
MicroRNA MultiTool: A Software for Identifying Modified and Unmodified Human microRNA Using Mass Spectrometry

Abstract
:1. Introduction
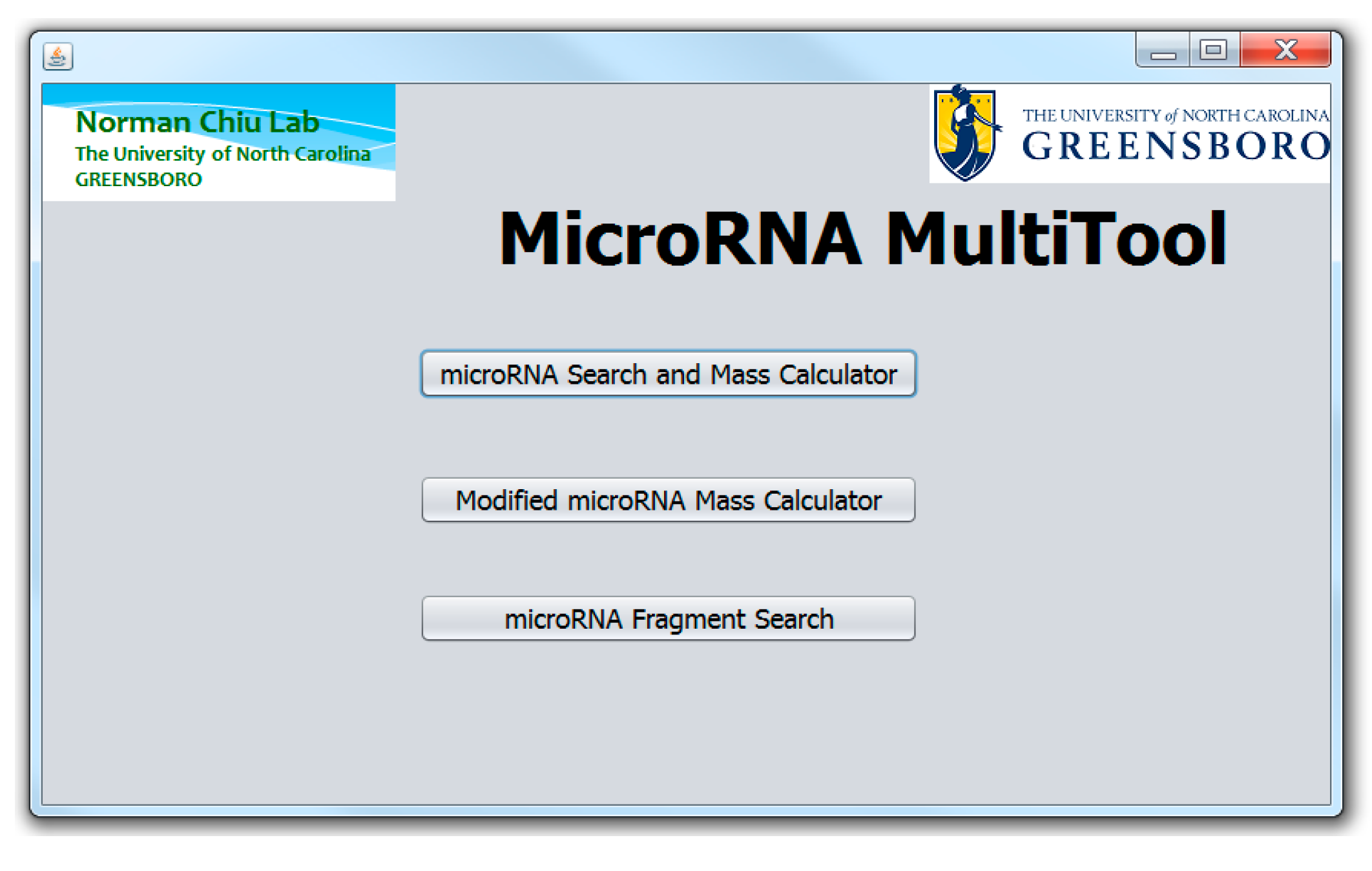
2. MicroRNA MultiTool Functionalities
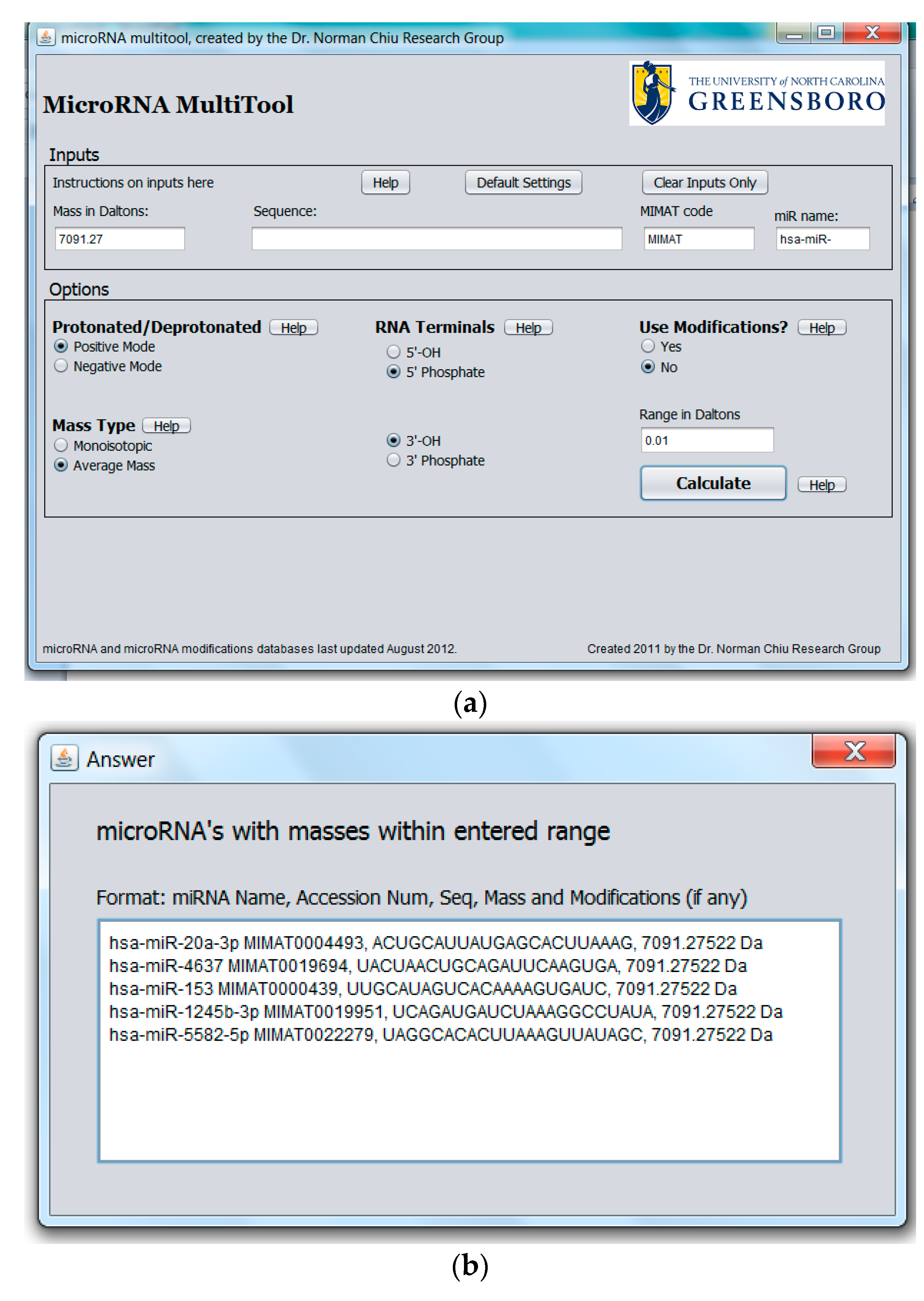
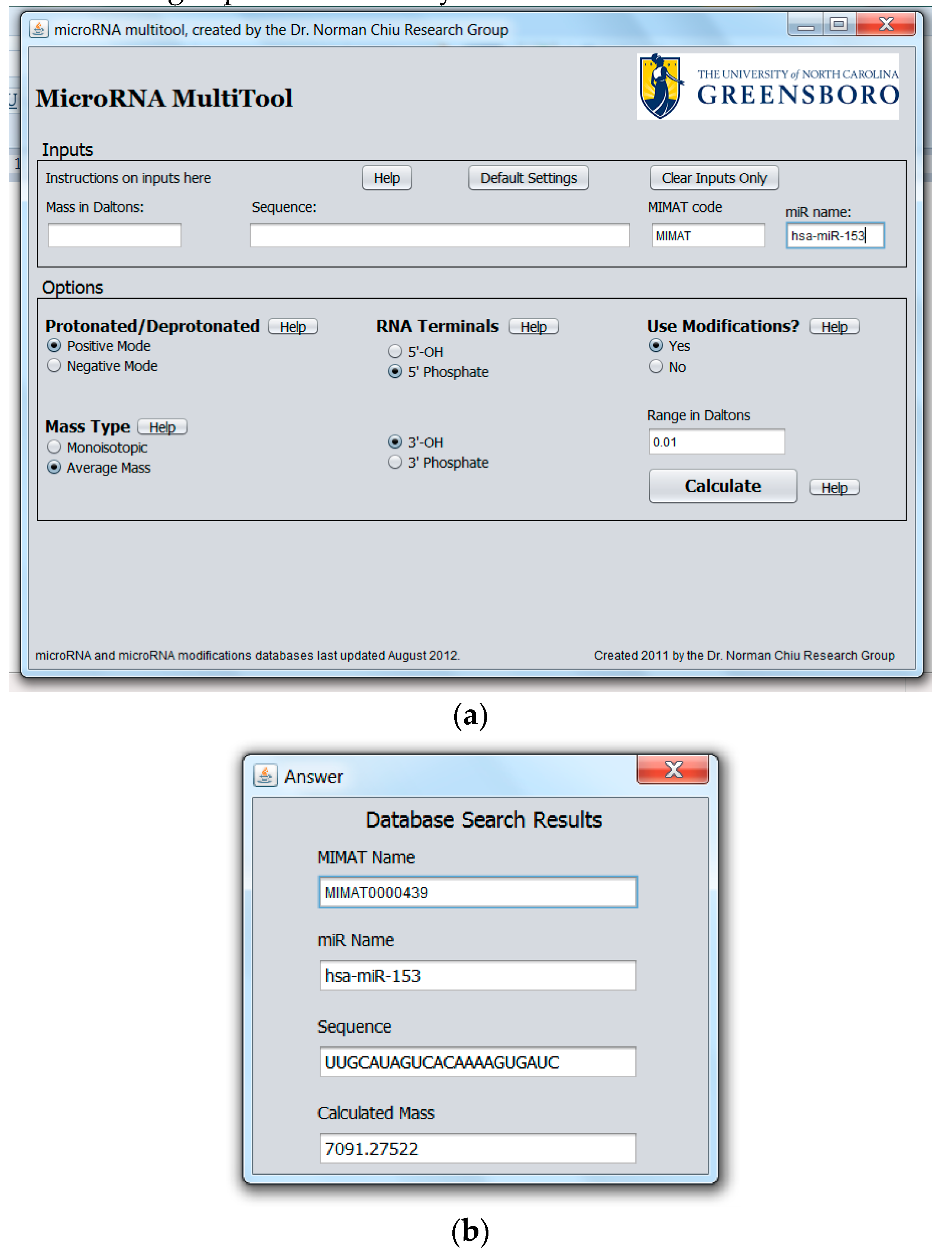
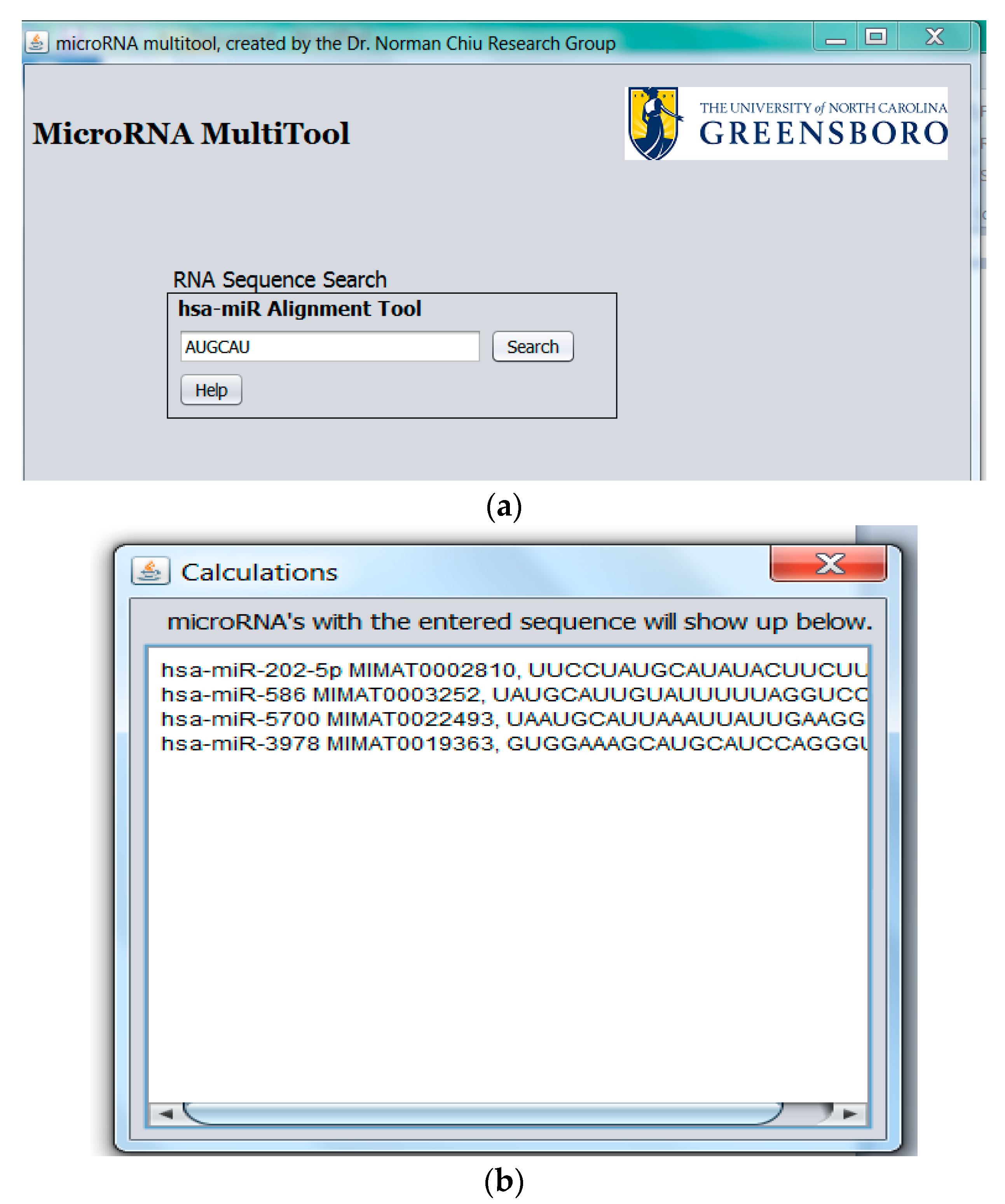
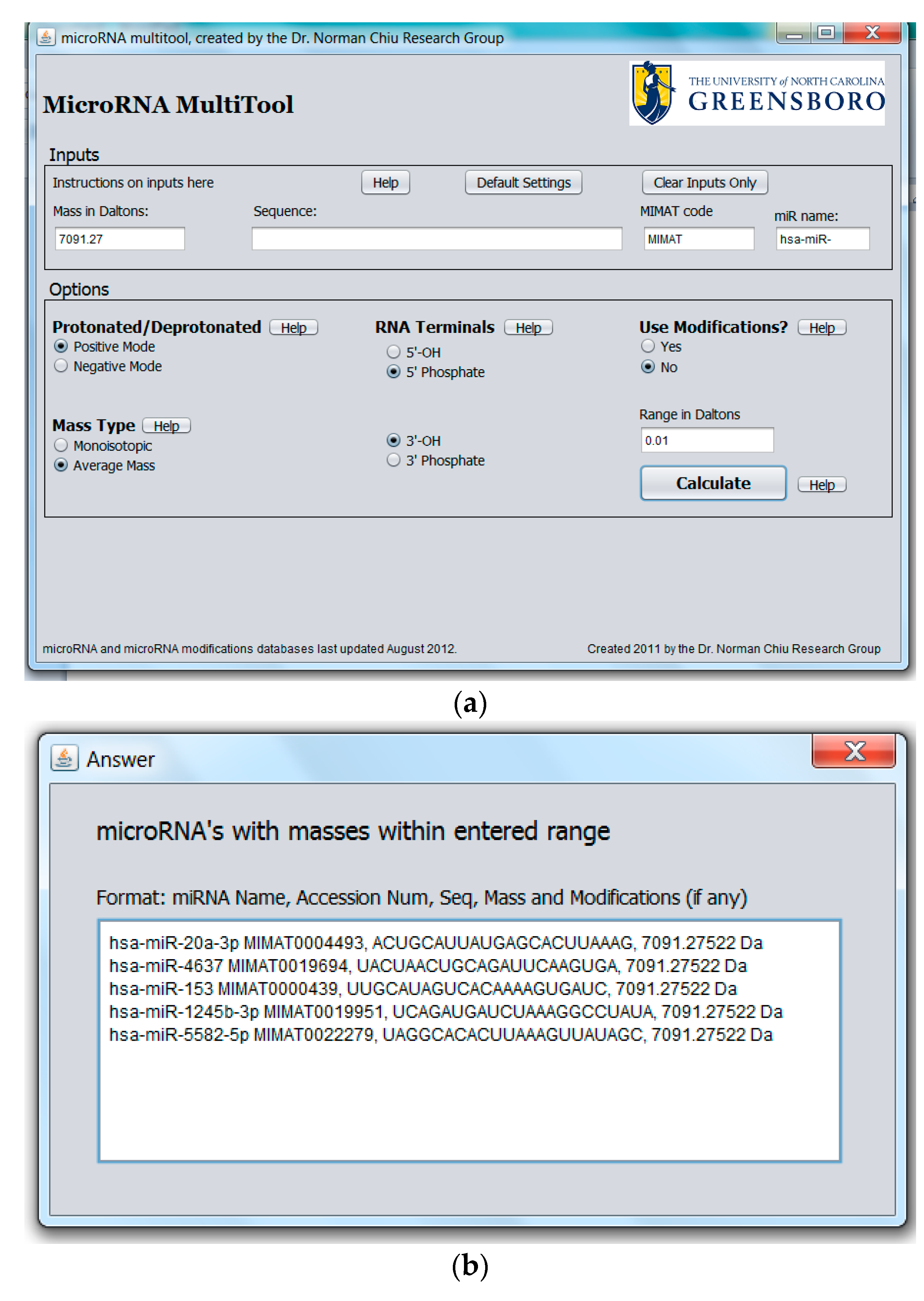
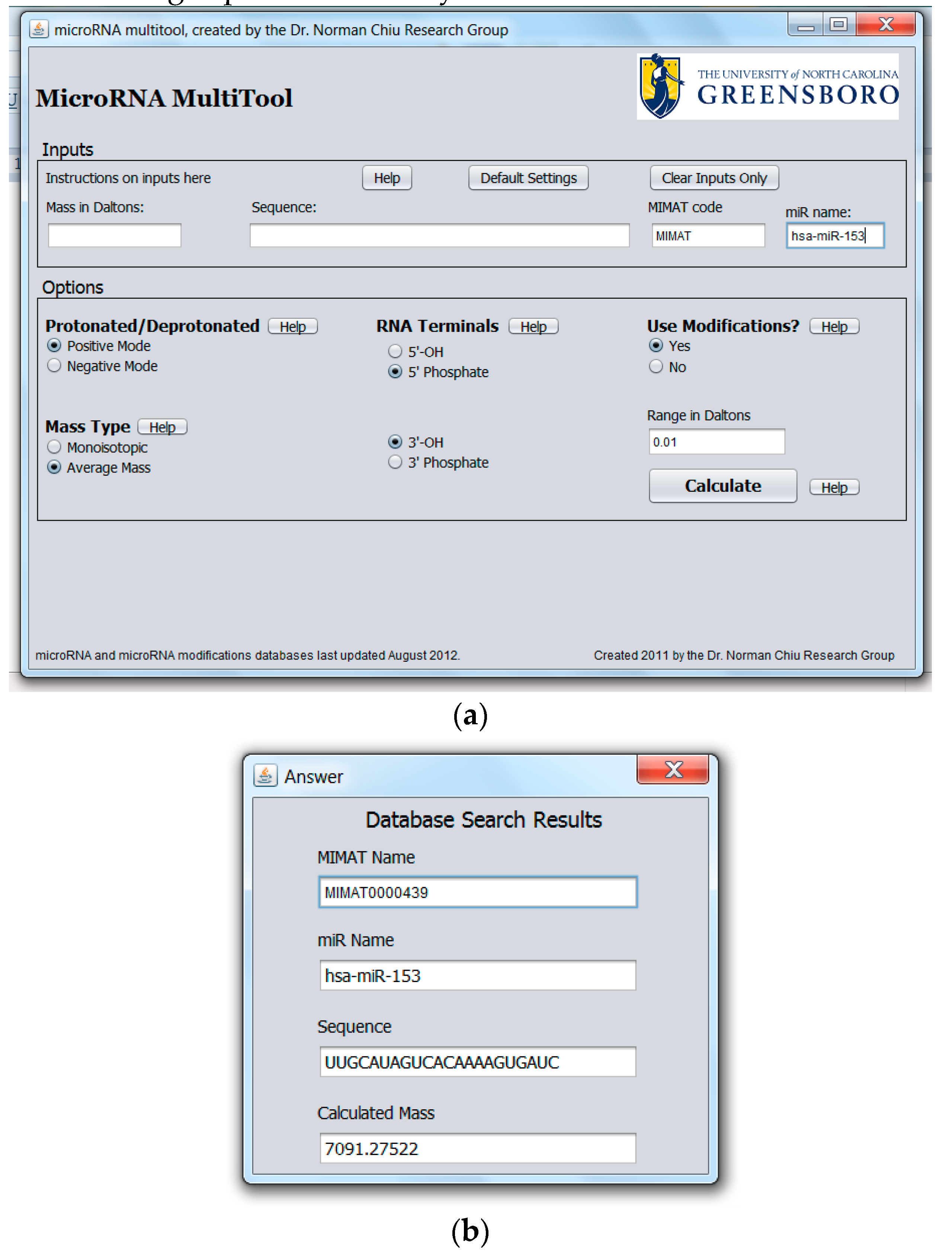
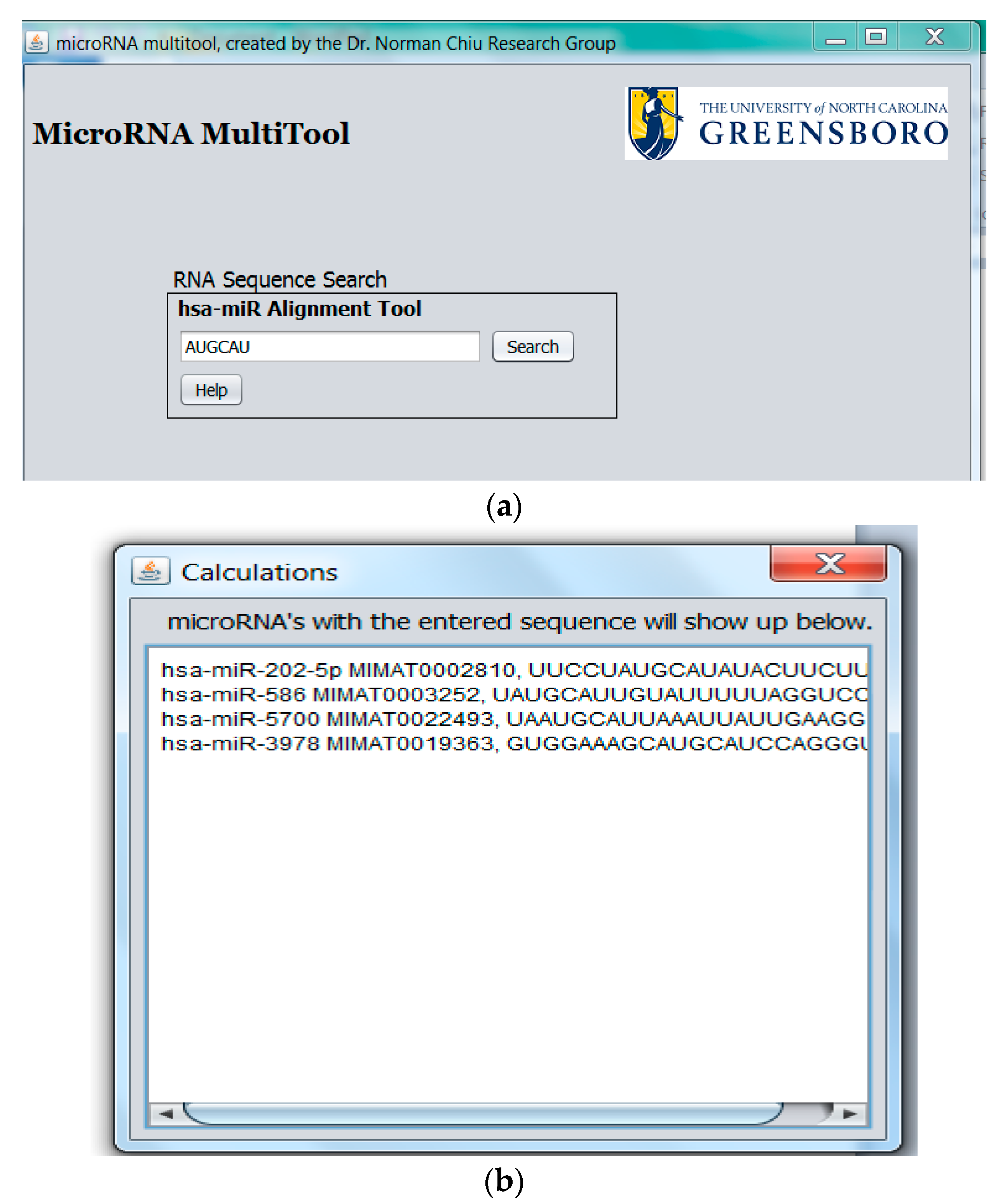
2.1. miRNA Search and Mass Calculator
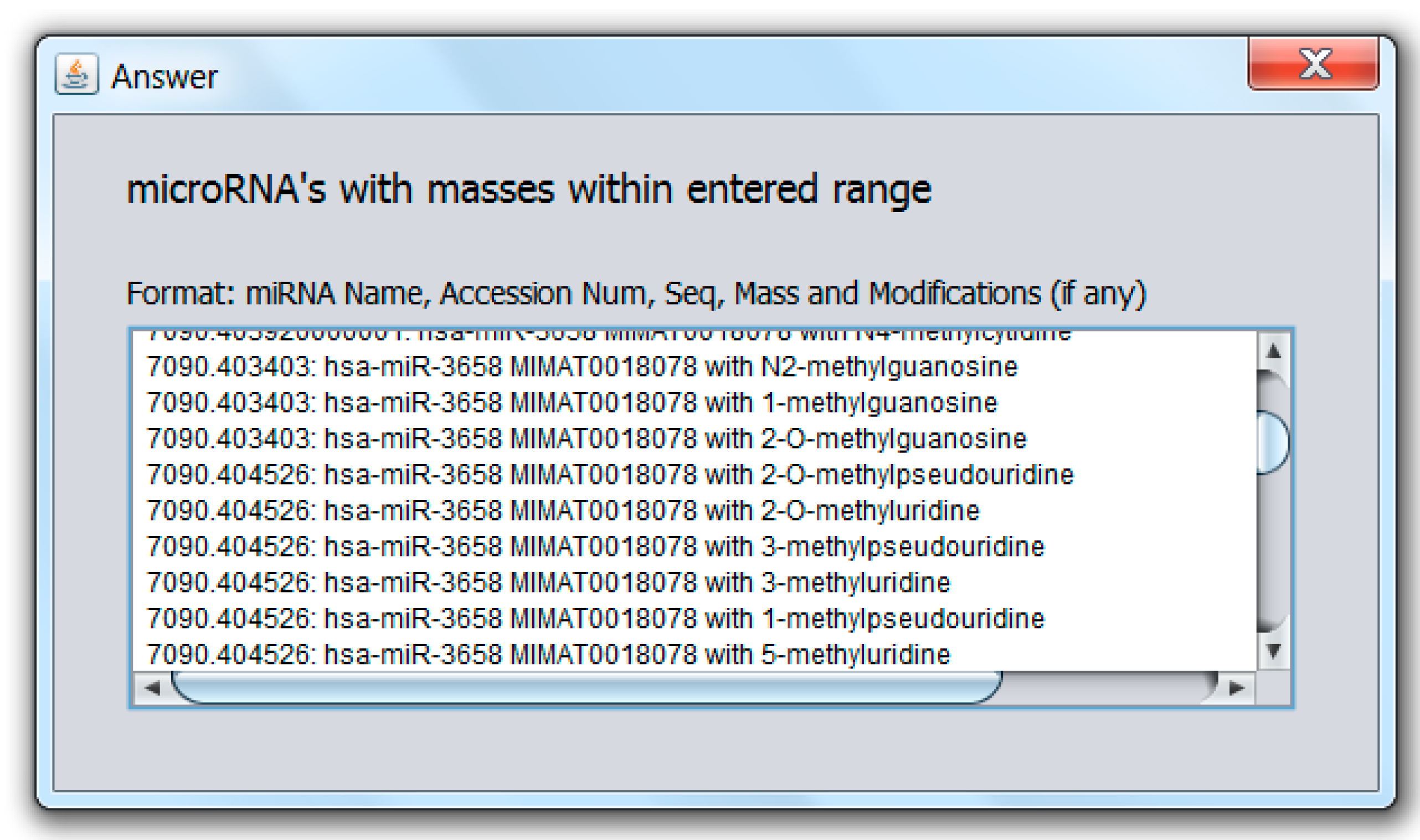
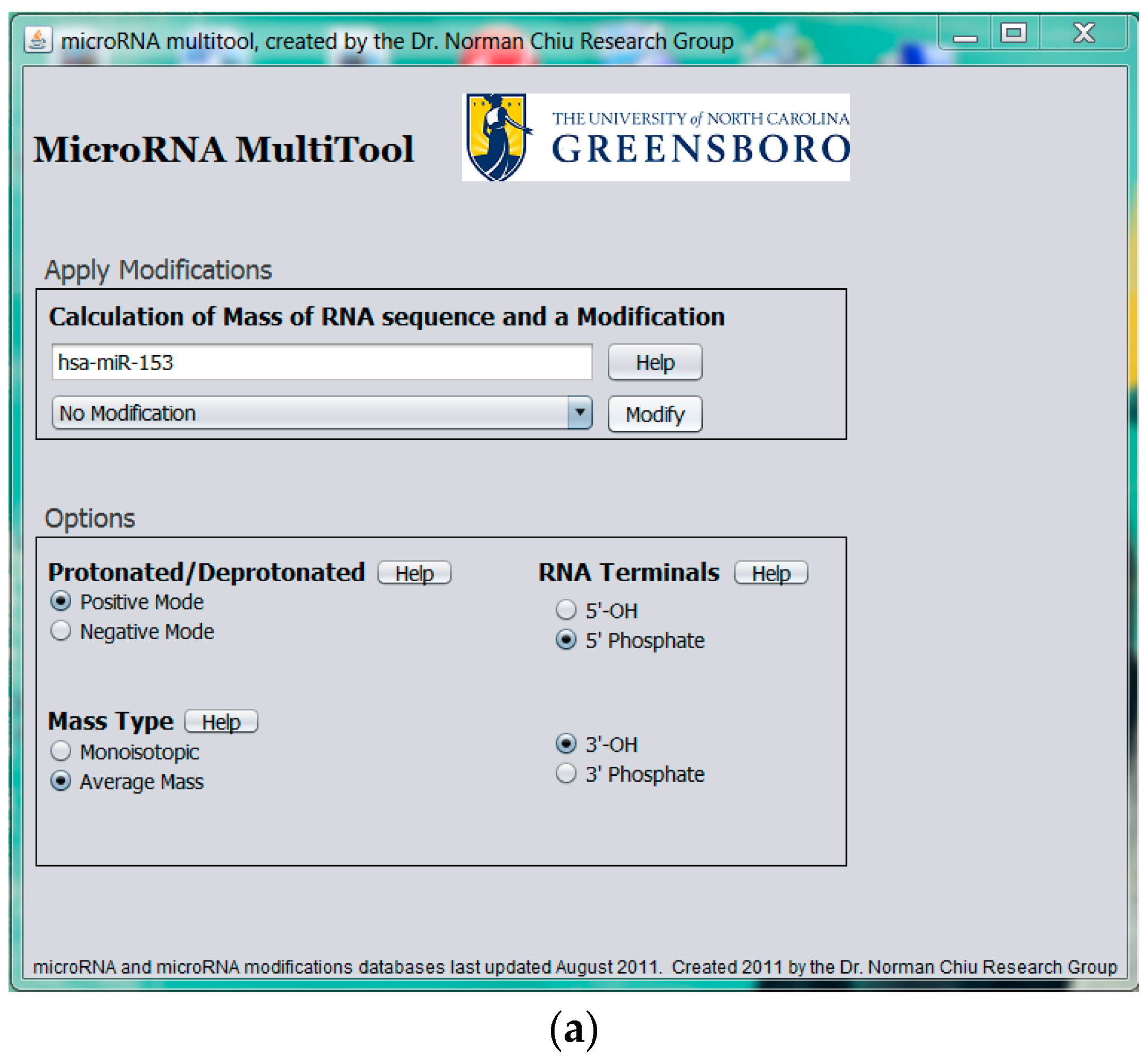
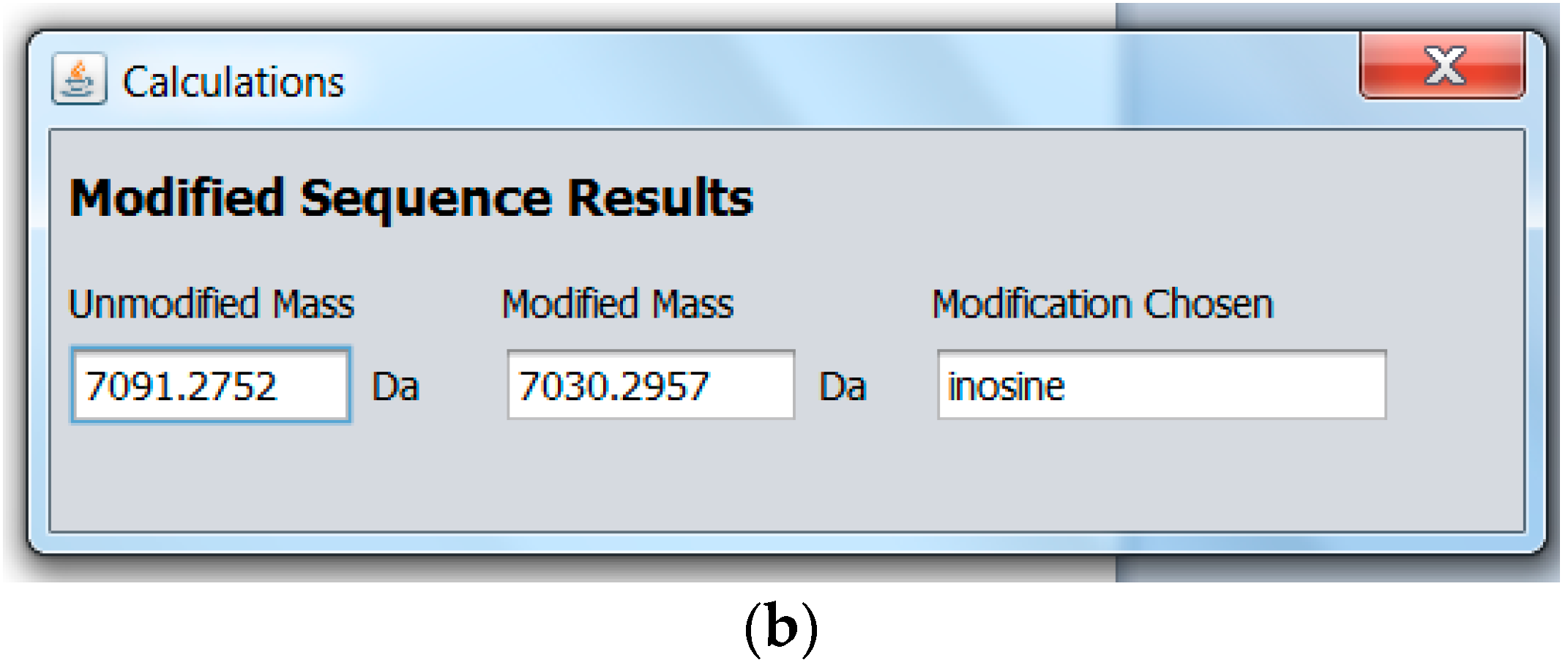
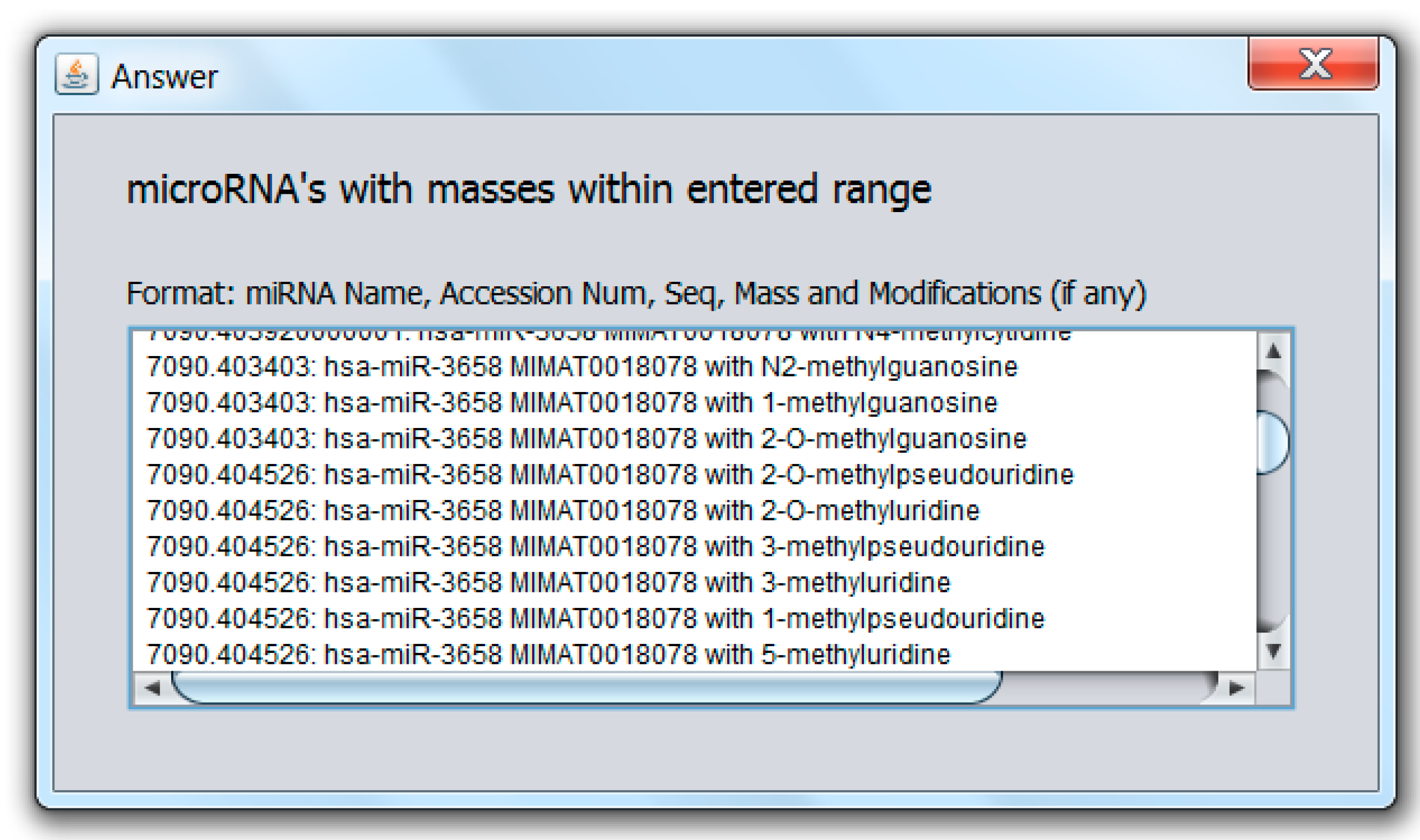
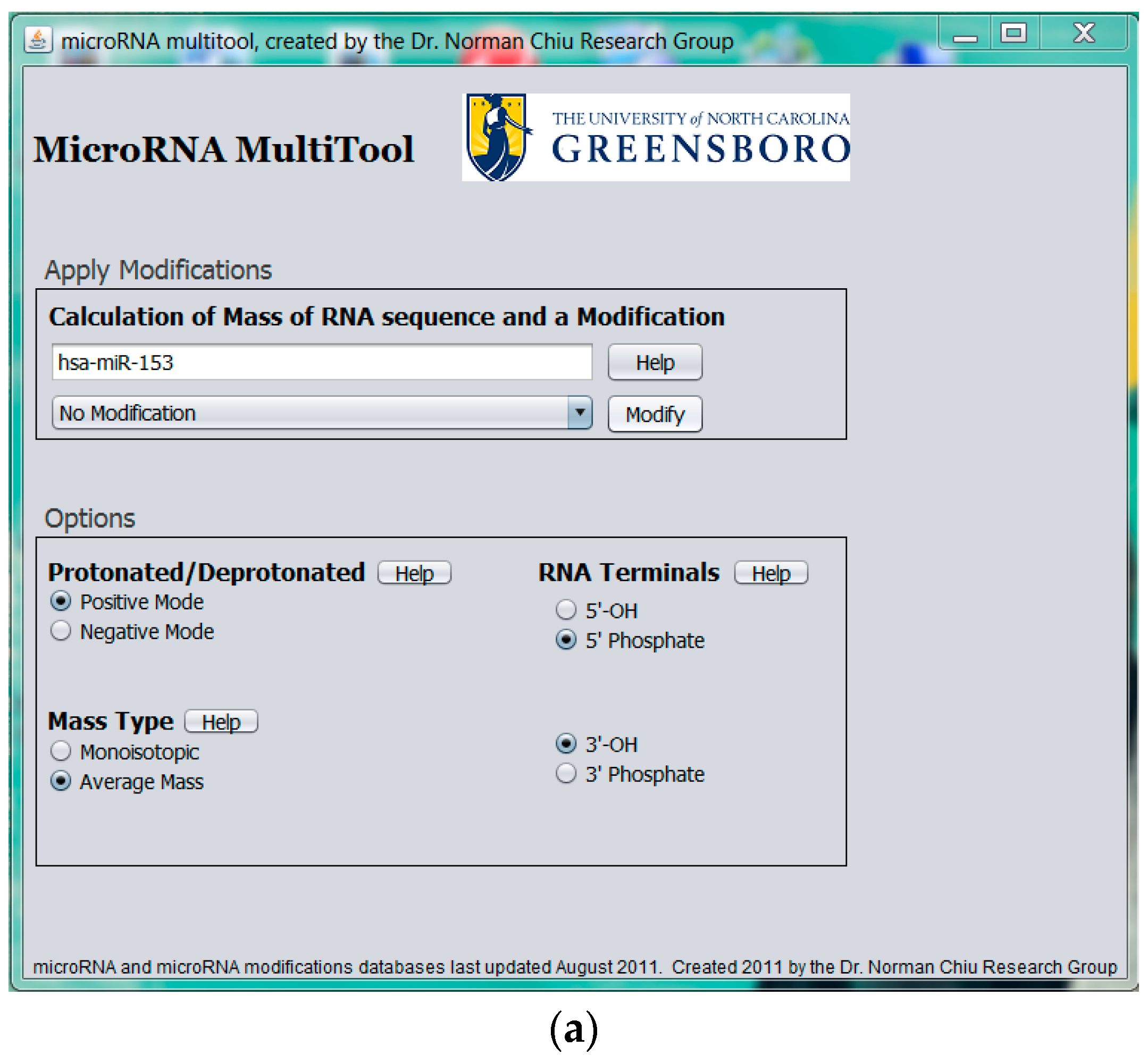
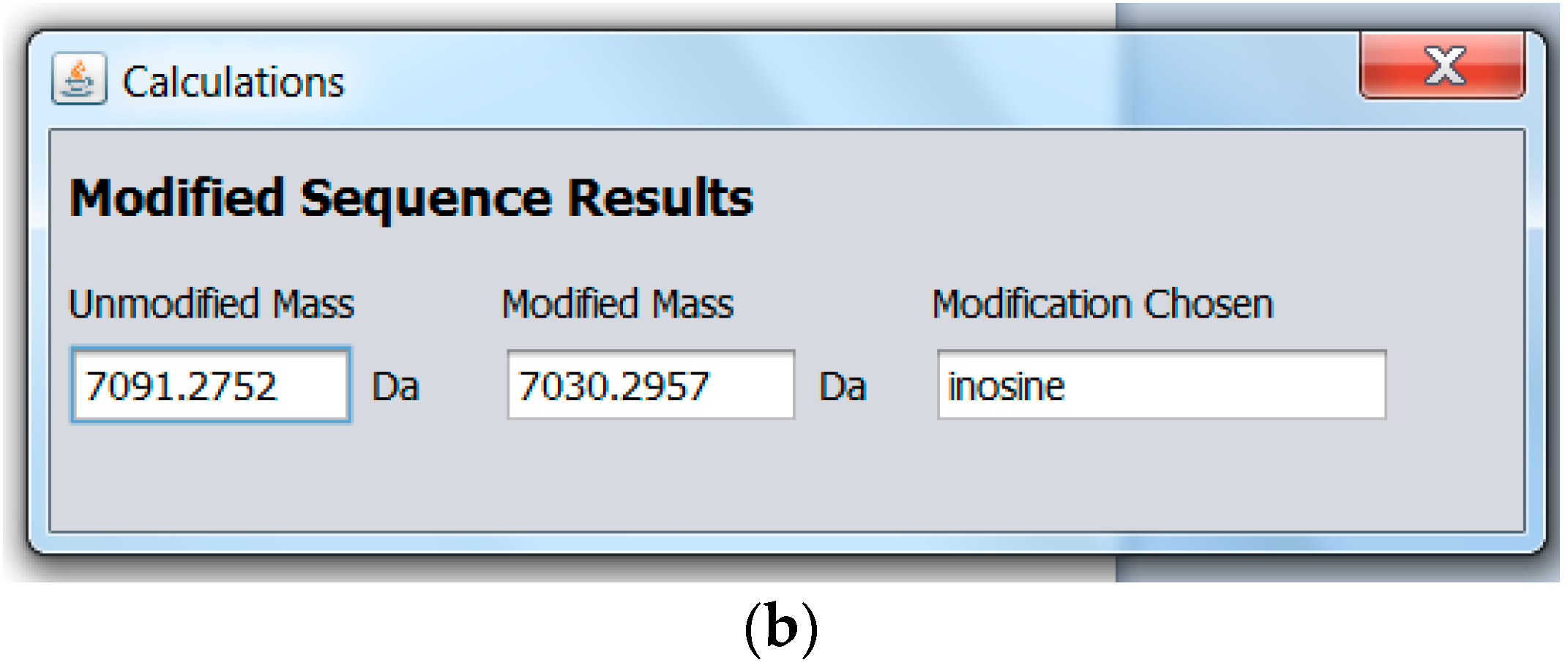
2.2. Modified miRNA Mass Calculator
2.3. miRNA Fragment Search
3. Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Griffiths-Jones, S.; Grocock, R.; van Dongen, S.; Bateman, A.; Enright, A. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Dutta, A. MicroRNAs in cancer. Annu. Rev. Pathol. 2009, 4, 199–227. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Esteller, M. Dysregulation of microRNAs in cancer: Playing with fire. FEBS Lett. 2011, 585, 2087–2099. [Google Scholar] [CrossRef] [PubMed]
