Enlightening the Association between Bicuspid Aortic Valve and Aortopathy

 ,
,
Abstract
:1. Introduction
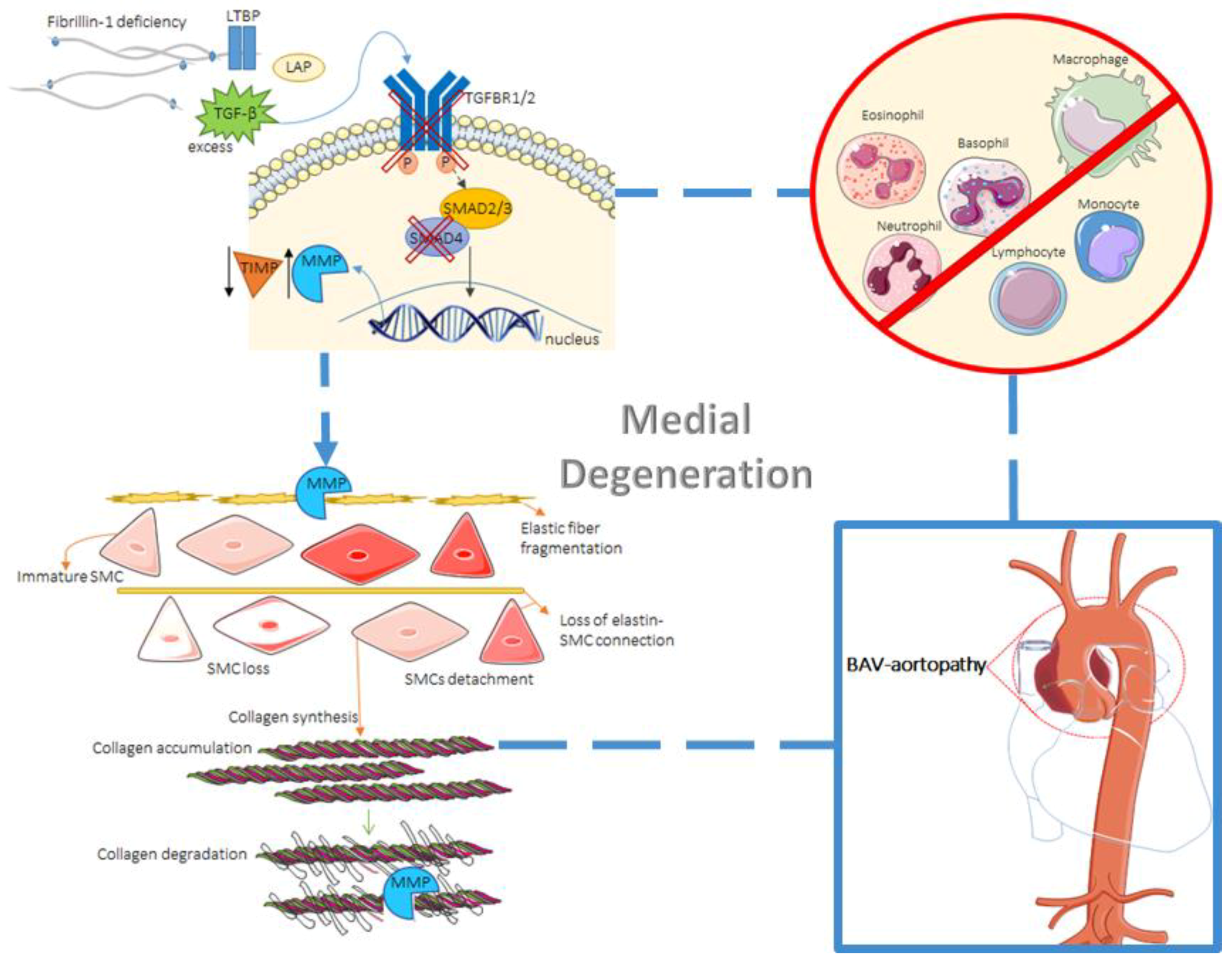
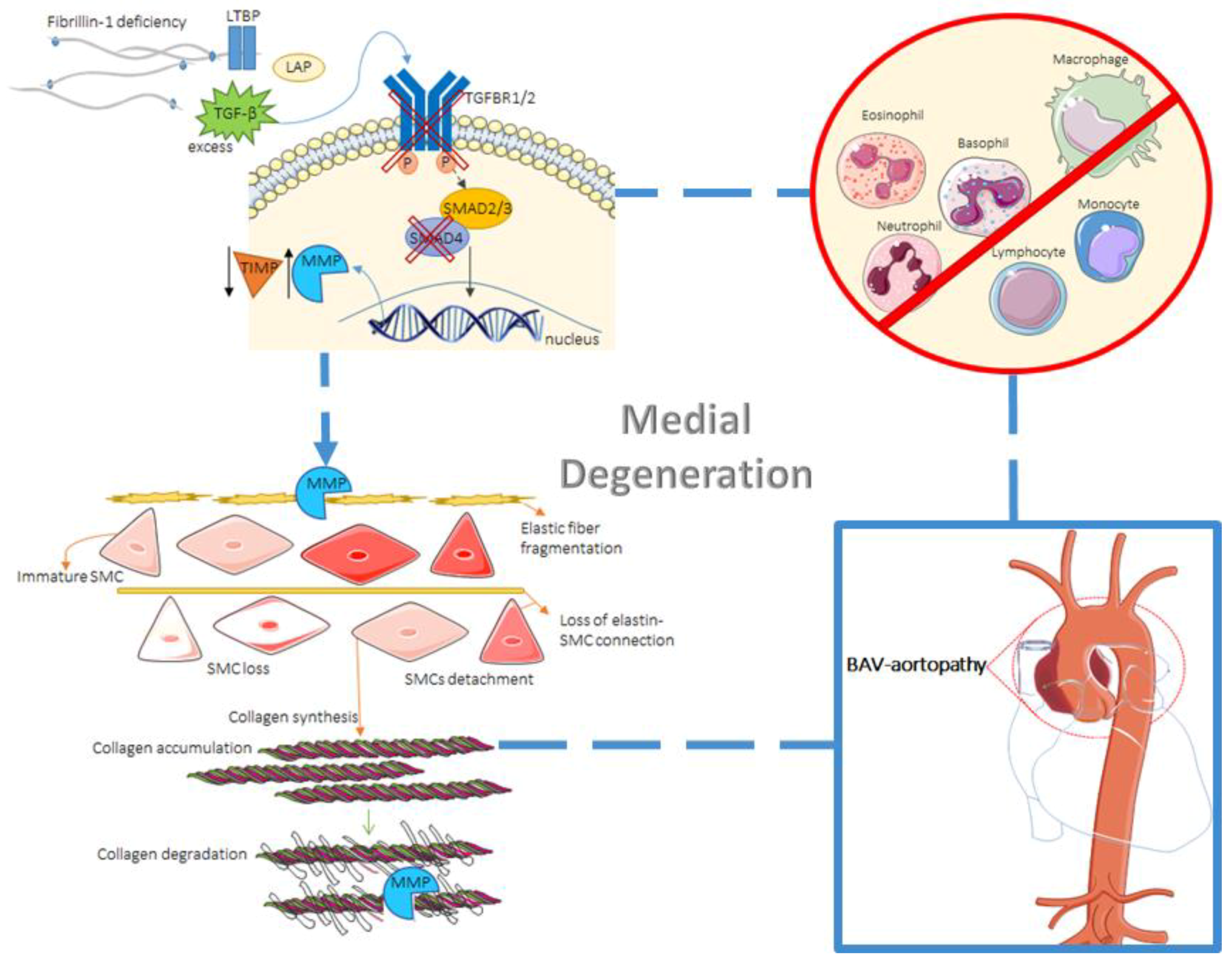
2. Tissue Biology
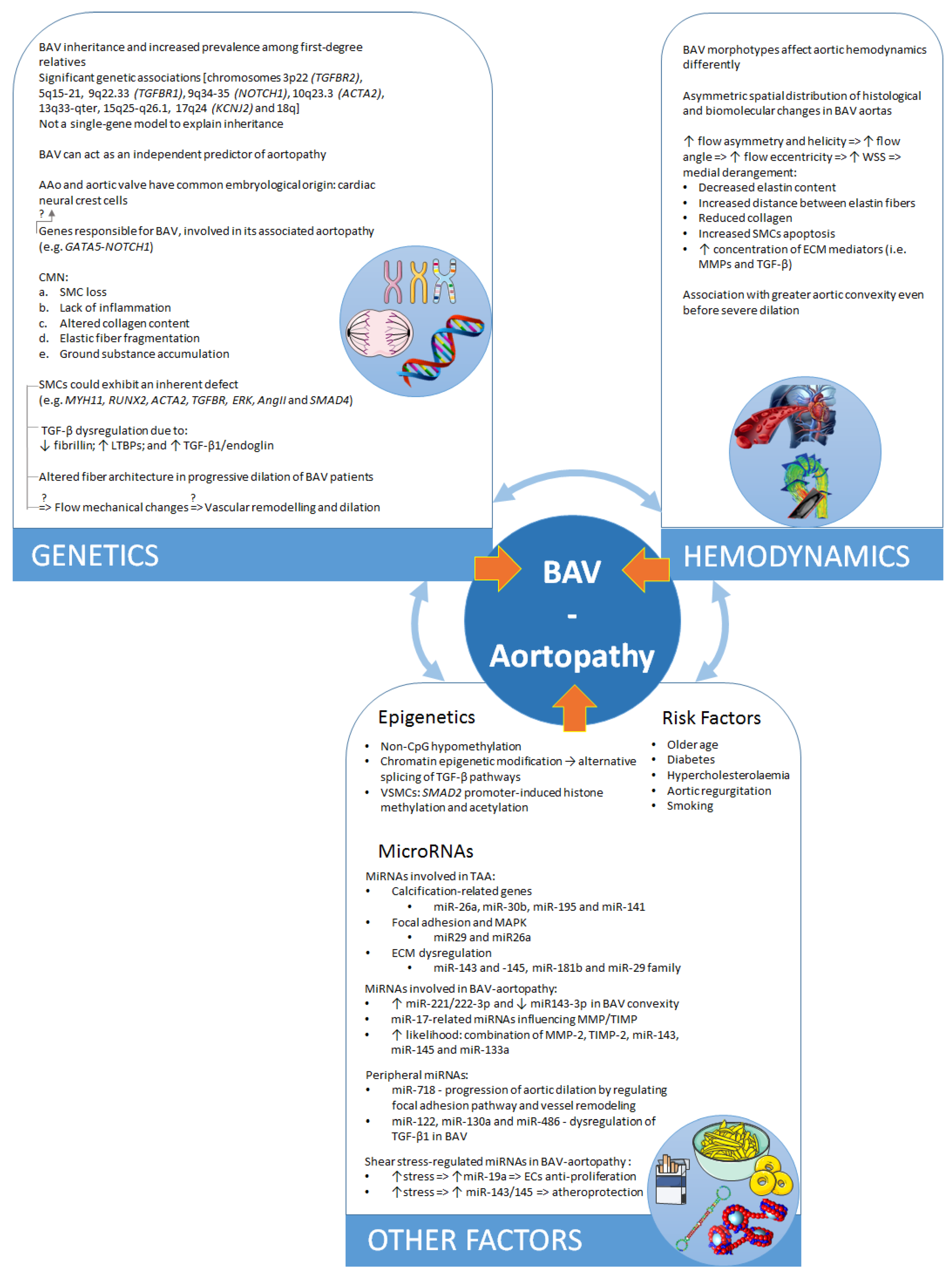
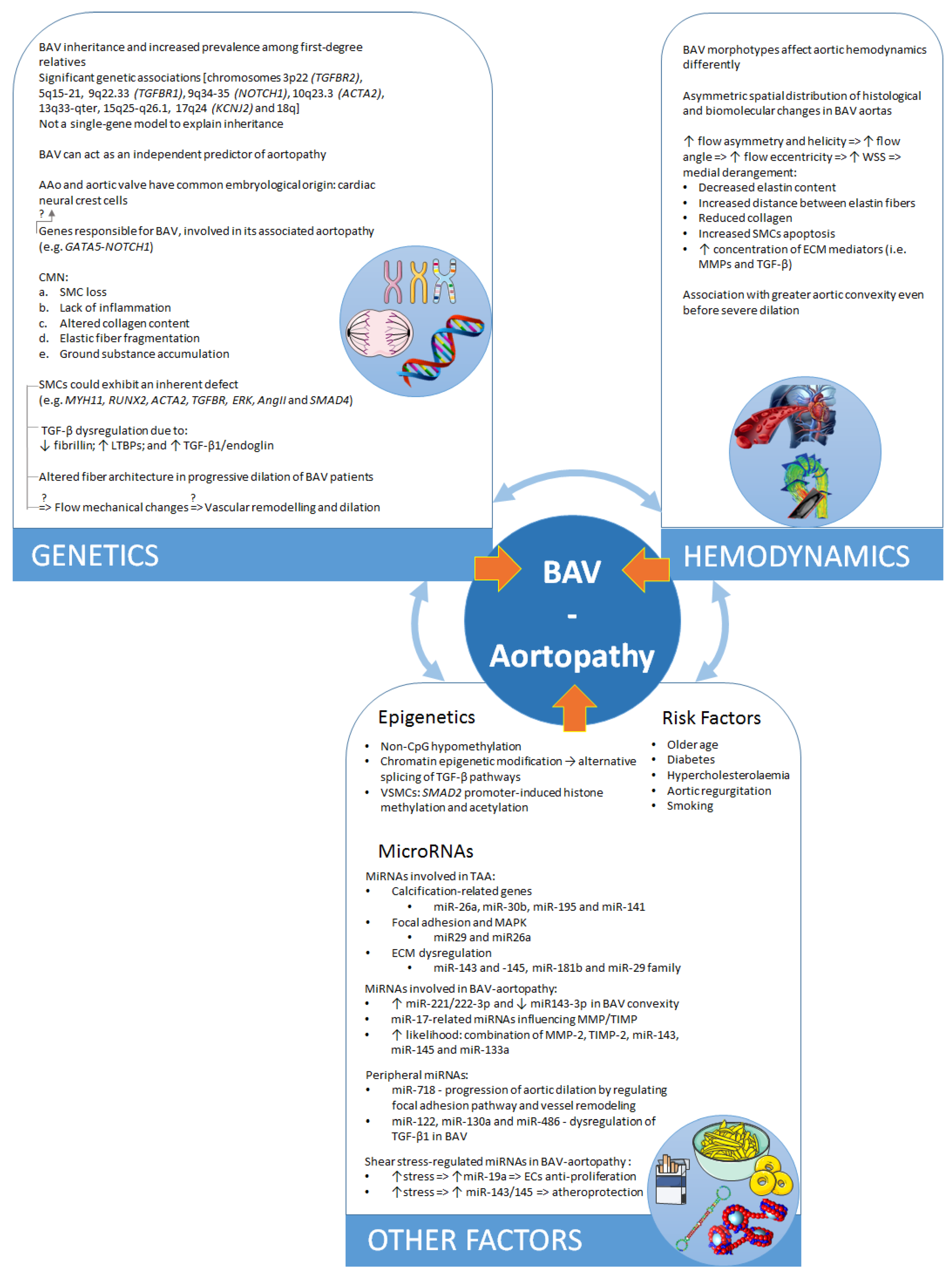
2.1. Distinct Genetic Aetiologies or Common Embryological Origin?
2.2. Histological Abnormalities
2.3. The Role of TGF-β
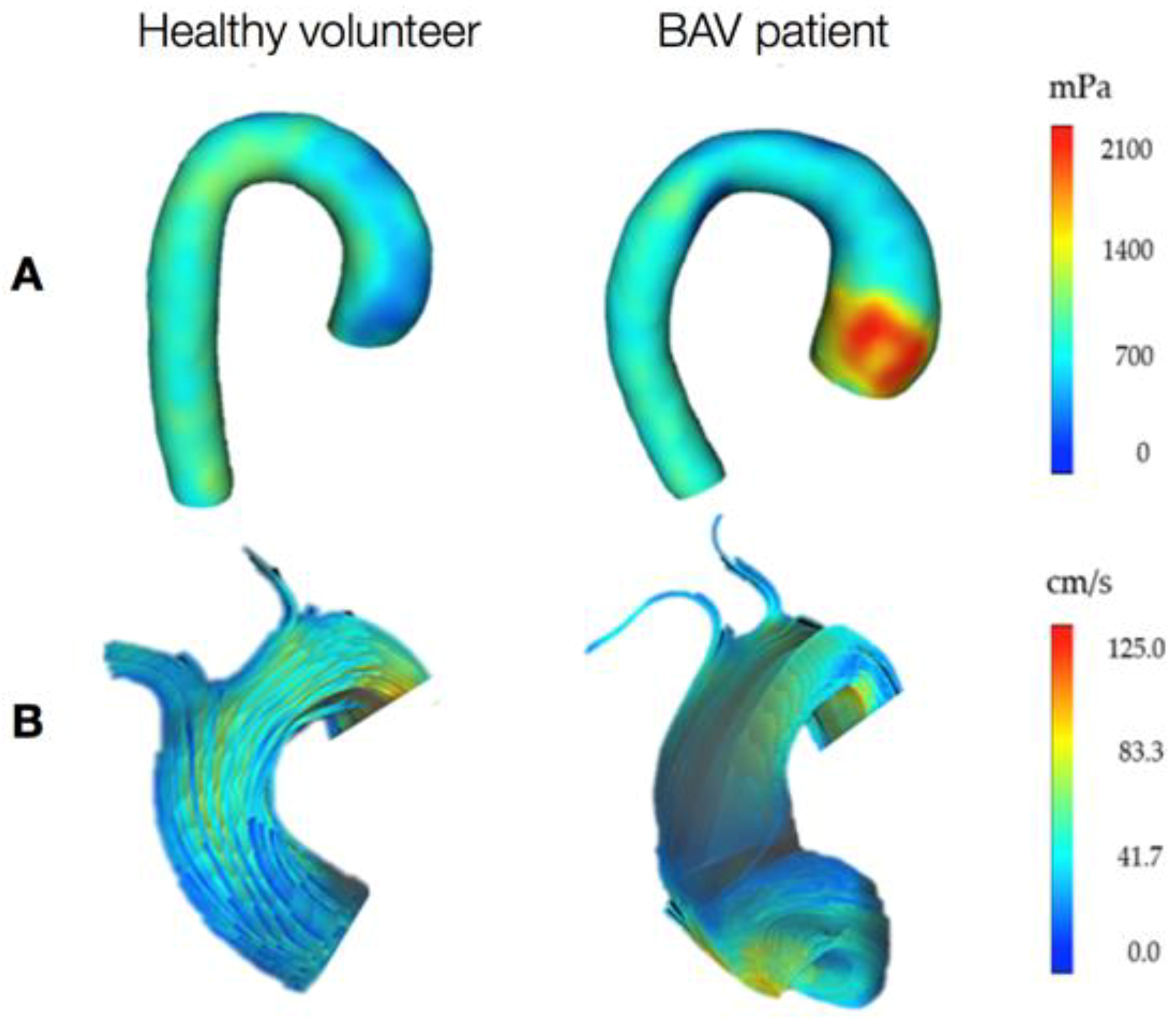
3. Hemodynamics
3.1. Two Theories on the Pathogenesis of BAV–Aortopathy
3.2. The Role of Hemodynamics
4. Environmental Impact
4.1. Environment and Risk Factors
4.2. Epigenetic Mechanisms
5. The Role of MicroRNAs
5.1. MicroRNAs Involved in BAV Disease and Valve Morphology
5.2. MicroRNAs Involved in Aortopathy
5.3. MicroRNAs Regulated by Changes in Shear Stress
5.4. MicroRNAs as Potential Therapeutic Targets and Biomarkers in BAV Aortopathy
6. Managing BAV Aortopathy
6.1. Clinical Management
6.2. Genetic Tools
7. Discussion
8. Conclusions
Conflicts of Interest
References
- Prakash, S.K.; Bosse, Y.; Muehlschlegel, J.D.; Michelena, H.I.; Limongelli, G.; Della Corte, A.; Pluchinotta, F.R.; Russo, M.G.; Evangelista, A.; Benson, D.W.; et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: Insights from the international bavcon (bicuspid aortic valve consortium). J. Am. Coll. Cardiol. 2014, 64, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, A.; Bancone, C.; Quarto, C.; Dialetto, G.; Covino, F.E.; Scardone, M.; Caianiello, G.; Cotrufo, M. Predictors of ascending aortic dilatation with bicuspid aortic valve: A wide spectrum of disease expression. Eur. J. Cardio-Thorac. Surg. 2007, 31, 397–405. [Google Scholar]
- Vallely, M.P.; Semsarian, C.; Bannon, P.G. Management of the ascending aorta in patients with bicuspid aortic valve disease. Heart Lung Circ. 2008, 17, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Padang, R.; Bannon, P.G.; Jeremy, R.; Richmond, D.R.; Semsarian, C.; Vallely, M.; Wilson, M.; Yan, T.D. The genetic and molecular basis of bicuspid aortic valve associated thoracic aortopathy: A link to phenotype heterogeneity. Ann. Cardiothorac. Surg. 2013, 2, 83–91. [Google Scholar] [PubMed]
- Losenno, K.L.; Goodman, R.L.; Chu, M.W. Bicuspid aortic valve disease and ascending aortic aneurysms: Gaps in knowledge. Cardiol. Res. Pract. 2012, 2012, 145202–145218. [Google Scholar] [CrossRef] [PubMed]
- Siu, S.C.; Silversides, C.K. Bicuspid aortic valve disease. J. Am. Coll. Cardiol. 2010, 55, 2789–2800. [Google Scholar] [CrossRef] [PubMed]
- Isselbacher, E.M.; Lino Cardenas, C.L.; Lindsay, M.E. Hereditary influence in thoracic aortic aneurysm and dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, J.M.; Mulder, B.J. Aortopathies in adult congenital heart disease and genetic aortopathy syndromes: Management strategies and indications for surgery. Heart 2017, 103, 952–966. [Google Scholar] [CrossRef] [PubMed]
- Phillippi, J.A.; Green, B.R.; Eskay, M.A.; Kotlarczyk, M.P.; Hill, M.R.; Robertson, A.M.; Watkins, S.C.; Vorp, D.A.; Gleason, T.G. Mechanism of aortic medial matrix remodeling is distinct in patients with bicuspid aortic valve. J. Thorac. Cardiovasc. Surg. 2014, 147, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.R.; Kaple, R.K.; Mandapati, D.; Gallo, A.; Botta, D.M., Jr.; Elefteriades, J.A.; Coady, M.A. Natural history of ascending aortic aneurysms in the setting of an unreplaced bicuspid aortic valve. Ann. Thorac. Surg. 2007, 83, 1338–1344. [Google Scholar] [CrossRef] [PubMed]
- Atkins, S.K.; Sucosky, P. Etiology of bicuspid aortic valve disease: Focus on hemodynamics. World J. Cardiol. 2014, 6, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Maegdefessel, L. Non-coding rna contribution to thoracic and abdominal aortic aneurysm disease development and progression. Front. Physiol. 2017, 8, 429. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Ramachandran, V.; Cripe, L.H.; Hinton, R.B.; Andelfinger, G.; Tabangin, M.; Shooner, K.; Keddache, M.; Benson, D.W. Evidence in favor of linkage to human chromosomal regions 18q, 5q and 13q for bicuspid aortic valve and associated cardiovascular malformations. Hum. Genet. 2007, 121, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Cripe, L.; Andelfinger, G.; Martin, L.J.; Shooner, K.; Benson, D.W. Bicuspid aortic valve is heritable. J. Am. Coll. Cardiol. 2004, 44, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Regalado, E. Heritable thoracic aortic disease overview. In Genereviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mefford, H.C., Stephens, K., Amemiya, A., Ledbetter, N., Eds.; Genereviews: Seattle, WA, USA, 1993. [Google Scholar]
- Martin, L.J.; Hinton, R.B.; Zhang, X.; Cripe, L.H.; Benson, D.W. Aorta measurements are heritable and influenced by bicuspid aortic valve. Front. Genet. 2011, 2, 61. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, P.; Bosse, Y.; Huggins, G.S.; Corte, A.D.; Pibarot, P.; Michelena, H.I.; Limongelli, G.; Boulanger, M.C.; Evangelista, A.; Bedard, E.; et al. The pathology and pathobiology of bicuspid aortic valve: State of the art and novel research perspectives. J. Pathol. Clin. Res. 2015, 1, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Yetman, A.T.; Graham, T. The dilated aorta in patients with congenital cardiac defects. J. Am. Coll. Cardiol. 2009, 53, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Grewal, N.; DeRuiter, M.C.; Jongbloed, M.R.; Goumans, M.J.; Klautz, R.J.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Normal and abnormal development of the aortic wall and valve: Correlation with clinical entities. Neth. Heart J. 2014, 22, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Rateri, D.L.; Moorleghen, J.J.; Majesky, M.W.; Daugherty, A. Smooth muscle cells derived from second heart field and cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta-brief report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1722–1726. [Google Scholar] [CrossRef] [PubMed]
- Harmon, A.W.; Nakano, A. Nkx2–5 lineage tracing visualizes the distribution of second heart field-derived aortic smooth muscle. Genesis 2013, 51, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Xiong, W.; Wang, L.C.; Yang, J.; Qiu, P.; Hirai, H.; Shao, L.N.; Milewicz, D.; Chen, Y.E.; Yang, B. Differentiation defect in neural crest-derived smooth muscle cells in patients with aortopathy associated with bicuspid aortic valves. Ebiomedicine 2016, 10, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Mead, T.J.; Yutzey, K.E. Notch pathway regulation of neural crest cell development in vivo. Dev. Dyn. 2012, 241, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Koenig, S.N.; Bosse, K.; Majumdar, U.; Bonachea, E.M.; Radtke, F.; Garg, V. Endothelial notch1 is required for proper development of the semilunar valves and cardiac outflow tract. J. Am. Heart Assoc. 2016, 5, e003075. [Google Scholar] [CrossRef] [PubMed]
