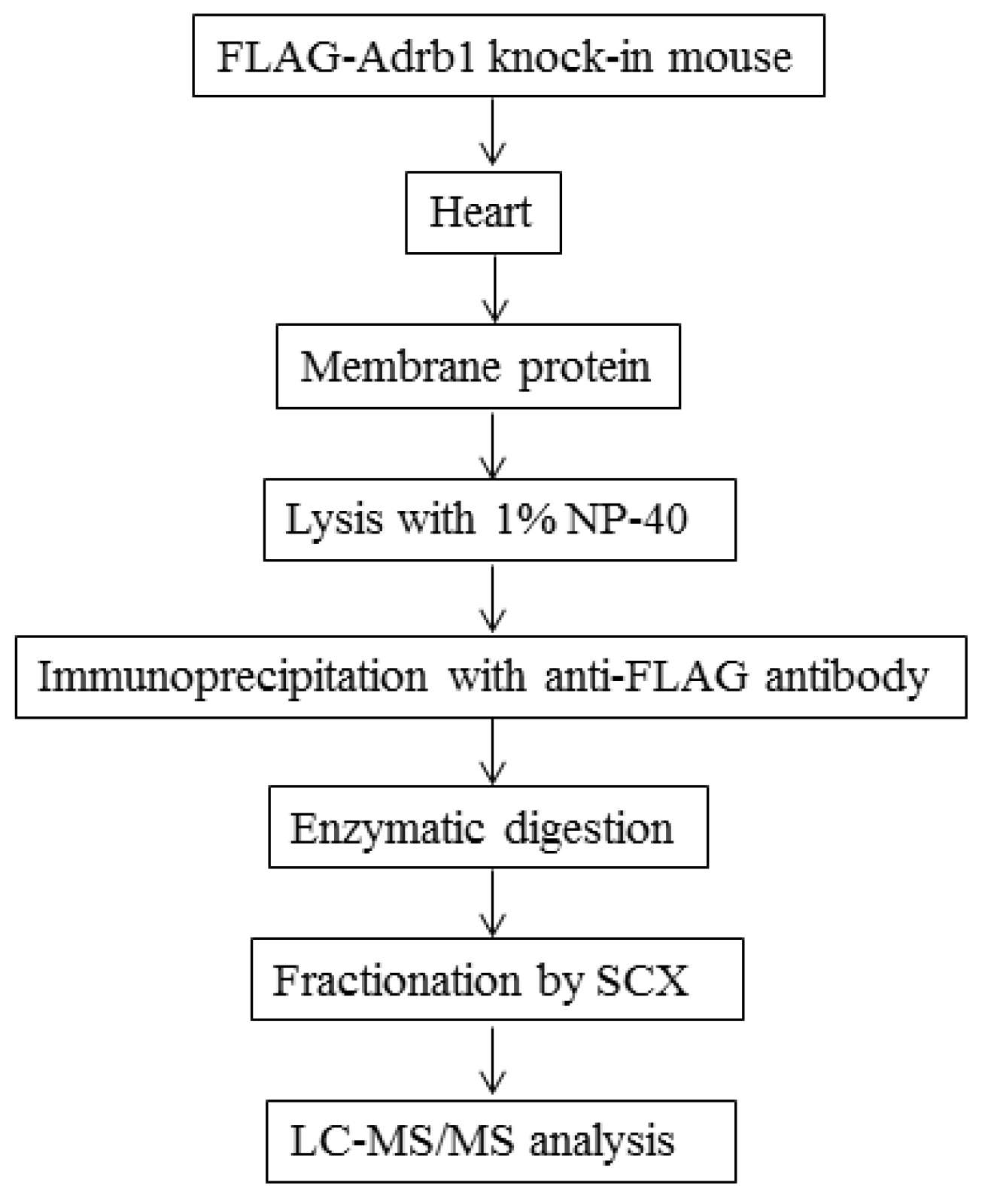
2.4. Preparation of Heart Membrane Proteins
2.5. IP of FLAG-Adrb1
2.6. Enzymatic Digestion of Immunoprecipitates
2.7. SCX Fractionation
2.8. Liquid Chromatography-Tandem Mass Spectrometry Analysis
2.9. Database Search
2.10. Statistical Analysis
3. Results
3.1. Generation of FLAG-β1AR KI Mice
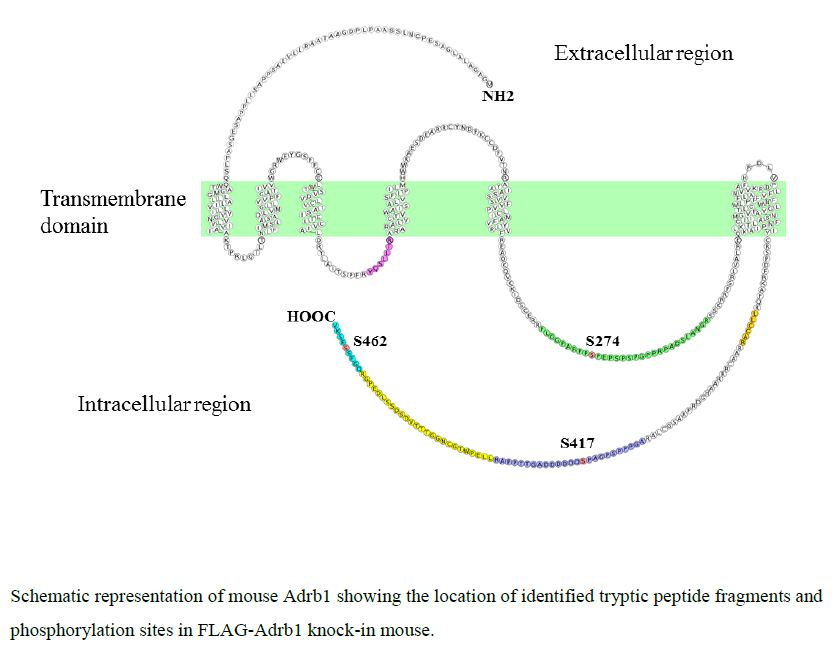
3.2. Identification and Quantitative Analysis of the Phosphorylation of Adrb1-Derived Tryptic Peptides in the Heart of KI Mice
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nobles, K.N.; Xiao, K.; Ahn, S.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; Shenoy, S.K.; et al. Distinct phosphorylation sites on the β (2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, I.; Spragg, E.J.; Poulin, B.; McWilliams, P.J.; Mistry, S.C.; Blaukat, A.; Tobin, A.B. Phosphorylation and regulation of a G protein-coupled receptor by protein kinase CK2. J. Cell Biol. 2007, 177, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Milting, H.; Scholz, C.; Arusoglu, L.; Freitag, M.; Cebulla, R.; Jaquet, K.; Körfer, R.; v. Lewinski, D.; Kassner, A.; Brodde, O.E.; et al. Selective upregulation of beta1-adrenergic receptors and dephosphorylation of troponin I in end-stage heart failure patients supported by ventricular assist devices. J. Mol. Cell. Cardiol. 2006, 41, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H. Novel therapeutic GPCRs for psychiatric disorders. Int. J. Mol. Sci. 2015, 16, 14109–14121. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Todd, N.; Thathiah, A. The role of GPCRs in neurodegenerative diseases: Avenues for therapeutic intervention. Curr. Opin. Pharmacol. 2017, 32, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z.; Brust, T.F.; Bohn, L.M. