Patient Susceptibility to Candidiasis—A Potential for Adjunctive Immunotherapy

Department of Internal Medicine and Radboud Center for Infectious diseases (RCI), Radboud University Medical Center, 6525 GA Nijmegen, The Netherlands
*
Author to whom correspondence should be addressed.
J. Fungi 2018, 4(1), 9; https://doi.org/10.3390/jof4010009
Submission received: 27 October 2017
/
Revised: 15 December 2017
/
Accepted: 30 December 2017
/
Published: 9 January 2018
(This article belongs to the Special Issue Translational Science: Approaches to Reducing Burden Caused by Fungal Diseases)
Abstract
:Candida spp. are colonizing fungi of human skin and mucosae of the gastrointestinal and genitourinary tract, present in 30–50% of healthy individuals in a population at any given moment. The host defense mechanisms prevent this commensal fungus from invading and causing disease. Loss of skin or mucosal barrier function, microbiome imbalances, or defects of immune defense mechanisms can lead to an increased susceptibility to severe mucocutaneous or invasive candidiasis. A comprehensive understanding of the immune defense against Candida is essential for developing adjunctive immunotherapy. The important role of underlying genetic susceptibility to Candida infections has become apparent over the years. In most patients, the cause of increased susceptibility to fungal infections is complex, based on a combination of immune regulation gene polymorphisms together with other non-genetic predisposing factors. Identification of patients with an underlying genetic predisposition could help determine which patients could benefit from prophylactic antifungal treatment or adjunctive immunotherapy. This review will provide an overview of patient susceptibility to mucocutaneous and invasive candidiasis and the potential for adjunctive immunotherapy.
1. Introduction
Candida spp., especially Candida albicans, are colonizing fungi of human skin and mucosae of the gastrointestinal and genitourinary tract of 30–50% of healthy individuals in any given human population at a certain moment, with the majority of the individuals being colonized during certain periods of their lives. Under normal conditions the human host defense prevents this commensal fungus from becoming a pathogen. Loss of skin or mucosal barrier function, microbiome imbalances, or defects of immune defense mechanisms can all lead to an increased susceptibility to severe mucocutaneous or invasive candidiasis [1].
Superficial candidiasis of mucosal membranes and skin is highly prevalent and occurs in immunocompromised as well as in apparently immunocompetent patients. In most cases these Candida infections of skin, nails, oropharyngeal mucosa, esophagus, and genital tract are occasional and not severe. Some patients, however, suffer from severe recurrent or persistent mucocutaneous Candida infections in the absence of commonly predisposing factors such as diabetes, and this condition has been termed chronic mucocutaneous candidiasis (CMC) [2].
Invasive candidiasis includes candidemia, deep-seated infections, and syndromes such as chronic disseminated (hepatosplenic) candidiasis. The mortality rate of invasive candidiasis is as high as 40%, which makes this a life-threatening condition. Known risk factors are indwelling vascular catheters, recent abdominal surgery, hematologic cancers, the administration of broad-spectrum antibiotic therapy, and multifocal C. albicans colonization. Interestingly, most patients admitted to the intensive care unit (ICU) have several of these risk factors but only a few develop invasive candidiasis [3].
Several genetic mutations and variations in immune regulation genes have been identified that confer susceptibility to Candida infections. Identification of patients with an underlying genetic predisposition could help determine which patients could benefit from prophylactic antifungal treatment or adjunctive immunotherapy [3,4]. This review will provide an overview of the mechanisms that increase patient susceptibility to mucocutaneous and invasive candidiasis and will provide a rationale of the potential for adjunctive immunotherapy.
2. The Host Immune Defense against Candida
2.1. Recognition of C. albicans
The first step of the host immune defence is recognition of an invading pathogen by the innate immune system. Recognition of microorganisms occurs through conserved chemical signatures, called pathogen-associated molecular patterns (PAMPs). These PAMPs are recognized by specific innate immune receptors known as pathogen recognition receptors (PRRs) [5,6]. The host is therefore able to orchestrate a pathogen-specific immune defense. The cell populations of the innate immune system involved in recognition of C. albicans are monocytes, macrophages, dendritic cells and neutrophils. These cells express PRRs in different patterns, rendering the host capable of a cell-type-specific immune defense against C. albicans [5].
The Candida cell wall consists of an inner layer of polysaccharides (chitin, 1,3-β-glucans and 1,6-β-glucans) and an outer layer of proteins glycosylated with mannan [1,5,6,7,8,9,10,11,12,13,14,15] constituting the PAMPs. The main PRRs that are involved in the recognition of C. albicans are the Toll-like receptors (TLRs) and the C-type lectin receptors (CLRs). TLR2 recognizes phospholipomannans, TLR4 recognizes O-linked mannans, the CLR dectin-1 recognizes β-glucans and the CLRs macrophage mannose receptor (MR) and dendritic cell (DC)-specific-ICAM3-grabbing non-integrin (DC-sign) recognize N-linked mannans [1,5,6,7,16,17,18,19,20,21,22,23,24,25,26,27]. The macrophage-inducible CLR (Mincle), expressed predominantly on macrophages, has been recognized as a receptor for C. albicans and plays a role in macrophage responses to C. albicans [28]. The ligand that binds to Mincle has not yet been identified. More recently, activation of the inflammasome through the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) pyrin domain-containing 3 (NLRP3) and the RigI-helicase receptor (RLR) melanoma differentiation-associated protein 5 (MDA5) has also been shown to be involved in anti-Candida host defense [29,30]. MDA5 is known for its recognition of viral RNA and its role in antiviral immunity, recently it has been suggested that MDA5 is also involved in antifungal immunity. Which ligands cause activation of MDA5 in Candida infection remains unclear [29,31].
The recognition of Candida species by innate immune cells is depicted in Figure 1.
2.2. Activation of Host Immune Defense
Ligation of the PAMPs with their corresponding PRR leads to activation of the innate immune system, inducing production of pro-inflammatory cytokines and chemokines, and activation of the NLRP3 inflammasome, which processes pro-interleukin (IL)-1β and pro-IL-18 into their biologically active forms, with subsequent activation of inflammation and a directed adaptive immune response. Subsequently, these pathways potentiate phagocytosis and killing of C. albicans, predominantly by neutrophils [1,5,6,7,32,33,34,35,36,37,38,39,40,41,42,43,44,45].
Antigen-presenting dendritic cells are important for the activation of T cell responses. Candida-specific T helper cells (TH-cells) consist of TH1-cells and TH17-cells. TH1-cells are induced by IL-18 and produce interferon-gamma (IFN-γ). TH17-cells are induced by IL-1β and produce IL-17 and IL-22. IFN-γ is important for the fungicidal activity of neutrophils and macrophages. IL-17 and IL-22 induce neutrophil recruitment and activate neutrophils as well as epithelial cells and induce the release of antifungal β-defensins [1,5,6,46,47,48,49,50,51]. Activation via the intracellular PRR MDA5 induces a signaling pathway leading to the production of type I interferons. Recent studies have led to the suggestion that these interferons skew the adaptive cytokine response, induced by C. albicans, from a TH17 response to a TH1 response [29,31].
The composition of the host microbiome likely has significant impact on Candida colonization, invasion and host defense against Candida. As an example, the skin microbiome of patients with immunodeficiency disorders as CMC and hyper IgE syndrome (HIES) differs substantially from healthy individuals [52,53]. This topic will be discussed in another chapter of this special issue.
3. Mucocutaneous Candidiasis
Most cases of oropharyngeal and esophageal candidiasis occur in the setting of well-known risk factors, are incidental, and not severe. In these patients, use of broad-spectrum antibiotic therapy leads to Candida colonization on epithelial surfaces of skin, oropharynx, and vagina [54,55]. Development of vulvovaginal candidiasis following antibiotic therapy occurred in 20–22% of patients previously colonized with Candida. Studies show that only patients that were previously colonized with Candida had an increased risk of developing vulvovaginal candidiasis after antibiotic therapy [56,57]. Inhalation corticosteroids and systemic prednisone increase the risk of Candida infections [58]. Patients with diabetes have higher rates of vaginal Candida colonization and vulvovaginal candidiasis [59,60].
In addition, disruption of epithelial function, microbiome imbalances and loss of T cell function increases susceptibility to mucocutaneous candidiasis. Radiation therapy of the oro-maxillo-facial area leads to mucositis and all patients develop oral candidiasis at some point during therapy [61]. Candida infection in HIV patients is one of the so-called AIDS-defining illnesses. In absence of antiretroviral therapy, 57% of patients develop oropharyngeal candidiasis, typically when T cell CD4+ count is below 200 cells/m3 [62].
3.1. Epithelial Function
A major player in the host immune defense against mucocutaneous candidiasis is the epithelium and its interaction with C. albicans. The epithelium lining the oropharynx, skin and vagina forms the first physical barrier for invading pathogens. In addition to their physical barrier function, epithelial cells have an important immune function. Epithelial cells recognize C. albicans through the pattern recognition receptor (PRR) TLR4 and are able to differentiate between colonization and invasion. Recognition leads to the activation of the protein complex nucleair factor-kappaB (NF-κB) and the proto-oncogene c-Jun (JUN). Only when Candida germinates and forms hyphae invading the epithelium, a second response is initiated by activation of mitogen-activated protein kinase 1 (MAPK1) and FOS protein signaling. This triggers the initiation of the host response by releasing pro-inflammatory cytokines [6,7]. Recently, a fungal peptide toxin called Candidalysin has been discovered, secreted by C. albicans hyphae, that damages the epithelium and leads to immune activation of the epithelium by recognition of the peptide [63].
3.2. Th17 Pathway
The adaptive immune system plays a major role in the host defense against mucocutaneous candidiasis. Th17 cells, a differentiated subset of the CD4+ T-helper lineage, are induced after recognition of C. albicans mannan by the macrophage mannose receptor (MR). The dectin-1/TLR2 pathway enhances this response [64]. Th17 cells are activated via pro-inflammatory cytokines IL-1β, IL-6, TGF-β and IL-23 produced by antigen-presenting cells [65]. Production of IL-1β is predominantly induced by Candida hyphae via activation of the NLRP3 inflammasome and caspase-1. This means the Th17 pathway is only fully induced when Candida is invading, in contrast to colonizing mucosal tissue [66]. When activated, the Th17 cells produce IL-17A, IL-17F and IL-22. At early stages of infection, before induction of the Th17 subset, mucosa-associated type 3 innate lymphoid cells (ILCs) secrete IL-17 and thereby support host defense against mucocutaneous candidiasis [67].
IL-22 acts on epithelial cells to induce the release of antimicrobial peptides such as β-defensins. These β-defensins have potent fungicidal activity enabling the epithelium to prevent invasion of C. albicans. IL-17A and IL-17F induce neutrophil recruitment from the bloodstream to the site of infection where these cells aid to prevent Candida dissemination [6,7,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82].