- McKellar, S.H.; Tester, D.J.; Yagubyan, M.; Majumdar, R.; Ackerman, M.J.; Sundt, T.M., III. Novel notch1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2007, 134, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Van de Pol, V.; Kurakula, K.; DeRuiter, M.C.; Goumans, M.J. Thoracic aortic aneurysm development in patients with bicuspid aortic valve: What is the role of endothelial cells? Front. Physiol. 2017, 8, 938. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.; Bernardo, A.S.; Trotter, M.W.B.; Pedersen, R.A.; Sinha, S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat. Biotechnol. 2012, 30, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Laforest, B.; Andelfinger, G.; Nemer, M. Loss of gata5 in mice leads to bicuspid aortic valve. J. Clin. Investig. 2011, 121, 2876–2887. [Google Scholar] [CrossRef] [PubMed]
- Padang, R.; Bagnall, R.D.; Richmond, D.R.; Bannon, P.G.; Semsarian, C. Rare non-synonymous variations in the transcriptional activation domains of gata5 in bicuspid aortic valve disease. J. Mol. Cell. Cardiol. 2012, 53, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.C.; Zhao, Y.D.; Courtman, D.W.; Stewart, D.J. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation 2000, 101, 2345–2348. [Google Scholar] [CrossRef] [PubMed]
- Pisano, C.; Maresi, E.; Balistreri, C.R.; Candore, G.; Merlo, D.; Fattouch, K.; Bianco, G.; Ruvolo, G. Histological and genetic studies in patients with bicuspid aortic valve and ascending aorta complications. Interact. Cardiovasc. Thorac. Surg. 2012, 14, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Bonderman, D.; Gharehbaghi-Schnell, E.; Wollenek, G.; Maurer, G.; Baumgartner, H.; Lang, I.M. Mechanisms underlying aortic dilatation in congenital aortic valve malformation. Circulation 1999, 99, 2138–2143. [Google Scholar] [CrossRef] [PubMed]
- Tadros, T.M.; Klein, M.D.; Shapira, O.M. Ascending aortic dilatation associated with bicuspid aortic valve: Pathophysiology, molecular biology, and clinical implications. Circulation 2009, 119, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Halushka, M.K.; Angelini, A.; Bartoloni, G.; Basso, C.; Batoroeva, L.; Bruneval, P.; Buja, L.M.; Butany, J.; d’Amati, G.; Fallon, J.T.; et al. Consensus statement on surgical pathology of the aorta from the society for cardiovascular pathology and the association for european cardiovascular pathology: Ii. Noninflammatory degenerative diseases-nomenclature and diagnostic criteria. Cardiovasc. Pathol. 2016, 25, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Nataatmadja, M.; West, M.; West, J.; Summers, K.; Walker, P.; Nagata, M.; Watanabe, T. Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm. Circulation 2003, 108, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Cotrufo, M.; Della Corte, A.; De Santo, L.S.; Quarto, C.; De Feo, M.; Romano, G.; Amarelli, C.; Scardone, M.; Di Meglio, F.; Guerra, G.; et al. Different patterns of extracellular matrix protein expression in the convexity and the concavity of the dilated aorta with bicuspid aortic valve: Preliminary results. J. Thorac. Cardiovasc. Surg. 2005, 130, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Matthias Bechtel, J.F.; Noack, F.; Sayk, F.; Erasmi, A.W.; Bartels, C.; Sievers, H.H. Histopathological grading of ascending aortic aneurysm: Comparison of patients with bicuspid versus tricuspid aortic valve. J. Heart Valve Dis. 2003, 12, 54–59. [Google Scholar] [PubMed]
- Grewal, N.; Gittenberger-de Groot, A.C. Pathogenesis of aortic wall complications in marfan syndrome. Cardiovasc. Pathol. 2018, 33, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Grewal, N.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; Klautz, R.J.; Lindeman, J.H.; Goumans, M.J.; Palmen, M.; Mohamed, S.A.; Sievers, H.H.; Bogers, A.J.; et al. Ascending aorta dilation in association with bicuspid aortic valve: A maturation defect of the aortic wall. J. Thorac. Cardiovasc. Surg. 2014, 148, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- LeMaire, S.A.; Wang, X.; Wilks, J.A.; Carter, S.A.; Wen, S.; Won, T.; Leonardelli, D.; Anand, G.; Conklin, L.D.; Wang, X.L.; et al. Matrix metalloproteinases in ascending aortic aneurysms: Bicuspid versus trileaflet aortic valves. J. Surg. Res. 2005, 123, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Bilen, E.; Akcay, M.; Bayram, N.A.; Kocak, U.; Kurt, M.; Tanboga, I.H.; Bozkurt, E. Aortic elastic properties and left ventricular diastolic function in patients with isolated bicuspid aortic valve. J. Heart Valve Dis. 2012, 21, 189–194. [Google Scholar] [PubMed]
- Nistri, S.; Grande-Allen, J.; Noale, M.; Basso, C.; Siviero, P.; Maggi, S.; Crepaldi, G.; Thiene, G. Aortic elasticity and size in bicuspid aortic valve syndrome. Eur. Heart J. 2008, 29, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.P.; Xu, C.; Plappert, T.; Desjardins, B.; Gorman, J.H., III; Bavaria, J.E.; Gorman, R.C.; Chandran, K.B.; Jackson, B.M. Increased ascending aortic wall stress in patients with bicuspid aortic valves. Ann. Thorac. Surg. 2011, 92, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Meierhofer, C.; Schneider, E.P.; Lyko, C.; Hutter, A.; Martinoff, S.; Markl, M.; Hager, A.; Hess, J.; Stern, H.; Fratz, S. Wall shear stress and flow patterns in the ascending aorta in patients with bicuspid aortic valves differ significantly from tricuspid aortic valves: A prospective study. Eur. Heart J. Cardiovasc. Imaging 2013, 14, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Pasta, S.; Rinaudo, A.; Luca, A.; Pilato, M.; Scardulla, C.; Gleason, T.G.; Vorp, D.A. Difference in hemodynamic and wall stress of ascending thoracic aortic aneurysms with bicuspid and tricuspid aortic valve. J. Biomech. 2013, 46, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Schmit, B.M.; Fu, C.H.; DeSart, K.; Oh, S.P.; Berceli, S.A.; Jiang, Z.H. Smooth muscle cell-specific tgfbr1 deficiency promotes aortic aneurysm formation by stimulating multiple signaling events. Sci. Rep. 2016, 6, 25444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hou, S.Y.; Chen, J.C.; Zhang, J.S.; Lin, F.Y.; Ju, R.J.; Cheng, X.; Ma, X.W.; Song, Y.; Zhang, Y.Y.; et al. Smad4 deficiency in smooth muscle cells initiates the formation of aortic aneurysm. Circ. Res. 2016, 118, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Ignatieva, E.; Kostina, D.; Irtyuga, O.; Uspensky, V.; Golovkin, A.; Gavriliuk, N.; Moiseeva, O.; Kostareva, A.; Malashicheva, A. Mechanisms of smooth muscle cell differentiation are distinctly altered in thoracic aortic aneurysms associated with bicuspid or tricuspid aortic valves. Front. Physiol. 