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett. 2016, 26, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Farran, B. An update on the physiological and therapeutic relevance of GPCR oligomers. Pharmacol. Res. 2017, 117, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Agüero, J.; Almenar, L.; D’Ocon, P.; Oliver, E.; Montó, F.; Moro, J.; Castelló, A.; Rueda, J.; Martínez-Dolz, L.; Sánchez-Lazaro, I.; et al. Correlation between beta-adrenoceptors and G-protein-coupled receptor kinases in pretransplantation heart failure. Transplant. Proc. 2008, 40, 3014–3016. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Gotou, M.; Matsui, T.; Imahashi, K.; Nishimoto, T.; Kobayashi, H. Identification of phosphorylation sites on β1-adrenergic receptor in the mouse heart. Biochem. Biophys. Res. 2017, 488, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Kozlosky, C.J.; Ericsson, L.H.; Daniel, T.O.; Cerretti, D.P.; Johnson, R.S. Studies of ligand-induced site-specific phosphorylation of epidermal growth factor receptor. J. Am. Soc. Mass Spectrom. 2003, 14, 1022–1031. [Google Scholar] [CrossRef]
- Steen, H.; Jebanathirajah, J.A.; Springer, M.; Kirschner, M.W. Stable isotope-free relative and absolute quantitation of protein phosphorylation stoichiometry by MS. Proc. Natl. Acad. Sci. USA 2005, 102, 3948–3953. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, I.; Aiba, A. Receptor knock-out and knock-in strategies. Methods Mol. Biol. 2004, 259, 379–390. [Google Scholar] [PubMed]