Induction of genes involved in neutrophil recruitment during oropharyngeal candidiasis is partly dependent on IL-17 receptor A activation [83]. Recently, it has been suggested that the role of IL-17 in neutrophil recruitment is dependent on the tissue environment [71]. In a mouse model of oropharyngeal candidiasis, the IL-17 pathway was not required for an adequate neutrophil response to C. albicans in the oral mucosa. Mice deficient in the IL-17 receptor A or depleted of IL-17A and IL-17F were able to establish normal neutrophil recruitment and function. Although the neutrophil response was normal, Candida colonization was persistent due to absent induction of epithelial antimicrobial peptides. These findings suggest that the main role of IL-17 in mucosal host defense against Candida is the stimulation of epithelial cells to release antimicrobial peptides, and to a lesser extent neutrophil recruitment [84]. Results from experimental psoriasis models in IL-17 receptor A deficient mice, support that neutrophil recruitment to the skin, is IL-17-independent [85].
Interestingly, while the Th17 pathway is important for defense against oropharyngeal candidiasis, it appears to be less important in the host defense against vulvovaginal candidiasis in mice [86]. Likewise, patients with genetic defects in their Th17 cell responses do not suffer from recurrent vulvovaginal candidiasis [2,87]. Major mechanisms involved in the pathogenesis of vulvovaginal candidiasis are local estrogen level, and imbalances in local microbiome [88]. In addition, IL-1β-induced hyperinflammation as a result of defects in the inflammasome seems to play a causal role in the pathogenesis of recurrent vulvovaginal candidiasis [30].
3.3. Genetic Susceptibility to Mucocutaneous Candidiasis
In the immunocompetent population, mucocutaneous candidiasis of skin, nails, oropharynx and esophagus occurs infrequent and is generally mild. Some patients, however, display a different clinical spectrum with severe and persistent Candida infections in the absence of risk factors. These syndromes comprise also the severe chronic mucocutaneous candidiasis (CMC) and hyper IgE syndrome (HIES). For most of these syndromes, the underlying genetic mutations or variation in immune regulation genes have now been identified.
3.3.1. Chronic Mucocutaneous Candidiasis (CMC)
Patients with CMC suffer from severe and persisting Candida infections of skin, nails and mucous membranes. There are several CMC phenotypes caused by specific genetic defects.
Autoimmune polyendocrinopathy, candidiasis, and ectodermal dystrophy (APECED) was first described in 1929. This disorder is also known under the name autoimmune polyendocrine syndrome type 1 (APS-1). Mutations in the autoimmune regulator (AIRE) gene were found responsible in 1997, with an autosomal recessive transmission [1,89,90,91]. In the general population, APECED accounts for up to 20–40% of CMC cases, while it is the predominant form of CMC in Finns, Sardinians and Iranian Jews [92,93]. The clinical phenotype consists of CMC and autoimmunity, primarily of endocrine organs. The classic triad of CMC, hypoparathyroidism and adrenal insufficiency affects more than 80–90% of patients. Autoimmunity can also involve the gonads and thyroid and in small amounts of patients non-endocrine organs such as liver, eye, kidney and intestine [93]. The AIRE gene is expressed in the thymus and secondary lymphoid organs and plays a key role in immune tolerance. As part of autoimmune processes, all patients have autoantibodies against at least one out of three IL-17 cytokines (IL-17A (41%), IL-17F (75%) and/or IL-22 (91%)), which are essential for host defense against mucocutaneous Candida infection. Patients may also have auto-antibodies against IFN-α and IFN-ω, but do not display an increased susceptibility to viral infections [74,89].
In 2011, signal transducer and activator of transcription 1 (STAT1) mutations were discovered to likely be the main cause of autosomal dominant CMC [2]. STAT1 is a signaling molecule downstream of the type I and type II IFN receptors and of the IL-12 and IL-23 receptors. Gain-of-function mutations in the CC domain, the DNA-binding domain and the SH2 domain of STAT1 cause disruption of the IL-12 and IL-23 pathways resulting in defective Th1 and Th17 cell responses and their consecutive production of IFN-γ, IL-17 and IL-22 [94]. Gain-of-function mutations of STAT1 are associated with a broad clinical phenotype and are the most common genetic cause of CMC [95]. In addition to mucocutaneous candidiasis, 74% of patients with STAT1 mutations suffer from cutaneous and respiratory bacterial infections (mainly Staphylococcus aureus), 38% suffer from viral skin infections (mainly Herpesviridae), 10% suffer from invasive fungal infections, and 6% suffer from mycobacterial infections [95]. Many patients with STAT1 mutations also display autoimmune manifestations (37%), mainly hypothyroidism [95]. Esophageal carcinoma and cerebral aneurysms have also been linked to gain-of-function STAT1 mutations [95,96]. The increased risk of invasive infections, squamous cell carcinoma and cerebral aneurysms make this disease potentially life-threatening.
Autosomal dominant IL-17F, autosomal recessive IL17-RA, autosomal recessive IL-17RC and autosomal recessive TRAF3IP2 (encodes ACT1) gene mutations have also been reported to cause CMC in patients without mutations in the AIRE or STAT1 genes. ACT1 is an adaptor molecule that interacts with the IL-17 receptor for downstream signaling response to IL-17A and IL-17F. Some of these patients also suffer from S. aureus skin infections [72,74,97].
Several gene mutations are associated with less severe manifestations of CMC. Autosomal recessive RAR Related Orphan Receptor C (RORC) gene mutations, causing RORγT deficiency affecting Th17 cell development, leads to mild CMC and severe mycobacterial infections. Autosomal recessive IL-12B or IL-12Rβ1 gene mutations, causing IL-12Rβ1 or IL-12p40 deficiency, affecting both IL-12 and IL-23 signaling pathways, leads to mycobacterial infections in 83% of patients, to Salmonella infections in 43% and CMC in 23% of patients [74].
3.3.2. Hyper IgE Syndrome (HIES)
HIES, also called Job syndrome, is characterized by recurrent staphylococcal skin abscesses, pulmonary aspergillosis, skeletal and dental abnormalities, eczema, eosinophilia, elevated serum immunoglobulin E concentrations, and mucocutaneous candidiasis [74].
Autosomal recessive inheritance of HIES is rare and caused mainly by mutations in dedicator of cytokinesis 8 (DOCK8) gene, encoding a protein involved in Th17 polarization [1,74]. Most cases of HIES are autosomal dominant and mainly caused by loss-of-function mutations in signal transducer and activator of transcription 3 (STAT3) [74]. STAT3 is a signaling molecule downstream of multiple cytokine receptors including IL-6, IL-10, IL-23, IL-17 and IL-22. The defective downstream signaling of the IL-23 receptor results in absent IL-17 production [74]. Of HIES patients with STAT3 mutations, 85% develop CMC [1,74].
In addition to the typical skeletal and dental abnormalities, the clinical phenotype of HIES differs from CMC mainly by including allergic manifestations. Recent research shows that HIES patients are able to induce normal Th2 responses, while CMC patients have total absent Th2 responses [98].
3.3.3. Recurrent Vulvovaginal Candidiasis (RVVC)
Vulvovaginal candidiasis is the most common form of mucocutaneous candidiasis in the immunocompetent host. A minority of women (5–8%) suffer from recurrent vulvovaginal candidiasis (RVVC), defined by recurrence of Candida infections more than three times a year, with many of these patients lacking any of the known clinical risk factors. Gene polymorphisms in PRRs, TLR2 and mannose-binding lectin (MBL), and in the NLRP3 inflammasome, and cytokine IL-4, have been discovered that play a role in the multifactorial susceptibility to RVVC.
The rs5743704 polymorphism in TLR2, increased the susceptibility to RVVC almost three-fold, in a study with 119 RVVC patients, functional assays suggested that this polymorphism reduces production of IL-17 and IFN-γ upon stimulation of peripheral blood mononuclear cells with C. albicans [99]. Polymorphisms in codon 54 in MBL2, has been linked to RVVC. This MBL2 gene codes for the soluble PRR mannose-binding lectin (MBL) which promotes complement activation and Candida killing [100]. In a group of 50 women with RVVC, 25% carried this polymorphism compared to 10.6% in controls [101].
The rs74163773 polymorphism in the NLRP3 inflammasome is associated with an increased susceptibility to RVVC, as studied in a group of 270 RVVC patients. This polymorphism leads to hyperinflammation by overproduction of IL-1β. Levels of IL-1β at the vaginal surface were higher in patients bearing this polymorphism, IL-1Ra levels were decreased [30].
The −589C/T polymorphism in the IL-4 gene is associated with an increased susceptibility to RVCC, in a group of 42 women with RVVC 59.5% had this polymorphism compared to 7.0% in controls. This polymorphism leads to an elevated concentration of IL-4 and decreased concentration of MBL and nitric oxide (NO) in vaginal fluid. IL-4 has been known to inhibit macrophage activation and NO production [102].
A very recent study has performed the first GWAS in patients with RVVC. In this study, the most important pathways regulating susceptibility to RVVC at a genetic level have been revealed, including cytokine production capacity, cellular morphogenesis and metabolism, as well as cell adhesion (Jaeger et al., personal communication).
3.3.4. Candida Colonization, Cutaneous Candidiasis and Onychomycosis
The Tyr238X polymorphism in an early stop codon in the dectin-1 gene, increases susceptibility to oral and gastrointestinal Candida colonization and onychomycosis, by defective β-glucan recognition and consecutive Th17 cell responses. Screening for dectin-1 polymorphism in patients undergoing hematopoietic stem cell transplantation, showed 10.6% of patients bearing this polymorphism, they were significantly more often colonized with Candida [103]. This polymorphism is present in up to 8% of Europeans and up to 40% of selected sub-Saharan African populations [103,104].
The L412F polymorphism in TLR3, is associated with chronic cutaneous candidiasis in patients without known genetic mutations conferring susceptibility to CMC [105]. Peripheral blood mononuclear cells of patients carrying this polymorphism, showed reduced IFN-γ and tumor necrosis factor alpha (TNF-α) secretion on response to stimulation with cytomegalovirus (CMV) and C. albicans. These patients seem to have an increased risk of CMV infection and autoimmune manifestations as well.
4. Invasive Candidiasis
Imbalances in microbiome caused by antibacterial agents, disruption of the barrier between the external and internal environment and loss of neutrophil function are well known risk factors for invasive candidiasis. Patients admitted to the ICU acquire Candida colonization in up to 80% of patients during the first 7 days [106]. Multifocal Candida colonization in combination with abdominal surgery or indwelling vascular catheters are major risk factors for the development of invasive candidiasis [3]. Of candidemia episodes, approximately 50% occur in the ICU, with 70–90% of patients having previously been exposed to broad-spectrum antibiotic therapy, 80–90% have an intravascular device in situ at the time of candidemia, and 40–50% have had recent surgery [107,108,109]. Remarkably, neutropenia is only present in less than 5% of patients with candidemia [108,109].
Candida colonizing the gut invades either through translocation, or through anastomotic leakage after laparotomy, and may cause localized, deep-seated infection (e.g., peritonitis), or candidemia via the portal circulation [3]. During candidemia, dissemination of Candida may lead to secondary metastatic lesions, e.g., in lung, liver, spleen, kidneys, bone and eye. Also indwelling intravascular catheters are likely to become colonized [3].
