Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System


1
Max Delbrück Center for Molecular Medicine Berlin (MDC), Berlin 13125, Germany
2
DZHK (German Centre for Cardiovascular Research), partner site Berlin 13347, Germany
*
Author to whom correspondence should be addressed.
J. Cardiovasc. Dev. Dis. 2018, 5(1), 14; https://doi.org/10.3390/jcdd5010014
Submission received: 31 January 2018
/
Revised: 16 February 2018
/
Accepted: 18 February 2018
/
Published: 20 February 2018
(This article belongs to the Special Issue Cyclic Nucleotide Signaling and the Cardiovascular System)
Abstract
:A-kinase anchoring proteins (AKAPs) and cyclic nucleotide phosphodiesterases (PDEs) are essential enzymes in the cyclic adenosine 3′-5′ monophosphate (cAMP) signaling cascade. They establish local cAMP pools by controlling the intensity, duration and compartmentalization of cyclic nucleotide-dependent signaling. Various members of the AKAP and PDE families are expressed in the cardiovascular system and direct important processes maintaining homeostatic functioning of the heart and vasculature, e.g., the endothelial barrier function and excitation-contraction coupling. Dysregulation of AKAP and PDE function is associated with pathophysiological conditions in the cardiovascular system including heart failure, hypertension and atherosclerosis. A number of diseases, including autosomal dominant hypertension with brachydactyly (HTNB) and type I long-QT syndrome (LQT1), result from mutations in genes encoding for distinct members of the two classes of enzymes. This review provides an overview over the AKAPs and PDEs relevant for cAMP compartmentalization in the heart and vasculature and discusses their pathophysiological role as well as highlights the potential benefits of targeting these proteins and their protein-protein interactions for the treatment of cardiovascular diseases.
1. Introduction
Cardiovascular diseases (CVD) represent the leading cause of death worldwide and hypertension is the main risk factor for such conditions [1]. Treatments targeting the causes of cardiovascular diseases such as hypertension or heart failure are rare [2].
The second messenger cyclic adenosine 3′-5′ monophosphate (cAMP) is ubiquitous and functions as a signal transducer of many extracellular cues [3]. It regulates a variety of biological processes that are essential for, among others, proper cardiac function and it is involved in disease [4,5]. cAMP exerts its effects via activation of downstream effector proteins, i.e., cAMP-dependent protein kinase A (PKA), exchange proteins activated by cAMP (Epac) and cyclic nucleotide-gated ion channels (CNG), hyperpolarization-activated cyclic nucleotide-gated channels (HCN) and the recently identified Popeye domain containing (POPDC) proteins [6,7,8].
The plethora of extracellular signals, the limited number of intracellular cAMP effectors and the requirement of a specific biological response to each of the external signals imply a tight control of the intracellular signaling. This is achieved through signaling in defined cellular compartments. Local cAMP pools are established by the interplay of essentially four processes within the cell, namely cAMP synthesis, its diffusion, formation of multi-protein signaling complexes and cAMP degradation. In the heart, stimulation of β-adrenoceptors (β-ARs) triggers the activation the α-subunits of the stimulatory G proteins (Gs), which in turn stimulate adenylyl cyclases (ACs) to convert ATP to cAMP [9]. The formation of multi-protein signaling complexes in the cAMP signaling pathway is orchestrated by the family of A-kinase anchoring proteins (AKAPs), which act as scaffolds and engage in direct protein-protein interactions, including with PKA, and target them to defined subcellular compartments [10,11,12,13,14,15]. AKAPs play essential roles both in the heart and vascular physiology by coordinating complexes involved in the regulation of various processes including endothelial-barrier function [16,17] cardiac contraction and relaxation [18,19,20,21] and action potential duration [22,23]. AKAPs apparently play a role in several pathophysiological conditions in the cardiovascular system, e.g., in the heart their dysregulation is associated with heart failure [24,25].
Termination of cAMP signaling is predominantly achieved by hydrolysis of the phosphodiester bond within the second messenger, a reaction catalyzed by cyclic nucleotide phosphodiesterases (PDEs) [26]. Various PDE families regulate different aspects of cardiac and vascular muscle functions [27], such as the endothelial barrier function [28,29], the Ca2+ handling and thus contractility [30] and the basal pacemaking activity [31]. PDEs are also involved in the pathological cardiac remodeling and dysfunction [4,32,33,34,35].
The aim of this review is to provide an overview over the AKAPs and PDEs that are relevant in the compartmentalization of cAMP signaling in the cardiovascular system, to discuss their role in physiology and pathophysiology and the potential of these proteins and their protein-protein interactions as pharmacological targets in cardiovascular diseases.
2. A-kinase Anchoring Proteins (AKAPs)
AKAPs are a family of over 40 different scaffolding proteins and are key players in the spatio-temporal control of cAMP-dependent signaling by targeting PKA and additional signaling proteins including ACs, PDEs, further protein kinases and phosphatases to specific subcellular compartments [11,36,37,38]. PKA is the major downstream effector of cAMP. It is a serine/threonine kinase with broad specificity that controls many cellular processes, e.g., metabolism, cell growth, cell division and cardiac myocyte contraction [39]. It is a heterotetramer that consists of two catalytic subunits (Cα, Cβ or Cγ) kept in an inactive state by two regulatory RI (RIα or RIβ) or RII (RIIα or RIIβ) subunits that are organized as homodimers in the holoenzyme [40]. Upon cAMP binding to the R subunits, the C subunits are released and thus activated and subsequently phosphorylate local substrates [39]. This view was recently confirmed by quantitative mass spectrometric analyses [41]. However, PKA holoenzyme can also be active, as indicated by early biochemical experiments [42,43]. This notion was supported by recent fluorescence resonance energy transfer (FRET) imaging-based experiments, which suggested that physiological cAMP levels promote only minimal dissociation of the C subunits from the holoenzyme, thereby limiting the range of PKA action to the substrates in the immediate proximity [44]. Thus, it appears that both PKA holoenzyme and/or the dissociated C subunits can be active.
The structural feature that all AKAPs share is their ability to bind PKA via their A-kinase binding domains (AKBs), a structurally conserved amphipathic helix of 14–18 amino acids that docks into the hydrophobic groove formed by the N-terminal dimerization/docking (D/D) domains upon R subunits’ dimerization (Figure 1) [45,46,47,48]. Despite the fact that most AKAPs bind to PKA-RII subunits [37,49], there are the so-called dual specific AKAPs [50] that can bind both RI and RII subunits as well as AKAPs that specifically bind RI subunits (e.g., sphingosine kinase interacting protein (SKIP) and small membrane (sm) AKAP) [51,52,53]. Recently, hydrophilic anchor points have been identified within and outside the amphipathic helix forming the AKB that are involved in determining the affinity of the binding between an AKAP and the D/D domain. These observations suggest that targeting the amino acids that act as anchor points could lower the binding affinity or even prevent the interaction, making them candidates for pharmacological targeting. Moreover, targeting the anchor points makes the development of selective inhibitors of specific AKAP-PKA interactions feasible [48]. Selective inhibitory agents would be valuable tools for the investigation of cellular functions of individual AKAP-PKA interactions and could be starting points for drug development efforts.
2.1. AKAP Subcellular Localization
Targeting of AKAPs to specific subcellular compartments is essential for a coordinated cAMP-dependent signaling response, including accurate PKA-catalyzed substrate phosphorylation [54]. AKAPs can be directed to various cellular compartments, including the plasma membrane (PM, e.g., AKAP18α, AKAP18β, AKAP79 [55,56,57]), the sarcoplasmic reticulum (SR, e.g., AKAP18δ [20]), the cytosol (e.g., SKIP, GSKIP [51,58,59,60,61]), the cytoskeleton (e.g., gravin, ezrin [62]), the mitochondria (e.g., D-AKAP1 [63]) and the nucleus (e.g., pericentrin and AKAP350 [64,65]).
2.2. AKAPs in the Cardiovascular System
Several AKAPs are expressed in the cardiovascular system (Table 1). They regulate a variety of processes and are key proteins in maintaining the homeostatic functioning of the heart and vasculature [66]. For instance, gravin and AKAP220 are involved in maintaining the vascular integrity [16,17]. Homeostasis of the vascular tone is achieved through tight control of the balance between contraction and relaxation of vascular smooth muscle cells (VSMC), processes in which AKAP79 is involved [67,68]. Ca2+ handling and thus cardiac myocyte contractility is regulated by several macromolecular protein complexes whose platforms are AKAPs, e.g., AKAP18α, γ and δ, mAKAPβ [19,20,21,69]. The AKAP Yotiao is the key player in cardiac myocyte repolarization that follows contraction [22]. Several AKAPs are involved in stress response-induced cardiac myocyte hypertrophy, including AKAP-Lbc and mAKAPβ [70,71]. AKAP79 and gravin are important for the recycling of β1-ARs and β2-ARs, respectively [72,73].
2.2.1. AKAPs Regulating the Endothelial Barrier Function
The vascular endothelium lining the intima of blood vessels consists of a layer of endothelial cells tightly adherent to each other through cell-cell junctions. A healthy endothelium plays an essential role in the proper functioning of the vascular system. It regulates macromolecular permeability and anti-inflammatory, anti-thrombotic and anti-hypertrophic responses. Inflammatory conditions trigger pathological changes in the vascular system that lead to endothelial dysfunction, a state in which pathologically activated endothelial cells lose their barrier properties and initiate expression of pro-inflammatory adhesion molecules on their surface [74]. This results in increased vascular permeability allowing the infiltration of various molecules such as lipoproteins into the sub-endothelial space, and of circulating immune cells (e.g., monocytes). Ultimately, this leads to severe pathological conditions including atherosclerosis, allergy and sepsis [75,76].
AKAP-mediated PKA compartmentalization is essential for the maintenance of proper endothelial barrier function [16,17,77]. The vascular endothelium integrity is mainly dependent on tight junctions (TJs), important in sealing space between adjacent cells, and on adherens junctions, which assure direct contacts with the actin cytoskeleton of neighboring cells, thus providing mechanical strength. AKAP220 associates with PKA, β-catenin and the endothelial adherens junctions protein VE-cadherin, tethering PKA in close proximity to the cell-cell junctions [16]. Gravin (also known as AKAP12 or AKAP250) promotes vascular integrity by regulating the actin cytoskeleton via p21-activated kinase family proteins 2 (PAK2), an actin cytoskeletal regulator and afadin (AF6), a linker of the actin cytoskeleton with intercellular adhesion molecules [17]. Rac1 is a member of the Rho family of small GTPases, which upon activation strengthens the adherens junctions and the cortical actin skeleton, thereby preserving the endothelial barrier [78]. Simultaneous depletion of gravin and AKAP220 inhibited cAMP-mediated Rac1 activation, underlining the importance of these AKAPs in preventing endothelial dysfunction [16].
One other member of the AKAP family is involved in maintaining vascular integrity, the long isoform of AKAP9. Following Epac1 activation, AKAP9 contributes to microtubule growth regulation and is essential for preserving the endothelial barrier [79].
2.2.2. AKAPs Regulating the Vascular Tone
Homeostasis of the vascular tone is maintained by a tight balance between dilation and constriction of blood vessel endothelium; the main regulator is the renin-angiotensin-aldosterone system (RAAS). The main effector molecule of this system is angiotensin II (AngII), which exerts most of its effects via angiotensin type I receptors (AT1R). For instance, arterial smooth muscle contraction is induced by AngII-dependent stimulation of AT1R, localized at the sarcolemma, and subsequent activation of phospholipase C (PLC), which catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG activates protein kinase C (PKC), which in turn phosphorylates L-type Ca2+ (CaV1.2) channels, thereby increasing their open probability and increasing Ca2+ entry into the cytosol [75].
In arterial smooth muscle cells, the activity of a specific subpopulation of CaV1.2 channels is regulated by AKAP79 (AKAP5/AKAP75/AKAP150)-dependent targeting of PKCα to the sarcolemma, which facilitates phosphorylation of the channels and increases their open probablility [80]. By affecting the opening probability of specific CaV1.2 channels, the AKAP79 complex regulates the so-called “CaV1.2 sparklets”, which refers to local elevations of intracellular Ca2+ pools that directly induce contraction of the VSMCs. The sparklets increase the vascular tone [67]. In addition, AKAP79 facilitates and most probably stabilizes the coupling of small clusters of adjacent CaV1.2 channels, which can then open synchronously and generate large CaV1.2 sparklets, thus increasing the contractile force [81,82]. Prolonged CaV1.2 channel activity and thus persistent CaV1.2 sparklets could lead to vascular dysfunction and eventually contribute to AngII-induced hypertension [80].
Transient receptor potential vanilloid 4 (TRPV4) channels are Ca2+ permeant channels that unlike the CaV1.2 channels, promote relaxation upon activation. Both CaV1.2- and TRPV4-mediated Ca2+ influxes activate adjacent ryanodine receptors (RyR), leading to release of Ca2+ from the SR into the cytosol in the form of Ca2+ sparks. While the CaV1.2-mediated Ca2+ influx increases contraction, the local TRPV4-generated Ca2+ sparks activate the large-conductance, Ca2+-activated K+ (BK) channels, which promote membrane hyperpolarization and closure of the CaV1.2 channels, ultimately resulting in relaxation [83,84]. In the arterial smooth muscle cells, AngII increased TRPV4 activity via PKC, which is tethered to the sarcolemma in close proximity of the channel by AKAP79, thus opposing the CaV1.2 channel-induced vasoconstriction [68].