- Wambua, D.M.; Ubukata, M.; Dane, J.; Cody, R.B.; Chiu, N.H.L. Bottom-up mass spectrometric sequencing of microRNA. Anal. Methods 2014, 6, 8829–8839. [Google Scholar] [CrossRef]
- Nakayama, H.; Yamauchi, Y.; Taoka, M.; Isobe, T. Direct identification of human cellular microRNAs by nanoflow liquid chromatography-high-resolution tandem mass spectrometry and database searching. Anal. Chem. 2015, 87, 2884–2891. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Xu, S.; Pan, C.; Ye, M.; Zou, H.; Guo, B. A matrix of 3,4-diaminobenzophenone for the analysis of oligonucleotides by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Nucleic Acids Res. 2006, 34, e94. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Saini, H.; van Dongen, S.; Enright, A. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed]
- Oberacher, H.; Pitterl, F. On the use of ESI-QqTOF-MS/MS for the comparative sequencing of nucleic acids. Biopolymers 2009, 91, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Jin, H.; Zhu, J.K. Bioinformatics analysis suggests base modifications of tRNAs and miRNAs in Arabidopsis thaliana. BMC Genom. 2009, 10, 155. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, A.M.; Ando, Y.; de Hoon, M.J.L.; Tomaru, Y.; Nishibu, T.; Ukekawa, R.; Funakoshi, T.; Kurokawa, T.; Suzuki, H.; Hayashizaki, Y.; et al. A comprehensive survey of 3′ animal miRNA modification events and a possible role for 3′ adenylation in modulating miRNA targeting effectiveness. Genome Res. 2010, 20, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Machnicka, M.A.; Milanowska, K.; Osman Oglou, O.; Purta, E.; Kurkowska, M.; Olchowik, A.; Januszewski, W.; Kalinowski, S.; Dunin-Horkawicz, S.; Rother, K.M.; et al. Modomics: A database of RNA modification pathways—2013 Update. Nucleic Acids Res. 2013, 41, D262–D267. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Takahashi, N.; Isobe, T. Informatics for mass spectrometry-based RNA analysis. Mass Spectrom. Rev. 2011, 30, 1000–1012. [Google Scholar] [CrossRef] [PubMed]