2017, 8, 536. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, A.; Quarto, C.; Bancone, C.; Castaldo, C.; Di Meglio, F.; Nurzynska, D.; De Santo, L.S.; De Feo, M.; Scardone, M.; Montagnani, S.; et al. Spatiotemporal patterns of smooth muscle cell changes in ascending aortic dilatation with bicuspid and tricuspid aortic valve stenosis: Focus on cell-matrix signaling. J. Thorac. Cardiovasc. Surg. 2008, 135, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta 2008, 1782, 197–228. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, V.; Warburton, D. Fibrillin controls tgf-beta activation. Nat. Genet. 2003, 33, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, S.S.; Cain, S.A.; Morgan, A.; Dallas, S.L.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 regulates the bioavailability of tgfbeta1. J. Cell. Biol. 2007, 176, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Rocchiccioli, S.; Cecchettini, A.; Panesi, P.; Farneti, P.A.; Mariani, M.; Ucciferri, N.; Citti, L.; Andreassi, M.G.; Foffa, I. Hypothesis-free secretome analysis of thoracic aortic aneurysm reinforces the central role of tgf-beta cascade in patients with bicuspid aortic valve. J. Cardiol. 2017, 69, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Fedak, P.W.; de Sa, M.P.; Verma, S.; Nili, N.; Kazemian, P.; Butany, J.; Strauss, B.H.; Weisel, R.D.; David, T.E. Vascular matrix remodeling in patients with bicuspid aortic valve malformations: Implications for aortic dilatation. J. Thorac. Cardiovasc. Surg. 2003, 126, 797–806. [Google Scholar] [CrossRef]
- Leme, M.P.; Butany, T.E.D.J.; Bastos, D.B.E.S.; Feitosa, S.C.P.L.A.; Murad, H.; Magnanini, M.M.F. Molecular evaluation of the great vessels of patients with bicuspid aortic valve disease. Braz. J. Cardiovasc. Surg. 2003, 18, 148–156. [Google Scholar] [CrossRef]
- Grewal, N.; Franken, R.; Mulder, B.J.; Goumans, M.J.; Lindeman, J.H.; Jongbloed, M.R.; DeRuiter, M.C.; Klautz, R.J.; Bogers, A.J.; Poelmann, R.E.; et al. Histopathology of aortic complications in bicuspid aortic valve versus marfan syndrome: Relevance for therapy? Heart Vessel. 2016, 31, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saharinen, J.; Taipale, J.; Monni, O.; Keski-Oja, J. Identification and characterization of a new latent transforming growth factor-beta-binding protein, ltbp-4. J. Biol. Chem. 1998, 273, 18459–18469. [Google Scholar] [CrossRef] [PubMed]
- Kurtovic, S.; Paloschi, V.; Folkersen, L.; Gottfries, J.; Franco-Cereceda, A.; Eriksson, P. Diverging alternative splicing fingerprints in the transforming growth factor-beta signaling pathway identified in thoracic aortic aneurysms. Mol. Med. 2011, 17, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Paloschi, V.; Kurtovic, S.; Folkersen, L.; Gomez, D.; Wagsater, D.; Roy, J.; Petrini, J.; Eriksson, M.J.; Caidahl, K.; Hamsten, A.; et al. Impaired splicing of fibronectin is associated with thoracic aortic aneurysm formation in patients with bicuspid aortic valve. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Folkersen, L.; Wagsater, D.; Paloschi, V.; Jackson, V.; Petrini, J.; Kurtovic, S.; Maleki, S.; Eriksson, M.J.; Caidahl, K.; Hamsten, A.; et al. Unraveling divergent gene expression profiles in bicuspid and tricuspid aortic valve patients with thoracic aortic dilatation: The asap study. Mol. Med. 2011, 17, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Forte, A.; Della Corte, A.; Grossi, M.; Bancone, C.; Provenzano, R.; Finicelli, M.; De Feo, M.; De Santo, L.S.; Nappi, G.; Cotrufo, M.; et al. Early cell changes and tgfbeta pathway alterations in the aortopathy associated with bicuspid aortic valve stenosis. Clin. Sci. 2013, 124, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.A.; Barbour, J.R.; Stroud, R.E.; Bouges, S.; Stephens, S.L.; Spinale, F.G.; Ikonomidis, J.S. Altered transforming growth factor-beta signaling in a murine model of thoracic aortic aneurysm. J. Vasc. Res. 2008, 45, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Forte, A.; Bancone, C.; Cobellis, G.; Buonocore, M.; Santarpino, G.; Fischlein, T.J.M.; Cipollaro, M.; De Feo, M.; Della Corte, A. A possible early biomarker for bicuspid aortopathy: Circulating transforming growth factor beta-1 to soluble endoglin ratio. Circ. Res. 2017, 120, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
- Girdauskas, E.; Borger, M.A.; Secknus, M.A.; Girdauskas, G.; Kuntze, T. Is aortopathy in bicuspid aortic valve disease a congenital defect or a result of abnormal hemodynamics? A critical reappraisal of a one-sided argument. Eur. J. Cardio-Thorac. 2011, 39, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Youssefi, P.; Gomez, A.; He, T.; Anderson, L.; Bunce, N.; Sharma, R.; Figueroa, C.A.; Jahangiri, M. Patient-specific computational fluid dynamics-assessment of aortic hemodynamics in a spectrum of aortic valve pathologies. J. Thorac. Cardiovasc. Surg. 2017, 153, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Den Reijer, P.M.; Sallee, D., III; Van der Velden, P.; Zaaijer, E.R.; Parks, W.J.; Ramamurthy, S.; Robbie, T.Q.; Donati, G.; Lamphier, C.; Beekman, R.P.; et al. Hemodynamic predictors of aortic dilatation in bicuspid aortic valve by velocity-encoded cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2010, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Grewal, N.; Girdauskas, E.; DeRuiter, M.C.; Goumans, M.J.; Lindeman, J.H.; Disha, K.; Wolterbeek, R.; Schoor, D.I.E.; Klautz, R.J.M.; Poelmann, R.E.; et al. The effects of hemodynamics on the inner layers of the aortic wall in patients with a bicuspid aortic valve. Integr. Mol. Med. 2017, 5, 7. [Google Scholar] [CrossRef]
- Sievers, H.H.; Schmidtke, C. A classification system for the bicuspid aortic valve from 304 surgical specimens. J. Thorac. Cardiovasc. Surg. 2007, 133, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Hope, M.D.; Hope, T.A.; Meadows, A.K.; Ordovas, K.G.; Urbania, T.H.; Alley, M.T.; Higgins, C.B. Bicuspid aortic valve: Four-dimensional mr evaluation of ascending aortic systolic flow patterns. Radiology 2010, 255, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, B.M.; Lewin, M.B.; Stout, K.K.; Gill, E.; Prueitt, A.; Byers, P.H.; Otto, C.M. The bicuspid aortic valve: An integrated phenotypic classification of leaflet morphology and aortic root shape. Heart 2008, 94, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, B.M.; Lewin, M.B.; Stout, K.K.; Byers, P.H.; Otto, C.M. Usefulness of bicuspid aortic valve phenotype to predict elastic properties of the ascending aorta. Am. J. Cardiol. 2007, 99, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Girdauskas, E.; Disha, K.; Borger, M.A.; Kuntze, T. Relation of bicuspid aortic valve morphology to the dilatation pattern of the proximal aorta: Focus on the transvalvular flow. Cardiol. Res. Pract. 2012, 2012, 478259. [Google Scholar] [CrossRef] [PubMed]