In conclusion, AKAP79 plays an essential role in the control of arterial vascular tone by regulating two opposing processes, contraction and relaxation of arterial myocytes.
2.2.3. AKAPs Controlling Excitation-Contraction Coupling
The cycling of Ca2+ between the cytosol and the SR is at the basis of cardiac contraction and relaxation. Key players in these processes are L-type Ca2+ CaV1.2 channels, RyR2, SR Ca2+ ATPase 2 (SERCA2) and the Na+/Ca2+ exchanger. More specifically, upon sarcolemma depolarization, CaV1.2 channels located at the T tubules open allowing Ca2+ influx into the cardiac myocyte. This causes Ca2+-induced Ca2+ release from the SR into the cytosol through RyR2 located at the SR. The Ca2+, upon interaction with troponin T located on the thin myofibers, promotes contraction. Relaxation occurs via SERCA2-mediated Ca2+ re-uptake into the SR and through Ca2+ transport out of the cell by Na+/Ca2+ exchangers [85]. SERCA2 is activated upon the phosphorylation and subsequent dissociation of phospholamban (PLN), a SR phosphoprotein [20,86,87].
β-ARs introduce a further layer into the regulation of cardiac myocyte contractility. Their stimulation induces PKA-dependent phosphorylation of several proteins involved in Ca2+ handling, e.g., the CaV1.2 channels, RyR2 and PLN. These phosphorylations are facilitated by distinct AKAPs.
AKAP18α is a membrane-associated scaffolding protein and is the smallest AKAP7 gene transcript, comprising 81 amino acids. AKAP18α promotes cardiac contractility by mediating the PKA-dependent phosphorylation of CaV1.2 channels at Serine 1928 (Ser1928) on its α subunit and at multiple sites on its β subunit, which enhances the open probability of the channel and increases the Ca2+ current [69,88]. The activity of a subset of CaV1.2 channels associated with caveolin-3 (Cav3) is regulated by PKA phosphorylation of the specific channel subpopulation mediated by an AKAP79 (AKAP5/AKAP75/AKAP150)-based macromolecular complex consisting of β-AR, PKA, AC5/6 and protein phosphatase calcineurin (PP2B) [89]. The muscle selective AKAP, mAKAPβ (a short version of mAKAP) associates with RyR2 at the SR and thereby facilitates the PKA phosphorylation of the channel, leading to enhanced opening of the channel and subsequent enhanced Ca2+ release from the SR into the cytosol [19]. In addition, mAKAPβ interacts with the Na+/Ca2+ exchanger 1 at the sarcolemma and promotes the PKA-dependent activation of the exchanger, resulting in increased Ca2+ efflux [90,91]. AKAP18δ (rat heart) and AKAP18γ (human heart) facilitate the PKA phosphorylation of PLN and promote its dissociation from SERCA2 and hence activation of the ATPase, thus enhancing the re-uptake of Ca2+ into the SR [20,21,92].
2.2.4. AKAPs Regulating Cardiac Repolarization
The cardiac repolarization phase is initiated by the slow heart potassium current (IKs) moving outwards through the IKs potassium channel, a macromolecular complex consisting of a pore-forming α subunit (KCNQ1) and a regulatory β subunit (KCNE1) along other intracellular proteins [93]. The AKAP Yotiao, the smallest transcript of the AKAP9 gene, is essential for cardiac repolarization since it mediates the PKA-dependent phosphorylation of KCNQ1 and therefore regulates the activity of the IKs potassium channel [22]. Mutations in the KCNQ1 subunit or Yotiao increase the duration of the action potential and lead to type I long-QT syndrome (LQT1), a channelopathy that can elicit fatal arrhythmia [94]. Another AKAP that contributes to the regulation of cardiac action potentials is the dual specific D-AKAP2 (AKAP10). A single-nucleotide polymorphism (SNP) in its PKA binding domain causes a decrease in the PR interval in the electrocardiogram, which in turn can cause arrhythmias and sudden cardiac death [54,95,96,97].
2.2.5. AKAPs Involved in Cardiac Stress Response
Cardiac hypertrophy is a stress-induced adaptation to maintain normal heart function [23,25]. At the cellular level, it is characterized by the upregulation of specific genes that promote the non-mitotic growth of cardiac myocytes [98]. AKAP-Lbc encodes in addition to its AKAP function for a guanine nucleotide exchange factor (GEF) that directly binds and activates the GTP-binding protein RhoA [99,100,101,102]. The interaction is involved in both cardiac development [103] and pathological cardiac myocyte hypertrophy [70]. α1-AR stimulation enhances the RhoGEF activity of AKAP-Lbc, which in turn activates RhoA, contributing to a pathological increase in the hypertrophic response [70]. PKA-mediated phosphorylation at Ser1565 of AKAP-Lbc leads to the recruitment of 14-3-3 proteins, which inhibit the Rho-GEF activity of the anchoring protein [104]. Also, an AKAP-Lbc-dependent signalosome mediates the activation and cytosolic release of activated protein kinase D (PKD), which has been shown to promote cardiac hypertrophy by facilitating the nuclear export of histone deacetylase 5 (HDAC5) [105,106].
Another AKAP that plays a central role in modulating stress signal-induced hypertrophic pathways is mAKAPβ. It coordinates a variety of cAMP-responsive enzymes. This anchoring protein is targeted to the nuclear envelope of cardiac myocytes via an interaction with nesprin-1α [107]. At the SR it can integrate and transduce a variety of hypertrophic signals [71]. For instance, mAKAPβ-mediated PKA phosphorylation and subsequent activation of RyR2 located at the nuclear envelope promotes the activation and nuclear translocation of the pro-hypertrophic transcription factor nuclear factor of activated T cells (NFAT) [108]. In addition, a mAKAPβ-based signalosome consisting of PKA, PDE4D3, Epac1, ERK5 and PP2A promotes ERK5-induced cardiomyocyte hypertrophy [71,109]. Cardiac remodeling can also be regulated by hypoxia, a process in which a mAKAP-based protein complex consisting of hypoxia-inducible factor 1α (HIF-1α), prolyl hydroxylase domain protein (PHD), the von Hippel-Lindau protein (pVHL) and the E3 ligase designated seven in absentia homolog 2 (Siah2) plays a role. More specifically, when oxygen levels are reduced, mAKAP promotes the degradation of PHD and thereby facilitates an increase in HIF-1α levels, which regulates transcription of genes that promote cell survival [110].
Other AKAPs that are thought to be involved in the cardiac stress response are D-AKAP1 and SKIP [111,112]. D-AKAP1 is a scaffolding protein of the outer mitochondrial membrane, which is protective against cardiac hypertrophy since its overexpression leads to cardiac myocyte cell size reduction and inhibition of the β-AR agonist isoproterenol-induced hypertrophy [111]. Moreover, D-AKAP1 expression maintains the mitochondrial structure and function in the heart and reduces the infarct size, cardiac remodeling and mortality under conditions of ischemia, i.e., after myocardial infarction [113].
SKIP plays an important role in the generation of the cardioprotective and anti-apoptotic lysophospholipid sphingosine-1-phosphate (S1P) produced upon myocardial ischemia-reperfusion injury [112]. It is involved in the regulation of sphingosine kinase type 1 (SPHK1), which upon activation phosphorylates sphingosine to form S1P [114].
2.2.6. AKAPs Involved in the β-ARs Desensitization/Resensitization Cycle
Upon activation, β-ARs are phosphorylated and subsequently bind β-arrestin, which prevents further ligand binding leading to receptor desensitization. The phosphorylated β-ARs are internalized and reach the early endosomes where they undergo resensitization after PP2A-mediated dephosphorylation. Upon resensitization, the non-phosphorylated receptors are recycled to the plasma membrane where they can bind further ligands. Therefore, β-AR desensitization and resensitization are essential processes in maintaining the proper functioning of the receptor [115].
Gravin and AKAP79 are important in the desensitization/resensitization cycle [72,73]. A gravin-based complex consisting of PKA, PKC, PP2B, β-arrestin and G protein-linked receptor kinase 2 (GRK2) is essential for the desensitization and resensitization of the β2-ARs, with which it interacts at their C-terminal tail [72]. AKAP79 mediates the PKA-dependent phosphorylation of the β1-ARs by also binding to the C terminus of the receptor, leading to their recycling and resensitization [73].
2.3. Aberrant cAMP Compartmentalization Can be Visualized
Dysregulation of local cAMP signaling is associated with cardiovascular diseases, e.g., maladaptive cardiac remodeling and heart failure [116,117]. FRET-based imaging using genetically encoded sensors (cAMP-binding and PKA activity reporters) is utilized to visualize local cAMP signaling components and real-time changes in cAMP levels with high spatio-temporal resolution [118,119,120,121]. Such sensors can be targeted to various subcellular locations including in cardiac myocytes in close proximity of sarcolemmal ion channels and SR proteins involved in Ca2+ handling, e.g., RyR2 and SERCA2a [122,123,124,125]. In addition, the FRET-based reporters can be used for cAMP imaging in intact cardiac tissue as well as in ex vivo and in vivo hearts [126]. For monitoring activities of signaling molecules in their cognate microdomains, FRET approaches have also been combined with other techniques such as scanning ion-conductance microscopy (SICM), a non-optical method that allows the imaging of both cell membrane morphology and functional parameters at resolutions in the nanometer range [127,128,129,130].
3. Cyclic Nucleotide Phosphodiesterases (PDEs)
Hydrolysis by PDEs is the main route for lowering of intracellular levels of cAMP and cGMP and is essential for the spatio-temporal regulation of cyclic nucleotide-dependent signaling [26]. The PDE superfamily consists of 21 genes that give rise to more than 100 proteins due to differential transcription initiation sites and alternative splicing. PDEs are classified into 11 families (PDE1–PDE11). They differ in their primary structures, substrate specificities, mechanisms of regulation and kinetic properties [131]. Some PDE families selectively hydrolyze cAMP or cGMP, while others, the so-called dual specific PDEs, catalyze the hydrolysis of both second messengers (Figure 2) [132].
PDEs display a common general structure consisting of three components: a family-specific N-terminal regulatory domain, a conserved catalytic domain (25–52 % homology) and a C-terminal domain that can be either phosphorylated by the mitogen-activated protein kinase (MAPK) or prenylated [131,133,134,135]. The regulatory domains contain various structural features involved in the regulation (i.e., sites for covalent modifications, e.g., phosphorylation), binding of regulatory molecules (e.g., Ca2+-binding protein calmodulin), localization (targeting domains and protein-protein interaction motifs, e.g., AKAP-binding motifs) and dimerization (e.g., GAF domains) of the enzymes [131,136,137]. The catalytic domains feature in the active site a Zn2+ binding motif and an additional divalent metal binding site that is most probably occupied by Mg2+, but could also correspond to Mn2+ and Co2+ [138].
3.1. PDE Subcellular Localization
The subcellular localization of PDEs is key in achieving compartmentalized cyclic nucleotide signaling and, therefore, in the generation of specific physiological responses [136]. PDEs are located at various intracellular locations, e.g., the cytosol (e.g., PDE3A3 and PDE5 [139,140]), plasma membrane (e.g., PDE2A, PDE3A1, PDE6α, PDE6β [139,141,142]), the Golgi–centrosome (e.g., PDE7A1 [143]) and nuclear regions (e.g., PDE9A1, PDE9A16, PDE9A17 [144]).
3.2. PDEs in the Cardiovascular System
Members from most PDE families are expressed in the cardiovascular system and regulate a variety of processes essential for the proper functioning of the heart and vasculature (Table 2). The PDE families 2, 3, 4 and 5, for example, regulate the endothelial barrier function and are, therefore, of utmost importance in maintaining vascular integrity [28,145,146]. Several members of the PDE families 2, 3, 4, 5 and 8 are involved in the control of cardiac contractility [30,120,147,148,149,150]. In addition, the basal pace-making activity of the sinoatrial (SA) node of the heart is regulated by two PDE families, namely PDE3 and PDE4 [151]. In addition, various PDE family members, PDE1A, PDE3A, PDE4B, PDE4D, PDE5 and PDE9A, are implicated in the cardiac stress response, which triggers pathological cardiac remodeling and ultimately cardiac dysfunction (e.g., heart failure and arrhythmias) [34,152,153,154,155,156].
3.2.1. PDE3A and Autosomal Dominant Hypertension with Brachydactyly (HTNB)
PDE3A along with PDE3B belongs to the PDE3 family, also known as the cGMP-inhibited cAMP PDE family, which is able to hydrolyze both cAMP and cGMP in a competitive manner. PDE3A is highly expressed and plays important roles in VSMCs, cardiac myocytes, platelets and oocytes, whereas PDE3B is mainly expressed in adipose and soft tissue. Upon alternative splicing, three PDE3A isoforms are generated, namely PDE3A1 (136 kDa), PDE3A2 (118 kDa) and PDE3A3 (94 kDa) (Figure 3A). They are located in different cellular compartments. PDE3A1 is the main isoform found in human cardiac myocytes and is predominantly located at membranes. It contains two N-terminal hydrophobic regions (NHR), of which the first one consists of four transmembrane domains. PDE3A2, which lacks the first but contains the second NHR can be both membrane-associated and cytosolic and is the main variant found in VSMCs. PDE3A3 is found only in the cytosol, since it lacks both previously mentioned hydrophobic regions. All three isoforms possess the same catalytic region and present high similarities regarding their catalytic activity and inhibitor sensitivity (Figure 3A) [157,158].
