Long Non-Coding RNAs in Neuronal Aging

1
University of Amsterdam, Swammerdam Institute for Life Sciences (SILS), 1098XH Amsterdam, The Netherlands
2
QIMR Berghofer Medical Research Institute, Herston, QLD, Brisbane, Australia
*
Authors to whom correspondence should be addressed.
Non-Coding RNA 2018, 4(2), 12; https://doi.org/10.3390/ncrna4020012
Submission received: 28 February 2018
/
Revised: 6 April 2018
/
Accepted: 10 April 2018
/
Published: 18 April 2018
(This article belongs to the Special Issue Non-Coding RNA in the Nervous System)
Abstract
:The expansion of long non-coding RNAs (lncRNAs) in organismal genomes has been associated with the emergence of sophisticated regulatory networks that may have contributed to more complex neuronal processes, such as higher-order cognition. In line with the important roles of lncRNAs in the normal functioning of the human brain, dysregulation of lncRNA expression has been implicated in aging and age-related neurodegenerative disorders. In this paper, we discuss the function and expression of known neuronal-associated lncRNAs, their impact on epigenetic changes, the contribution of transposable elements to lncRNA expression, and the implication of lncRNAs in maintaining the 3D nuclear architecture in neurons. Moreover, we discuss how the complex molecular processes that are orchestrated by lncRNAs in the aged brain may contribute to neuronal pathogenesis by promoting protein aggregation and neurodegeneration. Finally, this review explores the possibility that age-related disturbances of lncRNA expression change the genomic and epigenetic regulatory landscape of neurons, which may affect neuronal processes such as neurogenesis and synaptic plasticity.
1. Introduction
Aging of the human brain often leads to cognitive decline [1], reduced neurogenesis [2] and neurodegeneration [3]. Such neuronal vulnerability makes aging the primary risk factor for neurodegenerative diseases. Alterations in the aging brain include changes in the epigenetics [4,5] and transcription [6] of both coding and non-coding regions of the genome.
Among non-coding transcripts, long non-coding RNAs (lncRNAs) have recently emerged as key regulators of the molecular processes that underlie age-associated phenotypes [7,8]. lncRNAs are transcripts that are longer than 200 nucleotides in length with virtually no protein-coding capacity [9]. These transcripts are mostly uniquely expressed in cell types—both spatially and temporally—and are particularly enriched in the brain, where they play functional roles in neuroplasticity, cognition, and differentiation of neural stem cells [10,11]. Additionally, lncRNAs are known to orchestrate epigenetic processes through their interactions with epigenetic machinery [12]. Interestingly, differential expression of lncRNAs has been described not only in healthy aging [13,14], but also in developmental and neurodegenerative diseases [15], raising the question of whether lncRNAs play a role in the aging of the human brain.
This review proposes ways by which lncRNAs may contribute to neural aging and how their functions can be altered across the human lifespan. We discuss that antisense (AS) lncRNAs can regulate pathological protein aggregation and that subnuclear compartment specific (SCS) lncRNAs can regulate neuronal splicing, transcription, and sponging of ion channels in aging. Other pre- and post-transcriptional regulatory roles performed by lncRNAs are also discussed in the context of cognition, neurogenesis, and neurodegeneration in aging, including the possible influence of lncRNAs on the maintenance of the 3D nuclear architecture.
2. Long Non-coding RNAs in Adult Neurogenesis: Implications for Aging
Neurogenesis is the process by which new functional neurons are generated from neural stem cells (NSCs) throughout life. In the mammalian adult brain, NSCs persist in the subgranular zone of the dentate gyrus of the hippocampus and in the subventricular zone (SVZ) of the lateral ventricles [16,17]. The discovery of neurogenesis in the adult mammalian brain, and its widespread decline throughout aging [2,18,19,20,21], suggest that the loss of the capacity for neurogenesis is a possible cause of aging [2,22]. While the origin of this neurogenesis decline with age is not yet clear, studies performed on rodents show both a significant reduction in the numbers of NSCs [18,19] and their proliferative potential [20,21]. These studies are corroborated by the finding that the hippocampus of a human brain displays a decline in the turnover of both neuronal and non-neuronal cells during aging [23]. The persistence of neurogenesis in the adult brain is believed to attenuate age-related phenotypes in two ways: (1) a decreased neuronal turnover would compromise neuron replacement, which is required for repair mechanisms that are triggered by brain injury or age-related diseases [2] and (2) reduced hippocampal neurogenesis may be important in the age-related loss of cognitive ability, since newly generated neurons could enhance neuronal plasticity, learning, and memory. Indeed, a study by Drapeu et al., 2003, showed that the extent of memory dysfunction in aged rats is quantitatively related to their deficit in hippocampal neurogenesis [22].
Although lncRNAs have been found to play crucial roles in the developing mammalian brain, little is known about their function in post-natal and adult neurogenesis. It is conceivable that the same lncRNA pool that regulates NSC behavior in the embryonic stage is equally important in adults, since neuronal development pathways are highly conserved among embryonic, early post-natal, and adult neurogenesis [16]. A study conducted by Barry et al., 2015 [13], reported that lncRNAs previously linked to embryonic neurogenesis (e.g., MALAT1; BCYRN1; MIAT; SOX2-OT, TUG1, and RMST) [11] were also expressed in the SVZ of the human adult brain [13]. Dlx1as [24], Six3os [24] and Pnky [25] are amongst the first lncRNAs whose functionality has been confirmed in adult mouse neurogenesis. Although several other ncRNAs were found to be specifically expressed or enriched in the neurogenic regions of the brain, their exact function remains unknown [26,27].
Taken together, studies on the developing and adult mammalian brain suggest that the relative abundance of individual lncRNAs in the total NSC pool may determine the course of neurogenesis. lncRNAs are key modulators of NSC maintenance, lineage commitment/differentiation, and telomere maintenance (Figure 1). Therefore, since lncRNAs orchestrate temporally and spatially precise gene regulatory networks that are involved in neurogenesis, mild alterations in their expression in the aged SVZ may account for the neurogenesis decline.
2.1. lncRNAs in Neural Stem Cells: Self-Renewal, Amplification of Intermediate Progenitors, and Generation of Neuroblasts
In adult neurogenesis, activated NSCs give rise to transit-amplifying cells, which in turn generate neuroblasts [16]. lncRNAs play major stage-dependent roles, influencing not only the transition from one stage to the other, but also the number and type of cells generated in each stage. For instance, knockdown of the lncRNA Six3os in the SVZs of adult mice NSCs resulted in a two-fold decrease in Tuj1-positive cells (neuronal marker) and increased GFAP-positive cells (activated NSC marker) [24]. Moreover, Dlx1as knockdown in adult murine SVZ of NSCs caused a three-fold decrease in Tuj1-positive neuroblasts, an increase of nearly 60% in GFAP-positive cells, and a decrease in the expression of Dlx1 and Dlx2, which are two transcription factors that play major roles in neuronal development [24]. These results demonstrate that Six3os and Dlx1as are important for the amplification of intermediate progenitors and neuroblast generation. In contrast, Pnky knockdown in post-natal NSCs potentiates neuronal lineage commitment and expands the transit-amplifying cell population, increasing neuron production by several-fold [25]. Therefore, Pnky is important for NSC maintenance.
It is likely that subtle alterations in the levels of these lncRNAs have dramatic effects on destabilizing the dynamic balance that is established between NSC proliferation, intermediate amplification, and differentiation into neuroblasts, potentially leading to impaired neurogenesis in aging. Importantly, DLX1AS expression in the SVZ is not altered during aging [13]. However, a recent study showed that the expression of the lncRNAs MALAT1, GOMAFU, NEAT1, and TUG1 in the human SVZ significantly increases with age [13]. This may be related to their functions in non-neuronal cells, where they control cell-cycle [28] and senescence [29].
2.2. lncRNAs in Cell Lineage Commitment: Shifting from Neurogenesis to Oligodendrogenesis in Aging?
In addition to the neuronal cell lineage, NSCs also generate glial lineages, which give rise to astrocytes, oligodendrocytes, and ependymal cells [16]. Many lncRNAs exhibit dynamic expression patterns during neuronal–glial fate specification, suggesting roles in cell lineage commitment [30]. Transcript knockdown of Six3os lncRNA resulted in three-fold fewer cells expressing the oligodendrocyte marker OLIG2 [24], suggesting its involvement in the gliogenic specification of NSCs. Furthermore, long non-coding RNA-oligodendrocyte precursor cell (lnc-OPC) depletion resulted in a significant decrease in the expression of oligodendrocyte precursor cell (OPC) markers (MBP, PLP1, and CNP) and O4+ (oligodendrocyte surface marker), demonstrating that lnc-OPC plays a role in oligodendrogenesis [30].
lncRNAs may also direct a neurogenic fate in NSCs. For example, knockdown of the lncRNA RMST blocks neuronal differentiation and is required for the binding of SOX2 to promoter regions of neurogenic transcription factors [31]. Interestingly, a study conducted by Capilla-Gonzalez et al., 2013 reported that while the production of new neurons decreases during aging, the generation of oligodendroglial cells is not compromised in the murine SVZ [32]. The authors hypothesize that the preservation of oligodendrogenesis may be crucial for myelin maintenance in the aged brain [2].
2.3. Telomeric lncRNAs: Lying at the Root of Aged NSCs Survival?
Telomeres are repetitive DNA elements that cap the ends of chromosomes and protect their integrity. Throughout life, telomeres of somatic cells shorten at every round of DNA replication until the progressive and cumulative loss of telomere sequences ultimately triggers cellular senescence [33]. For this reason, telomere attrition is believed to be one of the main processes that determine the lifespan of somatic cells and organism aging. Several studies have demonstrated that aging can be delayed by telomerase activation and, accordingly, that pathological telomere dysfunction accelerates aging [34]. In germline and adult stem cells, telomere shortening can be countered by de novo addition of telomeric repeats by the TERT (telomerase reverse transcriptase) enzyme. In the adult mouse brain, telomerase activity is specific to NSCs isolated from the adult SVZ and hippocampus [35]. TERT expression is downregulated throughout aging in the SVZ of mice, leading to telomere shortening and strikingly disrupting neurogenesis and neuritogenesis [36]. Accumulating evidence suggests that telomere shortening is also an important cause of stem cell decline with aging in many other tissues [37,38]. Unsurprisingly, NSC functionality is highly dependent on telomere dynamics [36,39].
lncRNAs play key roles in telomere dynamics in stem cells. The lncRNA TERC (telomerase RNA component) forms a ribonucleoprotein complex with TERT, acting as a scaffold that brings the protein subunits of telomerase together, but also serves as a template for the synthesis of new telomeric repeats [40]. Both TERT and TERC are essential for telomere maintenance and elongation, as shown by their respective knockout mice models—which display short telomeres, instability, and premature aging [41,42]. Importantly, the lncRNA TERC is the limiting factor for telomerase activity as TERT heterozygote mice show no defects in telomere elongation, while TERC homozygotes do [42]. This finding suggests that the expression of the lncRNA TERC is not only crucial for telomerase activity, but also modulates it to promote and maintain telomere length. Interestingly, Klapper et al., 2001, observed that the temporal pattern of telomerase activity does not reflect the observed decrease in TERT transcript levels throughout pre- and post-natal neuronal development in mice [43]. Moreover, those same patterns of change occurred in association with decreased cell proliferation, differentiation, and natural cell death during early life neurogenesis. The authors propose a model in which the balance between TERT and TERC regulates neurogenesis, with high levels of TERT and TERC being responsible for NSC proliferation, low levels of TERC and high levels of TERT inducing differentiation, and low levels of both lncRNAs resulting in cell death. While the relative abundance of TERC and TERT in adult and aged NSCs remains unknown, this theory raises the question of whether a dysfunctional balance between TERC and TERT may trigger processes observed in aged NSCs, such as telomere de-protection (ultimately leading to senescence) or compromised cellular viability (leading to apoptosis).
Additionally, lncRNAs named TERRAs (telomeric repeat containing RNAs) are also key regulators of telomere dynamics. TERRA molecules are transcribed from the subtelomeric region of chromosomes and can be actively displaced to chromosome ends, the nucleosome [44,45], or the exterior of the cell, where they exist as components of inflammatory exosomes [46]. At chromosomal ends, TERRA transcripts can base-pair with complementary DNA, forming RNA:DNA hybrid structures that regulate telomere length [47,48]. In cells that display telomerase activity, the role of TERRA transcripts remains unclear. On one hand, in vitro experiments using TERRA-mimicking oligonucleotides suggest that TERRAs inhibit telomerase activity by directly binding to both TERT and TERC [49]. On the other hand, in yeast, TERRAs transcription is induced at short telomeres and form TERRA-telomerase RNA clusters in the early S phase. These are later recruited to short telomeres from which the TERRAs originated, triggering telomere elongation [45]. Interestingly, telomerase preferentially elongates short telomeres during the late S phase, a time point where TERRAs levels decline at telomeres [50]. Therefore, it appears that the dynamic balance of TERRA molecules throughout the cell cycle is a crucial factor that sustains and regulates telomere length [44,51]. Besides directly modulating telomerase activity, TERRAs are also proposed to regulate telomere length via heterochromatin formation at chromosome ends [52], capping of telomeres [53,54,55], and cellular differentiation [56]. Moreover, a recent study demonstrated that TERRA subtelomeric region knockouts in three human cell lines (HeLa, HCT116, and U2OS) are often lethal and lead to a dramatic loss of telomere sequences and a massive induction of the DNA damage response [55].