- Mengel-Jørgensen, J.; Kirpekar, F. Detection of pseudouridine and other modifications in tRNA by cyanoethylation and maldi mass spectrometry. Nucleic Acids Res. 2002, 30, e135. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, Y.; Mishima, T.; Yokomuro, S.; Arima, Y.; Kawahigashi, Y.; Shigehara, K.; Kanda, T.; Yoshida, H.; Uchida, E.; Tajiri, T.; et al. Sequencing and bioinformatics-based analyses of the microRNA transcriptome in hepatitis B-related hepatocellular carcinoma. PLoS ONE 2011, 6, e15304. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, K.; Qian, P.Y. Proteomics: Challenges, techniques and possibilities to overcome biological sample complexity. Hum Genom. Proteom. 2009, 2009, 239204. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. Mirbase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Czerwoniec, A.; Dunin-Horkawicz, S.; Purta, E.; Kaminska, K.; Kasprzak, J.; Bujnicki, J.; Grosjean, H.; Rother, K. Modomics: A database of RNA modification pathways. 2008 Update. Nucleic Acids Res. 2009, 37, D118–D121. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Takimura, S.; Yamaguchi, S.; Iida, J.; Bamba, T.; Fukusaki, E. Application of electrospray ionization ion trap/time-of-flight mass spectrometry for chemically-synthesized small RNAs. J. Biosci. Bioeng. 