- Hope, M.D.; Hope, T.A.; Crook, S.E.; Ordovas, K.G.; Urbania, T.H.; Alley, M.T.; Higgins, C.B. 4D flow CMR in assessment of valve-related ascending aortic disease. JACC: Cardiovasc. Imaging 2011, 4, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Ruzmetov, M.; Shah, J.J.; Fortuna, R.S.; Welke, K.F. The association between aortic valve leaflet morphology and patterns of aortic dilation in patients with bicuspid aortic valves. Ann. Thorac. Surg. 2015, 99, 2101–2108. [Google Scholar] [CrossRef] [PubMed]
- Jackson, V.; Petrini, J.; Caidahl, K.; Eriksson, M.J.; Liska, J.; Eriksson, P.; Franco-Cereceda, A. Bicuspid aortic valve leaflet morphology in relation to aortic root morphology: A study of 300 patients undergoing open-heart surgery. Eur. J. Cardio-Thorac. 2011, 40, E118–E124. [Google Scholar] [CrossRef] [PubMed]
- Sievers, H.H.; Stierle, U.; Hachmann, R.M.S.; Charitos, E.I. New insights in the association between bicuspid aortic valve phenotype, aortic configuration and valve haemodynamics. Eur. J. Cardio-Thorac. 2016, 49, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Habchi, K.M.; Ashikhmina, E.; Vieira, V.M.; Shahram, J.T.; Isselbacher, E.M.; Sundt, T.M.; Shekar, P.; Muehlschlegel, J.D.; Body, S.C.; Consortium, B.A.V. Association between bicuspid aortic valve morphotype and regional dilatation of the aortic root and trunk. Int. J. Cardiovasc. Imaging 2017, 33, 341–349. [Google Scholar] [CrossRef] [PubMed]
- McNally, A.; Madan, A.; Sucosky, P. Morphotype-dependent flow characteristics in bicuspid aortic valve ascending aortas: A benchtop particle image velocimetry study. Front. Physiol. 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Atkins, S.K.; McNally, A.; Liu, J.; Sucosky, P. Simulations of morphotype-dependent hemodynamics in non-dilated bicuspid aortic valve aortas. J. Biomech. 2017, 50, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Guzzardi, D.G.; Barker, A.J.; van Ooij, P.; Malaisrie, S.C.; Puthumana, J.J.; Belke, D.D.; Mewhort, H.E.; Svystonyuk, D.A.; Kang, S.; Verma, S.; et al. Valve-related hemodynamics mediate human bicuspid aortopathy: Insights from wall shear stress mapping. J. Am. Coll. Cardiol. 2015, 66, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Pasic, M.; Meyer, R.; Goetze, N.; Bauer, U.; Siniawski, H.; Hetzer, R. Morphometric analysis of aortic media in patients with bicuspid and tricuspid aortic valve. Ann. Thorac. Surg. 2002, 74, 58–62. [Google Scholar] [CrossRef]
- Ohno, M.; Cooke, J.P.; Dzau, V.J.; Gibbons, G.H. Fluid shear-stress induces endothelial transforming growth-factor-beta-1 transcription and production—Modulation by potassium channel blockade. J. Clin. Investig. 1995, 95, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- DeCampli, W.M. Ascending aortopathy with bicuspid aortic valve: More, but not enough, evidence for the hemodynamic theory. J. Thorac. Cardiovasc. Surg. 2017, 153, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.M.; Hess, A.T.; Biasiolli, L.; Glaze, S.J.; Loudon, M.; Pitcher, A.; Davis, A.; Prendergast, B.; Markl, M.; Barker, A.J.; et al. Aortic dilation in bicuspid aortic valve disease flow pattern is a major contributor and differs with valve fusion type. Circ.-Cardiovasc. Imaging 2013, 6, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Hope, T.A.; Markl, M.; Wigstrom, L.; Alley, M.T.; Miller, D.C.; Herfkens, R.J. Comparison of flow patterns in ascending aortic aneurysms and volunteers using four-dimensional magnetic resonance velocity mapping. J. Magn. Reson. Imaging 2007, 26, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Atkins, S.K.; Cao, K.; Rajamannan, N.M.; Sucosky, P. Bicuspid aortic valve hemodynamics induces abnormal medial remodeling in the convexity of porcine ascending aortas. Biomech. Model. Mechanobiol. 2014, 13, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Atkins, S.K.; Moore, A.N.; Sucosky, P. Bicuspid aortic valve hemodynamics does not promote remodeling in porcine aortic wall concavity. World J. Cardiol. 2016, 8, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Borger, M.A.; Preston, M.; Ivanov, J.; Fedak, F.W.M.; Davierwala, P.; Armstrong, S.; David, T.E. Should the ascending aorta be replaced more frequently in patients with bicuspid aortic valve disease? J. Thorac. Cardiovasc. Surg. 2004, 128, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.M.; Gallego, P.; Gonzalez, A.; Aroca, A.; Bret, M.; Mesa, J.M. Risk factors for aortic complications in adults with coarctation of the aorta. J. Am. Coll. Cardiol. 2004, 44, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.G.; Wiegers, S.E.; Plappert, T.; Pochettino, A.; Bavaria, J.E.; Sutton, M.G. Bicuspid aortic valves are associated with aortic dilatation out of proportion to coexistent valvular lesions. Circulation 2000, 102, 35–39. [Google Scholar] [CrossRef]
- Marsalese, D.L.; Moodie, D.S.; Lytle, B.W.; Cosgrove, D.M.; Ratliff, N.B.; Goormastic, M.; Kovacs, A. Cystic medial necrosis of the aorta in patients without marfan’s syndrome: Surgical outcome and long-term follow-up. J. Am. Coll. Cardiol. 1990, 16, 68–73. [Google Scholar] [CrossRef]
- Thanassoulis, G.; Yip, J.W.; Filion, K.; Jamorski, M.; Webb, G.; Siu, S.C.; Therrien, J. Retrospective study to identify predictors of the presence and rapid progression of aortic dilatation in patients with bicuspid aortic valves. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Valdar, W.; Solberg, L.C.; Gauguier, D.; Cookson, W.O.; Rawlins, J.N.; Mott, R.; Flint, J. Genetic and environmental effects on complex traits in mice. Genetics 2006, 174, 959–984. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Lai, H.; Shen, Y.; Breeze, C.; Beck, S.; Hong, T.; Wang, C.; Teschendorff, A.E. DNA methylome analysis reveals distinct epigenetic patterns of ascending aortic dissection and bicuspid aortic valve. Cardiovasc. Res. 2017, 113, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Gregory, S.G.; Krupp, D.; Feng, S.; Dorogi, A.; Haynes, C.; Grass, E.; Lin, S.S.; Hauser, E.R.; Kraus, W.E.; et al. Epigenetic profiling identifies novel genes for ascending aortic aneurysm formation with bicuspid aortic valves. Heart Surg. Forum 2015, 18, E134–E139. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Coyet, A.; Ollivier, V.; Jeunemaitre, X.; Jondeau, G.; Michel, J.B.; Vranckx, R. Epigenetic control of vascular smooth muscle cells in marfan and non-marfan thoracic aortic aneurysms. Cardiovasc. Res. 2011, 89, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Nigam, V.; Sievers, H.H.; Jensen, B.C.; Sier, H.A.; Simpson, P.C.; Srivastava, D.; Mohamed, S.A. Altered micrornas in bicuspid aortic valve: A comparison between stenotic and insufficient valves. J. Heart Valve Dis. 2010, 19, 459–465. [Google Scholar] [PubMed]