Taken together, these studies suggest that TERRAs are involved in multiple functions that mediate genomic instability, cell survival, and cellular senescence. As these functions are extensively affected through life, a role for TERRAs in aging can be reasonably inferred (e.g., [8,57]). Interestingly, the repression of general subtelomeric transcription by transiently activating mitochondrial reactive oxygen species in yeast was found to extend this organism’s lifespan [58]. In line with these findings, TERRA expression levels have been inversely correlated with telomere length [59,60], which implies that TERRAs are upregulated during aging. Human and murine induced pluripotent stem cells (iPSCs) have constantly elevated levels of TERRA transcripts [61,62]. Additionally, TERRA overexpression is found in proliferating progenitor cells in the developing mouse brain that exhibits TERRA foci [60]. This expression pattern supports the idea that high TERRA expression is coupled with cellular proliferation in progenitor cells. Therefore, TERRA expression in the presence of reduced telomerase activity—which is typical of aging—could lead to NSC cellular arrest and eventual senescence. The state of NSCs in aging is likely dependent on the interplay that is established between TERC, TERRA, and TERT. A shift in the abundance or activity of one of these molecules, both overall and in a cell-cycle-stage specific manner, could be implicated in the progression of aging.
3. lncRNAs in Cognitive Decline
Aging is associated with impairments in cognitive functions, including loss of memory and synaptic plasticity, and altered activation of the prefrontal cortex and hippocampus [1,63]. Cognitive decline seems to be independent of neuronal loss and may rather be a direct consequence of the alterations in synaptic connectivity [1]. The current understanding of learning, memory, and cognition is that neuronal activity is responsible for continuous changes in the synaptic connections that are established between neurons [1,64,65]. Modification of synaptic strength in the adult circuitry occurs through cellular mechanisms such as long-term potentiation (LTP) and long-term depression (LTD) [64,65]. Underlying these processes is a series of molecular events such as the activation, synthesis, and relocation of certain neurotransmitter receptors [64,65] and ion channels [65]. Likewise, local protein synthesis in dendrites [64,65,66] is regulated in response to neuronal activity, and is maintained by an asymmetric distribution of mRNAs and a control of their transcription, location, transport, and splicing [66]. The roles for lncRNAs in modulating the molecular processes on the basis of synaptic plasticity throughout life are just starting to emerge [67].
3.1. The Synaptic Coding/Non-Coding Interactome: Emerging Functions for lncRNAs on Synaptic Plasticity-Associated Genes, Transcripts, and Proteins
The regulation of synaptic plasticity by lncRNAs is a complex task that relies not only on their selective transport [68] to the dendrites of mature neurons [67,69,70], but also on the direct modulation of their expression levels by neuronal activity [71,72]. Thus, LTP studies in the dentate gyrus of living rats revealed dynamic expression profiles of lncRNAs that were highly correlated with synaptic plasticity-associated protein-coding genes [73]. Growing evidence suggests that, once in specific locations and at appropriate physiological levels, lncRNAs regulate the expression of such genes at both pre- and post-transcriptional levels, as discussed in more detail below (Figure 2). For instance, certain dendritic lncRNAs regulate local protein translation rates and the stability of protein-coding transcripts [74,75,76,77,78]. Furthermore, other nuclear-retained lncRNAs regulate the transcription of genes involved in the functioning of synapses, their splicing, and the nucleus-to-cytosol shuttling of ion channel subunits [72,79,80,81]. These lncRNA-mediated regulatory mechanisms may be involved in dynamic alterations in the synaptic connectivity and excitatory properties of a neuron.
3.2. lncRNAs Regulate Local Protein Translation Rates in Synapses
The regulation of local protein translation is partially carried out by the lncRNAs Bc1 and BC200, expressed in rodent and primate brains, respectively. In response to neuronal activity, Bc1 and BC200 are upregulated [82] and transported to dendrites [83], where they act as a scaffold that interacts with the translational machineries and represses local translation in synapses [74,75,76]. Bc1 gene knockout in mice results in neuronal hyperexcitability, convulsive seizures [84], anxiety, and exploratory behavioral defects [85]. Bc1 and BC200 activity is, therefore, crucial for normal neuronal activity and behavior. Importantly, BC200 levels in cortical areas are reduced by >60% between the ages of 49 and 86 years in healthy human individuals [86], raising the question of whether its expression changes may contribute to age-related cognitive decline.
3.3. Antisense lncRNAs Locally Regulate the Stability of Protein-Coding mRNAs Involved in Synaptic Plasticity
Among the classes of mRNAs expressed near synapses, natural sense/antisense transcript pairs are commonly found in the adult mouse forebrain [67]. Natural AS lncRNAs are transcripts that overlap—at least partially—with the mRNA of the coding gene. Given their significant sequence overlap, sense/antisense transcripts usually hybridize in an RNA duplex when in close proximity. This RNA structure regulates the stability of the coding mRNA and thus its protein level [87]. Importantly, some synaptic AS lncRNAs were found to downregulate the expression levels of proteins involved in neurite elaboration, such as BDNF [77,78], GDNF [77], and EPHB2 [77]. The core potassium channel subunit KCNA2 is also regulated by an AS lncRNA in response to peripheral nerve injury [88].
While some groundbreaking studies showed that antisense regulation of gene expression in synapses plays important roles in neuronal plasticity, its impact in aging remains unknown. Interestingly, in Aplysia sp., the regulation of sensorin (SRN), a gene involved in learning and long-term memory with no homologue in mammals, is performed by an AS lncRNA with an expression that is reduced in aging [89]. This study also reported that the distribution of SRN and SRN-AS within a neuron becomes asymmetric in aging. Hence, we hypothesize that a decreased generation of AS lncRNAs that are capable of dynamic gene regulation or the aberrant spatial distribution of sense/antisense transcripts in an aged neuron, might underlie local protein level defects and, consequently, synaptic plasticity deficiency.
3.4. Nuclear lncRNAs Dynamically Regulate the Transcription and Splicing of Coding Transcripts Involved in Synaptic Plasticity
lncRNAs also mediate the splicing of pre-mRNAs with retained introns. It has become increasingly clear that a dynamic regulation of splicing in the nervous system is critical for neuronal development and for the establishment and maintenance of neuronal networks [90]. A recent study by Traunmülle et al., 2016 was the first to elucidate the impact of splicing programs specifically in synapses [91]. The disruption of splicing patterns by interfering with a single RNA-binding protein, SLM2, resulted in defects in trans-synaptic protein complexes, impaired glutamatergic transmission, and synaptic plasticity [91]. Synaptic splicing programs are thus being regarded as creators of different, highly specific types of synapses, and thereby as mediators of neuronal plasticity. Interestingly, some lncRNAs are retained in the nucleus and predominantly localize to specific nuclear sub-compartments that are enriched in pre-mRNA splicing and processing factors, where they work as RNA-binding proteins (as discussed below).
As an example of lncRNAs located in subnuclear compartments, the lncRNA MALAT1 is specifically enriched in nuclear speckles, where it is proposed to act as a splicing factor sponge [92]. Accordingly, MALAT1 was shown to modulate the recruitment of SR family pre-mRNA-splicing factors to an active transcription site of a reporter gene locus [79]. Surprisingly, the depletion of MALAT1 in neuroblastoma cells affected not only the expression of genes involved in nuclear processes, but also that of genes in synapse function and dendrite development [79]. In line with this finding, MALAT1 knockdown in cultured hippocampal neurons significantly decreased the synaptic density, whereas its overexpression resulted in increased synaptic density [79]. Hence, MALAT1, in a similar way to the RNA-binding protein, SLM2, modulates the expression and splicing of genes involved in synapse function and maintenance. While this mechanism is not fully understood, further insights from studies of the lncRNA GOMAFU help to draw a possible working model.
Like MALAT1, GOMAFU is retained in the nucleus and is localized in another specific nuclear compartment [93], where it binds to splicing factors [72,94]. Loss-of-function mutations of GOMAFU in human iPSC-derived neurons lead to alternative splicing patterns [72] in the synaptic plasticity-related genes DISC1 [95,96,97,98], ERBB4 [99,100], and DRD2 [101,102]. Interestingly, upon depolarization of mouse primary cortical neurons and iPSC-derived neurons by KCl, GOMAFU transcript levels are downregulated while MALAT1 levels remain unchanged [72]. This suggests that GOMAFU regulates plasticity-related, activity-dependent alternative splicing. Thus, the authors suggest a model in which GOMAFU acts as a splicing factor scaffold in the nuclear compartments of an inactivated neuron. Upon neuronal activation, GOMAFU expression is downregulated, allowing for the release of splicing factors into the nucleoplasm, where they can modulate the splicing of transcripts involved in synaptic and dendritic growth, morphology, and function [72]. Although this is still a speculative hypothesis, it is likely that MALAT1 works in a similar way, but that its expression is regulated by stimuli other than the KCl-induced depolarization of neurons.
Taken together, these studies demonstrate that lncRNAs can act as RNA-binding proteins in specific subnuclear compartments and are dynamically regulated in response to neuronal activity, allowing for the splicing of genes involved in neuronal plasticity. Even though this mechanism remains poorly characterized, it would be very interesting to test these ideas in aging models in vivo.
3.5. Nuclear lncRNAs Regulate the Transcription and Nucleus-to-Cytosol Shuttling of Ion Channel Subunits in Response to Neuronal Activity
The lncRNA NEAT1 is retained in the nucleus, where it aggregates into paraspeckles structures [103]. NEAT1 expression level is dynamically regulated by neuronal activity and binds potassium channel-interacting proteins, including KCNAB2 and KCNIP [80]. The modulation of the stoichiometry of potassium channel protein subunits [104] and other ion channels [105] regulates neuronal excitability and thereby neuronal plasticity. Similarly, the shuttling of ion channel components from the nucleus to the cytosol may fine-tune the activity of ion channels, since these proteins can now interact with membrane channels. NEAT1 appears to be particularly important in this process as its transient downregulation in response to neuronal activity induces the release of potassium channel proteins, such as KCNAB2, from the nucleus into the cytosol [80]. Once in the cytosol, KCNAB2 is able to fine-tune the excitatory response, since knockdown of NEAT1 transcript induces a neuronal hyper-potentiation phenotype in iPSC-derived human cortical neurons [80]. NEAT1 is also involved in the transcriptional regulation of ion channel components as its knockdown in activated neurons drives a significant increase in ion channel gene expression [80]. Interestingly, the modulation of intrinsic neuronal excitability is a process that is severely affected in normal aging, and which may account for the learning impairment observed in normal aging subjects [106]. Additionally, dysregulation of NEAT1 activity may be involved in this phenotype.
3.6. lncRNAs Co-Expressed in the Nucleus and Cytoplasm Regulate the Trafficking of AMPA Receptors to the Plasma Membrane in Response to Glycine Stimulation
A recent study reported that the expression of a lncRNA cluster—consisting of MEG3, MEG8, MEG9, and RTL1-AS—in primary cortical neurons following glycine stimulation occurred in an N-Methyl-d-aspartate receptor (NMDAR)-dependent manner [81]. MEG3 knockdown blocked the glycine-induced increase of the GluA1 subunit of AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors at the plasma membrane by regulating its trafficking, which shows that MEG3 may have a role in the regulation of LTP [81].
3.7. Loss of Nuclear and Cytoskeleton Integrity as an Underlying Cause of Aging Synapses
While we have focused on alterations in the expression of lncRNAs that directly modulate the synaptic response, factors intrinsic to the cellular organization of an aged neuron may also modify lncRNA-associated synaptic activity. Nuclear and cytoplasmic [107] cellular organization of neurons contribute to modulate cell-autonomous processes, such as cytoskeletal protein transport [108,109] and nucleocytoplasmic compartmentalization [110,111]. The organization of the cytoskeleton in neurons suffers widespread alterations throughout aging [107,112] that impair protein transport [108], potentially compromising the transport of lncRNAs to dendrites and dysregulating the local dendritic processes that they orchestrate. The regulation of synaptic connectivity also depends on the selective exportation of a subset of lncRNAs from the nucleus to the cytoplasm. Since there is loss of cellular compartmentalization in aged neurons resulting from age-dependent nuclear pore deterioration [110], this transport may be compromised, and lncRNAs such as Bc1 and BC200 may leak into the cytoplasm, counteracting their neuronal activity-dependent regulation. The loss of cell nucleocytoplasmic compartmentation also poses a threat for nuclear lncRNAs. Firstly, cytoplasmic proteins and RNAs that leak into the nucleoplasm can compete with endogenous factors for binding sites in scaffolding lncRNAs, interfering with the specificity of their regulation. Secondly, the activity-dependent binding of ion channel-interacting proteins in the nucleus may also be compromised by their leakage into the cytoplasm. This would most likely give rise to hyperexcitability phenotypes, which have been found in aged CA3 pyramidal neurons [113]. Additionally, the nuclear lncRNAs that are discussed here exist in very specific subnuclear compartments, where they bind specific molecules and thus orchestrate the regulation of different, non-overlapping synaptic processes. A question that arises is whether genomic instability, re-organization of DNA architecture, and increased transcriptional noise in aging might cause non-specific binding of proteins and transcripts to different lncRNAs in different subnuclear domains. The abundance and spatial distribution of nuclear substructures in aging also remain elusive.