2012, 113, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Taucher, M.; Breuker, K. Top-down mass spectrometry for sequencing of larger (up to 61 nt) RNA by CAD and EDD. J. Am. Soc. Mass Spectrom. 2010, 21, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Taucher, M.; Rieder, U.; Breuker, K. Minimizing base loss and internal fragmentation in collisionally activated dissociation of multiply deprotonated RNA. J. Am. Soc. Mass Spectrom. 2010, 21, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Kharlamova, A.; Liu, J.; McLuckey, S.A. Ion trap collision-induced dissociation of multiply deprotonated RNA: C/y-ions versus (a-B)/w-ions. J. Am. Soc. Mass Spectrom. 2008, 19, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Cantara, W.A.; Crain, P.F.; Rozenski, J.; McCloskey, J.A.; Harris, K.A.; Zhang, X.; Vendeix, F.A.; Fabris, D.; Agris, P.F. The RNA modification database, RNAMDB: 2011 Update. Nucleic Acids Res. 2011, 39, D195–D201. [Google Scholar] [CrossRef] [PubMed]
- Rozenski, J.; McCloskey, J.A. SOS: A simple interactive program for ab initio oligonucleotide sequencing by mass spectrometry. J. Am. Soc. Mass Spectrom. 2002, 13, 200–203. [Google Scholar] [CrossRef]