Patients with hematologic malignancies receiving anticancer chemotherapy may develop extensive mucositis of the gastrointestinal tract, and hence be at risk for translocation of colonizing Candida to the blood stream. In patients with prolonged neutropenia, chronic disseminated candidiasis is a rare specific entity [110]. This form of invasive candidiasis primarily involves the liver and spleen, and often only becomes overt after neutrophil recovery. Immune reconstitution inflammatory syndrome (IRIS) plays a major part in the disease pathogenesis [110].
4.1. Th1 Pathway and Neutrophil Function
Th1 cell responses are crucial for protection against invasive candidiasis, and IFN-γ is the pivotal cytokine in anti-Candida host defense. The pro-inflammatory cytokine IL-18, processed by the inflammasome, induces Th1 cell responses such as IFN-γ production [43]. IFN-γ has a stimulating effect on the fungicidal activities of phagocytic cells mainly neutrophils and macrophages [48,49]. Natural killer cells (NK) further enhance this fungicidal activity by producing granulocyte -macrophage colony-stimulating factor (GM-CSF).
Neutrophil activation is essential for the clearance of Candida, as these are the most potent phagocytes to kill Candida, and the only host immune cell able to inhibit germination of yeasts into hyphae. After phagocytosis of Candida, neutrophils use oxidative as well as non-oxidative killing mechanisms. The production of reactive oxygen species (ROS), is mediated through a protein complex called nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. The enzyme myeloperoxidase (MPO) catalyses the conversion of hydrogen peroxide to hypohalous acid, which amplifies the toxicity of ROS. The fungicidal activity relies on toxicity of ROS and release of antifungal proteases. This ROS-dependent mechanism is essential for clearance of opsonized Candida, and depends on binding to the FcγRs receptor and protein kinase C activation. Non-oxidative killing proceeds by producing anti-microbial factors such as lysozyme, lactoferrin, elastase, β-defensins, gelatinases and cathepsin G. In addition to killing by phagocytosis, neutrophils can release chromatin fibers forming neutrophil extracellular traps (NETs). These NETS are able to kill Candida yeasts as well as hyphae, by binding them and releasing an antifungal peptide called calprotectin. This ROS-independent mechanism is essential for clearance of unopsonized Candida, and depends on binding to the CR3 receptor and CARD9 activation [6,7,111,112].
4.2. Genetic Susceptibility to Invasive Candidiasis
Even among patients with a combination of several risk factors for invasive candidiasis, the disease occurs in only a minority of patients. This suggests a role for underlying genetic variations, which, in combination with several risk factors, makes a patient prone to develop invasive candidiasis. Several polymorphisms in immune regulation genes have recently been described, leading to either an increased susceptibility to acquire or a decreased ability to clear invasive candidiasis.
4.2.1. Increased Susceptibility to Acquire Candidemia
The influence of polymorphisms in TLRs genes on susceptibility to candidemia was prospectively studied in a large case and control cohort consisting of European and North American hospitalized patients at risk for candidemia [113]. An association with increased susceptibility to acquiring candidemia was found for three polymorphisms in TLR1 gene. Patients with these genotypes display decreased proinflammatory cytokine release upon stimulation ex vivo. No association was found for polymorphisms in TLR2, TLR4, TLR6, TLR9, or their adaptor proteins myeloid differentiation primary response 88 (MyD88) or TIR domain containing adaptor protein (TIRAP) genes [113]. Three additional polymorphisms in CD58, LCE4A-C1orf68, and T-cell activation GTPase activating protein (TAGAP) loci associated with increased susceptibility to candidemia were identified in a genome-wide association study (GWAS) in the largest candidemia cohort to date. The risk of candidemia for patients on the ICU was increased by more than 19-fold when carrying at least two risk alleles from these loci. CD58 is important for Candida phagocytosis and inhibition of germination, TAGAP has a function in Candida-induced cytokine production, and LCE4A-C1orf68 locus contributes to mucosal integrity [114]. Recently, 18 novel susceptibility loci were validated, by integrating genotype data from a cohort of 217 candidemia patients with transcriptome changes in healthy human primary leucocytes induced by Candida. In these 18 loci, 31 candidate genes were identified [115].
4.2.2. Increased Susceptibility to Persistent Candidemia
Persistent candidemia is defined as positive blood cultures yielding Candida for more than five days despite adequate therapy. In a large prospective cohort of ICU patients with candidemia, polymorphisms in cytokine genes encoding IL-10 and IL12B were found to be associated with persistent candidemia [116]. This decreased capability to clear the blood stream from Candida is likely mediated by increased production of the anti-inflammatory cytokine IL-10 and decreased production of IL-12b, resulting in downregulation of IFN-γ production. Polymorphisms in IFN-γ, IL-18, IL-1β and IL-8 genes were not associated with persistent candidemia [116]. In a subsequent GWAS study, the CD58 polymorphism was also associated with persistent candidemia [114].
4.2.3. Increased Susceptibility to Candida Dissemination
Disseminated candidiasis is defined as the presence of Candida at normally sterile sites outside the bloodstream, and this condition may be either acute or chronic.
An association between genetic variants in IL-4 gene and susceptibility to chronic disseminated candidiasis (CDC) was found in a cohort of 90 patients with acute leukemia, of which 40 suffered from CDC [117]. The −1098T/−589C/−33C polymorphism in IL-4 gene was associated with increased susceptibility to CDC in acute leukemia patients whereas the −1098T/−589T/−33T polymorphism was associated with decreased susceptibility. IL-4 is an anti-inflammatory cytokine, induced by Th2 cell responses. The polymorphism that increased susceptibility to CDC was associated with a decreased IL-4 transcriptional activity [117].
The autosomal recessive inheritance of loss-of-function mutations in caspase recruitment domain-containing protein 9 (CARD9) gene, were first described in a large consanguineous Iranian family with recurrent mucocutaneous and invasive Candida infections [118]. Interestingly, this is the first, and as yet only, described genetic cause for a combined phenotype of mucocutaneous and invasive candidiasis. CARD9 is an intracellular adaptor molecule essential for dectin-1 signaling. This signaling pathway induces the production of IL-1β, IL-6, and IL-23, and consecutive Th17 cell responses. In the four affected family members, low numbers of circulating Th17 cells were found. Three affected family members died during adolescence, two died from Candida meningoencephalitis, and one died from presumed, but not confirmed, Candida cerebral involvement [118].
In a subsequent study, additional understanding was obtained on the invasive nature of candidiasis resulting from mutations in CARD9 gene [119]. Neutrophils of a patient diagnosed with Candida meningoencephalitis and CARD9 gene mutations, displayed a selective Candida killing defect, which was independent of ROS production. The underlying mechanism is not yet fully understood [119]. In five additional patients, with CARD9 gene mutations, who all were born to consanguineous parents of Arabic origin, an association of CARD9 gene mutations with tissue culture proven Candida colitis was described [120]. These observations support the role for CARD9 in anti-fungal immunity of the gut, as was previously found in murine models. CARD9-deficient mice displayed strong fungal colonization of the digestive tract with decreased numbers of colonic Th17 cells and innate lymphoid cells [120]. The earlier mentioned polymorphism in TAGAP gene is also associated with dissemination in organs [114]. An overview of genes involved in genetic susceptibility to Candida infections is depicted in Table 1.
5. Translating Knowledge into Clinical Practice
Morbidity and mortality associated with Candida disease remains substantial, despite advances in supportive care and novel antifungal agents, demanding improvement of antifungal therapy. The current knowledge on pattern recognition, Th1 and Th17 cell responses, and host effector mechanisms in candidiasis underlines the potential for adjunctive immunotherapy in antifungal treatment. Identifying patients within high-risk groups who bear genetic mutations or polymorphisms associated with specific immune pathway defects allows selection of subjects for whom antifungal prophylactic therapy, early empiric treatment, or host-directed adjunctive immunotherapy is expected to be most effective. Thus, more accurate definition of high-risk groups will, in light of overtreatment and emergence of fungal resistance, provide an additional benefit on a population level [3,121].
5.1. Prophylaxis
Antifungal prophylaxis on ICU is currently used only in specific high-risk groups, in which it has been shown to be effective, such as abdominal surgery with recurrent gastrointestinal anastomotic leakage, transplantation of the pancreas or small bowel, liver transplantation in selected patients who are at high risk of candidiasis, and extremely low-birth-weight neonates in settings with a high incidence of neonatal candidiasis [122].
Benefit of antifungal prophylaxis can, however, be expected in other ICU patient groups with additional risk factors, such as broad-spectrum antibiotic therapy use, gastrointestinal disease and indwelling vascular catheters. Studies to evaluate the value of antifungal prophylactic therapy have been based on defining these high-risk groups by clinical parameters. However, a recent randomized, placebo-controlled study in which patients admitted to ICU with a high risk of acquiring invasive candidiasis based on a clinical prediction rule, received antifungal prophylaxis, yielded no significant difference in candidemia rate or overall mortality [123]. Screening for genetic mutations and polymorphisms in immune regulation genes within these high-risk patient groups, may improve risk stratification to determine which patients need antifungal prophylaxis.
5.2. Immunostimulatory Therapy
5.2.1. Recombinant Cytokine Therapy
To date, recombinant cytokine therapy is the only clinically available form of adjunctive antifungal immunotherapy and relies on improving host immune effector functions. Colony-stimulating factors have been considered as antifungal adjunctive therapy [121,124,125]. These factors enhance phagocytosis and the release of ROS, prolong the survival of neutrophils by inhibiting programmed cell death and upregulate chitotriosidase promoting fungicidal activity. In addition, GM-CSF is known to stimulate upregulation of dectin-1 expression on macrophages. GM-CSF has been successfully used in HIV patients with refractory mucosal candidiasis [126,127]. No studies with GM-CSF have been performed in patients with invasive candidiasis. Granulocyte colony-stimulating factor (G-CSF) was shown to be effective in mice with disseminated candidiasis [128]. In a randomized placebo-controlled pilot study among non-neutropenic patients with disseminated candidiasis, adjunctive immunotherapy with G-CSF in combination with fluconazole showed a trend towards faster resolution of infection than fluconazole alone [129]. No follow-up studies have been done to date.
IFN-γ has been shown to enhance candidacidal activity of macrophages and neutrophils. Administration of IFN-γ to mice with disseminated candidiasis reduces fungal burden [130]. In an open-label prospective pilot study, patients with candidemia have received adjunctive IFN-γ or placebo. Adjunctive IFN-γ immunotherapy partially restored antifungal immune function in the setting of sepsis-induced immune paralysis, with improvement in leukocyte innate immune responses (IL-1β, TNFα), and increased production of T-lymphocyte cytokines (IL-17 and IL-22). This study was not powered to show effect on mortality [131].