4. lncRNA-Mediated Processes in the Pathogenesis of Neurodegenerative Disorders
The transcriptomes of mammalian brains show widespread changes during aging [6]. In addition to changes in transcript expression and the usage of alternative isoforms and promoters of protein-coding genes [114,115,116,117], quantitative and qualitative changes in the non-coding transcriptome also occur with age [115,118]. Epigenetic alterations inherent to the aging process, such as altered patterns of histone post-translational modifications and DNA methylation, have been considered as the basis for an aged transcriptome [4,8]. Extensive epigenetic rearrangements result in chromatin remodeling, leading to alterations in the local accessibility of the genetic material, and thus affecting gene expression [4].
We suggest that changes in the expression levels of lncRNAs, caused by age-dependent epigenetic alterations, may impair or attempt to compensate for processes of adult neurogenesis, cognition, and neurodegeneration. The insights from age-related neurodegenerative disorders (Table 1) imply that the relative abundance of specific lncRNAs in a neuron in a given space and time determines the narrow range in which lncRNA-mediated processes are beneficial, before becoming pathogenic (Figure 3).
4.1. Roles for AS lncRNAs in Neuronal Aging and Disease
More than half of mammalian coding genes have complementary non-coding AS transcription [119]. AS lncRNAs have emerged as important regulators of gene expression, being able to influence a myriad of processes from epigenetic regulation to splicing, stability, and translation of coding mRNAs [120].
In age-related neurodegenerative disorders, the dysregulation of AS lncRNAs plays crucial roles in pathological protein aggregation. In Alzheimer’s disease (AD), AS lncRNAs contribute to amyloid-beta (Aβ) aggregation by modulating the expression and/or splicing of proteins involved in the generation and trafficking of Aβ. For example, the lncRNA BACE1-AS hybridizes to BACE1 mRNA [121], a protein involved in Aβ processing, and inhibits its cytoplasmic miRNA-mediated decay. BACE1-AS is upregulated in the brains of AD patients, consequently leading to the overexpression of BACE1 and to an increase in Aβ generation [122] (Figure 3A). SORL1-AS, which is also upregulated in the brains of AD patients, induces the synthesis of pathogenic splicing isoforms of SORL1, which are associated with increased Aβ levels in cultured human neuronal cells [123]. Furthermore, the AS lncRNA UCHL1-AS targets UCHL1 mRNA to heavy polysomes for translation, resulting in increased UCHL1 protein levels [124]. UCHL1 is a Parkinson’s disease (PD) and AD risk gene that is believed to prevent pathological protein aggregation by promoting its (or its precursors) ubiquitination [125,126]. Interestingly, UCHL1-AS is downregulated in PD [127]. In the nucleus, antisense transcript expression can also control the transcription initiation of coding genes by sequestering chromatin-regulatory proteins. LRP1-AS directly binds to HMGB2, preventing it from enhancing LRP1 transcription, thereby reducing its expression [128] (Figure 3D). LRP1 modulates the integrity of dendritic spines, synapses, and neuronal viability, and is also involved in Aβ deposition [129]. LRP1 expression is downregulated with aging [130], compromising neuronal survival in the brains of aged mice [129]. Interestingly, LRP1-AS is upregulated in AD [128], thereby the sequestration of transcriptional activators such as HMGB2 may account for LRP1 downregulation with age and in neurodegenerative disorders.
Besides regulating protein aggregation, cytoplasmic AS lncRNAs can also regulate the stability of transcripts from genes linked to neurodegenerative disorders and cognition (e.g., PINK1 [131], GDNF, EPHB2 [77], and KCNA2 [88]). The binding of lncRNAs to protein-coding transcripts may affect their stability and either protect them from degradation (e.g., PINK1-AS) or cause their decay (e.g., GDNF-AS, EPHB2-AS, and KCNA2-AS). In the nucleus, besides sequestering chromatin regulatory molecules, AS expression may also recruit transcriptional repressors to chromatin-modifying complexes that are close to gene promoters, thus silencing gene expression. A recent study showed that 20% of lncRNAs (out of a cohort of 3300) often associate with the polycomb repressive complex 2 (PRC2), a histone methyltransferase that catalyzes repressive H3K27 methylation [132]. For example, BDNF-AS recruits PRC2 to the BDNF promoter, resulting in transcriptional repression [77], which possibly affects neurite elaboration in aging. The HOX transcript antisense RNA (HOTAIR) binds and targets PRC2 in trans to the HOXD cluster [133] and to multiple sites of the genome [134]. HOTAIR can also interact with multiple regulatory complexes simultaneously. HOTAIR binds both PRC2 and the LSD1/coREST/REST complex—which catalyzes H3K4 demethylation—into a single ribonucleic complex [135]. By functioning as a scaffold for selected chromatin-modifying enzymes, HOTAIR can specify the pattern of histone modifications on target genes (Figure 3E). AS lncRNA-mediated PRC2 repression of genes can also happen in cis. For example, the nascent lncRNA ANRIL—antisense to the INK4 locus—silences the CDKN2B–CDKN2A locus by recruiting PRC2, which induces H3K27 methylation and long-term promoter DNA methylation in the locus [136,137]. Interestingly, HOTAIR, ANRIL, and their targets (e.g., HOXD9, HOXD10, and CDKN2) are included in the ~2000 genes that are differentially expressed across aging in all human tissues [138]. Importantly, these lncRNAs are also associated with age-related neurodegenerative disorders, as HOTAIR is overexpressed in a mouse model of PD [139] and single-nucleotide polymorphisms in the CDKN2B–CDKN2A locus have been associated with AD pathology [140]. Furthermore, ANRIL regulates the expression of CDKN2B that accumulates in neurofibrillary tangles and amyloid plaques in the brains of AD patients [141].
4.2. Transposonable Elements as a Source of lncRNAs in Aging
Transposonable elements (TEs) are repetitive DNA elements that account for nearly 50% of the human genome [142]. TEs are present at transcriptional start sites of a significant number of lncRNAs and control their transcriptional regulation, functioning as promoters [143]. In certain families of TEs, such as the HERVH family, the regulation of lncRNA expression evolved in a tissue- and developmental phase-dependent way, highlighting the functional sophistication of lncRNAs [143]. For instance, HERVH-driven lncRNAs are required for pluripotency in human embryonic stem cells [143]. TEs are also a major source of genomic instability, thus eukaryotic genomes have evolved epigenetic “defense” mechanisms that keep the majority of these elements under strong repression, silencing their expression and preventing mobility [144,145]. Interestingly, mounting evidence from studies in the senescence and aging of multiple organisms suggests that alterations in the activity of TEs-associated repressors [146,147], and the loss of heterochromatin [148,149], underlie TE activation in aging [150,151]. As a result, de-repressed TEs harbor the potential to modify lncRNA expression levels. Age-dependent de-repression of TEs may also induce de novo lncRNA transcription. This was exemplified in a study in which it was found that expression of de novo lncRNAs from Alu TEs during adult human stem cell aging promotes senescence, whereas knockdown of Alu lncRNAs reverses senescence [152].
4.3. Chromatin Remodeling and Nuclear Architecture in Aging: the (Big) Impact of lncRNAs
Higher level chromatin organization within the nuclear space is termed nuclear architecture [153]. Organization of the nuclear architecture influences DNA stability and gene expression patterns [147] and, thereby, defines cell identity [154]. Changes in the nuclear architecture are a hallmark of aging and result in genomic instability and transcriptional deregulation. In fact, premature aging syndromes, such as the Hutchinson–Gilford progeria syndrome, the Werner syndrome, and ataxia telangiectasia, share dramatic disturbances in the nuclear architecture of cells, and defects in diverse sets of genes that are involved in their maintenance [153]. lncRNAs play crucial roles in organizing the nuclear architecture of neurons by three different, yet complementary, processes: (1) lncRNAs function as epigenetic modulators of chromatin states [12]; (2) SCS lncRNAs change the nuclear architecture and alter chromatin repositioning [155]; and (3) the act of lncRNA transcription defines/affects the nuclear architecture [156]. Each of these processes are discussed in detail below:
- (1)
- As previously mentioned, many lncRNAs bind to chromatin-modifying proteins and recruit their catalytic activity in cis or trans to specific gene loci, thereby modulating chromatin states and impacting gene expression [12]. Modulation of chromatin states occurs in several loci simultaneously and likely contributes to the overall nuclear architecture of the neuronal genome. Therefore, disturbances in the chromatin state of a single locus are probably sufficient to trigger genome-wide chromatin readjustments, not only because of the constrained nature of the human genome, but also because of its transcriptional output. This process is particularly relevant in loci that coordinate complex transcriptional programs. For instance, the INK4 and HOX loci coordinate the expression of genes involved in cell cycle regulation and developmental patterning, respectively. These loci also contain the lncRNAs ANRIL and HOTAIR, respectively, which have altered expression in human tissues during aging [138]. While ANRIL and HOTAIR may function in cis for loci regulation, and are likely to play roles in post-mitotic neuronal processes, their dysregulation may also trigger the aberrant re-activation of cell cycle and developmental transcriptional programs—changes that are typically found in neuronal aging [157,158].
- (2)
- lncRNAs influence the nuclear architecture directly by organizing the dynamic assembly and disassembly of subnuclear compartments in the periphery of active chromatin regions [155], and by altering chromatin repositioning. Since knockdown of MALAT1 causes differential expression of several genes that localize away from the MALAT1 locus [79], and because MALAT1-associated epigenetic-regulation of genes was found to happen exclusively in cis [159], it is tempting to speculate that the nuclear architecture is reorganized as a direct consequence of speckle assembly. Therefore, the assembly of subnuclear compartments might constrain chromatin into new locations in the 3D space, thereby affecting gene expression. Accordingly, knockdown of NEAT1 impairs paraspeckles’ assembly [103] and also affects gene expression [80]. In fact, insights from studies on another lncRNA, FIRRE, show that it forms punctate compartments in the nucleus that include not only its own locus, but also specific loci from several other chromosomes [160]. This finding raises the question of whether SCS lncRNAs have the ability to co-localize to specific genomic regions in close proximity with its subnuclear compartments. A recent study also showed that SCS lncRNAs can interact with molecules that are present in the promoters of genes and remodel their chromatin by repositioning the loci into actively-transcribed or repressed foci [161]. Another question is whether age-related disturbances and the abundance of SCS lncRNAs impact the location and assembly of subnuclear compartments, consequently dictating broader rearrangements in 3D chromatin organization and altered gene expression patterns.
- (3)
- An emerging view is that the act of lncRNA transcription possibly defines or affects the nuclear architecture [156]. This model suggests that the transcription of lncRNAs serves as a guide-post for shaping 3D genome organization and that, for this same reason, lncRNAs have low abundance and are tissue-specific [156]. It is plausible that qualitative and quantitative changes in lncRNA expression with aging can have major effects in the broad nuclear architecture of the cell and thus contribute to the loss of cellular identity.
It follows that long-term maintenance of the nuclear architecture by lncRNAs is vital for neuronal functioning, and that lncRNA-associated age-related disturbances have a broad impact on 3D nuclear architecture and may lead to neuronal dysfunction.
5. Perspective
In summary, we have discussed multiple examples whereby lncRNA activity is involved in aspects of neuronal function (Table 1), such as neurogenesis and synaptic function, and may allow for continuous reshaping of the nuclear architecture. In the context of aging, we also discuss how lncRNAs affect this genetic landscape through their involvement in transcriptional alterations of protein-coding genes, lncRNAs, and epigenetic alterations. Hence, the coding/non-coding interactome that sustains important processes of cognition and adult neurogenesis may become compromised during neuronal aging. It is not yet known whether changes in the transcription of lncRNAs are reactive, compensatory, or causative of aging. However, rapidly accumulating evidence supports the vital contribution of lncRNAs in neuronal aging.
Acknowledgments
This work was supported by an European Research Council Starting Grant to Frank M. J. Jacobs.