- Oberacher, H.; Parson, W.; Oefner, P.J.; Mayr, B.M.; Huber, C.G. Applicability of tandem mass spectrometry to the automated comparative sequencing of long-chain oligonucleotides. J. Am. Soc. Mass Spectrom. 2004, 15, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Akiyama, M.; Taoka, M.; Yamauchi, Y.; Nobe, Y.; Ishikawa, H.; Takahashi, N.; Isobe, T. Ariadne: A database search engine for identification and chemical analysis of RNA using tandem mass spectrometry data. Nucleic Acids Res. 2009, 37, e47. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, J.; Chiu, N. High percentage of isomeric human microRNA and their analytical challenges. Non-Coding RNA 2016, 2, 13. [Google Scholar] [CrossRef]
- Wambua, D.M.; Tannous, B.A.; Chiu, N.H.L. Creating mass signatures for the detection of microRNA. Anal. Methods 2012, 4, 3453–3459. [Google Scholar] [CrossRef]
- Gott, J.M. Methods in Enzymology; Academic Press: San Diego, CA, USA, 2007; Volume 425. [Google Scholar]
- Pantano, L.; Estivill, X.; Martí, E. Seqbuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res. 2010, 38, e34. [Google Scholar] [CrossRef] [PubMed]
- Ebhardt, H.; Tsang, H.; Dai, D.; Liu, Y.; Bostan, B.; Fahlman, R. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res. 2009, 37, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Galeano, F.; Tomaselli, S.; Locatelli, F.; Gallo, A. A-to-I RNA editing: The “ADAR” side of human cancer. Semin. Cell Dev. Biol. 2012, 23, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Kellner, S.; Burhenne, J.; Helm, M. Detection of RNA modifications. RNA Biol. 2010, 7, 237–247. [Google Scholar] [CrossRef]
- Taucher, M.; Ganisl, B.; Breuker, K. Identification, localization, and relative quantitation of pseudouridine in RNA by tandem mass spectrometry of hydrolysis products. Int. J. Mass Spectrom. 2011, 304, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Limbach, P.A. Mass spectrometry of RNA: Linking the genome to the proteome. Brief. Funct. Genom. Proteom. 2006, 5, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Bahr, U.; Aygün, H.; Karas, M. Sequencing of single and double stranded RNA oligonucleotides by acid hydrolysis and MALDI mass spectrometry. Anal. Chem. 2009, 81, 3173–3179. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5′ pUUGCAUAGUCACAAAAGUGAUC-OH 3′ (hsa-miR-153) | ||