- Nader, J.; Metzinger-Le Meuth, V.; Maitrias, P.; Humbert, J.R.; Brigant, B.; Tribouilloy, C.; Metzinger, L.; Caus, T. Mir-92a: A novel potential biomarker of rapid aortic valve calcification. J. Heart Valve Dis. 2017, 26, 327–333. [Google Scholar] [PubMed]
- Albanese, I.; Yu, B.; Al-Kindi, H.; Barratt, B.; Ott, L.; Al-Refai, M.; de Varennes, B.; Shum-Tim, D.; Cerruti, M.; Gourgas, O.; et al. Role of noncanonical wnt signaling pathway in human aortic valve calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, B.; Lovren, F.; Pan, Y.; Garg, V.; Quan, A.; Tang, G.; Singh, K.K.; Shukla, P.C.; Kalra, N.P.; Peterson, M.D.; et al. Mirna-141 is a novel regulator of bmp-2-mediated calcification in aortic stenosis. J. Thorac. Cardiovasc. Surg. 2012, 144, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Fullerton, D.A.; Ao, L.; Zheng, D.; Zhao, K.S.; Meng, X. Bmp-2 and tgf-beta1 mediate biglycan-induced pro-osteogenic reprogramming in aortic valve interstitial cells. J. Mol. Med. 2015, 93, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Fullerton, D.A.; Ao, L.H.; Zhao, K.S.; Meng, X.Z. An epigenetic regulatory loop controls pro-osteogenic activation by tgf-beta 1 or bone morphogenetic protein 2 in human aortic valve interstitial cells. J. Biol. Chem. 2017, 292, 8657–8666. [Google Scholar] [CrossRef] [PubMed]
- Kirk, M.D.; Kahn, A.J. Extracellular matrix synthesized by clonal osteogenic cells is osteoinductive in vivo and in vitro: Role of transforming growth factor-beta 1 in osteoblast cell-matrix interaction. J. Bone Miner. Res. 1995, 10, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Kapinas, K.; Kessler, C.B.; Delany, A.M. Mir-29 suppression of osteonectin in osteoblasts: Regulation during differentiation and by canonical wnt signaling. J. Cell. Biochem. 2009, 108, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Tokuzawa, Y.; Ninomiya, Y.; Yagi, K.; Yatsuka-Kanesaki, Y.; Suda, T.; Fukuda, T.; Katagiri, T.; Kondoh, Y.; Amemiya, T.; et al. Mir-210 promotes osteoblastic differentiation through inhibition of acvr1b. FEBS Lett. 2009, 583, 2263–2268. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Yagi, K.; Tokuzawa, Y.; Kanesaki-Yatsuka, Y.; Suda, T.; Katagiri, T.; Fukuda, T.; Maruyama, M.; Okuda, A.; Amemiya, T.; et al. Mir-125b inhibits osteoblastic differentiation by down-regulation of cell proliferation. Biochem. Biophys. Res. Commun. 2008, 368, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Luzi, E.; Marini, F.; Sala, S.C.; Tognarini, I.; Galli, G.; Brandi, M.L. Osteogenic differentiation of human adipose tissue-derived stem cells is modulated by the mir-26a targeting of the smad1 transcription factor. J. Bone Miner. Res. 2008, 23, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Bae, S.W.; Yu, S.S.; Bae, Y.C.; Jung, J.S. Mir-196a regulates proliferation and osteogenic differentiation in mesenchymal stem cells derived from human adipose tissue. J. Bone Miner. Res. 2009, 24, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, H.; Liu, W.; Hu, R.; Huang, B.; Tan, Y.F.; Xu, K.; Sheng, Z.F.; Zhou, H.D.; Wu, X.P.; et al. A novel microrna targeting hdac5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J. Clin. Investig. 2009, 119, 3666–3677. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Liu, W.; Li, H.; Yang, L.; Chen, C.; Xia, Z.Y.; Guo, L.J.; Xie, H.; Zhou, H.D.; Wu, X.P.; et al. A Runx2/miR-3960/miR-2861 regulatory feedback loop during mouse osteoblast differentiation. J. Biol. Chem. 2011, 286, 12328–12339. [Google Scholar] [CrossRef] [PubMed]
- Di Gregoli, K.; Mohamad Anuar, N.N.; Bianco, R.; White, S.J.; Newby, A.C.; George, S.J.; Johnson, J.L. MicroRNA-181b controls atherosclerosis and aneurysms through regulation of TIMP-3 and elastin. Circ. Res. 2017, 120, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Huang, A.; Ferruzzi, J.; Mecham, R.P.; Starcher, B.C.; Tellides, G.; Humphrey, J.D.; Giordano, F.J.; Niklason, L.E.; Sessa, W.C. Inhibition of microRNA-29 enhances elastin levels in cells haploinsufficient for elastin and in bioengineered vessels—Brief report. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Attia, R.; Mayr, U.; Gomes, R.S.; Phinikaridou, A.; Yin, X.; Langley, S.R.; Willeit, P.; Lu, R.; Fanshawe, B.; et al. Role of mir-195 in aortic aneurysmal disease. Circ. Res. 2014, 115, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Sudo, R.; Sato, F.; Azechi, T.; Wachi, H. MiR-29-mediated elastin down-regulation contributes to inorganic phosphorus-induced osteoblastic differentiation in vascular smooth muscle cells. Genes Cells 2015, 20, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zheng, R.; Xiao, F.; Zhang, S.; He, K.; Zhang, J.; Shao, Y. Downregulated microrna-195 in the bicuspid aortic valve promotes calcification of valve interstitial cells via targeting smad7. Cell. Physiol. Biochem. 2017, 44, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.M.; Fernandez Esmerats, J.; Khambouneheuang, L.; Kumar, S.; Simmons, R.; Jo, H. Mechanosensitive microrna-181b regulates aortic valve endothelial matrix degradation by targeting timp3. Cardiovasc. Eng. Technol. 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, S.; Deng, C.; Li, F.; Wang, Y.; Hu, X.; Shi, F.; Dong, N. Microrna-204 targets runx2 to attenuate bmp-2-induced osteoblast differentiation of human aortic valve interstitial cells. J. Cardiovasc. Pharmacol. 2015, 66, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhao, M.; Yang, Y.; Huang, Z.; Shi, C.; Hou, X.; Zhao, Y.; Chen, B.; Xiao, Z.; Liu, J.; et al. Microrna-449c-5p inhibits osteogenic differentiation of human vics through smad4-mediated pathway. Sci. Rep. 2017, 7, 8740. [Google Scholar] [CrossRef] [PubMed]