5.2.2. Vaccination and Antibodies
Passive immunization with anti-Candida antibodies could be of benefit in immunocompetent as well as immunodeficient patients. Screening among candidiasis patients for polymorphisms known to decrease the ability of Candida clearance, such as the polymorphism in IL-10 and IL-12B genes, may guide selection of patients most likely to benefit from adjunctive antibody therapy. Protective monoclonal antibodies against Candida have been developed and tested successful in murine models [124,132]. Anti-Candida antibodies are not clinically available yet.
Active vaccination for candidiasis would be the ultimate prevention method in immunocompetent patients undergoing elective procedures or treatments known to increase the risk of candidiasis. Extensive research over the years has provided several potential vaccines but none of these is clinically available yet. Two vaccines, containing recombinant C. albicans-derived proteins, have reached phase II trials. The NDV-3 vaccine, a recombinant alum-adjuvanted vaccine for Candida and Staphylococcus aureus, has been shown to protect mice against oropharyngeal, vulvovaginal, and invasive candidiasis as well as skin and soft tissue infections with S. aureus [133]. In phase I trials it has shown promising results in T en B cell responses [134]. A memory T-cell response was shown for IFN-γ in almost all participants, and for IL-17 in the majority of participants. The SAP2 vaccine, studied for its effect on vulvovaginal candidiasis, has shown to generate neutralizing vaginal antibodies [4,121]. Interestingly, studies have shown that by adding different adjuvants to vaccines, different cytokine profiles and Th cell responses are induced. By adding an adjuvant specific for mucocutaneous disease or for invasive disease, a vaccine may be expected to shape the adaptive immune response towards either a Th17 or a Th1 response [124].
5.2.3. Innate Cellular Immunotherapy
Granulocyte transfusion, as treatment for sepsis in neutropenic patients, was developed in the 1970s. Toxicity of the transfusion, development of haematologic growth factors, and antimicrobial and antimycotical agents have halted its use. The availability of recombinant GM-CSF and G-CSF, however, has inspired new trials based on the expected higher yield of donor granulocytes. No difference in survival was found in a randomized phase III trial in febrile neutropenia patients with fungal disease, receiving granulocyte transfusion adjunctive to standard of care [135]. Similarly, no difference in the overall success between adjunctive granulocyte transfusion or antibiotic therapy alone was found in a multicentre randomized trial in patients with febrile neutropenia and presumed infection [136]. In conclusion, the clinical efficacy of granulocyte transfusion has never been conclusively demonstrated. Another potential option for antifungal immunotherapy, yet to be studied in man, is dendritic cell-vaccination, in which dendritic cells are primed ex vivo with antigens that induce specific cytokine profiles that induce an anti-Candida host response, and are then infused in patients with candidiasis [121,124].
5.3. Immunosuppressive Therapy
In specific types of candidiasis, suppressing, rather than stimulating, host antifungal immune response may be beneficial. In chronic disseminated candidiasis, a hyperactive immune response occurs when neutrophil count returns to normal (IRIS). Adjunctive corticosteroid therapy, for at least three weeks up to one year, has been described to lead to resolution of symptoms and of inflammatory response [137]. Anakinra, a recombinant IL-1Ra and potent suppressor of inflammasome activity, has proven effective in mice studies of VVC [138]. Thus, based on the hyperinflammation as result of defects in the NLRP3 inflammasome and successive increase in IL-1β production in RVVC patients, a potential role for anakinra as antifungal immunotherapy has been suggested.
5.4. Future Perspectives
Increased knowledge about host immune response to Candida infections, and genetic mutations and variations in immune regulation genes conferring susceptibility to candidiasis has led to development of various forms of immunotherapy that show potential as adjunctive antifungal therapy. New development of immunotherapy could comprise recombinant cytokine IL-17 or IL-22, considering their value in antifungal immune responses.
Prospective studies are now warranted to investigate the efficacy of including genetic screening, within high-risk patient groups, in stratifying patient risk for candidiasis, and to evaluate the efficacy of antifungal prophylaxis and adjunctive antifungal host-directed immunotherapy when administered in this selection of patients.
Acknowledgments
We thank Mark Gresnigt for the help with drawing the figure. Mihai G. Netea was supported by an European Research Counsil (ERC) Consolidator Grant (#310372) and a Spinoza Grant of the Netherlands Organization for Scientific Research.
Author Contributions
All authors contributed equally to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Smeekens, S.P.; van de Veerdonk, F.L.; Kullberg, B.J.; Netea, M.G. Genetic susceptibility to Candida infections. EMBO Mol. Med. 2013, 5, 805–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Veerdonk, F.L.; Plantinga, T.S.; Hoischen, A.; Smeekens, S.P.; Joosten, L.A.; Gilissen, C.; Arts, P.; Rosentul, D.C.; Carmichael, A.J.; Smits-van der Graaf, C.A.; et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 2011, 365, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kullberg, B.J.; Arendrup, M.C. Invasive Candidiasis. N. Engl. J. Med. 2015, 373, 1445–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullberg, B.J.; van de Veerdonk, F.; Netea, M.G. Immunotherapy: A potential adjunctive treatment for fungal infection. Curr. Opin. Infect. Dis. 2014, 27, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Brown, G.D.; Kullberg, B.J.; Gow, N.A. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 2008, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.; van der Meer, J.W.; Kullberg, B.J.; van de Veerdonk, F.L. Immune defence against Candida fungal infections. Nat. Rev. Immunol. 2015, 15, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhang, L.; Xu, Z.; Zhang, J.; Jiang, Y.Y.; Cao, Y.; Yan, T. Innate immune cell response upon Candida albicans infection. Virulence 2016, 7, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Gow, N.A.; Hube, B. Importance of the Candida albicans cell wall during commensalism and infection. Curr. Opin. Microbiol. 2012, 15, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.A.; Gow, N.A. Mannosylation in Candida albicans: Role in cell wall function and immune recognition. Mol. Microbiol. 2013, 90, 1147–1161. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.F.; Prill, S.K. O-glycosylation. Med. Mycol. 2001, 39 (Suppl. S1), 67–74. [Google Scholar] [CrossRef] [PubMed]
- Cutler, J.E. N-glycosylation of yeast, with emphasis on Candida albicans. Med. Mycol. 2001, 39 (Suppl. S1), 75–86. [Google Scholar] [CrossRef] [PubMed]
- Klis, F.M.; Boorsma, A.; de Groot, P.W. Cell wall construction in Saccharomyces cerevisiae. Yeast 2006, 23, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Suzuki, A.; Kobayashi, H.; Okawa, Y. Chemical structure of the cell-wall mannan of Candida albicans serotype A and its difference in yeast and hyphal forms. Biochem. J. 2007, 404, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Lowman, D.W.; Greene, R.R.; Bearden, D.W.; Kruppa, M.D.; Pottier, M.; Monteiro, M.A.; Soldatov, D.V.; Ensley, H.E.; Cheng, S.C.; Netea, M.G.; et al. Novel structural features in Candida albicans hyphal glucan provide a basis for differential innate immune recognition of hyphae versus yeast. J. Biol. Chem. 2014, 289, 3432–3443. [Google Scholar] [CrossRef] [PubMed]
- Munro, C.A.; Schofield, D.A.; Gooday, G.W.; Gow, N.A. Regulation of chitin synthesis during dimorphic growth of Candida albicans. Microbiology 1998, 144 Pt 2, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Gordon, S. Immune recognition. A new receptor for β-glucans. Nature 2001, 413, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Cambi, A.; Gijzen, K.; de Vries, L.J.; Torensma, R.; Joosten, B.; Adema, G.J.; Netea, M.G.; Kullberg, B.J.; Romani, L.; Figdor, C.G. The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur. J. Immunol. 2003, 33, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Jouault, T.; Ibata-Ombetta, S.; Takeuchi, O.; Trinel, P.A.; Sacchetti, P.; Lefebvre, P.; Akira, S.; Poulain, D. Candida albicans phospholipomannan is sensed through toll-like receptors. J. Infect. Dis. 2003, 188, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Gow, N.A.; Munro, C.A.; Bates, S.; Collins, C.; Ferwerda, G.; Hobson, R.P.; Bertram, G.; Hughes, H.B.; Jansen, T.; et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J. Clin. Investig. 2006, 116, 1642–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gantner, B.N.; Simmons, R.M.; Underhill, D.M. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Wileman, T.E.; Lennartz, M.R.; Stahl, P.D. Identification of the macrophage mannose receptor as a 175-kDa membrane protein. Proc. Natl. Acad. Sci. USA 1986, 83, 2501–2505. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.D.; Shepherd, V.L. Purification of the human alveolar macrophage mannose receptor. Biochem. Biophys. Res. Commun. 1987, 148, 883–889. [Google Scholar] [CrossRef]
- Brown, G.D.; Gordon, S. Fungal β-glucans and mammalian immunity. Immunity 2003, 19, 311–315. [Google Scholar] [CrossRef]