Mutations in genes encoding for distinct PDE family members can have detrimental effects and cause specific human diseases. One such example is represented by the Mendelian syndrome Autosomal-dominant hypertension with brachydactyly type E (HTNB), caused by missense mutations in the gene encoding for PDE3A [159]. The syndrome is characterized by an age-dependent progressive hypertension, brachydactyly type E and blood vessel hyperplasia [159]. If untreated, blood pressure increases by 50 mm Hg and patients die from stroke before age 50 years. Surprisingly, hypertension-associated end organ damage such as cardiac hypertrophy, kidney damage or hypertensive retinopathy is low [160,161].
Eight mutations in PDE3A were discovered in eight unrelated families from Turkey, France, the United States, South Africa, Canada, Netherlands and Japan. All these mutations were missense, gain of function mutations and found in close proximity to each other. The identified mutations cause amino acid substitutions in a region between amino acids 445 and 449 and increases of PKA-mediated phosphorylation of serine residues 428 and 438 of PDE3A1 and PDE3A2. The region is not present in PDE3A3 (Figure 3A) [159]. The substitutions lead to increased cAMP affinity and hydrolytic activity of the enzymes (Figure 3B). In addition, the hyperactive enzyme is erroneously localized in microsomal fractions from HeLa cells, suggesting that aberrant compartmentalization is detrimental in the cardiovascular system [159,161]. VSMCs from patients expressing the hyperactive version of the enzyme with the T445N substitution display higher proliferation rates, explaining the vascular phenotype.
In the human heart, PKA-mediated phosphorylation of PDE3A1 induces its recruitment to an AKAP18-based signalosome in the heart that controls the Ca2+ reuptake into the SR and thereby participates in the control of cardiac relaxation [21]. An extended overview over functions of PDE3 in the heart was provided in recent reviews (e.g., [162,163]).
3.3. PDE Inhibitors
Due to their essential physiological and pathological roles in cyclic nucleotide signaling, PDEs are considered pharmacological targets for a variety of cardiovascular diseases, including atherosclerosis, hypertension, heart failure and intermittent claudication [26,164,165,166]. Several inhibitors of PDE3, 4 and 5 are approved as drugs, some of which are used for the treatment of cardiovascular diseases.
3.3.1. PDE3
The PDE3 inhibitor cilostazol is an antiplatelet agent with vasodilatory and antiproliferative properties. It has been widely studied in a number of cardiovascular diseases including coronary and peripheral artery diseases and cerebrovascular disease [167]. Cilostazol is administered for the treatment of peripheral arterial circulatory disorders and also used as an antiplatelet agent in patients that underwent carotid artery stenting [168,169]. In addition, it is also approved for the treatment of intermittent claudication-induced symptoms [170,171,172]. Cilostazol appears to be a promising therapeutic agent for secondary prevention of stroke and was shown to improve right ventricular systolic function as well as to decrease pulmonary artery pressure [167,173]. PDE3 inhibitors inhibit neointima formation in a rat balloon double-injury model displaying neither cytotoxicity nor effects on VSMC migration, and thus are considered targets in preventing acute re-occlusion after angioplasties, e.g., percutaneous transluminal coronary angioplasty (PTCA) [174].
Milrinone is another PDE3 inhibitor. It has inotropic and vasodilatory properties, and is widely used in patients with end-stage heart failure in order to temporarily improve cardiac contractility (positive inotropic effect) and decrease vascular resistance. Taking into account that long-term administration of milrinone can induce apoptosis of cardiac myocytes, cardiac arrhythmias, hypotension and increases cardiovascular mortality, it is only used in a selected group of patients [170,175,176,177]. A potential explanation for the long-term PDE3 inhibitor therapy-induced mortality could be the fact that PDE3A inhibition induces cardiac myocyte apoptosis via a PDE3A-inducible cAMP early repressor (ICER) feedback loop. More specifically, PDE3A inhibition leads to PKA activation and ICER protein stabilization, which, in turn, promotes cardiac myocyte apoptosis. Therefore, therapeutic strategies that would diminish PDE3A activity without affecting the PDE3A-ICER feedback loop could promote the beneficial effects while by-passing the side effects [152,162]. Current research aims at determining the effects of milrinone on pulmonary hypertension and right ventricular failure, where it is believed to be particularly helpful [176].
3.3.2. PDE4
The PDE4 family is encoded by four genes, PDE4A, PDE4B, PDE4C and PDE4D [178] and was shown to be involved in the excitation-contraction coupling regulation, especially in rodents. It has been recently suggested that PDE4 inhibitors could be beneficial in treating sepsis in infants with cardio-renal syndrome (CRS) since they are effective in improving cardiac function in a rat model suffering from sepsis-induced acute cardiac dysfunction and kidney injury [179]. In addition, PDE4 depletion stabilized the endothelial barrier by reducing the atrial natriuretic peptide (ANP)-induced vascular permeability and, therefore, was efficient in maintaining the plasma volume [180]. Despite the fact that PDE4A, PDE4B and PDE4D are expressed in the human and rodent heart, with PDE4D being the predominant isoform found in the human heart [181], there is no approval for a PDE4 inhibitor for the treatment of cardiovascular diseases. A highly selective PDE4 inhibitor, roflumilast, has been approved in various countries for the treatment of chronic obstructive pulmonary disease (COPD), a chronic inflammatory lung disease characterized by heavily breathing due to obstructive airflow from the lungs as well as a decline of lung function over time [182,183,184]. Another inhibitor, apremilast is employed for the treatment of psoriasis [185].
3.3.3. PDE5
PDE5A is the sole gene coding for the PDE5 family, which plays an essential role in the cardiovascular system. PDE5 expression is low in the healthy cardiac tissue, whereas it is upregulated in the diseased heart [155,186]. PDE5 inhibition counteracts cardiac remodeling and fibrosis of isolated cardiac fibroblasts via repression of transforming growth factor (TGF)-β1-induced Smad signaling [187]. PDE5 depletion inhibits left ventricular remodeling induced by hypertrophic and pro-fibrotic stimuli [188]. Reduction in PDE5 expression was beneficial for chronic heart failure patients by enhancing the endothelium-dependent, flow-mediated vasodilation [189]. In addition, high PDE5 expression was identified in the hypertrophic human right ventricle and its inhibition enhanced contractility, particularly important for pulmonary hypertension [186]. PDE5 inhibitors such as sildenafil, vardenafil and tadalafil are approved for the treatment of erectile dysfunction and pulmonary hypertension but are not yet approved for the treatment of other cardiovascular diseases [26]. Nevertheless, recent studies suggest potential therapeutic benefits for PDE5 inhibitors, i.e., sildenafil and tadalafil in the treatment of myocardial infarction, ischemia/reperfusion injury, endothelial dysfunction, cardiac hypertrophy and heart failure [190,191].
3.3.4. Potential for PDE1, PDE2, PDE8 and PDE9 Inhibitors
PDE1, PDE2, PDE8 and PDE9 inhibition is considered a therapeutic opportunity for the treatment of cardiovascular diseases but inhibitors are not approved.
The PDE1 family is encoded by three distinct genes, PDE1A, PDE1B and PDE1C, and is the only PDE family activated by calcium/calmodulin (Ca2+/CaM) binding [192,193]. Due to their potential to dilate coronary arteries, inhibition of PDE1 enzymes may be beneficial for the treatment of coronary artery disease (CAD) and angina pectoris [194]. Nuclear PDE1A is important for the proliferation of VSMCs and, therefore, could contribute to neointima formation in diseases, e.g., atherosclerosis and restenosis [195]. Thus, diminishing its expression could decrease pathological neointima development. In addition, inhibition of the PDE1 family might improve cardiopathy and pulmonary arterial hypertension since it decreases the structural remodeling process underlying these two conditions [196].
PDE2A is the only gene coding for the PDE2 family and plays a central role in the cardiac CaV1.2 current regulation. The expression is up-regulated in human failing hearts [197,198]. Inhibition of PDE2 had a positive inotropic effect in dogs and mice, whereas its overexpression decreased the heart rate in mice. Interestingly, in a heart-specific PDE2-transgenic mouse model, increased PDE2 abundance prevents ventricular arrhythmias by inhibiting Ca2+ leak from the SR and helps in maintaining the contractile function of the heart after myocardial infarction [199]. On the contrary, a recent study in patients that had experienced an acute myocardial infarction (AMI) suggests that inhibition of endothelial PDE2A could have a beneficial effect and improve the clinical outcome. Hypoxia and pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) promote PDE2A activation, which results in diminished submembrane cAMP levels and endothelial barrier disruption. This facilitates the extravasation of activated neutrophils and leads to inflammation in the early post-myocardial infarction phase [29].
The PDE8 family comprises two members, PDE8A and PDE8B and regulates excitation-contraction coupling in ventricular myocytes. More specifically, it has been suggested that PDE8A controls at least one cAMP pool involved in the cardiac myocyte-dependent Ca2+ cycling regulation. It was also observed that PDE8A deletion caused both increased RyR2 leak as well as enhanced Ca2+ refilling of the SR [150].
The PDE9 family is encoded by a single gene, PDE9A, and consists of more than 20 different splice variants. PDE9A expression was identified in human and rodent hearts, where its expression increased upon hypertrophy and heart failure development [156,200]. PDE9A depletion had a protective effect for the heart against pathological remodeling caused by pressure overload and it reversed a previously established heart disease without requiring the activity of NO synthase [156].
4. Concluding Remarks
Compartmentalized cyclic nucleotide signaling is found at the basis of precision of cellular signaling and its dysregulation is associated with various pathological conditions including several cardiovascular diseases. Local pools of cAMP are established by the interplay of cAMP synthesis, diffusion, degradation as well as positioning of the relevant signaling proteins. AKAPs and PDEs are essential players in these processes since they orchestrate the formation of multi-protein signaling complexes and terminate local cAMP signaling, respectively. This interplay ensures the spatio-temporal regulation of cyclic nucleotide-dependent signaling. Despite the fact that both molecules are key elements in the cAMP signaling pathway, very little is known with respect to their direct interaction or their interplay in the cardiovascular system. However, a few PDE-containing AKAP complexes have been identified; examples are the SERCA2/AKAP18 signalosome, which incorporates PDE3A1 upon its phosphorylation and is important for cardiac contractility, and the PDE4D3 containing mAKAPβ-based signalosome involved in cardiomyocyte hypertrophy regulation ([21,71,109]). Alterations in AKAP expression and their protein-protein interactions are associated with various cardiovascular diseases [12,24]. Hence the development of pharmacological agents targeting such dysregulated signaling components for evaluating their relevance as pharmacological targets is needed. First examples show that targeting AKAPs and their protein-protein interactions with small molecules is possible. For instance, an AKAP-PKA interaction inhibitor, FMP-API-1 [201] was identified. Recently, a novel small molecule, Scaff10-8, was developed, which inhibits the interaction of AKAP-Lbc and RhoA and prevents the AKAP-Lbc-mediated RhoA activation, an event pathologically activated in models of cardiac hypertrophy [102]. Further molecules directed against the AKAP-Lbc-RhoA interface have recently been identified and may serve to guide to further preclinical drug development efforts [202,203].
Approved inhibitors of PDEs target the catalytic activities of PDEs. However, the catalytic domains of the various members of individual families are identical and inhibition of one inhibits all. This lack of selectivity presumably explains PDE inhibitor therapy-associated side effects, which are frequent and dramatic over long-term administration [158]. PDE isoform-selective inhibition may be achieved through disruption of specific protein-protein interactions. and therefore the displacement of particular PDE isoforms from their subcellular compartments [204].
In conclusion, targeting proteins directing compartmentalized cAMP signaling, in particular AKAPs and PDEs, not only serves to understanding their role in heart and vascular physiology and pathophysiology but also has therapeutic potential for the treatment of a wide range of cardiovascular diseases.