- Faget, L.; Erbs, E.; Le Merrer, J.; Scherrer, G.; Matifas, A.; Benturquia, N.; Noble, F.; Decossas, M.; Koch, M.; Kessler, P.; et al. In vivo visualization of delta opioid receptors upon physiological activation uncovers a distinct internalization profile. J. Neurosci. 2012, 32, 7301–7310. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.A.; Tawfik, V.L.; Tipton, A.F.; Scherrer, G. In vivo techniques to investigate the internalization profile of opioid receptors. Methods Mol. Biol. 2015, 1230, 87–104. [Google Scholar] [PubMed]
- Ceredig, R.A.; Massotte, D. Fluorescent knock-in mice to decipher the physiopathological role of G protein-coupled receptors. Front. Pharmacol. 2015, 5, 289. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, A.; Brunori, G.; Mercatelli, D.; Wu, J.; Cippitelli, A.; Zou, B.; Xie, X.S.; Williams, M.; Zaveri, N.T.; Low, S.; et al. Knock-in mice with NOP-eGFP receptors identify receptor cellular and regional localization. J. Neurosci. 2015, 35, 11682–11693. [Google Scholar] [CrossRef] [PubMed]
- Bounoutas, G.S.; Tawfeek, H.; Fröhlich, L.F.; Chung, U.I.; Abou-Samra, A.B. Impact of impaired receptor internalization on calcium homeostasis in knock-in mice expressing a phosphorylation-deficient parathyroid hormone (PTH)/PTH-related peptide receptor. Endocrinology 2006, 147, 4674–4679. [Google Scholar] [CrossRef] [PubMed]
- Lemke, T.; Welling, A.; Christel, C.J.; Blaich, A.; Bernhard, D.; Lenhardt, P.; Hofmann, F.; Moosmang, S. Unchanged beta-adrenergic stimulation of cardiac L-type calcium channels in Ca v 1.2 phosphorylation site S1928A mutant mice. J. Biol. Chem. 2008, 283, 34738–34744. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.M.; Gao, E.; Fonseca, F.V.; Hayashi, H.; Shang, X.; Hoffman, N.E.; Chuprun, J.K.; Tian, X.; Tilley, D.G.; Madesh, M.; et al. Convergence of G protein-coupled receptor and S-nitrosylation signaling determines the outcome to cardiac ischemic injury. Sci. Signal 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Solari, F.A.; Dell'Aica, M.; Sickmann, A.; Zahedi, R.P. Why phosphoproteomics is still a challenge. Mol. Biosyst. 