- Cambi, A.; Netea, M.G.; Mora-Montes, H.M.; Gow, N.A.; Hato, S.V.; Lowman, D.W.; Kullberg, B.J.; Torensma, R.; Williams, D.L.; Figdor, C.G. Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J. Biol. Chem. 2008, 283, 20590–20599. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Reyes, C.N.; Becker, C.A.; Katsumoto, T.R.; Ma, J.; Wolf, A.J.; Bose, N.; Chan, A.S.; Magee, A.S.; Danielson, M.E.; et al. Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature 2011, 472, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Taylor, P.R.; Reid, D.M.; Willment, J.A.; Williams, D.L.; Martinez-Pomares, L.; Wong, S.Y.; Gordon, S. Dectin-1 is a major beta-glucan receptor on macrophages. J. Exp. Med. 2002, 196, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Drummond, R.A.; Brown, G.D. The role of Dectin-1 in the host defence against fungal infections. Curr. Opin. Microbiol. 2011, 14, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.A.; Salvage-Jones, J.A.; Li, X.; Hitchens, K.; Butcher, S.; Murray, R.Z.; Beckhouse, A.G.; Lo, Y.L.; Manzanero, S.; Cobbold, C.; et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J. Immunol. 2008, 180, 7404–7413. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.; van der Lee, R.; Cheng, S.C.; Johnson, M.D.; Kumar, V.; Ng, A.; Plantinga, T.S.; Smeekens, S.P.; Oosting, M.; Wang, X.; et al. The RIG-I-like helicase receptor MDA5 (IFIH1) is involved in the host defense against Candida infections. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.; Carvalho, A.; Cunha, C.; Plantinga, T.S.; van de Veerdonk, F.; Puccetti, M.; Galosi, C.; Joosten, L.A.; Dupont, B.; Kullberg, B.J.; et al. Association of a variable number tandem repeat in the NLRP3 gene in women with susceptibility to RVVC. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Smeekens, S.P.; Ng, A.; Kumar, V.; Johnson, M.D.; Plantinga, T.S.; van Diemen, C.; Arts, P.; Verwiel, E.T.; Gresnigt, M.S.; Fransen, K.; et al. Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. Nat. Commun. 2013, 4, 1342. [Google Scholar] [CrossRef] [PubMed]
- Heinsbroek, S.E.; Taylor, P.R.; Martinez, F.O.; Martinez-Pomares, L.; Brown, G.D.; Gordon, S. Stage-specific sampling by pattern recognition receptors during Candida albicans phagocytosis. PLoS Pathog. 2008, 4, e1000218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudkin, F.M.; Bain, J.M.; Walls, C.; Lewis, L.E.; Gow, N.A.; Erwig, L.P. Altered dynamics of Candida albicans phagocytosis by macrophages and PMNs when both phagocyte subsets are present. mBio 2013, 4, e00810–e00813. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [PubMed]
- Miramon, P.; Dunker, C.; Windecker, H.; Bohovych, I.M.; Brown, A.J.; Kurzai, O.; Hube, B. Cellular responses of Candida albicans to phagocytosis and the extracellular activities of neutrophils are critical to counteract carbohydrate starvation, oxidative and nitrosative stress. PLoS ONE 2012, 7, e52850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gantner, B.N.; Simmons, R.M.; Canavera, S.J.; Akira, S.; Underhill, D.M. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 2003, 197, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Herre, J.; Marshall, A.S.; Caron, E.; Edwards, A.D.; Williams, D.L.; Schweighoffer, E.; Tybulewicz, V.; Reis e Sousa, C.; Gordon, S.; Brown, G.D. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood 2004, 104, 4038–4045. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Willment, J.A.; Dorward, D.W.; Williams, D.L.; Brown, G.D.; DeLeo, F.R. Dectin-1 promotes fungicidal activity of human neutrophils. Eur. J. Immunol. 2007, 37, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.K.; Levitz, S.M. Interactions of fungi with phagocytes. Curr. Opin. Microbiol. 2002, 5, 359–365. [Google Scholar] [CrossRef]
- Reeves, E.P.; Lu, H.; Jacobs, H.L.; Messina, C.G.; Bolsover, S.; Gabella, G.; Potma, E.O.; Warley, A.; Roes, J.; Segal, A.W. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002, 416, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 2009, 5, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; van de Veerdonk, F.L.; Lenardon, M.; Stoffels, M.; Plantinga, T.; Smeekens, S.; Rizzetto, L.; Mukaremera, L.; Preechasuth, K.; Cavalieri, D.; et al. The dectin-1/inflammasome pathway is responsible for the induction of protective T-helper 17 responses that discriminate between yeasts and hyphae of Candida albicans. J. Leukoc. Biol. 2011, 90, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Lalor, S.J.; Dungan, L.S.; Sutton, C.E.; Basdeo, S.A.; Fletcher, J.M.; Mills, K.H. Caspase-1-processed cytokines IL-1β and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J. Immunol. 2011, 186, 5738–5748. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Joosten, L.A.; Shaw, P.J.; Smeekens, S.P.; Malireddi, R.K.; van der Meer, J.W.; Kullberg, B.J.; Netea, M.G.; Kanneganti, T.D. The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. Eur. J. Immunol. 2011, 41, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Wagener, J.; Wenzel, V.; Scarponi, C.; Pennino, D.; Albanesi, C.; Schaller, M.; Behrendt, H.; Ring, J.; Schmidt-Weber, C.B.; et al. IL-22 and TNF-α represent a key cytokine combination for epidermal integrity during infection with Candida albicans. Eur. J. Immunol. 2011, 41, 1894–1901. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, M.R.; Aggarwal, B.B.; Rinderknecht, E.; Svedersky, L.P.; Finkle, B.S.; Palladino, M.A., Jr. Activation of human polymorphonuclear neutrophil functions by interferon-γ and tumor necrosis factors. J. Immunol. 1985, 135, 2069–2073. [Google Scholar] [PubMed]
- Nathan, C.F.; Murray, H.W.; Wiebe, M.E.; Rubin, B.Y. Identification of interferon-γ as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J. Exp. Med. 1983, 158, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Na, L.; Fidel, P.L.; Schwarzenberger, P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 2004, 190, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Smeekens, S.P.; Huttenhower, C.; Riza, A.; van de Veerdonk, F.L.; Zeeuwen, P.L.; Schalkwijk, J.; van der Meer, J.W.; Xavier, R.J.; Netea, M.G.; Gevers, D. Skin microbiome imbalance in patients with STAT1/STAT3 defects impairs innate host defense responses. J. Innate Immun. 2014, 6, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Oever, J.T.; Netea, M.G. The bacteriome-mycobiome interaction and antifungal host defense. Eur. J. Immunol. 2014, 44, 3182–3191. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, F.; Laksmana, T.; Uribarri, A. Systemic antibiotics and the risk of superinfection in peri-implantitis. Arch. Oral Biol. 2016, 64, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.; Edlund, C.; Nord, C.E. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect. Dis. 2001, 1, 101–114. [Google Scholar] [CrossRef]
- Xu, J.; Schwartz, K.; Bartoces, M.; Monsur, J.; Severson, R.K.; Sobel, J.D. Effect of antibiotics on vulvovaginal candidiasis: A MetroNet study. J. Am. Board Fam. Med. 2008, 21, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, B.; Ferreira, C.; Alves, C.T.; Henriques, M.; Azeredo, J.; Silva, S. Vulvovaginal candidiasis: Epidemiology, microbiology and risk factors. Crit. Rev. Microbiol. 2016, 42, 905–927. [Google Scholar] [CrossRef] [PubMed]
- Ellepola, A.N.; Samaranayake, L.P. Inhalational and topical steroids, and oral candidosis: A mini review. Oral Dis. 2001, 7, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Gunther, L.S.; Martins, H.P.; Gimenes, F.; Abreu, A.L.; Consolaro, M.E.; Svidzinski, T.I. Prevalence of Candida albicans and non-albicans isolates from vaginal secretions: Comparative evaluation of colonization, vaginal candidiasis and recurrent vaginal candidiasis in diabetic and non-diabetic women. Sao Paulo Med. J. 2014, 132, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Van Boven, J.F.; de Jong-van den Berg, L.T.; Vegter, S. Inhaled corticosteroids and the occurrence of oral candidiasis: A prescription sequence symmetry analysis. Drug Saf. 2013, 36, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Ahadian, H.; Yassaei, S.; Bouzarjomehri, F.; Ghaffari Targhi, M.; Kheirollahi, K. Oral Complications of The Oromaxillofacial Area Radiotherapy. Asian Pac. J. Cancer Prev. 2017, 18, 721–725. [Google Scholar] [PubMed]
- Wulandari, E.A.T.; Saraswati, H.; Adawiyah, R.; Djauzi, S.; Wahyuningsih, R.; Price, P. Immunological and epidemiological factors affecting candidiasis in HIV patients beginning antiretroviral therapy in an Asian clinic. Arch. Oral Biol. 2017, 82, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Hofs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Marijnissen, R.J.; Kullberg, B.J.; Koenen, H.J.; Cheng, S.C.; Joosten, I.; van den Berg, W.B.; Williams, D.L.; van der Meer, J.W.; Joosten, L.A.; et al. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 2009, 5, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Gaffen, S.L. IL-17-Mediated Immunity to the Opportunistic Fungal Pathogen Candida albicans. J. Immunol. 2015, 195, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Gow, N.A.; van de Veerdonk, F.L.; Brown, A.J.; Netea, M.G. Candida albicans morphogenesis and host defence: Discriminating invasion from colonization. Nat. Rev. Microbiol. 2011, 10, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Gladiator, A.; Wangler, N.; Trautwein-Weidner, K.; LeibundGut-Landmann, S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J. Immunol. 2013, 190, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Weindl, G.; Wagener, J.; Schaller, M. Epithelial cells and innate antifungal defense. J. Dent. Res. 2010, 89, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Weindl, G.; Naglik, J.R.; Kaesler, S.; Biedermann, T.; Hube, B.; Korting, H.C.; Schaller, M. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J. Clin. Investig. 2007, 117, 3664–3672. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Joosten, L.A.; Kullberg, B.J.; Netea, M.G. Interplay between Candida albicans and the mammalian innate host defense. Infect. Immun. 2012, 80, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Sparber, F.; LeibundGut-Landmann, S. Interleukin 17-Mediated Host Defense against Candida albicans. Pathogens 2015, 4, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Soltesz, B.; Toth, B.; Sarkadi, A.K.; Erdos, M.; Marodi, L. The Evolving View of IL-17-Mediated Immunity in Defense Against Mucocutaneous Candidiasis in Humans. Int. Rev. Immunol. 2015, 34, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Puel, A.; Casanova, J.L.; Kobayashi, M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin. Transl. Immunol. 2016, 5, e114. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Santos, N.; Huppler, A.R.; Peterson, A.C.; Khader, S.A.; McKenna, K.C.; Gaffen, S.L. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol. 2013, 6, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Huppler, A.R.; Conti, H.R.; Hernandez-Santos, N.; Darville, T.; Biswas, P.S.; Gaffen, S.L. Role of neutrophils in IL-17-dependent immunity to mucosal candidiasis. J. Immunol. 2014, 192, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, K.; Dimartino, V.; Cavani, A. IL-17 and IL-22 in immunity: Driving protection and pathology. Eur. J. Immunol. 2017, 47, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Kolls, J.K. Th17 cells in mucosal immunity and tissue inflammation. Semin. Immunopathol. 2010, 32, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Marks, B.R.; Craft, J. Barrier immunity and IL-17. Semin. Immunol. 2009, 21, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Khader, S. Yin and yang of interleukin-17 in host immunity to infection. F1000Res 2017, 6, 741. [Google Scholar] [CrossRef] [PubMed]