- Keramati, A.R.; Sadeghpour, A.; Farahani, M.M.; Chandok, G.; Mani, A. The non-syndromic familial thoracic aortic aneurysms and dissections maps to 15q21 locus. BMC Med. Genet. 2010, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Barbour, J.R.; Spinale, F.G.; Ikonomidis, J.S. Proteinase systems and thoracic aortic aneurysm progression. J. Surg. Res. 2007, 139, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Patuzzo, C.; Pasquali, A.; Malerba, G.; Trabetti, E.; Pignatti, P.; Tessari, M.; Faggian, G. A preliminary microrna analysis of non syndromic thoracic aortic aneurysms. Balkan J. Med. Genet. 2012, 15, 51–55. [Google Scholar] [PubMed]
- Cammaerts, S.; Strazisar, M.; De Rijk, P.; Del Favero, J. Genetic variants in microrna genes: Impact on microrna expression, function, and disease. Front. Genet. 2015, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Duggirala, A.; Delogu, F.; Angelini, T.G.; Smith, T.; Caputo, M.; Rajakaruna, C.; Emanueli, C. Non coding rnas in aortic aneurysmal disease. Front. Genet. 2015, 6, 125. [Google Scholar] [CrossRef] [PubMed]
- Borghini, A.; Foffa, I.; Pulignani, S.; Vecoli, C.; Ait-Ali, L.; Andreassi, M.G. Mirnome profiling in bicuspid aortic valve-associated aortopathy by next-generation sequencing. Int. J. Mol. Sci. 2017, 18, 2948. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.F.; Zou, S.L.; Weng, J.F.; Hon, L.W.; Yang, L.; Zhao, Z.Q.; Bao, J.M.; Jing, Z.P. A microrna profile comparison between thoracic aortic dissection and normal thoracic aorta indicates the potential role of micrornas in contributing to thoracic aortic dissection pathogenesis. J. Vasc. Surg. 2011, 53, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.A.; Stroud, R.E.; O’Quinn, E.C.; Black, L.E.; Barth, J.L.; Elefteriades, J.A.; Bavaria, J.E.; Gorman, J.H., III; Gorman, R.C.; Spinale, F.G.; et al. Selective microrna suppression in human thoracic aneurysms: Relationship of mir-29a to aortic size and proteolytic induction. Circ. Cardiovasc. Genet. 2011, 4, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Seeger, T.; Heydt, S.; Fischer, A.; Hergenreider, E.; Horrevoets, A.J.; Vinciguerra, M.; Rosenthal, N.; Sciacca, S.; Pilato, M.; et al. Microrna-29 in aortic dilation: Implications for aneurysm formation. Circ. Res. 2011, 109, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Song, H.F.; Li, S.H.; Guo, J.; Tsang, K.; Tumiati, L.; Butany, J.; Yau, T.M.; Ouzounian, M.; Fu, S.B.; et al. Progressive aortic dilation is regulated by mir-17-associated mirnas. J. Am. Coll. Cardiol. 2016, 67, 2965–2977. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Tian, C.; Sun, X.; Qian, X.; Liu, P.; Liu, W.; Chang, Q. Overexpression of microrna-145 promotes ascending aortic aneurysm media remodeling through tgf-beta 1. Eur. J. Vasc. Endovasc. 2015, 49, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Albinsson, S.; Della Corte, A.; Alajbegovic, A.; Krawczyk, K.K.; Bancone, C.; Galderisi, U.; Cipollaro, M.; De Feo, M.; Forte, A. Patients with bicuspid and tricuspid aortic valve exhibit distinct regional microrna signatures in mildly dilated ascending aorta. Heart Vessel. 2017, 32, 750–767. [Google Scholar] [CrossRef] [PubMed]
- Elia, L.; Quintavalle, M.; Zhang, J.; Contu, R.; Cossu, L.; Latronico, M.V.; Peterson, K.L.; Indolfi, C.; Catalucci, D.; Chen, J.; et al. The knockout of mir-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ. 2009, 16, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Licholai, S.; Blaz, M.; Kapelak, B.; Sanak, M. Unbiased profile of microrna expression in ascending aortic aneurysm tissue appoints molecular pathways contributing to the pathology. Ann. Thorac. Surg. 2016, 102, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhang, Z.; Yang, H.; Lin, Q.; Han, C.; Qin, X. The involvement of mir-29b-3p in arterial calcification by targeting matrix metalloproteinase-2. Biomed. Res. Int. 2017, 2017, 6713606. [Google Scholar] [CrossRef] [PubMed]
- Albinsson, S.; Skoura, A.; Yu, J.; DiLorenzo, A.; Fernandez-Hernando, C.; Offermanns, S.; Miano, J.M.; Sessa, W.C. Smooth muscle mirnas are critical for post-natal regulation of blood pressure and vascular function. PLoS ONE 2011, 6, e18869. [Google Scholar] [CrossRef] [PubMed]
- Albinsson, S.; Sessa, W.C. Can micrornas control vascular smooth muscle phenotypic modulation and the response to injury? Physiol. Genom. 2011, 43, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Albinsson, S.; Suarez, Y.; Skoura, A.; Offermanns, S.; Miano, J.M.; Sessa, W.C. Micrornas are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, Y.S.; Nguyen, P.; Wang, K.C.; Weiss, A.; Kuo, Y.C.; Chiu, J.J.; Shyy, J.Y.; Chien, S. Regulation of vascular smooth muscle cell turnover by endothelial cell-secreted microrna-126: Role of shear stress. Cir. Res. 2013, 113, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Demolli, S.; Doddaballapur, A.; Devraj, K.; Stark, K.; Manavski, Y.; Eckart, A.; Zehendner, C.M.; Lucas, T.; Korff, T.; Hecker, M.; et al. Shear stress-regulated mir-27b controls pericyte recruitment by repressing sema6a and sema6d. Cardiovasc. Res. 2017, 113, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Hergenreider, E.; Heydt, S.; Treguer, K.; Boettger, T.; Horrevoets, A.J.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through mirnas. Nat. Cell Biol. 2012, 14, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Micaelo, N.; Beltran-Debon, R.; Baiges, I.; Faiges, M.; Alegret, J.M. Specific circulating microrna signature of bicuspid aortic valve disease. J. Transl. Med. 2017, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Kauppinen, S. Development of microrna therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.X.; Wang, Y.; Zheng, D.D.; Wang, T.; Pan, M.; Shi, J.H.; Zhu, J.H.; Li, X.F. Differential expression of micrornas in calcific aortic stenosis. Clin. Lab. 2017, 63, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, J.S.; Ivey, C.R.; Wheeler, J.B.; Akerman, A.W.; Rice, A.; Patel, R.K.; Stroud, R.E.; Shah, A.A.; Hughes, C.G.; Ferrari, G.; et al. Plasma biomarkers for distinguishing etiologic subtypes of thoracic aortic aneurysm disease. J. Thorac. Cardiovasc. Surg. 2013, 145, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Oury, C.; Servais, L.; Bouznad, N.; Hego, A.; Nchimi, A.; Lancellotti, P. Micrornas in valvular heart diseases: Potential role as markers and actors of valvular and cardiac remodeling. Int. J. Mol. Sci. 2016, 17, 1120. [Google Scholar] [CrossRef] [PubMed]