2015, 11, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Von Stechow, L.; Francavilla, C.; Olsen, J.V. Recent findings and technological advances in phosphoproteomics for cells and tissues. Expert Rev. Proteom. 2015, 12, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Anjum, R.; Kubota, K.; Rush, J.; Villen, J.; Gygi, S.P. A site-specific, multiplexed kinase activity assay using stable-isotope dilution and high-resolution mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Brönstrup, M. Absolute quantification strategies in proteomics based on mass spectrometry. Expert Rev. Proteom. 2004, 1, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Srivastava, A.K.; Zhou, M.; Veenstra, T. Quantitation of Met tyrosine phosphorylation using MRM-MS. Methods Mol. Biol. 2013, 1002, 181–193. [Google Scholar] [PubMed]
- Lu, R.; Li, Y.; Zhang, Y.; Chen, Y.; Shields, A.D.; Winder, D.G.; Angelotti, T.; Jiao, K.; Limbird, L.E.; Zhou, Y.; et al. Epitope-tagged receptor knock-in mice reveal that differential desensitization of alpha2-adrenergic responses is because of ligand-selective internalization. J. Biol. Chem. 2009, 284, 13233–13243. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hao, Y.; Du, Z.; Wang, Z.; Ewing, R.M. Identifying novel protein complexes in cancer cells using epitope-tagging of endogenous human genes and affinity-purification mass spectrometry. J. Proteome Res. 2012, 11, 5630–5641. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.L.; Mizuguchi, H.; Ishii-Watabe, A.; Uchida, E.; Mayumi, T.; Hayakawa, T. Optimization of transcriptional regulatory elements for constructing plasmid vectors. Gene 2001, 272, 149–156. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