|---|---|---|
| Partial Sequence | # of miRNAs with Partial Sequence | % Sequence Coverage |
| pUUGCAUAGUC | 1 | 45.4 |
| pUUGCAUAGU | 3 | 40.9 |
| pUUGCAUAG | 3 | 36.3 |
| pUUGCAUA | 5 | 31.8 |
| pUUGCAU | 17 | 27.2 |
| pUUGCA | 58 | 22.7 |
| pUUGC | 174 | 18.1 |
| pUUG | 597 | 13.6 |
| pUU | 1101 | 9.0 |
| pU | 1722 | 4.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, Z.; Chiu, N.H.L.; Wambua, D.M. MicroRNA MultiTool: A Software for Identifying Modified and Unmodified Human microRNA Using Mass Spectrometry. Non-Coding RNA 2017, 3, 13. https://doi.org/10.3390/ncrna3010013
Cui Z, Chiu NHL, Wambua DM. MicroRNA MultiTool: A Software for Identifying Modified and Unmodified Human microRNA Using Mass Spectrometry. Non-Coding RNA. 2017; 3(1):13. https://doi.org/10.3390/ncrna3010013
Chicago/Turabian StyleCui, Zhonghao, Norman H. L. Chiu, and Dickson M. Wambua. 2017. "MicroRNA MultiTool: A Software for Identifying Modified and Unmodified Human microRNA Using Mass Spectrometry" Non-Coding RNA 3, no. 1: 13. https://doi.org/10.3390/ncrna3010013