Acknowledgments
This work was supported by grants from the Else Kröner-Fresenius-Stiftung (2013_A145), the German-Israeli Foundation (G.I.F. I-1210-286.13/2012), the German Centre for Cardiovascular Research (DZHK 81X210012 and B18-005 SE), the Deutsche Forschungsgemeinschaft (DFG KL1415/7-1) and the Bundesministerium für Bildung und Forschung (BMBF; 16GW0179K) to EK.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lawes, C.M.; Vander Hoorn, S.; Rodgers, A.; Hypertension, I.S.o. Global burden of blood-pressure-related disease, 2001. Lancet 2008, 371, 1513–1518. [Google Scholar] [CrossRef]
- Bolívar, J.J. Essential hypertension: An approach to its etiology and neurogenic pathophysiology. Int. J. Hypertens. 2013, 2013, 547809. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Fischmeister, R.; Castro, L.R.; Abi-Gerges, A.; Rochais, F.; Jurevicius, J.; Leroy, J.; Vandecasteele, G. Compartmentation of cyclic nucleotide signaling in the heart: The role of cyclic nucleotide phosphodiesterases. Circ. Res. 2006, 99, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.K.; Nikolaev, V.O. Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol (Oxf) 2013, 207, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bertinetti, D.; Herberg, F.W. cAMP-dependent protein kinase and cGMP-dependent protein kinase as cyclic nucleotide effectors. Handb. Exp. Pharmacol. 2017, 238, 105–122. [Google Scholar] [PubMed]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP sensor epac proteins and their role in cardiovascular function and disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.; Schindler, R. New kids on the block: The popeye domain containing (popdc) protein family acting as a novel class of cAMP effector proteins in striated muscle. Cell Signal. 2017, 40, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Szaszák, M.; Christian, F.; Rosenthal, W.; Klussmann, E. Compartmentalized cAMP signalling in regulated exocytic processes in non-neuronal cells. Cell Signal. 2008, 20, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Skroblin, P.; Grossmann, S.; Schafer, G.; Rosenthal, W.; Klussmann, E. Mechanisms of protein kinase A anchoring. Int Rev. Cell Mol. Biol 2010, 283, 235–330. [Google Scholar] [PubMed]
- Dema, A.; Perets, E.; Schulz, M.S.; Deák, V.A.; Klussmann, E. Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell Signal. 2015, 27, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Pidoux, G.; Taskén, K. Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J. Mol. Endocrinol. 2010, 44, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Dessauer, C.W.; Taskén, K. Creating order from chaos: Cellular regulation by kinase anchoring. Annu Rev. Pharmacol Toxicol 2013, 53, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Zaccolo, M. Microdomains in the Cardiovascular System; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Radeva, M.Y.; Kugelmann, D.; Spindler, V.; Waschke, J. PKA compartmentalization via AKAP220 and AKAP12 contributes to endothelial barrier regulation. PLoS One 2014, 9, e106733. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.B.; Choi, Y.K.; Lim, J.J.; Kwon, S.H.; Her, S.; Kim, H.J.; Lim, K.J.; Ahn, J.C.; Kim, Y.M.; Bae, M.K.; et al. AKAP12 regulates vascular integrity in zebrafish. Exp. Mol. Med. 2012, 44, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.C.; Johnson, B.D.; Westenbroek, R.E.; Hays, L.G.; Yates, J.R.; Scheuer, T.; Catterall, W.A.; Murphy, B.J. Primary structure and function of an A kinase anchoring protein associated with calcium channels. Neuron 1998, 20, 1017–1026. [Google Scholar] [CrossRef]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. Pka phosphorylation dissociates fkbp12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Lygren, B.; Carlson, C.R.; Santamaria, K.; Lissandron, V.; McSorley, T.; Litzenberg, J.; Lorenz, D.; Wiesner, B.; Rosenthal, W.; Zaccolo, M.; et al. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007, 8, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Shen, W.; Vandeput, F.; Szabo-Fresnais, N.; Krall, J.; Degerman, E.; Goetz, F.; Klussmann, E.; Movsesian, M.; Manganiello, V. Regulation of sarcoplasmic reticulum Ca2+ atpase 2 (serca2) activity by phosphodiesterase 3a (pde3a) in human myocardium: Phosphorylation-dependent interaction of pde3a1 with serca2. J. Biol. Chem. 2015, 290, 6763–6776. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Kurokawa, J.; Reiken, S.; Motoike, H.; D’Armiento, J.; Marks, A.R.; Kass, R.S. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the kcnq1-kcne1 potassium channel. Science 2002, 295, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Katus, H.A.; Olson, E.N.; Hill, J.A. Hypertrophy of the heart: A new therapeutic target? Circulation 2004, 109, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Deák, V.A.; Klussmann, E. Pharmacological interference with protein-protein interactions of akinase anchoring proteins as a strategy for the treatment of disease. Curr. Drug Targets 2016, 17, 1147–1171. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Reggi, E.; Arambasic, M.; Caso, S.; Maric, D. Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochim. Biophys. Acta 2016, 1863, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.E.; Kass, D.A. Cardiac phosphodiesterases and their modulation for treating heart disease. Handb. Exp. Pharmacol. 2017, 243, 249–269. [Google Scholar] [PubMed]
- Surapisitchat, J.; Jeon, K.I.; Yan, C.; Beavo, J.A. Differential regulation of endothelial cell permeability by cgmp via phosphodiesterases 2 and 3. Circ. Res. 2007, 101, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Spitzl, A.; Mathes, D.; Nikolaev, V.O.; Werner, F.; Weirather, J.; Špiranec, K.; Röck, K.; Fischer, J.W.; Kämmerer, U.; et al. Endothelial actions of anp enhance myocardial inflammatory infiltration in the early phase after acute infarction. Circ. Res. 2016, 119, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Miller, C.L.; Abe, J. Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ. Res. 2007, 100, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Tovar, A.; Vargas, M.L.; Kaumann, A.J. Phosphodiesterases PDE3 and PDE4 jointly control the inotropic effects but not chronotropic effects of (-)-cgp12177 despite PDE4-evoked sinoatrial bradycardia in rat atrium. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 379, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Abe, J.I.; Wei, H.; Huang, Q.; Walsh, R.A.; Molina, C.A.; Zhao, A.; Sadoshima, J.; Blaxall, B.C.; Berk, B.C.; et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: Implication in heart failure. Circulation 2005, 111, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Abi-Gerges, A.; Richter, W.; Lefebvre, F.; Mateo, P.; Varin, A.; Heymes, C.; Samuel, J.L.; Lugnier, C.; Conti, M.; Fischmeister, R.; et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ. Res. 2009, 105, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Oikawa, M.; Cai, Y.; Wojtovich, A.P.; Nagel, D.J.; Xu, X.; Xu, H.; Florio, V.; Rybalkin, S.D.; Beavo, J.A.; et al. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ. Res. 2009, 105, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Bobin, P.; Belacel-Ouari, M.; Bedioune, I.; Zhang, L.; Leroy, J.; Leblais, V.; Fischmeister, R.; Vandecasteele, G. Cyclic nucleotide phosphodiesterases in heart and vessels: A therapeutic perspective. Arch. Cardiovasc. Dis. 2016, 109, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Rababa’h, A.; Singh, S.; Suryavanshi, S.V.; Altarabsheh, S.E.; Deo, S.V.; McConnell, B.K. Compartmentalization role of A-kinase anchoring proteins (AKAPs) in mediating protein kinase A (PKA) signaling and cardiomyocyte hypertrophy. Int. J. Mol. Sci. 2014, 16, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.J.; Jones, B.W.; Scott, J.D. Networking with AKAPs: Context-dependent regulation of anchored enzymes. Mol. Interv. 2010, 10, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Langeberg, L.K.; Scott, J.D. Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 2015, 16, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Corbin, J.D. Structure and function of cyclic nucleotide-dependent protein kinases. Annu. Rev. Physiol. 1994, 56, 237–272. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Ilouz, R.; Zhang, P.; Kornev, A.P. Assembly of allosteric macromolecular switches: Lessons from PKA. Nat. Rev. Mol. Cell Biol. 2012, 13, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Walker-Gray, R.; Stengel, F.; Gold, M.G. Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches. Proc. Natl. Acad. Sci. USA 2017, 114, 10414–10419. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Fletcher, W.H.; Johnson, D.A. Regulation of cAMP-dependent protein kinase: Enzyme activation without dissociation. Biochemistry 1995, 34, 6267–6271. [Google Scholar] [CrossRef] [PubMed]
- Kopperud, R.; Christensen, A.E.; Kjarland, E.; Viste, K.; Kleivdal, H.; Doskeland, S.O. Formation of inactive cAMP-saturated holoenzyme of cAMP-dependent protein kinase under physiological conditions. J. Biol. Chem. 2002, 277, 13443–13448. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.D.; Esseltine, J.L.; Nygren, P.J.; Veesler, D.; Byrne, D.P.; Vonderach, M.; Strashnov, I.; Eyers, C.E.; Eyers, P.A.; Langeberg, L.K.; et al. Local protein kinase A action proceeds through intact holoenzymes. Science 2017, 356, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Ruehr, M.L.; Zakhary, D.R.; Damron, D.S.; Bond, M. Cyclic amp-dependent protein kinase binding to A-kinase anchoring proteins in living cells by fluorescence resonance energy transfer of green fluorescent protein fusion proteins. J. Biol. Chem. 1999, 274, 33092–33096. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G.; Lygren, B.; Dokurno, P.; Hoshi, N.; McConnachie, G.; Tasken, K.; Carlson, C.R.; Scott, J.D.; Barford, D. Molecular basis of akap specificity for PKA regulatory subunits. Mol. Cell 2006, 24, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Kinderman, F.S.; Kim, C.; von Daake, S.; Ma, Y.; Pham, B.Q.; Spraggon, G.; Xuong, N.H.; Jennings, P.A.; Taylor, S.S. A dynamic mechanism for akap binding to rii isoforms of cAMP-dependent protein kinase. Mol. Cell 2006, 24, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Götz, F.; Roske, Y.; Schulz, M.S.; Autenrieth, K.; Bertinetti, D.; Faelber, K.; Zühlke, K.; Kreuchwig, A.; Kennedy, E.J.; Krause, G.; et al. AKAP18:Pka-riiα structure reveals crucial anchor points for recognition of regulatory subunits of PKA. Biochem. J. 2016, 473, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- McSorley, T.; Stefan, E.; Henn, V.; Wiesner, B.; Baillie, G.S.; Houslay, M.D.; Rosenthal, W.; Klussmann, E. Spatial organisation of AKAP18 and PDE4 isoforms in renal collecting duct principal cells. Eur. J. Cell. Biol. 2006, 85, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.J.; Durick, K.; Weiner, J.A.; Chun, J.; Taylor, S.S. Identification of a novel protein kinase A anchoring protein that binds both type i and type ii regulatory subunits. J. Biol. Chem. 1997, 272, 8057–8064. [Google Scholar] [CrossRef] [PubMed]
- Kovanich, D.; van der Heyden, M.A.; Aye, T.T.; van Veen, T.A.; Heck, A.J.; Scholten, A. Sphingosine kinase interacting protein is an A-kinase anchoring protein specific for type i cAMP-dependent protein kinase. Chembiochem 2010, 11, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Means, C.K.; Lygren, B.; Langeberg, L.K.; Jain, A.; Dixon, R.E.; Vega, A.L.; Gold, M.G.; Petrosyan, S.; Taylor, S.S.; Murphy, A.N.; et al. An entirely specific type i a-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, E1227–E1235. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P.P.; Ma, Y.; Margarucci, L.; Mackey, M.; van der Heyden, M.A.; Ellisman, M.; Scholten, A.; Taylor, S.S.; Heck, A.J. A small novel A-kinase anchoring protein (AKAP) that localizes specifically protein kinase A-regulatory subunit i (PKA-ri) to the plasma membrane. J. Biol. Chem. 2012, 287, 43789–43797. [Google Scholar] [CrossRef] [PubMed]