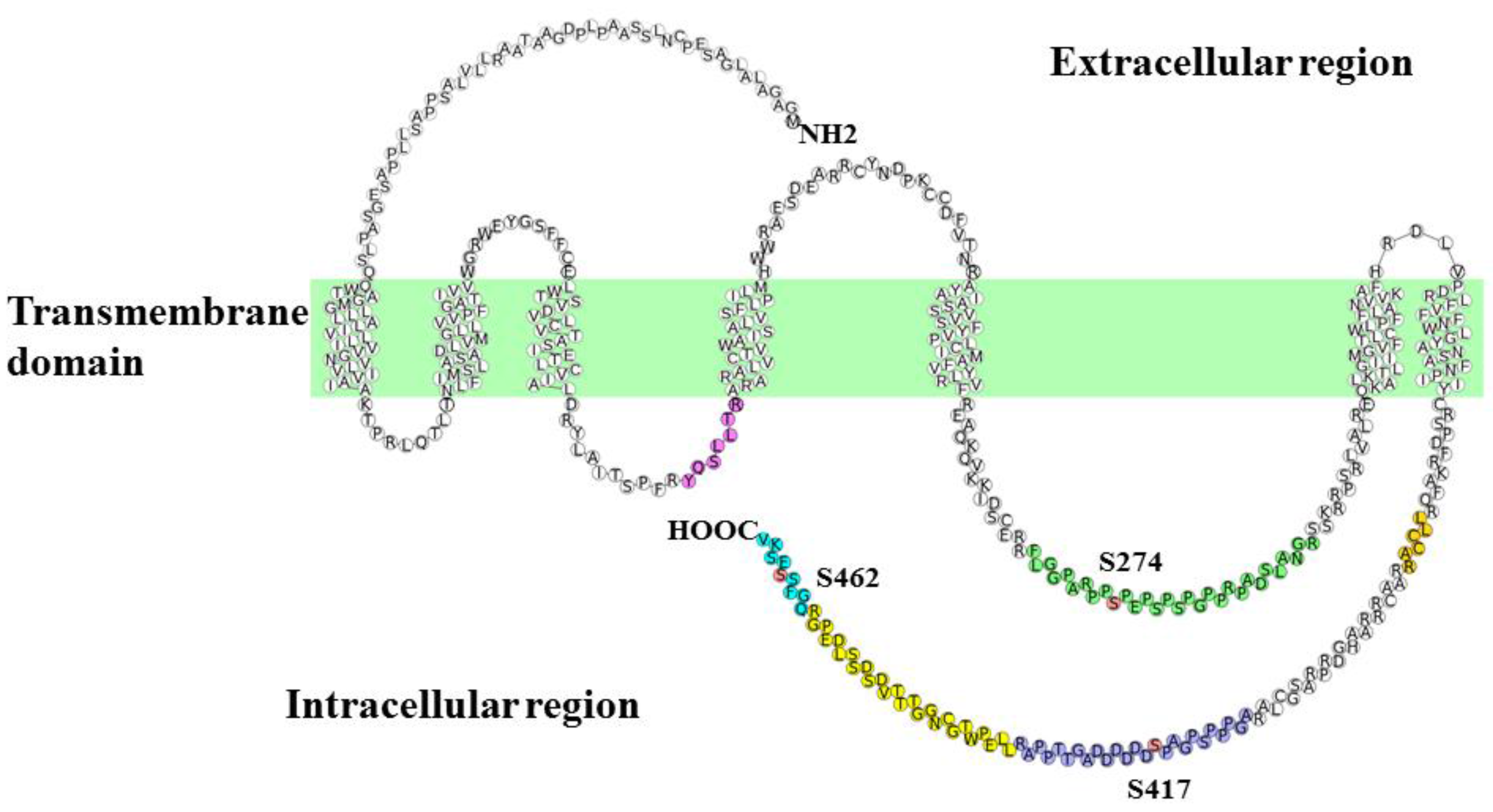{kind=link}
| Sequence | Start | End | Phos-Site | m/z | Charge | Phosphorylation ratio a (Phospho/Nonphospho) |
|---|---|---|---|---|---|---|
| FLGGPARPPpSPEPSPSPGPPRPADSLANGR | 265 | 294 | S274 | 765.38 | 4 | 0.25 ± 0.058 |
| AGPPPSPGAPpSDDDDDDAGTTPPAR | 407 | 431 | S417 | 819.34 | 3 | 0.56 ± 0.070 |
| QGFSpSESKV | 458 | 466 | S462 | 524.72 | 2 | 0.0023 b |
| YQSLLTR | 166 | 172 | - | 440.75 | 2 | - |
| LLCCAR | 379 | 384 | - | 396.70 | 2 | - |
| LLEPWTGCNGGTTTVDSDSSLDEPGR | 432 | 457 | - | 922.09 | 3 | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayashi, K.; Yamashita, R.; Takami, R.; Matsui, T.; Gotou, M.; Nishimoto, T.; Kobayashi, H. Strategy for Identification of Phosphorylation Levels of Low Abundance Proteins in Vivo for Which Antibodies Are not Available. J. Cardiovasc. Dev. Dis. 2017, 4, 17. https://doi.org/10.3390/jcdd4040017
Hayashi K, Yamashita R, Takami R, Matsui T, Gotou M, Nishimoto T, Kobayashi H. Strategy for Identification of Phosphorylation Levels of Low Abundance Proteins in Vivo for Which Antibodies Are not Available. Journal of Cardiovascular Development and Disease. 2017; 4(4):17. https://doi.org/10.3390/jcdd4040017
Chicago/Turabian StyleHayashi, Kozo, Ryo Yamashita, Ritsuko Takami, Toshikatsu Matsui, Masamitsu Gotou, Tomoyuki Nishimoto, and Hiroyuki Kobayashi. 2017. "Strategy for Identification of Phosphorylation Levels of Low Abundance Proteins in Vivo for Which Antibodies Are not Available" Journal of Cardiovascular Development and Disease 4, no. 4: 17. https://doi.org/10.3390/jcdd4040017