- Abdulkareem, N.; Smelt, J.; Jahangiri, M. Bicuspid aortic valve aortopathy: Genetics, pathophysiology and medical therapy. Interact. Cardiovasc. Thorac. Surg. 2013, 17, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Siu, S.C. Aortic dilatation in patients with bicuspid aortic valve. N. Engl. J. Med. 2014, 370, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- McGee, K.; Bollache, E.; Barker, A.J.; Carr, J.C.; Markl, M.; Kansal, P. Impact of beta-blocker, ace inhibitor, and arb therapy on thoracic aorta wall shear stress in bicuspid aortic valve patients. J. Cardiovasc. Magn. Reson. 2016, 18, 345. [Google Scholar] [CrossRef]
- Groenink, M.; den Hartog, A.W.; Franken, R.; Radonic, T.; de Waard, V.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Spijkerboer, A.M.; Marquering, H.A.; et al. Losartan reduces aortic dilatation rate in adults with marfan syndrome: A randomized controlled trial. Eur. Heart J. 2013, 34, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Yamamoto, H.; Iwata, K.; Goto, S.; Umemoto, T.; Group, A. Effects of statin therapy on abdominal aortic aneurysm growth: A meta-analysis and meta-regression of observational comparative studies. Eur. J. Vasc. Endovasc. Surg. 2012, 44, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.P.; McCarthy, P.; Andrei, A.C.; Li, Z.; Mcgee, E.; Malaisrie, S.C.; Clennon, C.; Puthumana, J. Statin use and aneurysm risk in patients with bicuspid aortic valve disease. J. Am. Coll. Cardiol. 2014, 63, A492. [Google Scholar] [CrossRef]
- Verma, S.; Yanagawa, B.; Kalra, S.; Ruel, M.; Peterson, M.D.; Yamashita, M.H.; Fagan, A.; Currie, M.E.; White, C.W.; Wai Sang, S.L.; et al. Knowledge, attitudes, and practice patterns in surgical management of bicuspid aortopathy: A survey of 100 cardiac surgeons. J. Thorac. Cardiovasc. Surg. 2013, 146, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Erbel, R.; Aboyans, V.; Boileau, C.; Bossone, E.; Bartolomeo, R.D.; Eggebrecht, H.; Evangelista, A.; Falk, V.; Frank, H.; Gaemperli, O.; et al. 2014 esc guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The task force for the diagnosis and treatment of aortic diseases of the european society of cardiology (esc). Eur. Heart J. 2014, 35, 2873–2926. [Google Scholar] [PubMed]
- Baumgartner, H.; Falk, V.; Bax, J.J.; De Bonis, M.; Hamm, C.; Holm, P.J.; Iung, B.; Lancellotti, P.; Lansac, E.; Munoz, D.R.; et al. 2017 ESC/EACTS guidelines for the management of valvular heart disease. Eur. Heart J. 2017, 38, 2739–2791. [Google Scholar] [CrossRef] [PubMed]
- Hiratzka, L.F.; Creager, M.A.; Isselbacher, E.M.; Svensson, L.G.; Nishimura, R.A.; Bonow, R.O.; Guyton, R.A.; Sundt, T.M., III. Surgery for aortic dilatation in patients with bicuspid aortic valves: A statement of clarification from the american college of cardiology/american heart association task force on clinical practice guidelines. J. Am. Coll. Cardiol. 2016, 67, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Greenland, P.; Alpert, J.S.; Beller, G.A.; Benjamin, E.J.; Budoff, M.J.; Fayad, Z.A.; Foster, E.; Hlatky, M.A.; Hodgson, J.M.; Kushner, F.G.; et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: Executive summary: A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Circulation 2010, 122, 2748–2764. [Google Scholar] [CrossRef] [PubMed]
- Wooten, E.C.; Iyer, L.K.; Montefusco, M.; Hedgepeth, A.K.; Payne, D.D.; Kapur, N.K.; Housman, D.E.; Mendelsohn, M.E.; Huggins, G.S. Application of gene network analysis techniques identifies axin1/pdia2 and endoglin haplotypes associated with bicuspid aortic valve. PLoS ONE 2010, 5, 8830. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.H. From genotypes to genometypes: Putting the genome back in genome-wide association studies. Eur. J. Hum. Genet. 2009, 17, 1205–1206. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhou, W.; Jiao, J.; Nielsen, J.B.; Mathis, M.R.; Heydarpour, M.; Lettre, G.; Folkersen, L.; Prakash, S.; Schurmann, C.; et al. Protein-altering and regulatory genetic variants near gata4 implicated in bicuspid aortic valve. Nat. Commun. 2017, 8, 15481. [Google Scholar] [CrossRef] [PubMed]
- Regalado, E.S.; Guo, D.C.; Villamizar, C.; Avidan, N.; Gilchrist, D.; McGillivray, B.; Clarke, L.; Bernier, F.; Santos-Cortez, R.L.; Leal, S.M.; et al. Exome sequencing identifies smad3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011, 109, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.; Kumar, A.A.; Luyckx, I.; Preuss, C.; Cannaerts, E.; van de Beek, G.; Wieschendorf, B.; Alaerts, M.; Bolar, N.; Vandeweyer, G.; et al. Candidate gene resequencing in a large bicuspid aortic valve-associated thoracic aortic aneurysm cohort: Smad6 as an important contributor. Front. Physiol. 2017, 8, 400. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.S.; Vasiliou, V.; Cooper, D.N. A new era in the discovery of de novo mutations underlying human genetic disease. Hum. Genom. 2012, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Hitz, M.P.; Lemieux-Perreault, L.P.; Marshall, C.; Feroz-Zada, Y.; Davies, R.; Yang, S.W.; Lionel, A.C.; D’Amours, G.; Lemyre, E.; Cullum, R.; et al. Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet. 2012, 8, e1002903. [Google Scholar] [CrossRef] [PubMed]
- Guala, D.; Sonnhammer, E.L.L. A large-scale benchmark of gene prioritization methods. Sci. Rep. 2017, 7, 46598. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, I.; Loeys, B.L. The genetic architecture of non-syndromic thoracic aortic aneurysm. Heart 2015, 101, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Scherer, S.; Liu, Y.; Presley, C.; Guo, D.; Estrera, A.L.; Safi, H.J.; Brasier, A.R.; et al. Myh11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin ii. Hum. Mol. Genet. 2007, 16, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.M.; Vranckx, R.; Van Kien, P.K.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.M.; Brunotte, F.O.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.Q.; Guo, D.C.; Prakash, S.K.; McDonald, M.L.N.; Johnson, R.J.; Wang, M.; Regalado, E.S.; Russell, L.; Cao, J.M.; Kwartler, C.; et al. Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet. 2011, 7, e1002118. [Google Scholar] [CrossRef] [PubMed]
- Pepe, G.; Nistri, S.; Giusti, B.; Sticchi, E.; Attanasio, M.; Porciani, C.; Abbate, R.; Bonow, R.O.; Yacoub, M.; Gensini, G.F. Identification of fibrillin 1 gene mutations in patients with bicuspid aortic valve (bav) without marfan syndrome. BMC Med. Genet. 2014, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Thompson, P.D.; Puffer, J.C.; McGrew, C.A.; Strong, W.B.; Douglas, P.S.; Clark, L.T.; Mitten, M.J.; Crawford, M.D.; Atkins, D.L.; et al. Cardiovascular preparticipation screening of competitive athletes: Addendum—An addendum to a statement for health professionals from the sudden death committee (council on clinical cardiology) and the congenital cardiac defects committee (council on cardiovascular disease in the young), american heart association. Circulation 1998, 97, 2294. [Google Scholar] [PubMed]