- Tröger, J.; Moutty, M.C.; Skroblin, P.; Klussmann, E. A-kinase anchoring proteins as potential drug targets. Br. J. Pharmacol. 2012, 166, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Fraser, I.D.; Tavalin, S.J.; Lester, L.B.; Langeberg, L.K.; Westphal, A.M.; Dean, R.A.; Marrion, N.V.; Scott, J.D. A novel lipid-anchored A-kinase anchoring protein facilitates cAMP-responsive membrane events. EMBO J. 1998, 17, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Trotter, K.W.; Fraser, I.D.; Scott, G.K.; Stutts, M.J.; Scott, J.D.; Milgram, S.L. Alternative splicing regulates the subcellular localization of A-kinase anchoring protein 18 isoforms. J. Cell Biol. 1999, 147, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Dell’Acqua, M.L.; Faux, M.C.; Thorburn, J.; Thorburn, A.; Scott, J.D. Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4,5-bisphosphate. EMBO J. 1998, 17, 2246–2260. [Google Scholar] [CrossRef] [PubMed]
- Hundsrucker, C.; Skroblin, P.; Christian, F.; Zenn, H.M.; Popara, V.; Joshi, M.; Eichhorst, J.; Wiesner, B.; Herberg, F.W.; Reif, B.; et al. Glycogen synthase kinase 3β interaction protein functions as an A-kinase anchoring protein. J. Biol. Chem. 2010, 285, 5507–5521. [Google Scholar] [CrossRef] [PubMed]
- Deák, V.A.; Skroblin, P.; Dittmayer, C.; Knobeloch, K.P.; Bachmann, S.; Klussmann, E. The A-kinase anchoring protein gskip regulates gsk3β activity and controls palatal shelf fusion in mice. J. Biol. Chem. 2016, 291, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Dema, A.; Schröter, M.F.; Perets, E.; Skroblin, P.; Moutty, M.C.; Deàk, V.A.; Birchmeier, W.; Klussmann, E. The A-kinase anchoring protein (AKAP) glycogen synthase kinase 3β interaction protein (gskip) regulates β-catenin through its interactions with both protein kinase a (PKA) and gsk3β. J. Biol. Chem. 2016, 291, 19618–19630. [Google Scholar] [CrossRef] [PubMed]
- Scholten, A.; Poh, M.K.; van Veen, T.A.; van Breukelen, B.; Vos, M.A.; Heck, A.J. Analysis of the cGMP/cAMP interactome using a chemical proteomics approach in mammalian heart tissue validates sphingosine kinase type 1-interacting protein as a genuine and highly abundant AKAP. J. Proteom. Res. 2006, 5, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Taskén, K.; Aandahl, E.M. Localized effects of cAMP mediated by distinct routes of protein kinase a. Physiol. Rev. 2004, 84, 137–167. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.J.; Wang, L.; Ma, Y.; Durick, K.; Perkins, G.; Deerinck, T.J.; Ellisman, M.H.; Taylor, S.S. Nh2-terminal targeting motifs direct dual specificity A-kinase-anchoring protein 1 (d-AKAP1) to either mitochondria or endoplasmic reticulum. J. Cell. Biol 1999, 145, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Langeberg, L.K.; Doxsey, S.J.; Scott, J.D. Pericentrin anchors protein kinase a at the centrosome through a newly identified rii-binding domain. Curr. Biol. 2000, 10, 417–420. [Google Scholar] [CrossRef]
- Gillingham, A.K.; Munro, S. The pact domain, a conserved centrosomal targeting motif in the coiled-coil proteins AKAP450 and pericentrin. EMBO Rep. 2000, 1, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Santana, L.F. A-kinase anchoring proteins: Getting to the heart of the matter. Circulation 2010, 121, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Navedo, M.F.; Santana, L.F. Cav1.2 sparklets in heart and vascular smooth muscle. J. Mol. Cell. Cardiol. 2013, 58, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Mercado, J.; Baylie, R.; Navedo, M.F.; Yuan, C.; Scott, J.D.; Nelson, M.T.; Brayden, J.E.; Santana, L.F. Local control of trpv4 channels by AKAP150-targeted PKC in arterial smooth muscle. J. Gen. Physiol. 2014, 143, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Hulme, J.T.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac cav1.2 channels during β1-adrenergic regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 16574–16579. [Google Scholar] [CrossRef] [PubMed]
- Appert-Collin, A.; Cotecchia, S.; Nenniger-Tosato, M.; Pedrazzini, T.; Diviani, D. The A-kinase anchoring protein (AKAP)-lbc-signaling complex mediates alpha1 adrenergic receptor-induced cardiomyocyte hypertrophy. Proc. Natl Acad Sci USA 2007, 104, 10140–10145. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.L.; Soughayer, J.; Pare, G.C.; Carlisle Michel, J.J.; Langeberg, L.K.; Kapiloff, M.S.; Scott, J.D. The protein kinase a anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 2005, 437, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Shumay, E.; Wang, H.; Malbon, C.C. The scaffold protein gravin (cAMP-dependent protein kinase-anchoring protein 250) binds the β2-adrenergic receptor via the receptor cytoplasmic Arg-329 to Leu-413 domain and provides a mobile scaffold during desensitization. J. Biol. Chem. 2001, 276, 24005–24014. [Google Scholar] [CrossRef] [PubMed]
- Gardner, L.A.; Tavalin, S.J.; Goehring, A.S.; Scott, J.D.; Bahouth, S.W. AKAP79-mediated targeting of the cyclic AMP-dependent protein kinase to the β1-adrenergic receptor promotes recycling and functional resensitization of the receptor. J. Biol. Chem. 2006, 281, 33537–33553. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Cinel, I.; Dellinger, R.P. Advances in pathogenesis and management of sepsis. Curr. Opin. Infect. Dis. 2007, 20, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Kim, J.H.; Kim, W.J.; Lee, H.Y.; Park, J.A.; Lee, S.W.; Yoon, D.K.; Kim, H.H.; Chung, H.; Yu, Y.S.; et al. AKAP12 regulates human blood-retinal barrier formation by downregulation of hypoxia-inducible factor-1α. J. Neurosci. 2007, 27, 4472–4481. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, N.; Waschke, J. cAMP with other signaling cues converges on rac1 to stabilize the endothelial barrier—A signaling pathway compromised in inflammation. Cell Tissue Res. 2014, 355, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Sehrawat, S.; Ernandez, T.; Cullere, X.; Takahashi, M.; Ono, Y.; Komarova, Y.; Mayadas, T.N. AKAP9 regulation of microtubule dynamics promotes epac1-induced endothelial barrier properties. Blood 2011, 117, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Navedo, M.F.; Nieves-Cintron, M.; Amberg, G.C.; Yuan, C.; Votaw, V.S.; Lederer, W.J.; McKnight, G.S.; Santana, L.F. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin ii-induced hypertension. Circ. Res. 2008, 102, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Navedo, M.F.; Cheng, E.P.; Yuan, C.; Votaw, S.; Molkentin, J.D.; Scott, J.D.; Santana, L.F. Increased coupled gating of l-type Ca2+ channels during hypertension and timothy syndrome. Circ. Res. 2010, 106, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.E.; Cheng, E.P.; Mercado, J.L.; Santana, L.F. L-type Ca2+ channel function during timothy syndrome. Trends Cardiovasc. Med. 2012, 22, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Heppner, T.J.; Nelson, M.T.; Brayden, J.E. Trpv4 forms a novel Ca2+ signaling complex with ryanodine receptors and bkca channels. Circ. Res. 2005, 97, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Pauyo, T.; Drapp, R.; Tavares, M.J.; Liedtke, W.; Brayden, J.E. Trpv4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1096–H1102. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Szentesi, P.; Pignier, C.; Egger, M.; Kranias, E.G.; Niggli, E. Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+-induced Ca2+ release refractoriness in heart muscle. Circ. Res. 2004, 95, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/serca2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Bünemann, M.; Gerhardstein, B.L.; Gao, T.; Hosey, M.M. Functional regulation of l-type calcium channels via protein kinase A-mediated phosphorylation of the β2 subunit. J. Biol. Chem. 1999, 274, 33851–33854. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.B.; Rossow, C.F.; Navedo, M.F.; Westenbroek, R.E.; Catterall, W.A.; Santana, L.F.; McKnight, G.S. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of l-type calcium channels. Circ. Res. 2010, 107, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Schulze, D.H.; Muqhal, M.; Lederer, W.J.; Ruknudin, A.M. Sodium/calcium exchanger (ncx1) macromolecular complex. J. Biol. Chem. 2003, 278, 28849–28855. [Google Scholar] [CrossRef] [PubMed]
- Ruknudin, A.; He, S.; Lederer, W.J.; Schulze, D.H. Functional differences between cardiac and renal isoforms of the rat Na+-Ca2+ exchanger ncx1 expressed in xenopus oocytes. J. Physiol. 2000, 529, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Nicodemus-Johnson, J.; Carnegie, G.K.; Danziger, R.S. Molecular evolution of A-kinase anchoring protein (AKAP)-7: Implications in comparative PKA compartmentalization. BMC Evol. Biol. 2012, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M.; Kass, R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005, 85, 1205–1253. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.T.; Kass, R.S. Recent progress in congenital long qt syndrome. Curr. Opin. Cardiol. 2010, 25, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, S.; Burns-Hamuro, L.L.; Ma, Y.; Hamon, S.C.; Canaves, J.M.; Shi, M.M.; Nelson, M.R.; Sing, C.F.; Cantor, C.R.; Taylor, S.S.; et al. Amino acid variant in the kinase binding domain of dual-specific a kinase-anchoring protein 2: A disease susceptibility polymorphism. Proc. Natl. Acad. Sci. USA 2003, 100, 4066–4071. [Google Scholar] [CrossRef] [PubMed]
- Tingley, W.G.; Pawlikowska, L.; Zaroff, J.G.; Kim, T.; Nguyen, T.; Young, S.G.; Vranizan, K.; Kwok, P.Y.; Whooley, M.A.; Conklin, B.R. Gene-trapped mouse embryonic stem cell-derived cardiac myocytes and human genetics implicate AKAP10 in heart rhythm regulation. Proc. Natl. Acad. Sci. USA 2007, 104, 8461–8466. [Google Scholar] [CrossRef] [PubMed]
- Łoniewska, B.; Kaczmarczyk, M.; Clark, J.S.; Gorący, I.; Horodnicka-Józwa, A.; Ciechanowicz, A. Association of functional genetic variants of A-kinase anchoring protein 10 with qt interval length in full-term polish newborns. Arch. Med. Sci. 2015, 11, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Soderling, J.; Scott, J.D. AKAP-lbc anchors protein kinase A and nucleates Gα12-selective rho-mediated stress fiber formation. J. Biol. Chem. 2001, 276, 44247–44257. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, E.; Edemir, B.; Pepperle, B.; Tamma, G.; Henn, V.; Klauschenz, E.; Hundsrucker, C.; Maric, K.; Rosenthal, W. Ht31: The first protein kinase A anchoring protein to integrate protein kinase A and rho signaling. FEBS Lett. 2001, 507, 264–268. [Google Scholar] [CrossRef]
- Abdul Azeez, K.R.; Knapp, S.; Fernandes, J.M.; Klussmann, E.; Elkins, J.M. The crystal structure of the rhoa-AKAP-lbc dh-ph domain complex. Biochem. J. 2014, 464, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Schrade, K.; Tröger, J.; Eldahshan, A.; Zühlke, K.; Abdul Azeez, K.R.; Elkins, J.M.; Neuenschwander, M.; Oder, A.; Elkewedi, M.; Jaksch, S.; et al. An AKAP-lbc-rhoa interaction inhibitor promotes the translocation of aquaporin-2 to the plasma membrane of renal collecting duct principal cells. PLoS One 2018, 13, e0191423. [Google Scholar] [CrossRef] [PubMed]
- Mayers, C.M.; Wadell, J.; McLean, K.; Venere, M.; Malik, M.; Shibata, T.; Driggers, P.H.; Kino, T.; Guo, X.C.; Koide, H.; et al. The rho guanine nucleotide exchange factor AKAP13 (brx) is essential for cardiac development in mice. J. Biol. Chem. 2010, 285, 12344–12354. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Abuin, L.; Cotecchia, S.; Pansier, L. Anchoring of both PKA and 14-3-3 inhibits the rho-gef activity of the AKAP-lbc signaling complex. EMBO J. 2004, 23, 2811–2820. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Smith, F.D.; McConnachie, G.; Langeberg, L.K.; Scott, J.D. AKAP-lbc nucleates a protein kinase D activation scaffold. Mol. Cell 2004, 15, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Soughayer, J.; Smith, F.D.; Pedroja, B.S.; Zhang, F.; Diviani, D.; Bristow, M.R.; Kunkel, M.T.; Newton, A.C.; Langeberg, L.K.; et al. AKAP-lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell 2008, 32, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Pare, G.C.; Easlick, J.L.; Mislow, J.M.; McNally, E.M.; Kapiloff, M.S. Nesprin-1alpha contributes to the targeting of mAKAP to the cardiac myocyte nuclear envelope. Exp. Cell Res. 2005, 303, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Malik, S.; Kelley, G.G.; Kapiloff, M.S.; Smrcka, A.V. Phospholipase c epsilon scaffolds to muscle-specific a kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J. Biol. Chem. 2011, 286, 23012–23021. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.L.; Bauman, A.; Mayer, N.; Henson, E.; Heredia, L.; Ahn, J.; McAvoy, T.; Nairn, A.C.; Kapiloff, M.S. Camp-stimulated protein phosphatase 2a activity associated with muscle a kinase-anchoring protein (mAKAP) signaling complexes inhibits the phosphorylation and activity of the cAMP-specific phosphodiesterase pde4d3. J. Biol. Chem. 2010, 285, 11078–11086. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Goehring, A.S.; Kapiloff, M.S.; Langeberg, L.K.; Scott, J.D. MAKAP compartmentalizes oxygen-dependent control of hif-1α. Sci. Signal. 2008, 1, ra18. [Google Scholar] [CrossRef] [PubMed]