Author Contributions
Conception and drafting of the work: D.P.F., G.B.; manuscript review: D.P.F., M.B., F.M.J.J., G.B.; conception and drafting of the figures: D.P.F.; figure review: D.P.F., M.B., F.M.J.J., G.B.; Supervision: F.M.J.J., G.B.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Morrison, J.H.; Baxter, M.G. The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nat. Rev. Neurosci. 2012, 13, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Capilla-Gonzalez, V.; Herranz-Pérez, V.; García-Verdugo, J.M. The aged brain: Genesis and fate of residual progenitor cells in the subventricular zone. Front. Cell. Neurosci. 2015, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, J.D. Epigenetics and cognitive aging. Science 2010, 328, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Mather, K.A.; Thalamuthu, A.; Baune, B.T.; Sachdev, P.S. Gene expression in the aging human brain: An overview. Curr. Opin. Psychiatry 2016, 29, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Kour, S.; Rath, P.C. Long noncoding RNAs in aging and age-related diseases. Ageing Res. Rev. 2016, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Grammatikakis, I.; Panda, A.C.; Abdelmohsen, K.; Gorospe, M. Long noncoding RNAs (lncRNAs) and the molecular hallmarks of aging. Aging 2014, 6, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Knauss, J.L.; Sun, T. Regulatory mechanisms of long noncoding RNAs in vertebrate central nervous system development and function. Neuroscience 2013, 235, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Barry, G. Integrating the roles of long and small non-coding RNA in brain function and disease. Mol. Psychiatry 2014, 19, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Wolvetang, E.J.; Mattick, J.S.; Rinn, J.L.; Barry, G. Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron 2015, 88, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Barry, G.; Guennewig, B.; Fung, S.; Kaczorowski, D.; Weickert, C.S. Long non-coding RNA expression during aging in the human subependymal zone. Front. Neurol. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kour, S.; Rath, P.C. Differential Expression of Long Noncoding RNA in the Rat Brain during Aging; Springer: Berlin, Germany, 2017; ISBN 978-981-10-2154-1. [Google Scholar]
- Wan, P.; Su, W.; Zhuo, Y. The role of long noncoding RNAs in neurodegenerative diseases. Mol. Neurobiol. 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.L.; Song, H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Frisén, J. Adult neurogenesis in humans- common and unique traits in mammals. PLoS Biol. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Maslov, A.Y.; Barone, T.A.; Plunkett, R.J.; Pruitt, S.C. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J. Neurosci. 2004, 24, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Encinas, J.M.; Michurina, T.V.; Peunova, N.; Park, J.H.; Tordo, J.; Peterson, D.A.; Fishell, G.; Koulakov, A.; Enikolopov, G. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell 2011, 8, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.G.; Dickinson-Anson, H.; Gage, F.H. Neurogenesis in the dentate gyrus of the adult rat: Age-related decrease of neuronal progenitor proliferation. J. Neurosci. 1996, 16, 2027–2033. [Google Scholar] [CrossRef] [PubMed]
- Capilla-Gonzalez, V.; Cebrian-Silla, A.; Guerrero-Cazares, H.; Garcia-Verdugo, J.M.; Quiñones-Hinojosa, A. Age-related changes in astrocytic and ependymal cells of the subventricular zone. Glia 2014, 62, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Drapeau, E.; Mayo, W.; Aurousseau, C.; Le Moal, M.; Piazza, P.-V.; Abrous, D.N. Spatial memory performances of aged rats in the water maze predict levels of hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 14385–14390. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.; et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227, S1–S11. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.D.; Diaz, A.; Nellore, A.; Delgado, R.N.; Park, K.Y.; Gonzales-Roybal, G.; Oldham, M.C.; Song, J.S.; Lim, D.A. Integration of genome-wide approaches identifies lncRNAs of adult neural stem cells and their progeny in vivo. Cell Stem Cell 2013, 12, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.D.; Andersen, R.E.; Liu, S.J.; Nowakowski, T.J.; Hong, S.J.; Gertz, C.C.; Salinas, R.D.; Zarabi, H.; Kriegstein, A.R.; Lim, D.A. The long noncoding RNA Pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell 2015, 16, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Goff, L.A.; Groff, A.F.; Sauvageau, M.; Trayes-Gibson, Z.; Sanchez-Gomez, D.B.; Morse, M.; Martin, R.D.; Elcavage, L.E.; Liapis, S.C.; Gonzalez-Celeiro, M.; et al. Spatiotemporal expression and transcriptional perturbations by long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2015, 112, 6855–6862. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Sun, S.; Lu, M.; Xia, Y. Regulation of neuronal-glial fate specification by long non-coding RNAs. Rev. Neurosci. 2016, 27. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Shen, Z.; Chakraborty, A.; Giri, S.; Freier, S.M.; Wu, X.; Zhang, Y.; Gorospe, M.; Prasanth, S.G.; Lal, A.; et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B.-MYB. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Panda, A.; Kang, M.J.; Xu, J.; Selimyan, R.; Yoon, J.H.; Martindale, J.L.; De, S.; Wood, W.H.; Becker, K.G.; et al. Senescence-associated lncRNAs: Senescence-associated long noncoding RNAs. Aging Cell 2013, 12, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Chen, K.; Cuevas-Diaz Duran, R.; You, Y.; Sloan, S.A.; Zhang, Y.; Zong, S.; Cao, Q.; Barres, B.A.; Wu, J.Q. Comprehensive identification of long non-coding RNAs in purified cell types from the brain reveals functional lncRNA in OPC fate determination. PLoS Genet. 2015, 11, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Bogu, G.K.; Soh, B.; Stanton, L.W. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol. Cell 2013, 51, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Capilla-Gonzalez, V.; Cebrian-Silla, A.; Guerrero-Cazares, H.; Garcia-Verdugo, J.M.; Quiñones-Hinojosa, A. The generation of oligodendroglial cells is preserved in the rostral migratory stream during aging. Front. Cell. Neurosci. 2013, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, G.L.; Lim, D.A.; Alvarez-Buylla, A.; Chao, M.V. Telomerase activity in the subventricular zone of adult mice. Mol. Cell. Neurosci. 2003, 23, 693–702. [Google Scholar] [CrossRef]
- Ferrón, S.; Mira, H.; Franco, S.; Cano-Jaimez, M.; Bellmunt, E.; Ramírez, C.; Fariñas, I.; Blasco, M.A. Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development 2004, 131, 4059–4070. [Google Scholar] [CrossRef] [PubMed]
- Flores, I.; Cayuela, M.L.; Blasco, M.A. Effects of telomerase and telomere length on epidermal stem cell behavior. Science 2005, 309, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; DePinho, R.A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 2007, 8, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Ferrón, S.R.; Marqués-Torrejón, M.A.; Mira, H.; Flores, I.; Taylor, K.; Blasco, M.A.; Fariñas, I. Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J. Neurosci. 2009, 29, 14394–14407. [Google Scholar] [CrossRef] [PubMed]
- Kathleen, C. Investigations of the constitutive overexpression of CYP6D1 in the permethrin resistantLPR strain of house fly (Musca domestica). Psychiatry Interpers. Biol. Process. 2009, 162, 214–220. [Google Scholar] [CrossRef]
- Samper, E.; Flores, J.M.; Blasco, M.A. Restoration of telomerase activity rescues chromosomal instability and premature aging in Terc-/- mice with short telomeres. EMBO Rep. 2001, 2, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.J.; Hemann, M.T.; Hathcock, K.S.; Tessarollo, L.; Feigenbaum, L.; Hahn, W.C.; Hodes, R.J. Expression of telomerase RNA template, but not telomerase reverse transcriptase, is limiting for telomere length maintenance in vivo. Mol. Cell. Biol. 2004, 24, 7024–7031. [Google Scholar] [CrossRef] [PubMed]
- Klapper, W.; Shin, T.; Mattson, M.P. Differential regulation of telomerase activity and TERT expression during brain development in mice. J. Neurosci. Res. 2001, 64, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Cusanelli, E.; Chartrand, P. Telomeric repeat-containing RNA TERRA: A noncoding RNA connecting telomere biology to genome integrity. Front. Genet. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cusanelli, E.; Romero, C.; Chartrand, P. Telomeric noncoding RNA TERRA is induced by telomere shortening to nucleate telomerase molecules at short telomeres. Mol. Cell 2013, 51, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Deng, Z.; Dahmane, N.; Tsai, K.; Wang, P.; Williams, D.R.; Kossenkov, A.V.; Showe, L.C.; Zhang, R.; Huang, Q.; et al. Telomeric repeat-containing RNA (TERRA) constitutes a nucleoprotein component of extracellular inflammatory exosomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6293–E6300. [Google Scholar] [CrossRef] [PubMed]
- Balk, B.; Maicher, A.; Dees, M.; Klermund, J.; Luke-Glaser, S.; Bender, K.; Luke, B. Telomeric RNA–DNA hybrids affect telomere-length dynamics and senescence. Nat. Struct. Mol. Biol. 2013, 20, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef] [PubMed]
- Redon, S.; Reichenbach, P.; Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res. 2010, 38, 5797–5806. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Feuerhahn, S.; Reichenbach, P.; Lingner, J. Molecular dissection of telomeric repeat-containing RNA biogenesis unveils the presence of distinct and multiple regulatory pathways. Mol. Cell. Biol. 2010, 30, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, L.; Lu, S. Role of TERRA in the regulation of telomere length. Int. J. Biol. Sci. 2015, 11, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Norseen, J.; Wiedmer, A.; Riethman, H.; Lieberman, P.M. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol. Cell 2009, 35, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Feuerhahn, S.; Lingner, J. TERRA-Reinforced Association of LSD1 with MRE11 promotes processing of uncapped telomeres. Cell Rep. 2014, 6, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Centore, R.C.; Sullivan, R.J.O.; Rai, R.; Tse, A.; Songyang, Z.; Chang, S.; Karlseder, J.; Zou, L. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 2011, 471, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.J.; López de Silanes, I.; Graña, O.B.M.A. Telomeric RNAs are essential to maintain telomeres. Nat. Commun. 2016, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.F.; Ogawa, Y.; Ahn, J.Y.; Namekawa, S.H.; Silva, S.S.; Lee, J.T. Telomeric RNAs mark sex chromosomes in stem cells. Genetics 2009, 182, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, U.; Lei, S. Role of lncRNAs in cellular aging. Front. Endocrinol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, E.A.; Raimundo, N.; Shadel, G.S. Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 2013, 17, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, N.; Van Beneden, A.; Decottignies, A. Telomere length regulates TERRA levels through increased trimethylation of telomeric H3K9 and HP1α. Nat. Struct. Mol. Biol. 2012, 19, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, Z.; Xiang, C.; Molczan, A.; Baubet, V.; Conejo-Garcia, J.; Xu, X.; Lieberman, P.M.; Dahmane, N. Formation of telomeric repeat-containing RNA (TERRA) foci in highly proliferating mouse cerebellar neuronal progenitors and medulloblastoma. J. Cell Sci. 2012, 125, 4383–4394. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Tejera, A.; Schoeftner, S.; Ortega, S.; Serrano, M.; Blasco, M.A. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 2009, 4, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Yehezkel, S.; Rebibo-Sabbah, A.; Segev, Y.; Tzukerman, M.; Shaked, R.; Huber, I.; Gepstein, L.; Skorecki, K.; Selig, S. Reprogramming of telomeric regions during the generation of human induced pluripotent stem cells and subsequent differentiation into fibroblast-like derivatives. Epigenetics 2011, 6, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Yankner, B.A.; Lu, T.; Loerch, P. The aging brain. Annu. Rev. Pathol. 2008, 3, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Poo, M.M.; Pignatelli, M.; Ryan, T.J.; Tonegawa, S.; Bonhoeffer, T.; Martin, K.C.; Rudenko, A.; Tsai, L.H.; Tsien, R.W.; Fishell, G.; et al. What is memory? The present state of the engram. BMC Biol. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Amaya, V. Molecular mechanisms of synaptic plasticity underlying long-term memory formation. In Neural Plasticity and Memory: From Genes to Brain Imaging; Bermúdez-Rattoni, F., Ed.; RC Press/Taylor & Francis: Boca Raton, FL, USA, 2007; pp. 20–22. [Google Scholar]