- Staudt, D.; Stainier, D. Uncovering the molecular and cellular mechanisms of heart development using the zebrafish. Annu. Rev. Genet. 2012, 46, 397–418. [Google Scholar] [CrossRef] [PubMed]
- Nigam, V.; Srivastava, D. Notch1 represses osteogenic pathways in aortic valve cells. J. Mol. Cell. Cardiol. 2009, 47, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Ipe, J.; Swart, M.; Burgess, K.S.; Skaar, T.C. High-throughput assays to assess the functional impact of genetic variants: A road towards genomic-driven medicine. Cts-Clin. Transl. Sci. 2017, 10, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.; McGarry, M.P.; Lee, N.A.; Lee, J.J. The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic. Res. 2012, 21, 327–349. [Google Scholar] [CrossRef] [PubMed]
- Igoucheva, O.; Alexeev, V.; Halabi, C.M.; Adams, S.M.; Stoilov, I.; Sasaki, T.; Arita, M.; Donahue, A.; Mecham, R.P.; Birk, D.E.; et al. Fibulin-4 e57k knock-in mice recapitulate cutaneous, vascular and skeletal defects of recessive cutis laxa 1b with both elastic fiber and collagen fibril abnormalities. J. Biol. Chem. 2015, 290, 21443–21459. [Google Scholar] [CrossRef] [PubMed]
- Bick, A.G.; Flannick, J.; Ito, K.; Cheng, S.; Vasan, R.S.; Parfenov, M.G.; Herman, D.S.; DePalma, S.R.; Gupta, N.; Gabriel, S.B.; et al. Burden of rare sarcomere gene variants in the framingham and jackson heart study cohorts. Am. J. Hum. Genet. 2012, 91, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Greenway, S.C.; Pereira, A.C.; Lin, J.C.; DePalma, S.R.; Israel, S.J.; Mesquita, S.M.; Ergul, E.; Conta, J.H.; Korn, J.M.; McCarroll, S.A.; et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of fallot. Nat. Genet. 2009, 41, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.M.; Wakimoto, H.; Seidman, J.G.; Seidman, C.E. Allele-specific silencing of mutant myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science 2013, 342, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Fedak, P.W.; Verma, S.; David, T.E.; Leask, R.L.; Weisel, R.D.; Butany, J. Clinical and pathophysiological implications of a bicuspid aortic valve. Circulation 2002, 106, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Padang, R.; Bagnall, R.D.; Semsarian, C. Genetic basis of familial valvular heart disease. Circ.-Cardiovasc. Gene. 2012, 5, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Muller, J.; Daugherty, A.; Norman, P. Abdominal aortic aneurysm: Pathogenesis and implications for management. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2605–2613. [Google Scholar] [CrossRef] [PubMed]
- Schanzer, A.; Simons, J.P.; Flahive, J.; Durgin, J.; Aiello, F.A.; Doucet, D.; Steppacher, R.; Messina, L.M. Outcomes of fenestrated and branched endovascular repair of complex abdominal and thoracoabdominal aortic aneurysms. J. Vasc. Surg. 2017, 66, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.R.; Brooke, B.S. Effectiveness of open versus endovascular abdominal aortic aneurysm repair in population settings: A systematic review of statewide databases. Surgery 2017, 162, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, Z.; Allen, B.D.; Garcia, J.; Jarvis, K.B.; Markl, M. 4D flow imaging with MRI. Cardiovasc. Diagn. Ther. 2014, 4, 173–192. [Google Scholar] [PubMed]
- Della Corte, A.; Body, S.C.; Booher, A.M.; Schaefers, H.J.; Milewski, R.K.; Michelena, H.I.; Evangelista, A.; Pibarot, P.; Mathieu, P.; Limongelli, G.; et al. Surgical treatment of bicuspid aortic valve disease: Knowledge gaps and research perspectives. J. Thorac. Cardiovasc. Surg. 2014, 147, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Park, C.B.; Greason, K.L.; Suri, R.M.; Michelena, H.I.; Schaff, H.V.; Sundt, T.M. Fate of nonreplaced sinuses of valsalva in bicuspid aortic valve disease. J. Thorac. Cardiovasc. Surg. 2011, 142, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Schafers, H.J.; Kunihara, T.; Fries, P.; Brittner, B.; Aicher, D. Valve-preserving root replacement in bicuspid aortic valves. J. Thorac. Cardiovasc. Surg. 2010, 140, S36–S40. [Google Scholar] [CrossRef] [PubMed]
- Fesler, A.; Jiang, J.; Zhai, H.; Ju, J. Circulating microrna testing for the early diagnosis and follow-up of colorectal cancer patients. Mol. Diagn. Ther. 2014, 18, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Li, C.; Ge, H.; Jiang, Y.; Li, Y. Circulating microrna: A novel potential biomarker for early diagnosis of intracranial aneurysm rupture a case control study. J. Transl. Med. 2013, 11, 296. [Google Scholar] [CrossRef] [PubMed]
- Michelena, H.I.; Prakash, S.K.; Della Corte, A.; Bissell, M.M.; Anavekar, N.; Mathieu, P.; Bosse, Y.; Limongelli, G.; Bossone, E.; Benson, D.W.; et al. Bicuspid aortic valve: Identifying knowledge gaps and rising to the challenge from the international bicuspid aortic valve consortium (bavcon). Circulation 2014, 129, 2691–2704. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Full Name |
|---|---|
| AAo | Ascending aorta |
| ACE | Angiotensin-converting enzyme |
| BAV | Bicuspid aortic valve |
| ECM | Extracellular matrix |
| GWA | Genome wide association |
| LLC | Large latent complex |
| LTBP | Latent transforming growth factor beta binding protein |
| miRNA | microRNA |
| MMP | Matrix metalloproteinase |
| TAA | Thoracic aortic aneurysm |
| TAV | Tricuspid aortic valve |
| TGF | Transforming growth factor |
| TIMP | Tissue inhibitor matrix metalloproteinase |
| SLC | Small latent complex |
| SMC | Smooth muscle cells |
| WSS | Wall shear stress |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sophocleous, F.; Milano, E.G.; Pontecorboli, G.; Chivasso, P.; Caputo, M.; Rajakaruna, C.; Bucciarelli-Ducci, C.; Emanueli, C.; Biglino, G. Enlightening the Association between Bicuspid Aortic Valve and Aortopathy. J. Cardiovasc. Dev. Dis. 2018, 5, 21. https://doi.org/10.3390/jcdd5020021
Sophocleous F, Milano EG, Pontecorboli G, Chivasso P, Caputo M, Rajakaruna C, Bucciarelli-Ducci C, Emanueli C, Biglino G. Enlightening the Association between Bicuspid Aortic Valve and Aortopathy. Journal of Cardiovascular Development and Disease. 2018; 5(2):21. https://doi.org/10.3390/jcdd5020021
Chicago/Turabian StyleSophocleous, Froso, Elena Giulia Milano, Giulia Pontecorboli, Pierpaolo Chivasso, Massimo Caputo, Cha Rajakaruna, Chiara Bucciarelli-Ducci, Costanza Emanueli, and Giovanni Biglino. 2018. "Enlightening the Association between Bicuspid Aortic Valve and Aortopathy" Journal of Cardiovascular Development and Disease 5, no. 2: 21. https://doi.org/10.3390/jcdd5020021