- Abrenica, B.; AlShaaban, M.; Czubryt, M.P. The a-kinase anchor protein AKAP121 is a negative regulator of cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol 2009, 46, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Means, C.K.; Xiao, C.Y.; Li, Z.; Zhang, T.; Omens, J.H.; Ishii, I.; Chun, J.; Brown, J.H. Sphingosine 1-phosphate s1p2 and s1p3 receptor-mediated akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2944–H2951. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Cattaneo, F.; Pironti, G.; Magliulo, F.; Carotenuto, G.; Pirozzi, M.; Polishchuk, R.; Borzacchiello, D.; Paolillo, R.; Oliveti, M.; et al. AKAP1 deficiency promotes mitochondrial aberrations and exacerbates cardiac injury following permanent coronary ligation via enhanced mitophagy and apoptosis. PLoS One 2016, 11, e0154076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacaná, E.; Maceyka, M.; Milstien, S.; Spiegel, S. Cloning and characterization of a protein kinase a anchoring protein (AKAP)-related protein that interacts with and regulates sphingosine kinase 1 activity. J. Biol. Chem. 2002, 277, 32947–32953. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, N.T.; Mohan, M.L.; Goswami, S.K.; Naga Prasad, S.V. Regulation of β-adrenergic receptor function: An emphasis on receptor resensitization. Cell Cycle 2011, 10, 3684–3691. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What is the role of β-adrenergic signaling in heart failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G.; Gonen, T.; Scott, J.D. Local cAMP signaling in disease at a glance. J. Cell Sci. 2013, 126, 4537–4543. [Google Scholar] [CrossRef] [PubMed]
- Berisha, F.; Nikolaev, V.O. Cyclic nucleotide imaging and cardiovascular disease. Pharmacol. Ther. 2017, 175, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.; Nikolaev, V.O. Imaging alterations of cardiomyocyte cAMP microdomains in disease. Front. Pharmacol. 2015, 6, 172. [Google Scholar] [CrossRef] [PubMed]
- Pavlaki, N.; Nikolaev, V.O. Imaging of PDE2- and PDE3-mediated cGMP-to-cAMP cross-talk in cardiomyocytes. J. Cardiovasc. Dev. Dis. 2018, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Musheshe, N.; Schmidt, M.; Zaccolo, M. cAMP: From long-range second messenger to nanodomain signalling. Trends Pharmacol. Sci. 2017, 39, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Thestrup, T.; Litzlbauer, J.; Bartholomäus, I.; Mues, M.; Russo, L.; Dana, H.; Kovalchuk, Y.; Liang, Y.; Kalamakis, G.; Laukat, Y.; et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and t lymphocytes. Nat. Methods 2014, 11, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Nikolaev, V.O. Biophysical techniques for detection of cAMP and cGMP in living cells. Int J. Mol. Sci. 2013, 14, 8025–8046. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.K.; Sprenger, J.U.; Steinbrecher, J.H.; Hübscher, D.; Lehnart, S.E.; Abesser, M.; Schuh, K.; El-Armouche, A.; Nikolaev, V.O. Microdomain switch of cGMP-regulated phosphodiesterases leads to anp-induced augmentation of β-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ. Res. 2015, 116, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat. Commun. 2015, 6, 6965. [Google Scholar] [CrossRef] [PubMed]
- Jungen, C.; Scherschel, K.; Eickholt, C.; Kuklik, P.; Klatt, N.; Bork, N.; Salzbrunn, T.; Alken, F.; Angendohr, S.; Klene, C.; et al. Disruption of cardiac cholinergic neurons enhances susceptibility to ventricular arrhythmias. Nat. Commun. 2017, 8, 14155. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alonso, J.L.; Bhargava, A.; O’Hara, T.; Glukhov, A.V.; Schobesberger, S.; Bhogal, N.; Sikkel, M.B.; Mansfield, C.; Korchev, Y.E.; Lyon, A.R.; et al. Microdomain-specific modulation of l-type calcium channels leads to triggered ventricular arrhythmia in heart failure. Circ. Res. 2016, 119, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; Rieken, F.; Wagner, J.; Sungkaworn, T.; Zabel, U.; Borzi, A.; Cocucci, E.; Zürn, A.; Lohse, M.J. Single-molecule analysis of fluorescently labeled g-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc. Natl Acad Sci USA 2013, 110, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Moshkov, A.; Novak, P.; Shevchuk, A.; Nikolaev, V.O.; El-Hamamsy, I.; Potter, C.M.; Wright, P.; Kadir, S.H.; Lyon, A.R.; et al. Scanning ion conductance microscopy: A convergent high-resolution technology for multi-parametric analysis of living cardiovascular cells. J. R Soc. Interface 2011, 8, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Palmer, D.; Tilley, D.G.; Dunkerley, H.A.; Netherton, S.J.; Raymond, D.R.; Elbatarny, H.S.; Jimmo, S.L. Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol. Pharmacol. 2003, 64, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterases (PDE) and peptide motifs. Curr. Pharm. Des. 2010, 16, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Anant, J.S.; Ong, O.C.; Xie, H.Y.; Clarke, S.; O’Brien, P.J.; Fung, B.K. In vivo differential prenylation of retinal cyclic gmp phosphodiesterase catalytic subunits. J. Biol. Chem. 1992, 267, 687–690. [Google Scholar] [PubMed]
- Baillie, G.S.; MacKenzie, S.J.; McPhee, I.; Houslay, M.D. Sub-family selective actions in the ability of erk2 map kinase to phosphorylate and regulate the activity of PDE4 cyclic AMP-specific phosphodiesterases. Br. J. Pharmacol. 2000, 131, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of pdes and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Wang, H. Crystal structures of phosphodiesterases and implications on substrate specificity and inhibitor selectivity. Curr. Top. Med. Chem. 2007, 7, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, J.; Choi, Y.H.; Krall, J.; Ahmad, F.; Manganiello, V.C.; Movsesian, M.A. Isoforms of cyclic nucleotide phosphodiesterase PDE3a in cardiac myocytes. J. Biol. Chem. 2002, 277, 38072–38078. [Google Scholar] [CrossRef] [PubMed]
- Senzaki, H.; Smith, C.J.; Juang, G.J.; Isoda, T.; Mayer, S.P.; Ohler, A.; Paolocci, N.; Tomaselli, G.F.; Hare, J.M.; Kass, D.A. Cardiac phosphodiesterase 5 (cGMP-specific) modulates β-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J. 2001, 15, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Rosman, G.J.; Martins, T.J.; Sonnenburg, W.K.; Beavo, J.A.; Ferguson, K.; Loughney, K. Isolation and characterization of human cdnas encoding a cGMP-stimulated 3′,5′-cyclic nucleotide phosphodiesterase. Gene 1997, 191, 89–95. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, X.H.; Zhang, K.; Chen, C.K.; Frederick, J.M.; Prestwich, G.D.; Baehr, W. Photoreceptor cgmp phosphodiesterase delta subunit (pdedelta) functions as a prenyl-binding protein. J. Biol. Chem. 2004, 279, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Sonati, P.; Rubin, C.; Michaeli, T. Pde7a1, a cAMP-specific phosphodiesterase, inhibits cAMP-dependent protein kinase by a direct interaction with c. J. Biol. Chem. 2006, 281, 15050–15057. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, P.; Egan, R.W.; Billah, M.M. Identification and characterization of a new human type 9 cGMP-specific phosphodiesterase splice variant (PDE9a5). Differential tissue distribution and subcellular localization of PDE9a variants. Gene 2003, 314, 15–27. [Google Scholar] [CrossRef]
- Sayner, S.; Stevens, T. Soluble adenylate cyclase reveals the significance of compartmentalized cAMP on endothelial cell barrier function. Biochem. Soc. Trans. 2006, 34, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Creighton, J.; Zhu, B.; Alexeyev, M.; Stevens, T. Spectrin-anchored phosphodiesterase 4d4 restricts cAMP from disrupting microtubules and inducing endothelial cell gap formation. J. Cell. Sci. 2008, 121, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; Tocchetti, C.G.; Terrin, A.; Lissandron, V.; Cheung, Y.F.; Dostmann, W.R.; Pozzan, T.; Kass, D.A.; Paolocci, N.; Houslay, M.D.; et al. Compartmentalized phosphodiesterase-2 activity blunts β-adrenergic cardiac inotropy via an no/cGMP-dependent pathway. Circ. Res. 2006, 98, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Helli, P.B.; Simpson, J.A.; Zhao, D.; Farman, G.P.; Jones, P.; Tian, X.; Wilson, L.S.; Ahmad, F.; Chen, S.R.W.; et al. Phosphodiesterase 4d regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of l-type Ca2+ current. Circ. Res. 2011, 109, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Melenovsky, V.; Marhin, T.; Fitzgerald, P.; Kass, D.A. Sildenafil inhibits β-adrenergic-stimulated cardiac contractility in humans. Circulation 2005, 112, 2642–2649. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, E.; Albergine, M.S.; Santana, L.F.; Beavo, J.A. Phosphodiesterase 8a (PDE8a) regulates excitation-contraction coupling in ventricular myocytes. J. Mol. Cell. Cardiol. 2010, 49, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Tovar, A.; Kaumann, A.J. Phosphodiesterase-4 blunts inotropism and arrhythmias but not sinoatrial tachycardia of (−)-adrenaline mediated through mouse cardiac β(1)-adrenoceptors. Br. J. Pharmacol. 2008, 153, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Abe, J.; Wei, H.; Xu, H.; Che, W.; Aizawa, T.; Liu, W.; Molina, C.A.; Sadoshima, J.; Blaxall, B.C.; et al. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (icer) leads to cardiomyocyte apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 14771–14776. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Richter, W.; Mika, D.; Castro, L.R.; Abi-Gerges, A.; Xie, M.; Scheitrum, C.; Lefebvre, F.; Schittl, J.; Mateo, P.; et al. Phosphodiesterase 4b in the cardiac l-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias in mice. J. Clin. Invest. 2011, 121, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.; Conti, M.; Marks, A.R. Phosphodiesterase 4d deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Pokreisz, P.; Vandenwijngaert, S.; Bito, V.; Van den Bergh, A.; Lenaerts, I.; Busch, C.; Marsboom, G.; Gheysens, O.; Vermeersch, P.; Biesmans, L.; et al. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation 2009, 119, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9a controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, R.; Krall, J.; Tikishvili, E.; Honeggar, M.; Ahmad, F.; Manganiello, V.C.; Movsesian, M.A. Isoforms of cyclic nucleotide phosphodiesterase pde3 and their contribution to cAMP hydrolytic activity in subcellular fractions of human myocardium. J. Biol. Chem. 2005, 280, 39168–39174. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Murata, T.; Shimizu, K.; Degerman, E.; Maurice, D.; Manganiello, V. Cyclic nucleotide phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2015, 21, e25–e50. [Google Scholar] [CrossRef] [PubMed]
- Maass, P.G.; Aydin, A.; Luft, F.C.; Schächterle, C.; Weise, A.; Stricker, S.; Lindschau, C.; Vaegler, M.; Qadri, F.; Toka, H.R.; et al. PDE3a mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet. 2015, 47, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Hattenbach, L.O.; Toka, H.R.; Toka, O.; Schuster, H.; Luft, F.C. Absence of hypertensive retinopathy in a turkish kindred with autosomal dominant hypertension and brachydactyly. Br. J. Ophthalmol. 1998, 82, 1363–1365. [Google Scholar] [CrossRef] [PubMed]
- Toka, O.; Tank, J.; Schächterle, C.; Aydin, A.; Maass, P.G.; Elitok, S.; Bartels-Klein, E.; Hollfinger, I.; Lindschau, C.; Mai, K.; et al. Clinical effects of phosphodiesterase 3a mutations in inherited hypertension with brachydactyly. Hypertension 2015, 66, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Movsesian, M. Novel approaches to targeting PDE3 in cardiovascular disease. Pharmacol. Ther. 2016, 163, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Movsesian, M.; Ahmad, F.; Hirsch, E. Functions of PDE3 isoforms in cardiac muscle. J. Cardiovasc. Dev. Dis 2018, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Conti, M.; Houslay, M.D. Handbook of Experimental Pharmacology 204. In Phosphodiesterases as Drug Targets; Springer International Publishing: Cham, Switzerland, 2011. [Google Scholar]