- Bramham, C.R.; Wells, D.G. Dendritic mRNA: Transport, translation and function. Nat. Rev. Neurosci. 2007, 8, 776–789. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R. The RNA-centred view of the synapse: Non-coding RNAs and synaptic plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Puthanveettil, S.V.; Antonov, I.; Kalachikov, S.; Rajasethupathy, P.; Choi, Y.-B.; Kohn, A.B.; Citarella, M.; Yu, F.; Karl, K.A.; Kinet, M.; et al. A strategy to capture and characterize the synaptic transcriptome. Proc. Natl. Acad. Sci. USA 2013, 110, 7464–7469. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Sunkin, S.M.; Mehler, M.F.; Mattick, J.S. Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Earls, L.R.; Westmoreland, J.J.; Zakharenko, S.S. Non-coding RNA regulation of synaptic plasticity and memory: Implications for aging. Ageing Res. Rev. 2014, 17, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Lipovich, L.; Dachet, F.; Cai, J.; Bagla, S.; Balan, K.; Jia, H.; Loeb, J.A. Activity-dependent human brain coding/noncoding gene regulatory networks. Genetics 2012, 192, 1133–1148. [Google Scholar] [CrossRef] [PubMed]
- Barry, G.; Briggs, J.A.; Vanichkina, D.P.; Poth, E.M.; Beveridge, N.J.; Ratnu, V.S.; Nayler, S.P.; Nones, K.; Hu, J.; Bredy, T.W.; et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol. Psychiatry 2014, 19, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Maag, J.L.V.; Panja, D.; Sporild, I.; Patil, S.; Kaczorowski, D.C.; Bramham, C.R.; Dinger, M.E.; Wibrand, K. Dynamic expression of long noncoding RNAs and repeat elements in synaptic plasticity. Front. Neurosci. 2015, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Iacoangeli, A.; Popp, S.; Muslimov, I.A.; Imataka, H.; Sonenberg, N.; Lomakin, I.B.; Tiedge, H. Dendritic BC1 RNA: Functional role in regulation of translation initiation. J. Neurosci. 2002, 22, 10232–10241. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Pestova, T.V.; Hellen, C.U.T.; Tiedge, H. Translational control by a small RNA: Dendritic BC1 RNA targets the eukaryotic initiation factor 4A helicase mechanism. Mol. Cell. Biol. 2008, 28, 3008–3019. [Google Scholar] [CrossRef] [PubMed]
- Kondrashov, A.V.; Kiefmann, M.; Ebnet, K.; Khanam, T.; Muddashetty, R.S.; Brosius, J. Inhibitory effect of naked neural BC1 RNA or BC200 RNA on eukaryotic in vitro translation systems is reversed by poly(A)-binding protein (PABP). J. Mol. Biol. 2005, 353, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Modarresi, F.; Faghihi, M.A.; Lopez-Toledano, M.A.; Fatemi, R.P.; Magistri, M.; Brothers, S.P.; van der Brug, M.P.; Wahlestedt, C. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 2012, 30, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Modarresi, F.; Faghihi, M.A.; Lopez-Toledano, M.A.; Fatemi, R.P.; Magistri, M.; Brothers, S.P.; Van Der Brug, M.P.; Wahlestedt, C. Natural antisense inhibition results in transcriptional de-repression and gene upregulation. Nat. Biotechnol. 2012, 30, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Prasanth, K.V.; Tripathi, V.; Colasse, S.; Nakamura, T.; Xuan, Z.; Zhang, M.Q.; Sedel, F.; Jourdren, L.; Coulpier, F.; et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010, 29, 3082–3093. [Google Scholar] [CrossRef] [PubMed]
- Barry, G.; Briggs, J.A.; Hwang, D.W.; Nayler, S.P.; Fortuna, P.R.J.; Jonkhout, N.; Dachet, F.; Maag, J.L.V.; Mestdagh, P.; Singh, E.M.; et al. The long non-coding RNA NEAT1 is responsive to neuronal activity and is associated with hyperexcitability states. Sci. Rep. 2017, 7, 40127. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.C.; Widagdo, J.; Chau, Y.Q.; Zhu, T.; Wong, J.J.-L.; Cheung, A.; Anggono, V. The activity-induced long non-coding RNA Meg3 modulates AMPA receptor surface expression in primary cortical neurons. Front. Cell. Neurosci. 2017, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Muslimov, I.A.; Banker, G.; Brosius, J.; Tiedge, H. Activity-dependent regulation of dendritic BC1 RNA in hippocampal neurons in culture. J. Cell Biol. 1998, 141, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Muslimov, I.A.; Santi, E.; Homel, P.; Perini, S.; Higgins, D.; Tiedge, H. RNA transport in dendrites: A cis-acting targeting element is contained within neuronal BC1 RNA. J. Neurosci. 1997, 17, 4722–4733. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Chuang, S.-C.; Bianchi, R.; Zhao, W.; Lee, H.; Fenton, A.A.; Wong, R.K.S.; Tiedge, H. BC1 regulation of metabotropic glutamate receptor-mediated neuronal excitability. J. Neurosci. 2009, 29, 9977–9986. [Google Scholar] [CrossRef] [PubMed]
- Lewejohann, L.; Skryabin, B.V.; Sachser, N.; Prehn, C.; Heiduschka, P.; Thanos, S.; Jordan, U.; Dell’Omo, G.; Vyssotski, A.L.; Pleskacheva, M.G.; et al. Role of a neuronal small non-messenger RNA: Behavioural alterations in BC1 RNA-deleted mice. Behav. Brain Res. 2004, 154, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Mus, E.; Hof, P.R.; Tiedge, H. Dendritic BC200 RNA in aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 10679–10684. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Wahlestedt, C. Regulatory roles of natural antisense transcripts. Nat. Rev. Mol. Cell Biol. 2009, 10, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Tang, Z.; Zhang, H.; Atianjoh, F.E.; Zhao, J.-Y.; Liang, L.; Wang, W.; Guan, X.; Kao, S.-C.; Tiwari, V.; et al. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat. Neurosci. 2013, 16, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Kadakkuzha, B.M.; Liu, X.A.; Narvaez, M.; Kaye, A.; Akhmedov, K.; Puthanveettil, S.V. Asymmetric localization of natural antisense RNA of neuropeptide sensorin in Aplysia sensory neurons during aging and activity. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Raj, B.; Blencowe, B.J. Alternative splicing in the mammalian nervous system: recent insights into mechanisms and functional roles. Neuron 2015, 87, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Traunmuller, L.; Gomez, A.M.; Nguyen, T.-M.; Scheiffele, P. Control of neuronal synapse specification by a highly dedicated alternative splicing program. Science 2016, 352, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Sone, M.; Hayashi, T.; Tarui, H.; Agata, K.; Takeichi, M.; Nakagawa, S. The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J. Cell Sci. 2007, 120, 2498–2506. [Google Scholar] [CrossRef] [PubMed]
- Tsuiji, H.; Yoshimoto, R.; Hasegawa, Y.; Furuno, M.; Yoshida, M.; Nakagawa, S. Competition between a noncoding exon and introns: Gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes Cells 2011, 16, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Takagi, A.; Takaki, M.; Graziane, N.; Seshadri, S.; Murdoch, H.; Dunlop, A.J.; Makino, Y.; Seshadri, A.J.; Ishizuka, K.; Srivastava, D.P.; et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 2010, 13, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Unda, B.K.; Kwan, V.; Singh, K.K. Neuregulin-1 regulates cortical inhibitory neuron dendrite and synapse growth through DISC1. Neural Plast. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Graziane, N.M.; Gu, Z.; Yan, Z. DISC1 Regulates GABAA Receptor Trafficking and Inhibitory Synaptic Transmission in Cortical Neurons. J. Biol. Chem. 2015, 290, jbc.M115.656173. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Thevathasan, J.V.; Lin, Q.; Lim, K.B.; Kuroda, K.; Kaibuchi, K.; Bilger, M.; Soong, T.W.; Fivaz, M. Stimulation of synaptic vesicle exocytosis by the mental disease gene DISC1 is mediated by N-type voltage-gated calcium channels. Front. Synaptic Neurosci. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Krivosheya, D.; Tapia, L.; Levinson, J.N.; Huang, K.; Kang, Y.; Hines, R.; Ting, A.K.; Craig, A.M.; Mei, L.; Bamji, S.X.; et al. ErbB4-neuregulin signaling modulates synapse development and dendritic arborization through distinct mechanisms. J. Biol. Chem. 2008, 283, 32944–32956. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Woo, R.S.; Mei, L.; Malinow, R. The Neuregulin-1 Receptor ErbB4 controls glutamatergic synapse maturation and plasticity. Neuron 2007, 54, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Cazorla, M.; Shegda, M.; Ramesh, B.; Harrison, N.L.; Kellendonk, C. Striatal D2 receptors regulate dendritic morphology of medium spiny neurons via Kir2 channels. J. Neurosci. 2012, 32, 2398–2409. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.F.; Christensen, C.H.; Hazelwood, L.A.; Dobi, A.; Bock, R.; Sibley, D.R.; Mateo, Y.; Alvarez, V.A. Dopamine D2 Receptor Overexpression Alters Behavior and Physiology in Drd2-EGFP Mice. J. Neurosci. 2011, 31, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Stühmer, W.; Ruppersberg, J.P.; Schröter, K.H.; Sakmann, B.; Stocker, M.; Giese, K.P.; Perschke, A.; Baumann, A.; Pongs, O. Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J. 1989, 8, 3235–3244. [Google Scholar] [PubMed]
- Voglis, G.; Tavernarakis, N. The role of synaptic ion channels in synaptic plasticity. EMBO Rep. 2006, 7, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Disterhoft, J.F.; Oh, M.M. Alterations in intrinsic neuronal excitability during normal aging. Aging Cell 2007, 6, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Trojanowski, J.Q.; Schmidt, M.L.; Otvos, L., Jr.; Arai, H.; Hill, W.D.; Lee, V.M. Vulnerability of the neuronal cytoskeleton in aging and Alzheimer disease: Widespread involvement of all three major filament systems. Annu. Rev. Gerontol. Geriatr. 1990, 10, 167–182. [Google Scholar] [PubMed]
- Niewiadomska, G.; Baksalerska-Pazera, M.; Riedel, G. Cytoskeletal transport in the aging brain: Focus on the cholinergic system. Rev. Neurosci. 2006, 17, 581–618. [Google Scholar] [CrossRef] [PubMed]
- Milde, S.; Adalbert, R.; Elaman, M.H.; Coleman, M.P. Axonal transport declines with age in two distinct phases separated by a period of relative stability. Neurobiol. Aging 2015, 36, 971–981. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.A.; Raices, M.; Panowski, S.H.; Hetzer, M.W. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 2009, 136, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.K.; DeLorenzo, R.J. Calcium and neuronal cytoskeletal proteins: Alterations with aging. Neurobiol. Aging 1987, 8, 359–361. [Google Scholar] [CrossRef]