- Tomalka, J.; Azodi, E.; Narra, H.P.; Patel, K.; O’Neill, S.; Cardwell, C.; Hall, B.A.; Wilson, J.M.; Hise, A.G. Beta-Defensin 1 plays a role in acute mucosal defense against Candida albicans. J. Immunol. 2015, 194, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunology 2010, 129, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Trautwein-Weidner, K.; Gladiator, A.; Nur, S.; Diethelm, P.; LeibundGut-Landmann, S. IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol. 2015, 8, 221–231. [Google Scholar] [CrossRef] [PubMed]
- El Malki, K.; Karbach, S.H.; Huppert, J.; Zayoud, M.; Reissig, S.; Schuler, R.; Nikolaev, A.; Karram, K.; Munzel, T.; Kuhlmann, C.R.; et al. An alternative pathway of imiquimod-induced psoriasis-like skin inflammation in the absence of interleukin-17 receptor a signaling. J. Investig. Dermatol. 2013, 133, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Kolls, J.K.; Happel, K.I.; Wormley, F.; Wozniak, K.L.; Fidel, P.L., Jr. The acute neutrophil response mediated by S100 alarmins during vaginal Candida infections is independent of the Th17-pathway. PLoS ONE 2012, 7, e46311. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.M.; DeLeo, F.R.; Elloumi, H.Z.; Hsu, A.P.; Uzel, G.; Brodsky, N.; Freeman, A.F.; Demidowich, A.; Davis, J.; Turner, M.L.; et al. STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 2007, 357, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Sobel, J.D. Recurrent vulvovaginal candidiasis. Am. J. Obstet. Gynecol. 2016, 214, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Doffinger, R.; Natividad, A.; Chrabieh, M.; Barcenas-Morales, G.; Picard, C.; Cobat, A.; Ouachee-Chardin, M.; Toulon, A.; Bustamante, J.; et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 2010, 207, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Bjorses, P.; Aaltonen, J.; Horelli-Kuitunen, N.; Yaspo, M.L.; Peltonen, L. Gene defect behind APECED: A new clue to autoimmunity. Hum. Mol. Genet. 1998, 7, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, K.; Peterson, P.; Scott, H.S.; Kudoh, J.; Minoshima, S.; Heino, M.; Krohn, K.J.; Lalioti, M.D.; Mullis, P.E.; Antonarakis, S.E.; et al. Positional cloning of the APECED gene. Nat. Genet. 1997, 17, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Ahonen, P.; Myllarniemi, S.; Sipila, I.; Perheentupa, J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N. Engl. J. Med. 1990, 322, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Ferre, E.M.; Rose, S.R.; Rosenzweig, S.D.; Burbelo, P.D.; Romito, K.R.; Niemela, J.E.; Rosen, L.B.; Break, T.J.; Gu, W.; Hunsberger, S.; et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight 2016, 1, e88782. [Google Scholar] [CrossRef] [PubMed]
- Meesilpavikkai, K.; Dik, W.A.; Schrijver, B.; Nagtzaam, N.M.; van Rijswijk, A.; Driessen, G.J.; van der Spek, P.J.; van Hagen, P.M.; Dalm, V.A. A Novel Heterozygous Mutation in the STAT1 SH2 Domain Causes Chronic Mucocutaneous Candidiasis, Atypically Diverse Infections, Autoimmunity, and Impaired Cytokine Regulation. Front. Immunol. 2017, 8, 274. [Google Scholar] [CrossRef] [PubMed]
- Toubiana, J.; Okada, S.; Hiller, J.; Oleastro, M.; Lagos Gomez, M.; Aldave Becerra, J.C.; Ouachee-Chardin, M.; Fouyssac, F.; Girisha, K.M.; Etzioni, A.; et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 2016, 127, 3154–3164. [Google Scholar] [CrossRef] [PubMed]
- Dadak, M.; Jacobs, R.; Skuljec, J.; Jirmo, A.C.; Yildiz, O.; Donnerstag, F.; Baerlecken, N.T.; Schmidt, R.E.; Lanfermann, H.; Skripuletz, T.; et al. Gain-of-function STAT1 mutations are associated with intracranial aneurysms. Clin. Immunol. 2017, 178, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Marodi, L.; Abel, L.; Picard, C.; Casanova, J.L. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.L.; Rosler, B.; Wang, X.; Lachmandas, E.; Kamsteeg, M.; Jacobs, C.W.; Joosten, L.A.; Netea, M.G.; van de Veerdonk, F.L. Th2 and Th9 responses in patients with chronic mucocutaneous candidiasis and hyper-IgE syndrome. Clin. Exp. Allergy 2016, 46, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Rosentul, D.C.; Delsing, C.E.; Jaeger, M.; Plantinga, T.S.; Oosting, M.; Costantini, I.; Venselaar, H.; Joosten, L.A.; van der Meer, J.W.; Dupont, B.; et al. Gene polymorphisms in pattern recognition receptors and susceptibility to idiopathic recurrent vulvovaginal candidiasis. Front. Microbiol. 2014, 5, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babula, O.; Lazdane, G.; Kroica, J.; Ledger, W.J.; Witkin, S.S. Relation between recurrent vulvovaginal candidiasis, vaginal concentrations of mannose-binding lectin, and a mannose-binding lectin gene polymorphism in Latvian women. Clin. Infect. Dis. 2003, 37, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, P.C.; Babula, O.; Goncalves, A.K.; Linhares, I.M.; Amaral, R.L.; Ledger, W.J.; Witkin, S.S. Mannose-binding lectin gene polymorphism, vulvovaginal candidiasis, and bacterial vaginosis. Obstet. Gynecol. 2007, 109, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Babula, O.; Lazdane, G.; Kroica, J.; Linhares, I.M.; Ledger, W.J.; Witkin, S.S. Frequency of interleukin-4 (IL-4) -589 gene polymorphism and vaginal concentrations of IL-4, nitric oxide, and mannose-binding lectin in women with recurrent vulvovaginal candidiasis. Clin. Infect. Dis. 2005, 40, 1258–1262. [Google Scholar] [CrossRef] [PubMed]
- Plantinga, T.S.; van der Velden, W.J.; Ferwerda, B.; van Spriel, A.B.; Adema, G.; Feuth, T.; Donnelly, J.P.; Brown, G.D.; Kullberg, B.J.; Blijlevens, N.M.; et al. Early stop polymorphism in human DECTIN-1 is associated with increased candida colonization in hematopoietic stem cell transplant recipients. Clin. Infect. Dis. 2009, 49, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferwerda, B.; Ferwerda, G.; Plantinga, T.S.; Willment, J.A.; van Spriel, A.B.; Venselaar, H.; Elbers, C.C.; Johnson, M.D.; Cambi, A.; Huysamen, C.; et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 2009, 361, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Nahum, A.; Dadi, H.; Bates, A.; Roifman, C.M. The biological significance of TLR3 variant, L412F, in conferring susceptibility to cutaneous candidiasis, CMV and autoimmunity. Autoimmun. Rev. 2012, 11, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Lagunes, L.; Rello, J. Invasive candidiasis: From mycobiome to infection, therapy, and prevention. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Kullberg, B.J.; Sobel, J.D.; Ruhnke, M.; Pappas, P.G.; Viscoli, C.; Rex, J.H.; Cleary, J.D.; Rubinstein, E.; Church, L.W.; Brown, J.M.; et al. Voriconazole versus a regimen of amphotericin B followed by fluconazole for candidaemia in non-neutropenic patients: A randomised non-inferiority trial. Lancet 2005, 366, 1435–1442. [Google Scholar] [CrossRef]
- Das, I.; Nightingale, P.; Patel, M.; Jumaa, P. Epidemiology, clinical characteristics, and outcome of candidemia: Experience in a tertiary referral center in the UK. Int. J. Infect. Dis. 2011, 15, e759–e763. [Google Scholar] [CrossRef] [PubMed]
- Reboli, A.C.; Rotstein, C.; Pappas, P.G.; Chapman, S.W.; Kett, D.H.; Kumar, D.; Betts, R.; Wible, M.; Goldstein, B.P.; Schranz, J.; et al. Anidulafungin versus fluconazole for invasive candidiasis. N. Engl. J. Med. 2007, 356, 2472–2482. [Google Scholar] [CrossRef] [PubMed]
- Rammaert, B.; Desjardins, A.; Lortholary, O. New insights into hepatosplenic candidosis, a manifestation of chronic disseminated candidosis. Mycoses 2012, 55, e74–e84. [Google Scholar] [CrossRef] [PubMed]
- Lionakis, M.S.; Netea, M.G. Candida and host determinants of susceptibility to invasive candidiasis. PLoS Pathog. 2013, 9, e1003079. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D. Innate antifungal immunity: The key role of phagocytes. Annu. Rev. Immunol. 2011, 29, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Plantinga, T.S.; Johnson, M.D.; Scott, W.K.; van de Vosse, E.; Velez Edwards, D.R.; Smith, P.B.; Alexander, B.D.; Yang, J.C.; Kremer, D.; Laird, G.M.; et al. Toll-like receptor 1 polymorphisms increase susceptibility to candidemia. J. Infect. Dis. 2012, 205, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Cheng, S.C.; Johnson, M.D.; Smeekens, S.P.; Wojtowicz, A.; Giamarellos-Bourboulis, E.; Karjalainen, J.; Franke, L.; Withoff, S.; Plantinga, T.S.; et al. Immunochip SNP array identifies novel genetic variants conferring susceptibility to candidaemia. Nat. Commun. 2014, 5, 4675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzaraki, V.; Gresnigt, M.S.; Jaeger, M.; Ricano-Ponce, I.; Johnson, M.D.; Oosting, M.; Franke, L.; Withoff, S.; Perfect, J.R.; Joosten, L.A.B.; et al. An integrative genomics approach identifies novel pathways that influence candidaemia susceptibility. PLoS ONE 2017, 12, e0180824. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Plantinga, T.S.; van de Vosse, E.; Velez Edwards, D.R.; Smith, P.B.; Alexander, B.D.; Yang, J.C.; Kremer, D.; Laird, G.M.; Oosting, M.; et al. Cytokine gene polymorphisms and the outcome of invasive candidiasis: A prospective cohort study. Clin. Infect. Dis. 2012, 54, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Foster, C.B.; Taylor, J.G.; Erichsen, H.C.; Chen, R.A.; Walsh, T.J.; Anttila, V.J.; Ruutu, T.; Palotie, A.; Chanock, S.J. Association between chronic disseminated candidiasis in adult acute leukemia and common IL4 promoter haplotypes. J. Infect. Dis. 2003, 187, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Glocker, E.O.; Hennigs, A.; Nabavi, M.; Schaffer, A.A.; Woellner, C.; Salzer, U.; Pfeifer, D.; Veelken, H.; Warnatz, K.; Tahami, F.; et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009, 361, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Drewniak, A.; Gazendam, R.P.; Tool, A.T.; van Houdt, M.; Jansen, M.H.; van Hamme, J.L.; van Leeuwen, E.M.; Roos, D.; Scalais, E.; de Beaufort, C.; et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 2013, 121, 2385–2392. [Google Scholar] [CrossRef] [PubMed]