- Knight, W.; Yan, C. Therapeutic potential of pde modulation in treating heart disease. Future Med. Chem. 2013, 5, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shakur, Y.; Kambayashi, J. Phosphodiesterases as targets for intermittent claudication. Handb Exp. Pharmacol. 2011, 211–236. [Google Scholar]
- Rogers, K.C.; Oliphant, C.S.; Finks, S.W. Clinical efficacy and safety of cilostazol: A critical review of the literature. Drugs 2015, 75, 377–395. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, W.R.; Money, S.R.; Brass, E.P. Long-term safety of cilostazol in patients with peripheral artery disease: The castle study (cilostazol: A study in long-term effects). J. Vasc. Surg. 2008, 47, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, T.; Matsumaru, Y.; Hayakawa, M.; Nemoto, S.; Matsumura, A. Cilostazol reduces restenosis after carotid artery stenting. J. Vasc. Surg. 2010, 51, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cone, J.; Wang, S.; Tandon, N.; Fong, M.; Sun, B.; Sakurai, K.; Yoshitake, M.; Kambayashi, J.; Liu, Y. Comparison of the effects of cilostazol and milrinone on intracellular cAMP levels and cellular function in platelets and cardiac cells. J. Cardiovasc. Pharmacol. 1999, 34, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Faxon, D.P.; Creager, M.A.; Smith, S.C.; Pasternak, R.C.; Olin, J.W.; Bettmann, M.A.; Criqui, M.H.; Milani, R.V.; Loscalzo, J.; Kaufman, J.A.; et al. Atherosclerotic vascular disease conference: Executive summary: Atherosclerotic vascular disease conference proceeding for healthcare professionals from a special writing group of the american heart association. Circulation 2004, 109, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Momi, S.; Falcinelli, E. Anti-platelet therapy: Phosphodiesterase inhibitors. Br. J. Clin. Pharmacol. 2011, 72, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Sahin, M.; Alizade, E.; Pala, S.; Alici, G.; Ozkan, B.; Akgun, T.; Emiroglu, Y.; Demir, S.; Yazicioglu, M.V.; Turkmen, M.M. The effect of cilostazol on right heart function and pulmonary pressure. Cardiovasc. Ther. 2013, 31, e88–e93. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Toga, K.; Sudo, T.; Tachibana, K.; Tochizawa, S.; Kimura, Y.; Yoshida, Y.; Hidaka, H. Suppression of arterial intimal hyperplasia by cilostamide, a cyclic nucleotide phosphodiesterase 3 inhibitor, in a rat balloon double-injury model. Br. J. Pharmacol. 2000, 130, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, C.B.; Dzavík, V. Inotropes and vasopressors: Review of physiology and clinical use in cardiovascular disease. Circulation 2008, 118, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Tariq, S.; Aronow, W.S. Use of inotropic agents in treatment of systolic heart failure. Int J. Mol. Sci. 2015, 16, 29060–29068. [Google Scholar] [CrossRef] [PubMed]
- Movsesian, M. New pharmacologic interventions to increase cardiac contractility: Challenges and opportunities. Curr. Opin. Cardiol. 2015, 30, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, E. Protein-protein interactions of PDE4 family members—Functions, interactions and therapeutic value. Cell Signal. 2016, 28, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Sims, C.R.; Singh, S.P.; Mu, S.; Gokden, N.; Zakaria, D.; Nguyen, T.C.; Mayeux, P.R. Rolipram improves outcome in a rat model of infant sepsis-induced cardiorenal syndrome. Front. Pharmacol. 2017, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Samardzic, H.; Adamson, R.H.; Renkin, E.M.; Clark, J.F.; Reed, R.K.; Curry, F.R. Phosphodiesterase 4 inhibition attenuates atrial natriuretic peptide-induced vascular hyperpermeability and loss of plasma volume. J. Physiol. 2011, 589, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Xie, M.; Scheitrum, C.; Krall, J.; Movsesian, M.A.; Conti, M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 2011, 106, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br. J. Pharmacol. 2011, 163, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Tenor, H.; Hatzelmann, A.; Beume, R.; Lahu, G.; Zech, K.; Bethke, T.D. Pharmacology, clinical efficacy, and tolerability of phosphodiesterase-4 inhibitors: Impact of human pharmacokinetics. Handb Exp. Pharmacol. 2011, 85–119. [Google Scholar]
- Sakkas, L.I.; Mavropoulos, A.; Bogdanos, D.P. Phosphodiesterase 4 inhibitors in immune-mediated diseases: Mode of action, clinical applications, current and future perspectives. Curr. Med. Chem. 2017, 24, 3054–3067. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Apremilast: A review in psoriasis and psoriatic arthritis. Drugs 2017, 77, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Nagendran, J.; Archer, S.L.; Soliman, D.; Gurtu, V.; Moudgil, R.; Haromy, A.; St Aubin, C.; Webster, L.; Rebeyka, I.M.; Ross, D.B.; et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 2007, 116, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Yan, M.; Chen, J.; Chaugai, S.; Chen, C.; Wang, D. Chronic inhibition of cyclic guanosine monophosphate-specific phosphodiesterase 5 prevented cardiac fibrosis through inhibition of transforming growth factor β-induced smad signaling. Front. Med. 2014, 8, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; van Heerebeek, L.; Paulus, W.J. Phosphodiesterase-5 inhibition in heart failure with preserved ejection fraction: Trading therapy for prevention. Eur J. Heart Fail. 2017, 19, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Balidemaj, K.; Homma, S.; Wu, H.; Wang, J.; Maybaum, S. Acute type 5 phosphodiesterase inhibition with sildenafil enhances flow-mediated vasodilation in patients with chronic heart failure. J. Am. Coll. Cardiol. 2000, 36, 845–851. [Google Scholar] [CrossRef]
- Kukreja, R.C.; Salloum, F.N.; Das, A.; Koka, S.; Ockaili, R.A.; Xi, L. Emerging new uses of phosphodiesterase-5 inhibitors in cardiovascular diseases. Exp. Clin. Cardiol. 2011, 16, e30–35. [Google Scholar] [PubMed]
- Pofi, R.; Gianfrilli, D.; Badagliacca, R.; Di Dato, C.; Venneri, M.A.; Giannetta, E. Everything you ever wanted to know about phosphodiesterase 5 inhibitors and the heart (but never dared ask): How do they work? J. Endocrinol. Invest. 2016, 39, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, W.K.; Seger, D.; Beavo, J.A. Molecular cloning of a cdna encoding the “61-kda” Calmodulin-stimulated cyclic nucleotide phosphodiesterase. Tissue-specific expression of structurally related isoforms. J. Biol. Chem. 1993, 268, 645–652. [Google Scholar] [PubMed]
- Goraya, T.A.; Cooper, D.M. Ca2+-calmodulin-dependent phosphodiesterase (pde1): Current perspectives. Cell Signal. 2005, 17, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Saeki, T.; Adachi, H.; Takase, Y.; Yoshitake, S.; Souda, S.; Saito, I. A selective type v phosphodiesterase inhibitor, e4021, dilates porcine large coronary artery. J. Pharmacol. Exp. Ther. 1995, 272, 825–831. [Google Scholar] [PubMed]
- Nagel, D.J.; Aizawa, T.; Jeon, K.I.; Liu, W.; Mohan, A.; Wei, H.; Miano, J.M.; Florio, V.A.; Gao, P.; Korshunov, V.A.; et al. Role of nuclear Ca2+/calmodulin-stimulated phosphodiesterase 1a in vascular smooth muscle cell growth and survival. Circ. Res. 2006, 98, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Pullamsetti, S.S.; Kwapiszewska, G.; Dumitrascu, R.; Tian, X.; Weissmann, N.; Ghofrani, H.A.; Kaulen, C.; Dunkern, T.; Schudt, C.; et al. Phosphodiesterase 1 upregulation in pulmonary arterial hypertension: Target for reverse-remodeling therapy. Circulation 2007, 115, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Fischmeister, R.; Castro, L.; Abi-Gerges, A.; Rochais, F.; Vandecasteele, G. Species- and tissue-dependent effects of no and cyclic gmp on cardiac ion channels. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2005, 142, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Mehel, H.; Emons, J.; Vettel, C.; Wittköpper, K.; Seppelt, D.; Dewenter, M.; Lutz, S.; Sossalla, S.; Maier, L.S.; Lechêne, P.; et al. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts β-adrenergic responses in cardiomyocytes. J. Am. Coll. Cardiol. 2013, 62, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Vettel, C.; Lindner, M.; Dewenter, M.; Lorenz, K.; Schanbacher, C.; Riedel, M.; Lämmle, S.; Meinecke, S.; Mason, F.E.; Sossalla, S.; et al. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ. Res. 2017, 120, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Rentero, C.; Monfort, A.; Puigdomènech, P. Identification and distribution of different mrna variants produced by differential splicing in the human phosphodiesterase 9a gene. Biochem. Biophys. Res. Commun. 2003, 301, 686–692. [Google Scholar] [CrossRef]
- Christian, F.; Szaszák, M.; Friedl, S.; Drewianka, S.; Lorenz, D.; Goncalves, A.; Furkert, J.; Vargas, C.; Schmieder, P.; Götz, F.; et al. Small molecule AKAP-protein kinase a (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J. Biol. Chem. 2011, 286, 9079–9096. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Munir, M.; Aiman, S.; Wadood, A.; Khan, A.U. The in silico identification of small molecules for protein-protein interaction inhibition in AKAP-lbc-rhoa signaling complex. Comput. Biol. Chem. 2017, 67, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Raimondi, F.; Del Vescovo, C.D.; Dreyer, E.; Reggi, E.; Osman, H.; Ruggieri, L.; Gonano, C.; Cavin, S.; Box, C.L.; et al. Small-molecule protein-protein interaction inhibitor of oncogenic rho signaling. Cell Chem. Biol. 2016, 23, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Serrels, B.; Sandilands, E.; Serrels, A.; Baillie, G.; Houslay, M.D.; Brunton, V.G.; Canel, M.; Machesky, L.M.; Anderson, K.I.; Frame, M.C. A complex between fak, rack1, and PDE4d5 controls spreading initiation and cancer cell polarity. Curr. Biol. 2010, 20, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of A-kinase anchoring proteins (AKAP)-protein kinase A (PKA) interactions displayed at two different angles. The amphipathic AKB helix of AKAPs docks into the hydrophobic groove formed by the dimers of the N-terminal D/D domains of regulatory subunits of PKA. AKB A-kinase-binding domain; D/D dimerization and docking domain.
Figure 1.
Schematic representation of A-kinase anchoring proteins (AKAP)-protein kinase A (PKA) interactions displayed at two different angles. The amphipathic AKB helix of AKAPs docks into the hydrophobic groove formed by the dimers of the N-terminal D/D domains of regulatory subunits of PKA. AKB A-kinase-binding domain; D/D dimerization and docking domain.
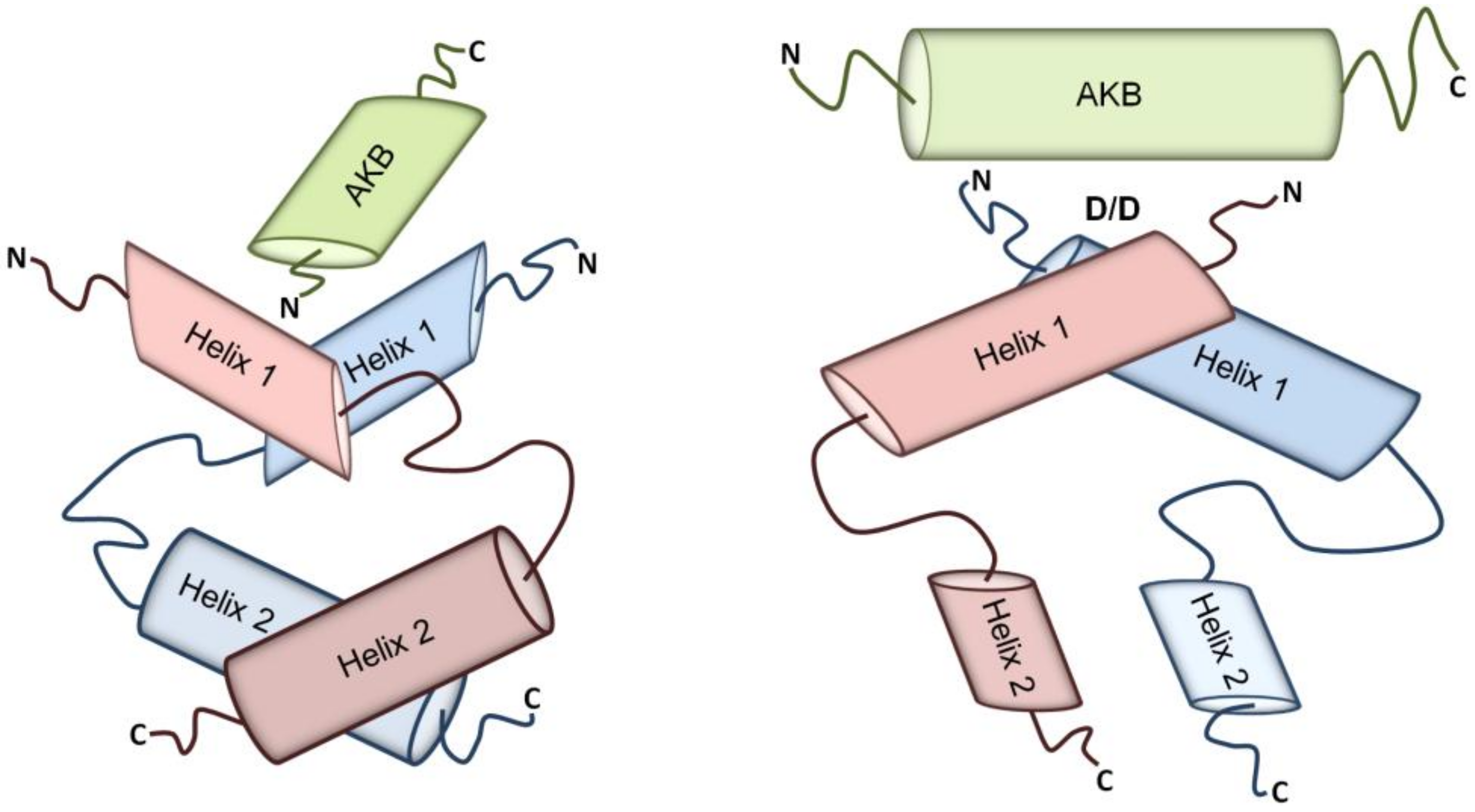
Figure 2.
Substrate specificity of individual phosphodiesterase (PDE) families.

Figure 3.
Schematic representation of the PDE3A gene, PDE3A protein isoforms and the hyperphosphorylation caused by the identified mutations. (A) Eight mutations have been identified in families from the countries indicated by the flags. The mutations cluster within a region of the gene encoding amino acids 445 and 449. The mutations cause hyperphosphorylations of Ser428 and Ser438. The N-terminal hydrophobic region (NHR) 1 of PDE3A1 comprises four transmembrane domains, while NHR2 contains no typical transmembrane region but a cluster of hydrophobic amino acids. PDE3A2 contains only NHR2 and PDE3A3 lacks all N-terminal hydrophobic regions (for details see text). (B) The hyperphosphorylation increases cyclic adenosine 3′-5′ monophosphate (cAMP) hydrolysis, causing low cAMP levels.
Figure 3.
Schematic representation of the PDE3A gene, PDE3A protein isoforms and the hyperphosphorylation caused by the identified mutations. (A) Eight mutations have been identified in families from the countries indicated by the flags. The mutations cluster within a region of the gene encoding amino acids 445 and 449. The mutations cause hyperphosphorylations of Ser428 and Ser438. The N-terminal hydrophobic region (NHR) 1 of PDE3A1 comprises four transmembrane domains, while NHR2 contains no typical transmembrane region but a cluster of hydrophobic amino acids. PDE3A2 contains only NHR2 and PDE3A3 lacks all N-terminal hydrophobic regions (for details see text). (B) The hyperphosphorylation increases cyclic adenosine 3′-5′ monophosphate (cAMP) hydrolysis, causing low cAMP levels.