- Simkin, D.; Hattori, S.; Ybarra, N.; Musial, T.F.; Buss, E.W.; Richter, H.; Oh, M.M.; Nicholson, D.A.; Disterhoft, J.F. Aging-related hyperexcitability in CA3 pyramidal neurons is mediated by enhanced A-type K+ channel function and expression. J. Neurosci. 2015, 35, 13206–13218. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.H.; Craig, T.; Li, Y.; Merry, B.; De Magalhães, J.P. Whole transcriptome sequencing of the aging rat brain reveals dynamic RNA changes in the dark matter of the genome. Age 2013, 35, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.M.; Rizzu, P.; Francescatto, M.; Vitezic, M.; Leday, G.G.R.; Sanchez, J.S.; Khamis, A.; Takahashi, H.; van de Berg, W.D.J.; Medvedeva, Y.A.; et al. Regional differences in gene expression and promoter usage in aged human brains. Neurobiol. Aging 2013, 34, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.H.; Tsien, J.Z.; Schultz, P.G.; Hu, Y. The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc. Natl. Acad. Sci. USA 2001, 98, 1930–1934. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Ueberham, U.; Mills, J.D.; Kirazov, L.; Kirazov, E.; Knobloch, M.; Bochmann, J.; Jendrek, R.; Takenaka, K.; Bliim, N.; et al. RNA sequencing reveals pronounced changes in the noncoding transcriptome of aging synaptosomes. Neurobiol. Aging 2017, 56, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S. Antisense Transcription in the Mammalian Transcriptome. Science 2005, 309, 1564–1566. [Google Scholar] [CrossRef] [PubMed]
- Pelechano, V.; Steinmetz, L.M. Gene regulation by antisense transcription. Nat. Rev. Genet. 2013, 14, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; Van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G.; Wahlestedt, C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010, 11, R56. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G.; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Ciarlo, E.; Massone, S.; Penna, I.; Nizzari, M.; Gigoni, A.; Dieci, G.; Russo, C.; Florio, T.; Cancedda, R.; Pagano, A. An intronic ncRNA-dependent regulation of SORL1 expression affecting Aβ formation is upregulated in post-mortem Alzheimer’s disease brain samples. Dis. Model. Mech. 2013, 6, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cai, F.; Zhang, S.; Zhang, S.; Song, W. Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer’s progression in vivo. Sci. Rep. 2015, 4, 7298. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Jangir, D.K.; Verma, G.; Shekhar, S.; Hanpude, P.; Kumar, S.; Kumari, R.; Singh, N.; Sarovar Bhavesh, N.; et al. S-nitrosylation of UCHL1 induces its structural instability and promotes α-synuclein aggregation. Sci. Rep. 2017, 7, 44558. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, C.; Forrest, A.R.R.; Santoro, C.; Persichetti, F.; Carninci, P.; Zucchelli, S.; Gustincich, S. Expression analysis of the long non-coding RNA antisense to Uchl1 (AS Uchl1) during dopaminergic cells’ differentiation in vitro and in neurochemical models of Parkinson’s disease. Front. Cell. Neurosci. 2015, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, Y.; Faghihi, M.A.; Magistri, M.; Alvarez-Garcia, O.; Lotz, M.; Wahlestedt, C. Antisense RNA controls LRP1 sense transcript expression through interaction with a chromatin-associated protein, HMGB2. Cell Rep. 2015, 11, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Trotter, J.; Zhang, J.; Peters, M.M.; Cheng, H.; Bao, J.; Han, X.; Weeber, E.J.; Bu, G. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J. Neurosci. 2010, 30, 17068–17078. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.E.; Pietrzik, C.U.; Baum, L.; Chevallier, N.; Merriam, D.E.; Kounnas, M.Z.; Wagner, S.L.; Troncoso, J.C.; Kawas, C.H.; Katzman, R.; et al. Modulation of amyloid β-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J. Clin. Investig. 2000, 106, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Scheele, C.; Petrovic, N.; Faghihi, M.A.; Lassmann, T.; Fredriksson, K.; Rooyackers, O.; Wahlestedt, C.; Good, L.; Timmons, J.A. The human PINK1 locus is regulated in vivo by a non-coding natural antisense RNA during modulation of mitochondrial function. BMC Genom. 2007, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Gius, D.; Onyango, P.; Muldoon-Jacobs, K.; Karp, J.; Feinberg, A.P.; Cui, H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 2008, 451, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cui, B.; Dai, Z.X.; Shi, P.K.; Wang, Z.H.; Guo, Y.Y. Long Non-coding RNA HOTAIR Promotes Parkinson’s Disease Induced by MPTP Through up-regulating the Expression of LRRK2. Curr. Neurovasc. Res. 2016, 13, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; De Strooper, B. Noncoding RNAs in neurodegeneration. Nat. Rev. Neurosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Holzer, M.; Gärtner, U. Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J. Neural Transm. 1998, 105, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ayarpadikannan, S.; Kim, H. Role of transposable elements in genomic rearrangement, evolution, gene regulation and epigenetics in primates. Genes Genet. Syst. 2016, 90, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Rinn, J.L. Transposable elements reveal a stem cell specific class of long noncoding RNAs. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, F.M.J.; Greenberg, D.; Nguyen, N.; Haeussler, M.; Ewing, A.D.; Katzman, S.; Paten, B.; Salama, S.R.; Haussler, D. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 2014, 516, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Van Meter, M.; Kashyap, M.; Rezazadeh, S.; Geneva, A.J.; Morello, T.D.; Seluanov, A.; Gorbunova, V. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 2014, 5, 5011. [Google Scholar] [CrossRef] [PubMed]
- Oberdoerffer, P.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, Sa.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; et al. DNA damage-induced alterations in chromatin contribute to genomic integrity and age-related changes in gene expression. Cell 2008, 135, 907–918. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.A.; Neretti, N.; Sedivy, J.M. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 2013, 12, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.H.; Chen, C.C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014, 28, 396–408. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Criscione, S.W.; Peterson, A.L.; Neretti, N.; Sedivy, J.M.; Kreiling, J.A. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging 2013, 5, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Burhans, W.C.; Curcio, M.J. Retrotransposition is associated with genome instability during chronological aging. Proc. Natl. Acad. Sci. USA 2011, 108, 20376–20381. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Geesman, G.J.; Hostikka, S.L.; Atallah, M.; Blackwell, B.; Lee, E.; Cook, P.J.; Pasaniuc, B.; Shariat, G.; Halperin, E.; et al. Inhibition of activated pericentromeric SINE/Alu repeat transcription in senescent human adult stem cells reinstates self-renewal. Cell Cycle 2011, 10, 3016–3030. [Google Scholar] [CrossRef] [PubMed]
- Oberdoerffer, P.; Sinclair, D.A. The role of nuclear architecture in genomic instability and ageing. Nat. Rev. Mol. Cell Biol. 2007, 8, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Ollikainen, N.; Guttman, M. Long non-coding RNAs: Spatial amplifiers that control nuclear structure and gene expression. Nat. Rev. Mol. Cell Biol. 2016, 17, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Melé, M.; Rinn, J.L. “Cat’s cradling” the 3D genome by the act of lncRNA transcription. Mol. Cell 2016, 62, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K.; Yang, Y. Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat. Rev. Neurosci. 2007, 8, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Dönertaş, H.M.; İzgi, H.; Kamacıoğlu, A.; He, Z.; Khaitovich, P.; Somel, M. Gene expression reversal toward pre-adult levels in the aging human brain and age-related loss of cellular identity. Sci. Rep. 2017, 7, 5894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Arun, G.; Mao, Y.S.; Lazar, Z.; Hung, G.; Bhattacharjee, G.; Xiao, X.; Booth, C.J.; Wu, J.; Zhang, C.; et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012, 2, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Lin, C.; Liu, W.; Zhang, J.; Ohgi, K.A.; Grinstein, J.D.; Dorrestein, P.C.; Rosenfeld, M.G. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 2011, 147, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Dajas-Bailador, F.; Bonev, B.; Garcez, P.; Stanley, P.; Guillemot, F.; Papalopulu, N. microRNA-9 regulates axon extension and branching by targeting Map1b in mouse cortical neurons. Nat. Neurosci. 2012, 15, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, M.; Yang, H.; Mao, L.; He, Q.; Jin, H.; Ye, Z.M.; Luo, X.Y.; Xia, Y.P.; Hu, B. LncRNA TUG1 sponges microRNA-9 to promote neurons apoptosis by up-regulated Bcl2l11 under ischemia. Biochem. Biophys. Res. Commun. 2017, 485, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, R.A.; Liu, Q.; Kanekiyo, T.; Zhang, J.; Bu, G. Low density lipoprotein receptor-related protein 1 promotes anti-apoptotic signaling in neurons by activating Akt survival pathway. J. Biol. Chem. 2009, 284, 34045–34053. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Župunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Q.; Zhang, J.; Pan, W.; Zhao, J.; Xu, Y. Long non-coding RNA MALAT1 contributes to cell apoptosis by sponging miR-124 in Parkinson disease. Cell Biosci. 2017, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Stilling, R.M.; Benito, E.; Gertig, M.; Barth, J.; Capece, V.; Burkhardt, S.; Bonn, S.; Fischer, A. De-regulation of gene expression and alternative splicing affects distinct cellular pathways in the aging hippocampus. Front. Cell. Neurosci. 2014, 8, 373. [Google Scholar] [CrossRef] [PubMed]
- Huh, C.J.; Zhang, B.; Victor, M.B.; Dahiya, S.; Batista, L.F.Z.; Horvath, S.; Yoo, A.S. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Massone, S.; Vassallo, I.; Fiorino, G.; Castelnuovo, M.; Barbieri, F.; Borghi, R.; Tabaton, M.; Robello, M.; Gatta, E.; Russo, C.; et al. 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol. Dis. 2011, 41, 308–317. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Long non-coding RNAs (lncRNAs) orchestrate temporally and spatially precise gene regulatory networks involved in the course of adult neurogenesis. (A) lncRNAs influence neural stem cells’ (NSCs) proliferation, expansion of transit-amplifying cells and differentiation into neuroblasts. In aging, mild alterations in lncRNA expression in the subventricular zone (SVZ) may compromise these processes, thus accounting for a decline in neurogenesis. (B) lncRNAs participate in lineage commitment and cell maturation. In the aged SVZ, the balance between lncRNAs involved in glial and neuronal fate specification may determine the cell fate of NSCs, leading to alterations in long-term neuronal/glial turnover. (C) lncRNAs are critical for telomere homeostasis. It is likely that an interplay between the telomeric lncRNAs TERRA (telomeric repeat containing RNA) and TERC (telomerase RNA component) regulates telomerase activity and the survival of NSCs during aging.
Figure 1.
Long non-coding RNAs (lncRNAs) orchestrate temporally and spatially precise gene regulatory networks involved in the course of adult neurogenesis. (A) lncRNAs influence neural stem cells’ (NSCs) proliferation, expansion of transit-amplifying cells and differentiation into neuroblasts. In aging, mild alterations in lncRNA expression in the subventricular zone (SVZ) may compromise these processes, thus accounting for a decline in neurogenesis. (B) lncRNAs participate in lineage commitment and cell maturation. In the aged SVZ, the balance between lncRNAs involved in glial and neuronal fate specification may determine the cell fate of NSCs, leading to alterations in long-term neuronal/glial turnover. (C) lncRNAs are critical for telomere homeostasis. It is likely that an interplay between the telomeric lncRNAs TERRA (telomeric repeat containing RNA) and TERC (telomerase RNA component) regulates telomerase activity and the survival of NSCs during aging.
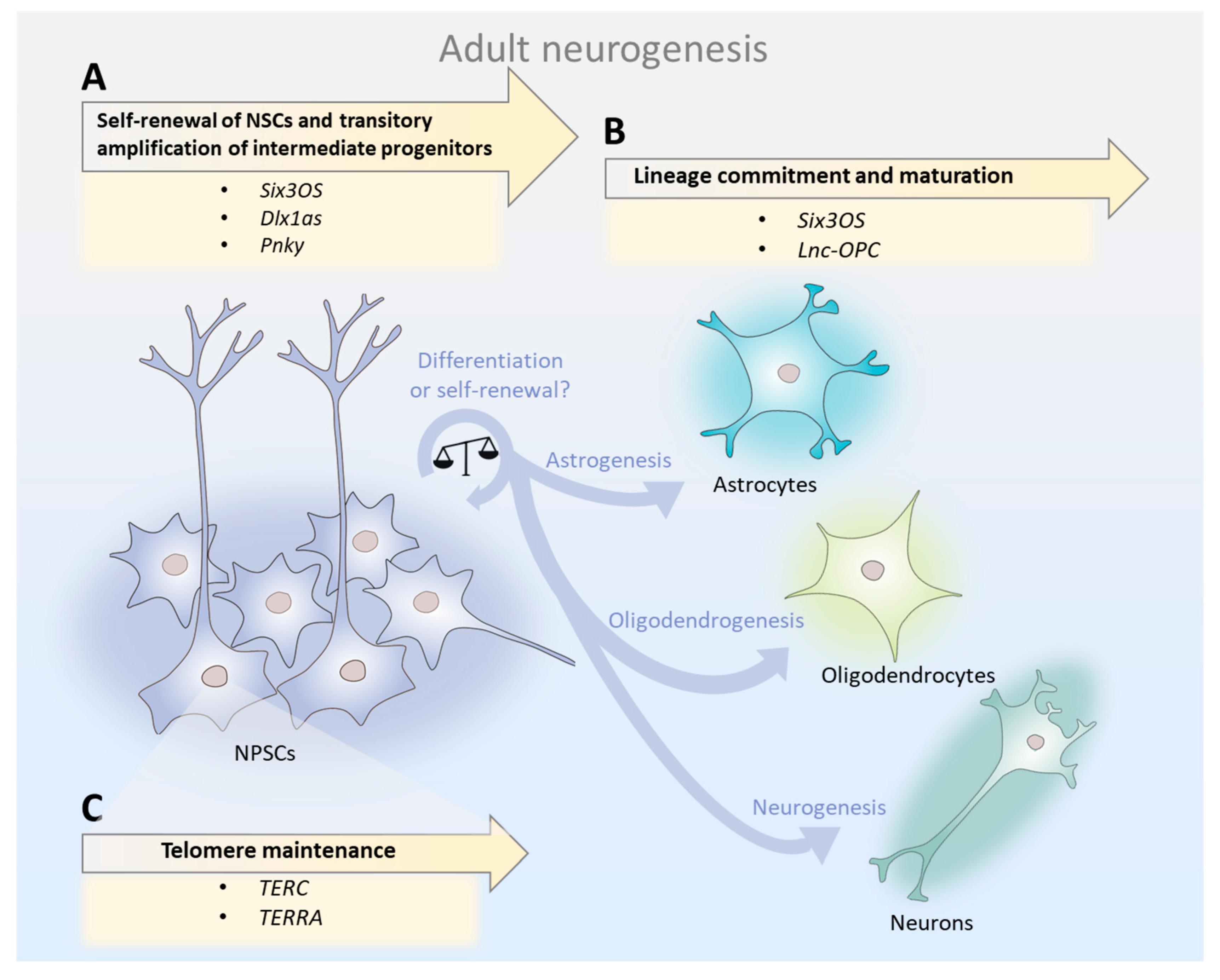
Figure 2.