- Lanternier, F.; Mahdaviani, S.A.; Barbati, E.; Chaussade, H.; Koumar, Y.; Levy, R.; Denis, B.; Brunel, A.S.; Martin, S.; Loop, M.; et al. Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species-induced meningoencephalitis, colitis, or both. J. Allergy Clin. Immunol. 2015, 135, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Armstrong-James, D.; Brown, G.D.; Netea, M.G.; Zelante, T.; Gresnigt, M.S.; van de Veerdonk, F.L.; Levitz, S.M. Immunotherapeutic approaches to treatment of fungal diseases. Lancet Infect. Dis. 2017. [Google Scholar] [CrossRef]
- Charlier, C.; Hart, E.; Lefort, A.; Ribaud, P.; Dromer, F.; Denning, D.W.; Lortholary, O. Fluconazole for the management of invasive candidiasis: Where do we stand after 15 years? J. Antimicrob. Chemother. 2006, 57, 384–410. [Google Scholar] [CrossRef] [PubMed]
- Ostrosky-Zeichner, L.; Shoham, S.; Vazquez, J.; Reboli, A.; Betts, R.; Barron, M.A.; Schuster, M.; Judson, M.A.; Revankar, S.G.; Caeiro, J.P.; et al. MSG-01: A randomized, double-blind, placebo-controlled trial of caspofungin prophylaxis followed by preemptive therapy for invasive candidiasis in high-risk adults in the critical care setting. Clin. Infect. Dis. 2014, 58, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Netea, M.G.; Joosten, L.A.; van der Meer, J.W.; Kullberg, B.J. Novel strategies for the prevention and treatment of Candida infections: The potential of immunotherapy. FEMS Microbiol. Rev. 2010, 34, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.E.; Silva, S.; Azeredo, J.; Henriques, M. Novel strategies to fight Candida species infection. Crit. Rev. Microbiol. 2016, 42, 594–606. [Google Scholar] [PubMed]
- Vazquez, J.A.; Gupta, S.; Villanueva, A. Potential utility of recombinant human GM-CSF as adjunctive treatment of refractory oropharyngeal candidiasis in AIDS patients. Eur. J. Clin. Microbiol. Infect. Dis. 1998, 17, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, J.A.; Hidalgo, J.A.; de Bono, S. Use of sargramostim (rh-GM-CSF) as adjunctive treatment of fluconazole-refractory oropharyngeal candidiasis in patients with AIDS: A pilot study. HIV Clin. Trials 2000, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Graybill, J.R.; Bocanegra, R.; Luther, M. Antifungal combination therapy with granulocyte colony-stimulating factor and fluconazole in experimental disseminated candidiasis. Eur. J. Clin. Microbiol. Infect. Dis. 1995, 14, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Kullberg, B.J.; Oude Lashof, A.M.; Netea, M.G. Design of efficacy trials of cytokines in combination with antifungal drugs. Clin. Infect. Dis. 2004, 39 (Suppl. S4), S218–S223. [Google Scholar] [CrossRef] [PubMed]
- Kullberg, B.J.; van’t Wout, J.W.; Hoogstraten, C.; van Furth, R. Recombinant interferon-gamma enhances resistance to acute disseminated Candida albicans infection in mice. J. Infect. Dis. 1993, 168, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Delsing, C.E.; Gresnigt, M.S.; Leentjens, J.; Preijers, F.; Frager, F.A.; Kox, M.; Monneret, G.; Venet, F.; Bleeker-Rovers, C.P.; van de Veerdonk, F.L.; et al. Interferon-γ as adjunctive immunotherapy for invasive fungal infections: A case series. BMC Infect. Dis. 2014, 14, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torosantucci, A.; Bromuro, C.; Chiani, P.; de Bernardis, F.; Berti, F.; Galli, C.; Norelli, F.; Bellucci, C.; Polonelli, L.; Costantino, P.; et al. A novel glyco-conjugate vaccine against fungal pathogens. J. Exp. Med. 2005, 202, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; Luo, G.; Gebremariam, T.; Lee, H.; Schmidt, C.S.; Hennessey, J.P., Jr.; French, S.W.; Yeaman, M.R.; Filler, S.G.; Edwards, J.E., Jr. NDV-3 protects mice from vulvovaginal candidiasis through T- and B-cell immune response. Vaccine 2013, 31, 5549–5556. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.S.; White, C.J.; Ibrahim, A.S.; Filler, S.G.; Fu, Y.; Yeaman, M.R.; Edwards, J.E., Jr.; Hennessey, J.P., Jr. NDV-3, a recombinant alum-adjuvanted vaccine for Candida and Staphylococcus aureus, is safe and immunogenic in healthy adults. Vaccine 2012, 30, 7594–7600. [Google Scholar] [CrossRef] [PubMed]
- Seidel, M.G.; Peters, C.; Wacker, A.; Northoff, H.; Moog, R.; Boehme, A.; Silling, G.; Grimminger, W.; Einsele, H. Randomized phase III study of granulocyte transfusions in neutropenic patients. Bone Marrow Transpl. 2008, 42, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Price, T.H.; Boeckh, M.; Harrison, R.W.; McCullough, J.; Ness, P.M.; Strauss, R.G.; Nichols, W.G.; Hamza, T.H.; Cushing, M.M.; King, K.E.; et al. Efficacy of transfusion with granulocytes from G-CSF/dexamethasone-treated donors in neutropenic patients with infection. Blood 2015, 126, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Legrand, F.; Lecuit, M.; Dupont, B.; Bellaton, E.; Huerre, M.; Rohrlich, P.S.; Lortholary, O. Adjuvant corticosteroid therapy for chronic disseminated candidiasis. Clin. Infect. Dis. 2008, 46, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Borghi, M.; de Luca, A.; Puccetti, M.; Jaeger, M.; Mencacci, A.; Oikonomou, V.; Pariano, M.; Garlanda, C.; Moretti, S.; Bartoli, A.; et al. Pathogenic NLRP3 Inflammasome Activity during Candida Infection Is Negatively Regulated by IL-22 via Activation of NLRC4 and IL-1Ra. Cell Host Microbe 2015, 18, 198–209. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Recognition of Candida species by innate immune cells. Ligand binding to extracellular Toll-like receptors (TLRs), such as TLR2 and TLR4, leads to the production of pro-inflammatory cytokines during Candida infections. The intracellular TLRs that recognize nucleic acids—namely, TLR3 and TLR9—might also have a role in anti-Candida host responses. Chitin from C. albicans has been proposed to induce the production of interleukin-10 (IL-10) via a nucleotide-binding oligomerization domain-containing protein 2 (NOD2)-dependent mechanism and in this way may contribute to dampening pro-inflammatory host responses during fungal infection. The pattern recognition receptors (PRRs) dectin 1, dectin 2 and dectin 3, and Fc receptors for IgG (FcγRs), induce responses in a spleen tyrosine kinase (SYK)-dependent manner, whereas the signalling pathways engaged by the mannose receptor remain unknown. Dectin 1 can interact with TLR2 and can induce intracellular signalling via SYK- and RAF proto-oncogene serine/threonine-protein kinase (RAF1)-dependent pathways. Complement receptor 3 (CR3) is important for the recognition of unopsonized Candida, whereas FcγRs are important for recognition of opsonized Candida by neutrophils. Dendritic cell (DC)-specific -ICAM3-grabbing non-integrin (DC-SIGN) recognizes N-linked mannans of Candida and has a role in inducing T helper (TH) cell responses. There is no known Candida-derived ligand that triggers the macrophage-inducible C-type lectin receptor (MINCLE), whereas β-mannans from Candida are recognized by galectin 3. Although a role for melanoma differentiation-associated protein 5 (MDA5) in anti-Candida host responses has been described, it remains to be determined what ligand induces MDA5 activation. Together, these signalling pathways induce the secretion of cytokines and chemokines and initiate phagocytosis to clear Candida infections. CARD9, caspase activation and recruitment domain-containing 9; C. glabrata, Candida glabrata; NF-κB, nuclear factor-κB; PAMP, pathogen-associated molecular pattern; PKCδ, protein kinase Cδ; ROS, reactive oxygen species.
Figure 1.
Recognition of Candida species by innate immune cells. Ligand binding to extracellular Toll-like receptors (TLRs), such as TLR2 and TLR4, leads to the production of pro-inflammatory cytokines during Candida infections. The intracellular TLRs that recognize nucleic acids—namely, TLR3 and TLR9—might also have a role in anti-Candida host responses. Chitin from C. albicans has been proposed to induce the production of interleukin-10 (IL-10) via a nucleotide-binding oligomerization domain-containing protein 2 (NOD2)-dependent mechanism and in this way may contribute to dampening pro-inflammatory host responses during fungal infection. The pattern recognition receptors (PRRs) dectin 1, dectin 2 and dectin 3, and Fc receptors for IgG (FcγRs), induce responses in a spleen tyrosine kinase (SYK)-dependent manner, whereas the signalling pathways engaged by the mannose receptor remain unknown. Dectin 1 can interact with TLR2 and can induce intracellular signalling via SYK- and RAF proto-oncogene serine/threonine-protein kinase (RAF1)-dependent pathways. Complement receptor 3 (CR3) is important for the recognition of unopsonized Candida, whereas FcγRs are important for recognition of opsonized Candida by neutrophils. Dendritic cell (DC)-specific -ICAM3-grabbing non-integrin (DC-SIGN) recognizes N-linked mannans of Candida and has a role in inducing T helper (TH) cell responses. There is no known Candida-derived ligand that triggers the macrophage-inducible C-type lectin receptor (MINCLE), whereas β-mannans from Candida are recognized by galectin 3. Although a role for melanoma differentiation-associated protein 5 (MDA5) in anti-Candida host responses has been described, it remains to be determined what ligand induces MDA5 activation. Together, these signalling pathways induce the secretion of cytokines and chemokines and initiate phagocytosis to clear Candida infections. CARD9, caspase activation and recruitment domain-containing 9; C. glabrata, Candida glabrata; NF-κB, nuclear factor-κB; PAMP, pathogen-associated molecular pattern; PKCδ, protein kinase Cδ; ROS, reactive oxygen species.