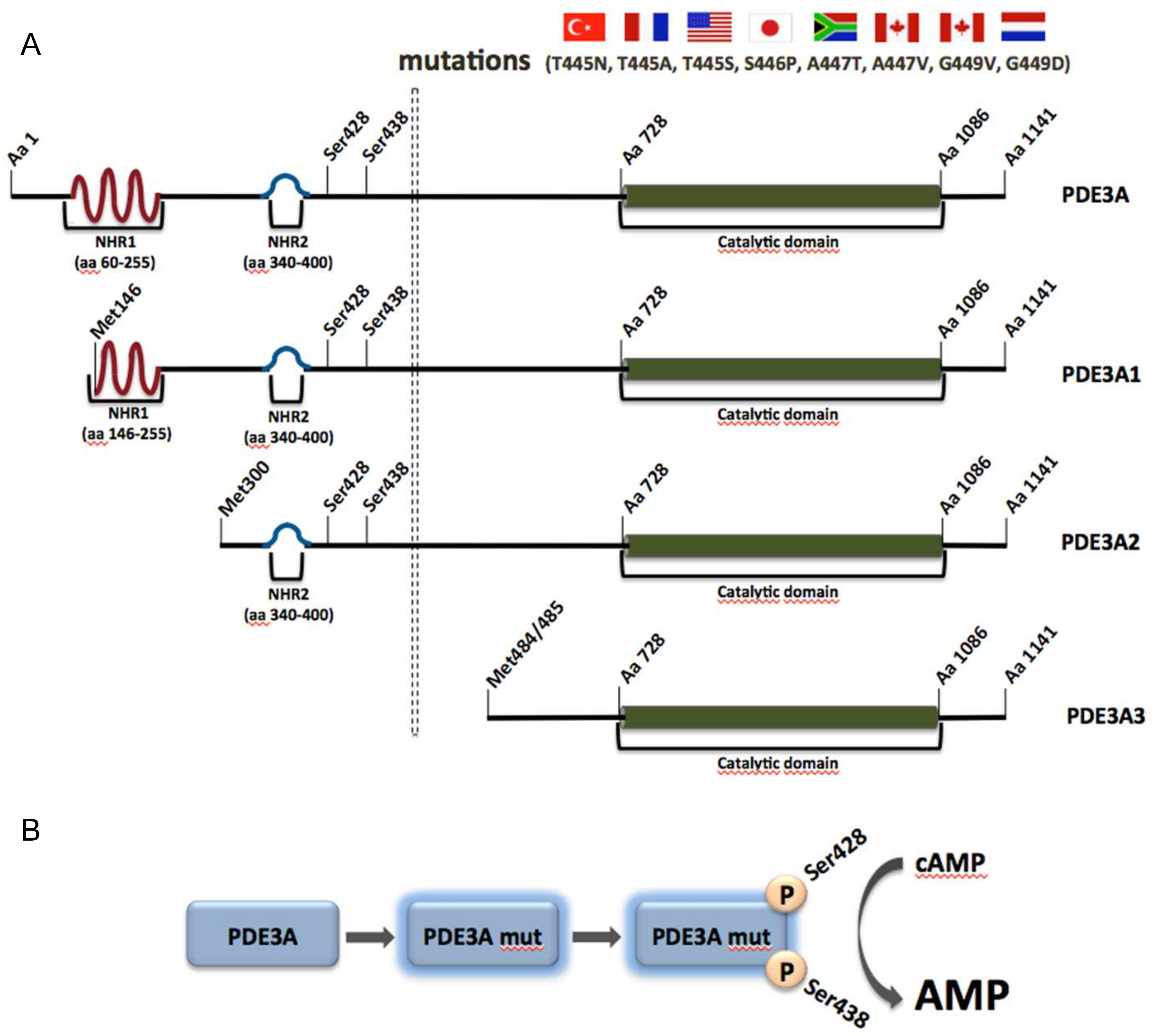

{kind=link}
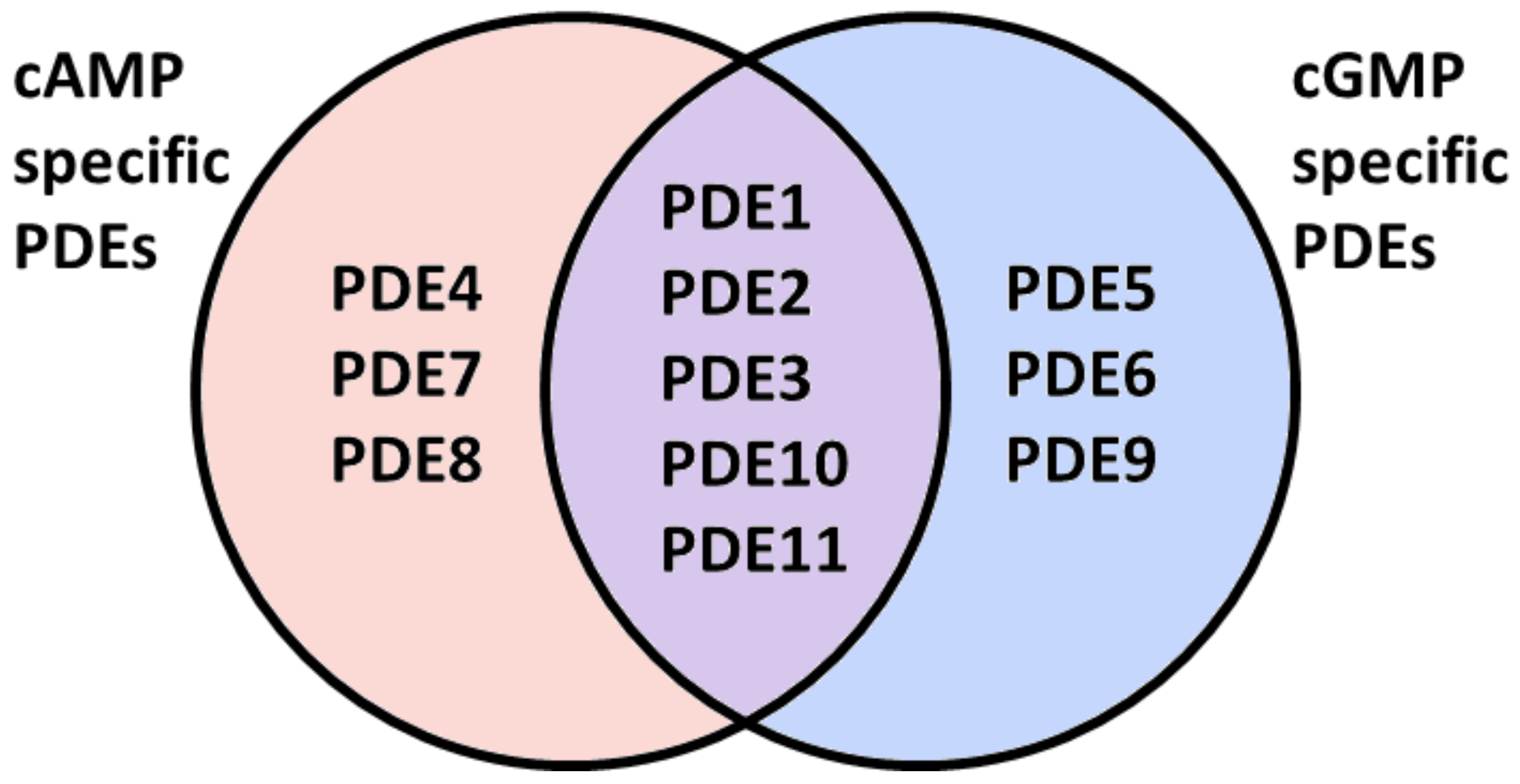{kind=link}
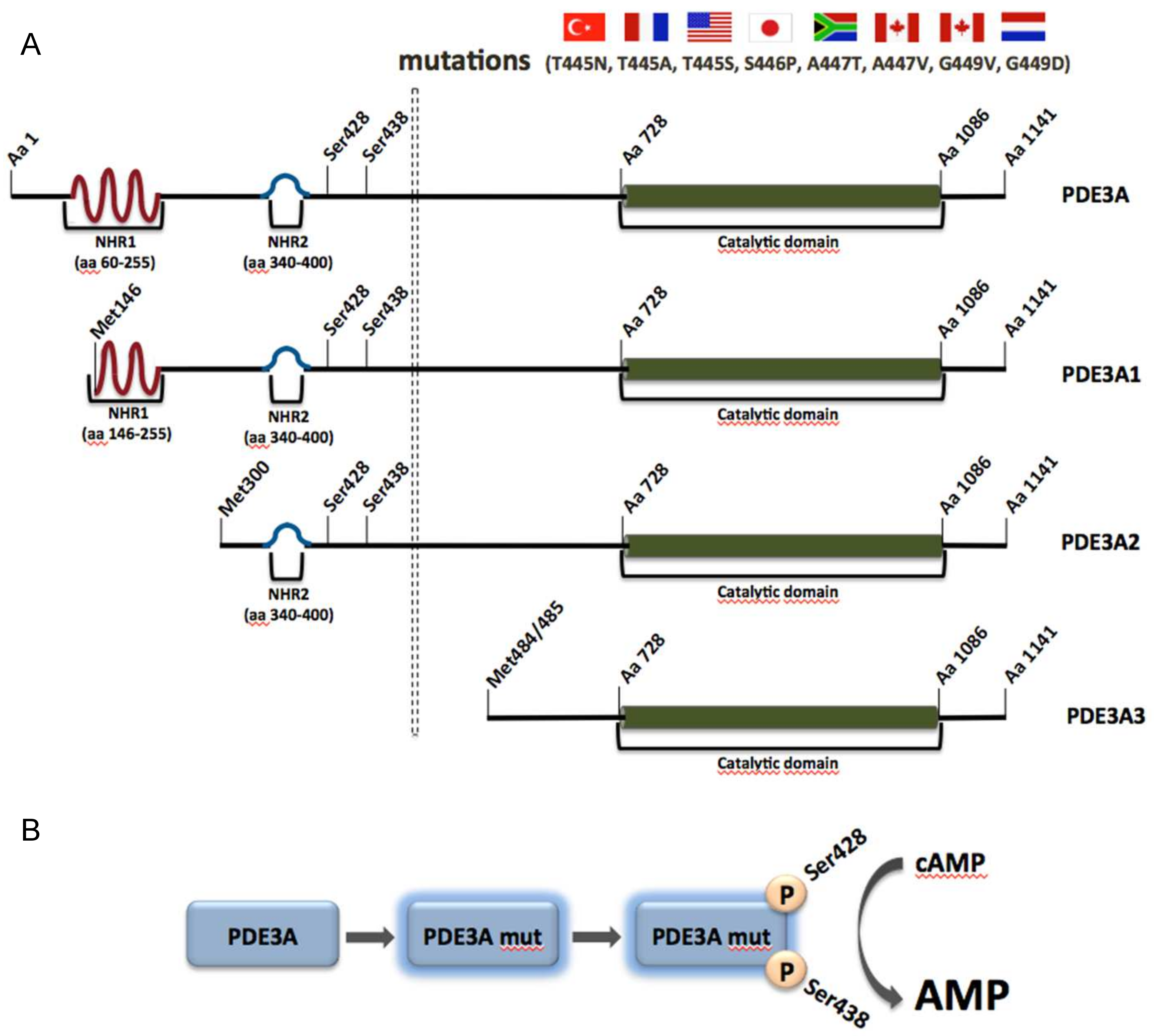{kind=link}
Table 1.
Overview of AKAPs expressed in the heart and vasculature and of the cardiovascular processes that they regulate.
Table 1.
Overview of AKAPs expressed in the heart and vasculature and of the cardiovascular processes that they regulate.
| Common Name | Gene Name | Alternative Name | Regulated Cardiovascular Process |
|---|---|---|---|
| D-AKAP1 | AKAP1 | AKAP121/ AKAP149/AKAP84 | Cardiac stress response |
| D-AKAP2 | AKAP10 | - | Cardiac repolarization |
| AKAP9 (long isoform) | AKAP9 | - | Endothelial barrier function |
| AKAP18α AKAP18γ AKAP18δ | AKAP7 | - | Excitation-contraction coupling |
| AKAP79 | AKAP5 | AKAP75/AKAP150 | Vascular tone; Excitation-contraction coupling; β-AR desensitization/resensitization cycle |
| AKAP220 | AKAP11 | - | Endothelial barrier function |
| AKAP-Lbc | AKAP13 | Brx-1/Proto-Lbc/ Ht31 | Cardiac stress response |
| mAKAPβ | AKAP6 | AKAP100 | Excitation-contraction coupling; Cardiac stress response |
| AKAP Yotiao | AKAP9 | GC-NAP | Cardiac repolarization |
| Gravin | AKAP12 | AKAP250 | Endothelial barrier function; β-AR desensitization/resensitization cycle |
| SKIP | SPHKAP | - | Cardiac stress response |
Table 2.
Overview of the PDE families expressed in the cardiovascular system and the corresponding cardiovascular processes that they regulate.
Table 2.
Overview of the PDE families expressed in the cardiovascular system and the corresponding cardiovascular processes that they regulate.
| PDE Family | PDE Gene | Substrate Specificity | Regulated Cardiovascular Process |
|---|---|---|---|
| PDE1 | PDE1A | cAMP, cGMP | Cardiac stress response |
| PDE1B | |||
| PDE1C | |||
| PDE2 | PDE2A | cAMP, cGMP | Endothelial barrier function; Excitation-contraction coupling |
| PDE3 | PDE3A | cAMP, cGMP | Endothelial barrier function; Excitation-contraction coupling; Basal pacemaking activity of the SA node; Cardiac stress response |
| PDE3B | |||
| PDE4 | PDE4A | cAMP | Endothelial barrier function; Excitation-contraction coupling; Basal pacemaking activity of the SA node; Cardiac stress response |
| PDE4B | |||
| PDE4C | |||
| PDE4D | |||
| PDE5 | PDE5A | cGMP | Endothelial barrier function; Excitation-contraction coupling; Cardiac stress response |
| PDE8 | PDE8A | cAMP | Excitation-contraction coupling |
| PDE8B | |||
| PDE9 | PDE9A | cGMP | Cardiac stress response |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ercu, M.; Klussmann, E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 14. https://doi.org/10.3390/jcdd5010014
AMA Style
Ercu M, Klussmann E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. Journal of Cardiovascular Development and Disease. 2018; 5(1):14. https://doi.org/10.3390/jcdd5010014
Chicago/Turabian StyleErcu, Maria, and Enno Klussmann. 2018. "Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System" Journal of Cardiovascular Development and Disease 5, no. 1: 14. https://doi.org/10.3390/jcdd5010014
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.