Mechanisms underlying the lncRNA function in synaptic plasticity. lncRNAs respond to neuronal activity and not only regulate the expression of genes involved in neurite outgrowth, but also modulate ion channel stoichiometry, thereby altering the synaptic connectivity and excitatory properties of a neuron. (A) Upon neuronal activity, certain lncRNAs (e.g., BC200, Bc1) are transcribed and transported to dendrites, where they regulate local protein translation rates. (B) The stability of protein-coding transcripts involved in synaptic plasticity may be controlled by their antisense pairs (e.g., BDNF-AS, GDNF-AS, EPHB2-AS) in dendrites. (C) In response to glycine stimulation, some cytosolic lncRNAs (e.g., MEG3) regulate the trafficking of AMPA receptors to the plasma membrane, modifying the excitatory landscape of a neuron. (D) Nuclear-retained lncRNAs (e.g., GOMAFU) control the activity-dependent release of splicing factors to regulate gene expression and splice variant distributions that influence dendritic growth, morphology, and function. (E) Nuclear lncRNAs (e.g., NEAT1) also control the activity-dependent transcription and nucleus-to-cytosol shuttling of ion channel subunits, altering the excitatory properties of a neuron. FMRP, fragile X mental retardation protein.
Figure 2.
Mechanisms underlying the lncRNA function in synaptic plasticity. lncRNAs respond to neuronal activity and not only regulate the expression of genes involved in neurite outgrowth, but also modulate ion channel stoichiometry, thereby altering the synaptic connectivity and excitatory properties of a neuron. (A) Upon neuronal activity, certain lncRNAs (e.g., BC200, Bc1) are transcribed and transported to dendrites, where they regulate local protein translation rates. (B) The stability of protein-coding transcripts involved in synaptic plasticity may be controlled by their antisense pairs (e.g., BDNF-AS, GDNF-AS, EPHB2-AS) in dendrites. (C) In response to glycine stimulation, some cytosolic lncRNAs (e.g., MEG3) regulate the trafficking of AMPA receptors to the plasma membrane, modifying the excitatory landscape of a neuron. (D) Nuclear-retained lncRNAs (e.g., GOMAFU) control the activity-dependent release of splicing factors to regulate gene expression and splice variant distributions that influence dendritic growth, morphology, and function. (E) Nuclear lncRNAs (e.g., NEAT1) also control the activity-dependent transcription and nucleus-to-cytosol shuttling of ion channel subunits, altering the excitatory properties of a neuron. FMRP, fragile X mental retardation protein.

Figure 3.
lncRNA-associated mechanisms in the healthy brain and in age-related neurodegenerative disorders. A shift in lncRNA abundance triggers alterations in the pre-transcriptional (blue columns) or post-transcriptional (yellow columns) regulation of neuronal genes, increasing susceptibility to disease and cognitive decline. (A) Certain lncRNAs inhibit or promote the translation of specific target mRNAs. The lncRNA BC200 represses translation by recruiting translational machinery. (B) Natural sense/antisense transcripts hybridize and regulate the stability of coding (sense) mRNAs involved in synaptic plasticity and neurodegeneration. Hybridization of BACE1-AS with BACE1 mRNA inhibits BACE1 transcript miRNA-mediated decay, ultimately leading to overexpression of BACE1 in the brains of Alzheimer’s disease patients and accounting for amyloid-beta (Aβ) pathological aggregation. (C) In the cytosol, lncRNAs can also act as a sponge for miRNAs in order to sequester them and prevent them from binding to target transcripts. The lncRNA TUG1 traps miRNA-9, preventing it from binding to the 3’UTR of BCL2L11 mRNA, where it inhibits BCL2L11 translation. As a result, the pro-apoptotic factor BCL2L11 is overexpressed and induces apoptosis in ischemia. (D) In the nucleus, lncRNAs can also act as a sponge to ubiquitous chromatin-regulatory proteins, fine-tuning their activity. The lncRNA LRP1-AS binds to HMGB2, preventing it from enhancing LRP1 transcription in cis, and thereby reducing its expression. (E) Nuclear lncRNAs have affinity for chromatin-regulatory proteins, allowing for the assembly of complexes that base new histone modifications in cis or trans. The lncRNA HOTAIR silences the HOXD locus in trans by recruiting Polycomb proteins. Additionally, it can interact with multiple regulatory complexes simultaneously, having a genome-wide effect. (F) Subnuclear compartment specific (SCS) lncRNAs shape nuclear architecture by acting as epigenetic modulators of chromatin states in cis or trans, both by organizing the dynamic assembly and disassembly of subnuclear compartments in the periphery of active chromatin regions and by influencing the splicing or sponging of ion channels.
Figure 3.