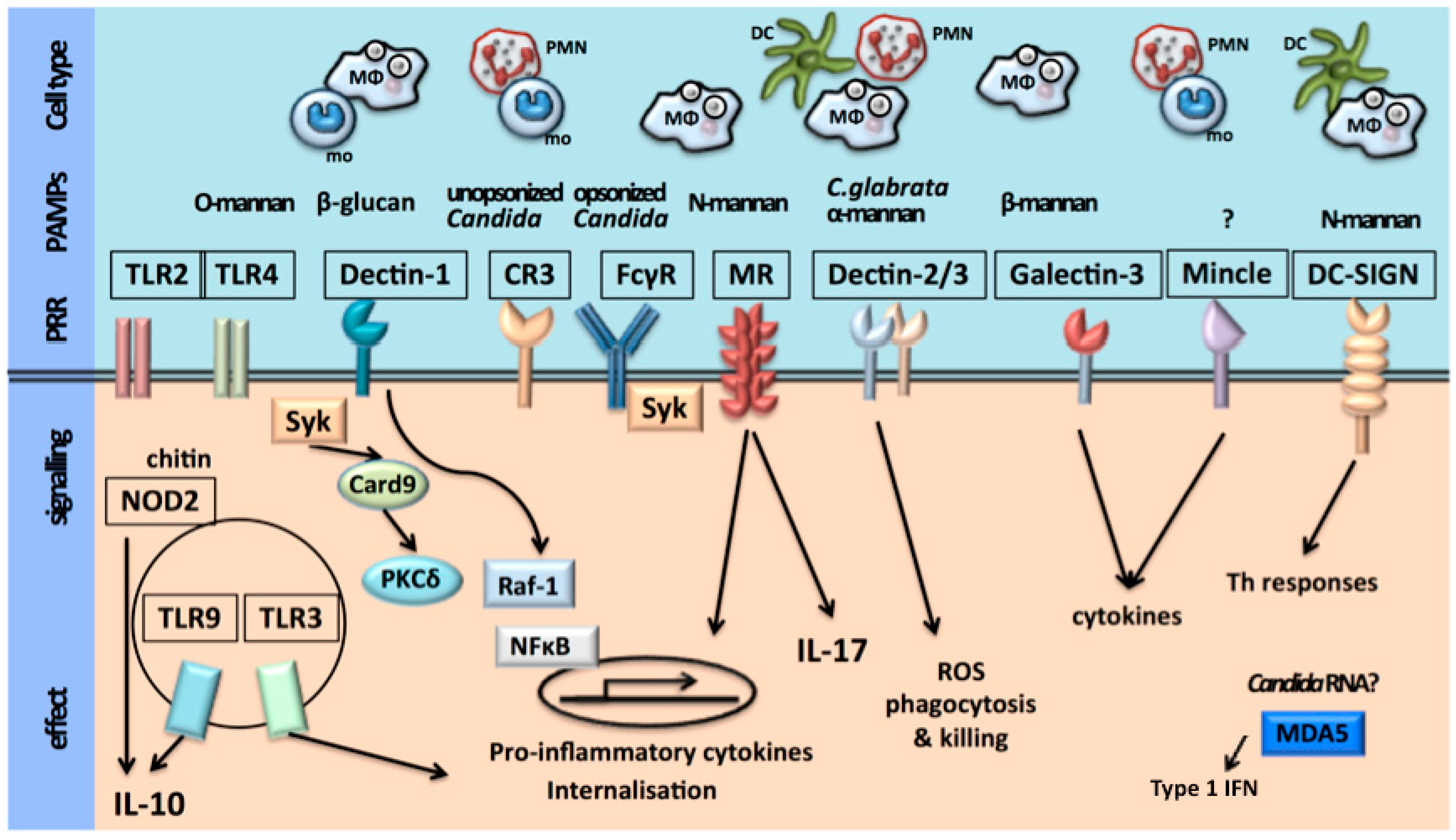

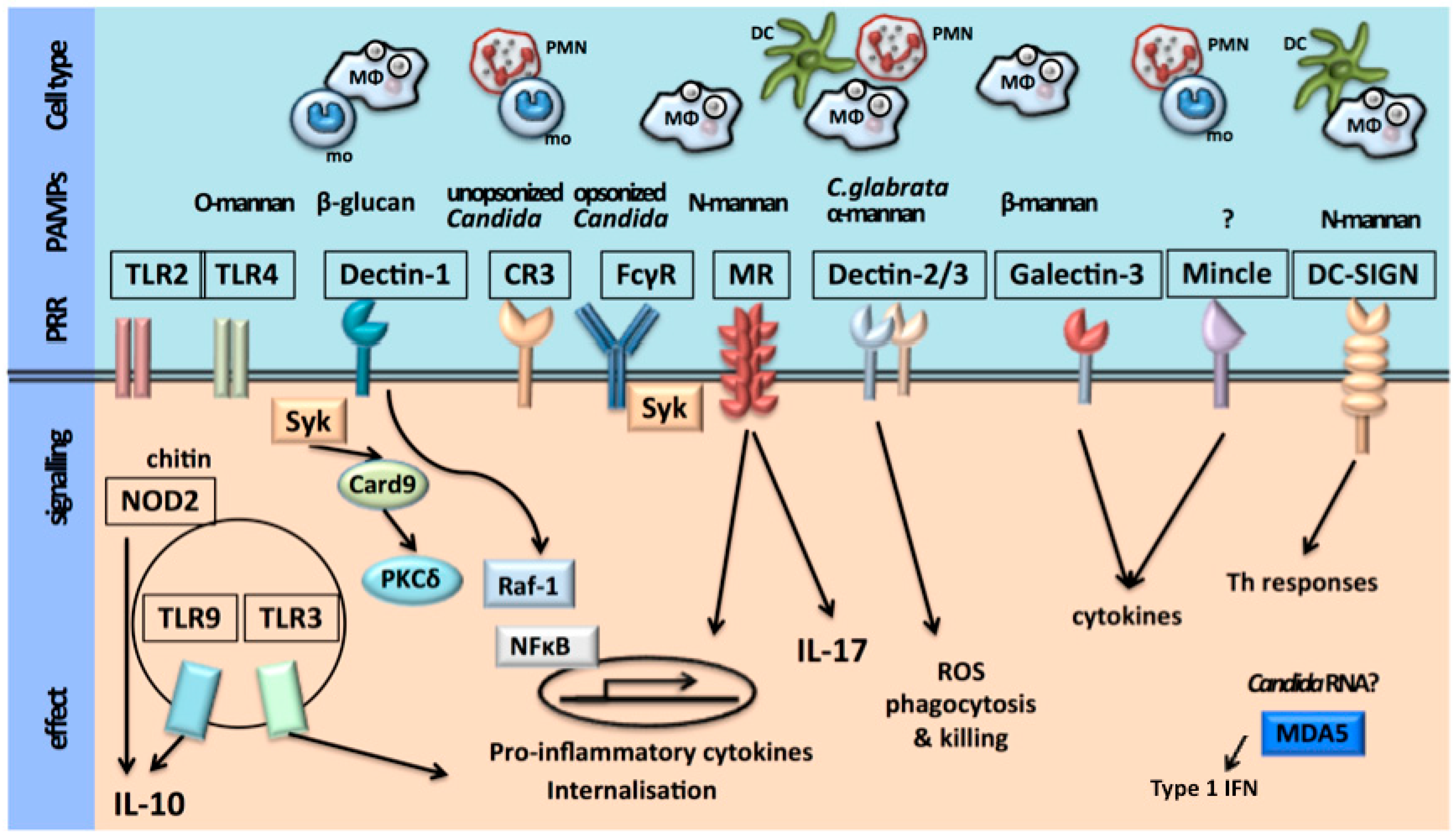{kind=link}
Table 1.
Genes involved in genetic susceptibility to Candida infections.
| Disease | Gene | Immune Modification | Infectious Phenotype | Non-Infectious Phenotype | References |
|---|---|---|---|---|---|
| CMC | AIRE | Autoantibodies against at least one out of three IL-17 cytokines; IL-17A (41%), IL-17F (75%) and/or IL-22 (91%) | CMC | Autoimmune manifestations: hypoparathyroidism and adrenal insufficiency | [89,90,91,92,93] |
| STAT1 (gain-of-function) | Disruption of the IL-12 and IL-23 pathways resulting in defective Th1 and Th17 cell responses and their consecutive production of IFN-γ, IL-17 and IL-22 | CMC, cutaneous and respiratory bacterial infections (mainly Staphylococcus aureus), viral skin infections (mainly Herpesviridae), invasive fungal infections | Autoimmune manifestations (hypothyroidism, autoimmune hemolytic anemia, etc), esophageal carcinoma, cerebral aneurysms | [2,94,95,96] | |
| IL-17RA, IL-17F, IL-17RC, TRAF3IP2 (encodes ACT1) | Deficiency of IL-17RA, IL-17F , IL-17RC, ACT1 causing disruption of the downstream signaling response to IL-17A and IL-17F | CMC and S. aureus skin infections | - | [72,74,97] | |
| RORC | RORγT deficiency affecting Th17 cell development | Mild CMC and severe mycobacterial infections | - | [74] | |
| IL-12B or IL-12Rβ1 | IL-12Rβ1 or IL-12p40 deficiency, affecting both IL-12 and IL-23 signaling pathways | CMC, mycobacterial infections, and Salmonella infections | - | [74] | |
| HIES | STAT3 | Defective downstream signaling of the IL-23 receptor resulting in absent IL-17 production | Mucocutaneous candidiasis, recurrent staphylococcal skin abscesses and pulmonary aspergillosis | Skeletal and dental abnormalities, pneumatoceles, eczema, eosinophilia, and elevated serum immunoglobulin E concentrations | [74] |
| DOCK8 | Disruption in Th17 differentiation | Mucocutaneous candidiasis, recurrent staphylococcal skin abscesses and pulmonary aspergillosis | Eczema, eosinophilia and, elevated serum immunoglobulin E concentrations | [1,74] | |
| RVVC | TLR2 | Reduced production of IL-17 and IFN-γ | RVVC | - | [99] |
| IL-4 | Elevated concentration of IL-4 and decreases concentration of MBL and nitric oxide (NO) in vaginal fluid | RVVC | - | [102] | |
| MBL2 | Reduced complement activation and Candida killing | RVVC | - | [100] | |
| NLPR3 | Hyper-inflammation by overproduction of IL-1β, high levels of IL-1β and low levels of IL-1Ra at the vaginal surface | RVVC | - | [30] | |
| Onychomycosis | Dectin-1 | Defective β-glucan recognition and consecutive Th17 cell responses | Onychomycosis and Candida colonization oral and gastrointestinal | - | [103,104] |
| Cutaneous candidiasis | TLR3 | Reduced CMV and Candida-induced IFN-γ and TNF-α production | Cutaneous candidiasis and CMV infection | Autoimmune manifestations: hypothyroidism, hypogonadism, idiopathic thrombocytopenic purpura, pancytopenia, alopecia, enteritis | [105] |
| Candidemia | IL-10 | Increased production of the anti-inflammatory cytokine IL-10 | Increased susceptibility to persistent candidemia | - | [116] |
| IL-12B | Decreased production of the pro-inflammatory cytokine IL-12b, resulting in downregulation of IFN-γ production. | Increased susceptibility to persistent candidemia | - | [116] | |
| TLR1 | Decreased Candida-induced cytokine production | Increased susceptibility to acquire candidemia | - | [113] | |
| CD58 | Disruption of Candida phagocytosis and loss of inhibition of germination | Increased susceptibility to acquire candidemia | - | [114] | |
| LCE4A-C1orf68 | Disruption of mucosal integrity | Increased susceptibility to acquire candidemia | - | [114] | |
| TAGAP | Decreased Candida-induced cytokine production | Increased susceptibility to acquire candidemia | - | [114] | |
| CDC | CARD9 | Low numbers of ciruculating Th17 cells and ROS-independent selective Candida killing effect neutrophils | Recurrent mucocutaneous and invasive Candida infections (meningoencephalitis, colitis) | - | [118,119,120] |
| IL-4 | Decreased IL-4 transcriptional activity | Increased susceptibility to CDC in acute leukemia patients | - | [117] | |
| TAGAP | Decreased Candida-induced cytokine production | Increased susceptibility to Candida dissemination into organs | - | [114] |
CMC: chronic mucocutaneous candidiasis; HIES: hyper IgE syndrome; RVVC: recurrent vulvovaginal candidiasis; CDC: chronic disseminated candidiasis
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Davidson, L.; Netea, M.G.; Kullberg, B.J. Patient Susceptibility to Candidiasis—A Potential for Adjunctive Immunotherapy. J. Fungi 2018, 4, 9. https://doi.org/10.3390/jof4010009
AMA Style
Davidson L, Netea MG, Kullberg BJ. Patient Susceptibility to Candidiasis—A Potential for Adjunctive Immunotherapy. Journal of Fungi. 2018; 4(1):9. https://doi.org/10.3390/jof4010009
Chicago/Turabian StyleDavidson, Linda, Mihai G. Netea, and Bart Jan Kullberg. 2018. "Patient Susceptibility to Candidiasis—A Potential for Adjunctive Immunotherapy" Journal of Fungi 4, no. 1: 9. https://doi.org/10.3390/jof4010009
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.