lncRNA-associated mechanisms in the healthy brain and in age-related neurodegenerative disorders. A shift in lncRNA abundance triggers alterations in the pre-transcriptional (blue columns) or post-transcriptional (yellow columns) regulation of neuronal genes, increasing susceptibility to disease and cognitive decline. (A) Certain lncRNAs inhibit or promote the translation of specific target mRNAs. The lncRNA BC200 represses translation by recruiting translational machinery. (B) Natural sense/antisense transcripts hybridize and regulate the stability of coding (sense) mRNAs involved in synaptic plasticity and neurodegeneration. Hybridization of BACE1-AS with BACE1 mRNA inhibits BACE1 transcript miRNA-mediated decay, ultimately leading to overexpression of BACE1 in the brains of Alzheimer’s disease patients and accounting for amyloid-beta (Aβ) pathological aggregation. (C) In the cytosol, lncRNAs can also act as a sponge for miRNAs in order to sequester them and prevent them from binding to target transcripts. The lncRNA TUG1 traps miRNA-9, preventing it from binding to the 3’UTR of BCL2L11 mRNA, where it inhibits BCL2L11 translation. As a result, the pro-apoptotic factor BCL2L11 is overexpressed and induces apoptosis in ischemia. (D) In the nucleus, lncRNAs can also act as a sponge to ubiquitous chromatin-regulatory proteins, fine-tuning their activity. The lncRNA LRP1-AS binds to HMGB2, preventing it from enhancing LRP1 transcription in cis, and thereby reducing its expression. (E) Nuclear lncRNAs have affinity for chromatin-regulatory proteins, allowing for the assembly of complexes that base new histone modifications in cis or trans. The lncRNA HOTAIR silences the HOXD locus in trans by recruiting Polycomb proteins. Additionally, it can interact with multiple regulatory complexes simultaneously, having a genome-wide effect. (F) Subnuclear compartment specific (SCS) lncRNAs shape nuclear architecture by acting as epigenetic modulators of chromatin states in cis or trans, both by organizing the dynamic assembly and disassembly of subnuclear compartments in the periphery of active chromatin regions and by influencing the splicing or sponging of ion channels.
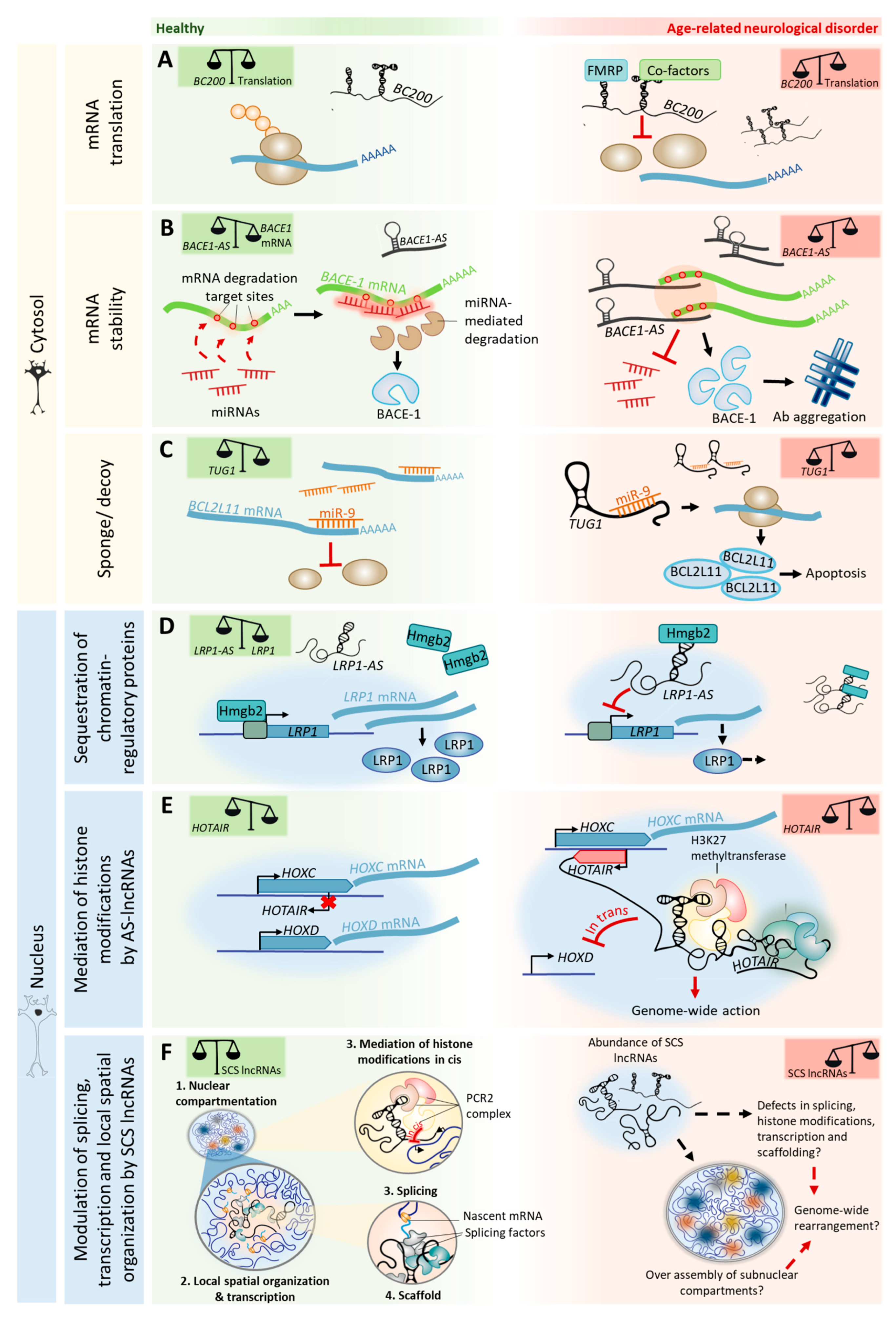

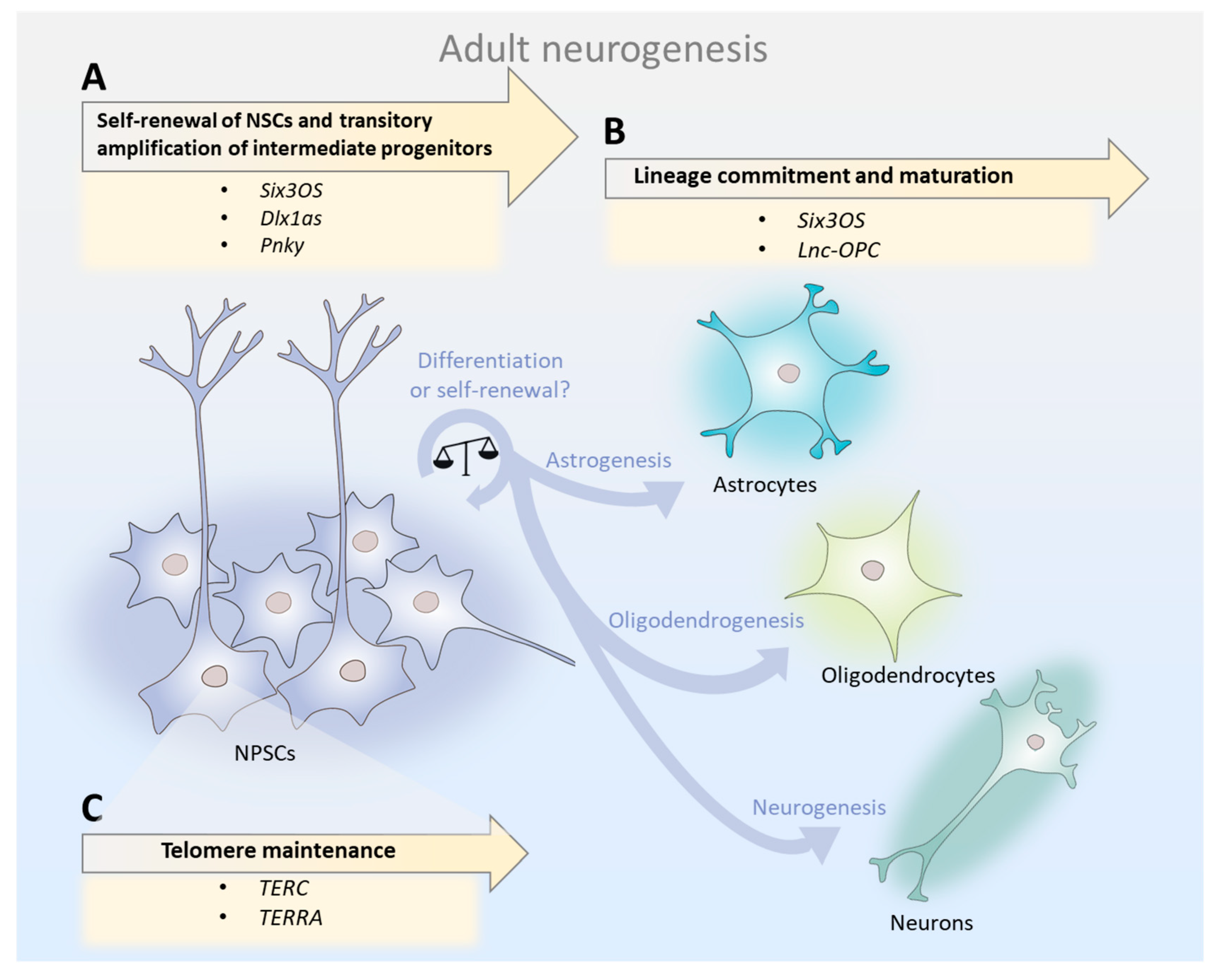{kind=link}
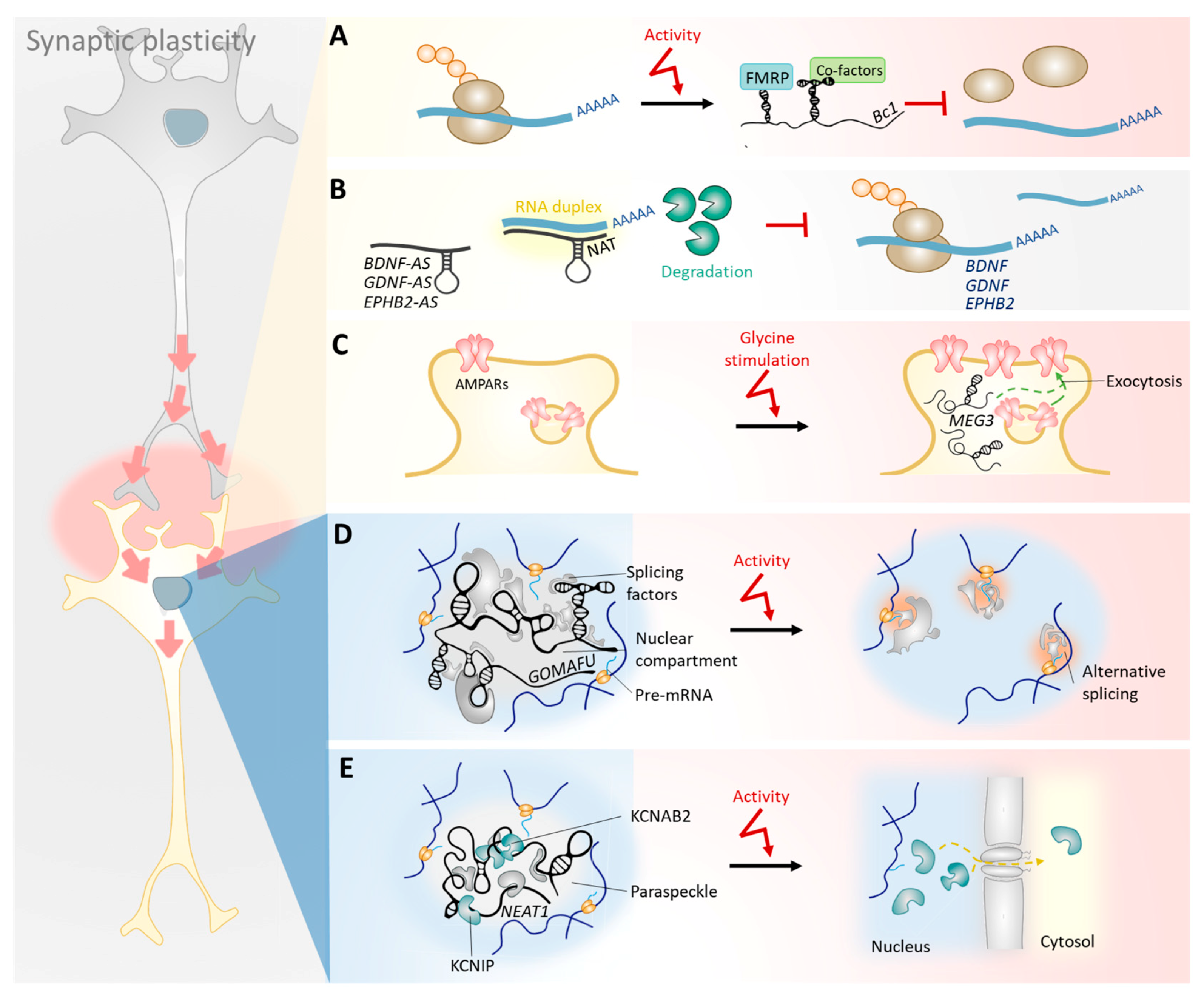{kind=link}
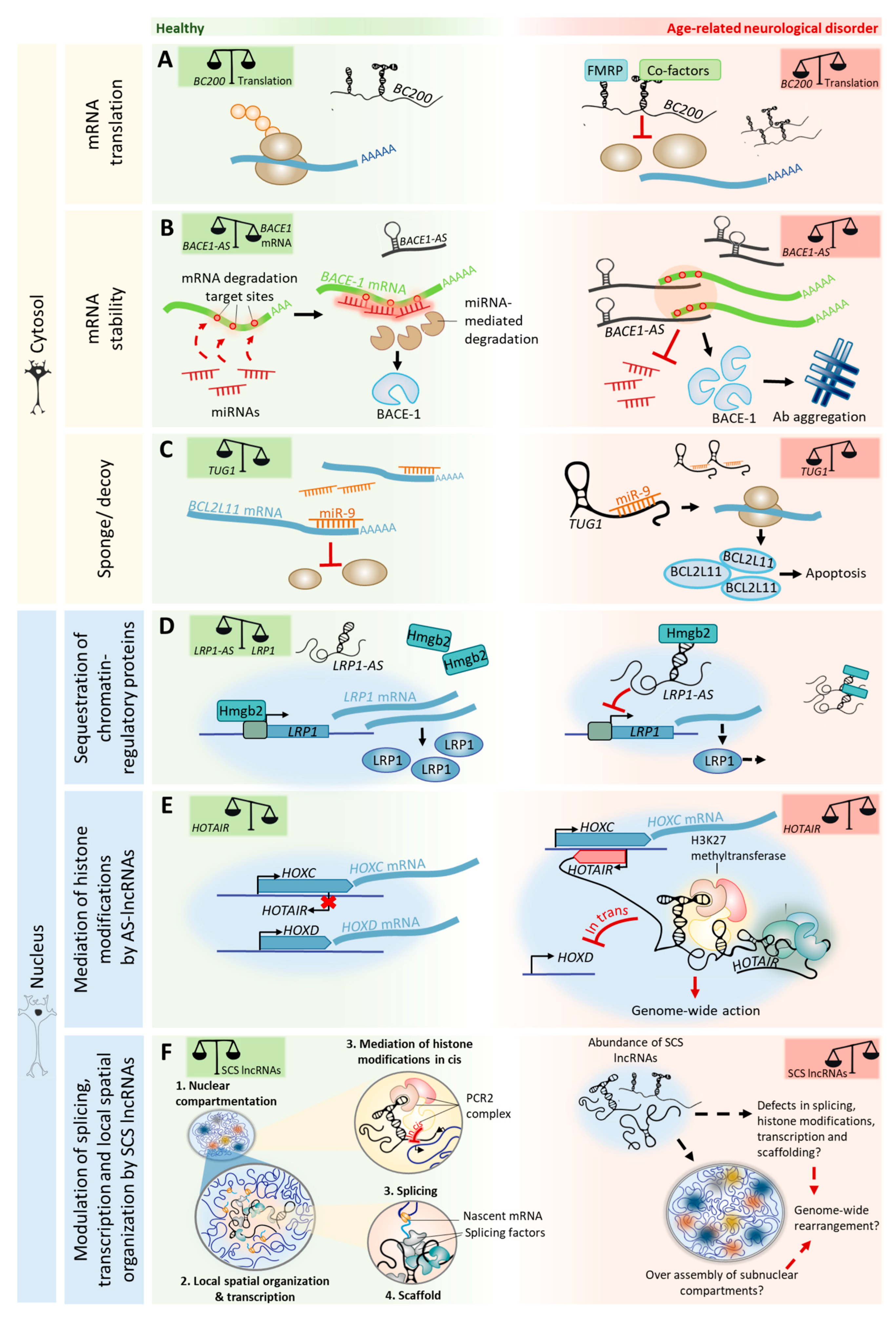{kind=link}
Table 1.
Mechanisms underlying lncRNAs neuronal functions and their affected expression levels in aging and age-related neurodegenerative disorders.
Table 1.
Mechanisms underlying lncRNAs neuronal functions and their affected expression levels in aging and age-related neurodegenerative disorders.
| Mechanisms Underlying lncRNA Activity | lncRNA | Implication in Aging/Age-Related Neurodegenerative Disorders | Affected Neuronal Process | ||
|---|---|---|---|---|---|
| Cytoplasm (post-transcriptional modulation of gene expression) | mRNA translation | Bc1, BC200 | Downregulated in aging; upregulated in Alzheimer’s disease (AD) [86] | Cognitive decline | |
| UCHL1-AS [124] | Downregulated in Parkinson’s disease (PD) [127] | Neurodegeneration | |||
| mRNA stability | BACE1-AS [121,122] | Upregulated in AD [122] | Protein aggregation in neurons; Possible role in cognitive decline | ||
| PINK1-AS [131] | Unknown | Neurodegeneration | |||
| GDNF-AS [77], EPHB2-AS [77], CNA2-AS | Unknown | Cognitive decline | |||
| Sponge/decoy | TUG1 [162] | Upregulated in human subventricular zone (SVZ) with aging [13]; upregulated in brain ischemia [163] | Adult neurogenesis decline; Possible role in cognitive decline and neurodegeneration | ||
| Nucleus (pre-transcriptional regulation of gene expression) | Transcription repression | by sequestration of chromatin-regulatory proteins | LRP1-AS [128] | Upregulated in AD [128] | Neurodegeneration [164]; Possible role in protein aggregation |
| by affecting histone modifications | BDNF-AS [77] | Unknown | Cognitive decline; neurodegeneration | ||
| HOTAIRM | Altered expression in all human tissues assayed in aging [138]; overexpressed in a mouse model of PD [139]. | Possible role in neurodegeneration [139] | |||
| ANRIL | Variants have been associated with AD [140]; altered expression in all human tissues assayed in aging [138]. | Neurodegeneration | |||
| Scaffold for proteins and RNAs in subnuclear compartments | NEAT1 | Dysregulated expression in a temporal lobe epilepsy mouse model [80]; upregulated in the human SVZ with age [13]; upregulated in Frontotemporal Dementia (FTLD) and Amyotrophic Lateral Sclerosis (ALS) [165]; upregulated in the hippocampus of old mice [166]. | Adult neurogenesis decline; cognitive decline | ||
| MALAT-1 | Upregulated in human SVZ with aging [13]; upregulated in FTLD-ALS [165]; upregulated in the hippocampus of old mice [167]; upregulated in PD mouse model [166]. | Adult neurogenesis decline; cognitive decline; neurodegeneration | |||
| GOMAFU | Upregulated in SVZ with aging [13]; downregulated in the grey matter of schizophrenia patients [72]; upregulated in the hippocampus of old mice [167]. | Cognitive decline; | |||
| Unclear mechanisms | Six3OS, Dlx1A-S, Lnc-OPC | Unknown | Adult neurogenesis | ||
| LNC00657 | Downregulated in the human SVZ with age | Adult neurogenesis | |||
| Meg3 | Upregulated in the hippocampus of old mice [167]; downregulated in old induced striatal medium spiny neurons [168]. | Cognitive decline | |||
| SORL1-AS [123] | Upregulated in AD | Protein aggregation; Possible role in cognitive decline | |||
| 17A [169] | Upregulated in AD | Cognitive decline | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pereira Fernandes, D.; Bitar, M.; Jacobs, F.M.J.; Barry, G. Long Non-Coding RNAs in Neuronal Aging. Non-Coding RNA 2018, 4, 12. https://doi.org/10.3390/ncrna4020012
AMA Style
Pereira Fernandes D, Bitar M, Jacobs FMJ, Barry G. Long Non-Coding RNAs in Neuronal Aging. Non-Coding RNA. 2018; 4(2):12. https://doi.org/10.3390/ncrna4020012
Chicago/Turabian StylePereira Fernandes, Diana, Mainá Bitar, Frank M. J. Jacobs, and Guy Barry. 2018. "Long Non-Coding RNAs in Neuronal Aging" Non-Coding RNA 4, no. 2: 12. https://doi.org/10.3390/ncrna4020012
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.