
DFT Study on the Interaction of the Smallest Fullerene C20 with Lithium Ions and Atoms

Division of Applied Chemistry, Graduate School of Engineering, Hokkaido University, Sapporo 060-8628, Japan
*
Author to whom correspondence should be addressed.
C 2017, 3(2), 15; https://doi.org/10.3390/c3020015
Submission received: 28 March 2017
/
Revised: 26 April 2017
/
Accepted: 28 April 2017
/
Published: 10 May 2017
(This article belongs to the Special Issue Chemical Bond Formation for Nanocarbon-Based Composites)
Abstract
:The smallest fullerene C20 with positive electron affinity is considered to be a new organic nano-electronic material. The binding structures and electronic states of lithium ions and atoms (Li+ and Li) trapped on the surface of C20 have been investigated by means of density functional theory (DFT) calculation to elucidate the nature of their interaction. It was found that a Li+ can bind to only one site of C20. This is, specifically, on top of the site where Li+ binds to the carbon atom of C20. On the other hand, in the case of a Li atom, two structures were obtained besides the on-top structure. One was pentagonal structure which included a Li atom on a five-membered ring of C20. The other was a triangular structure in which the Li atom bind to the the carbon–carbon bond of C20. Finally, the nature of the interactions between Li ions or atoms and the C20 cluster was discussed on the basis of theoretical results.
1. Introduction
Currently, the diameter of the thinnest carbon nanotube synthesized is 4 Å, and its tip is expected to have a structure similar to that of a C20 fullerene [1]. It has been reported by photoelectron spectroscopy that C20 is predicted to be the smallest fullerene that is synthesized from vapor growth, and that its electron affinity is positive [2,3]. Its structure is very simple and highly symmetrical, consisting of 12 five-membered rings only. Its diameter is about 4 Å, which is less than half of that of C60 fullerene. It is also known to form one-dimensional chains, the key to a controlled structure, via vapor deposition using an arc plasma gun [4]. C20 and its derivatives are considered to be not only new n-type materials, but also ultimate organic nanomaterials capable of controlling size and orientation.
In order to utilize this organic molecule as an electronic material, knowledge of fundamental physical properties as to how its electronic state is changed by doping is very important. The interaction of C60 with alkali metal is investigated by various methods such as spectroscopy and electrochemical methods [5,6,7,8,9,10]. In particular, superconductivity is reported to occur in systems doped with K atoms [10]. It is also well known that alkali metal-doped nanostructures are promising for technical applications such as gas storage and the mobilization of small organic molecules [11,12]. For these reasons, it is important to know the exact structure and electronic properties of the systems interacting with alkali metals. We are particularly interested in chemical stability, reactivity of C20, and the change of electronic state by doping. However, basic data on the doping state of C20 is rarely found in experimental research as well as in theoretical research.
Recently, we studied the structures and electronic states of alkyl and hydroxyl radical functionalized C20 fullerene [13,14]. We also studied the binding structures and electronic states of sodium ions and atoms trapped on the surface of C20 [15]. However, the interaction between C20 and Li+ ions or Li atoms, as well as its reaction mechanisms have never been studied so far.
Tachikawa theoretically investigated the diffusion dynamics of an Li+ ion on the surface of C60 fullerene by the direct dynamics method [16,17]. This study revealed two transition states (TSs) of Li+ ion diffusion. It also revealed that a Li+ ion diffuses along the node faces of the highest occupied molecular orbital (HOMO) of C60 and graphene [17,18]. However, the interaction between C20 and other alkali metals, the transition states of the reaction, and the migration path on the surface have not yet been elucidated.
In this study, the interactions of Li+ ions and Li atoms with C20 were investigated by means of density functional theory (DFT) to clarify their binding site and TS.
2. Results and Discussion
2.1. C20–Li+
2.1.1. Optimized Structure of the C20–Li+ System
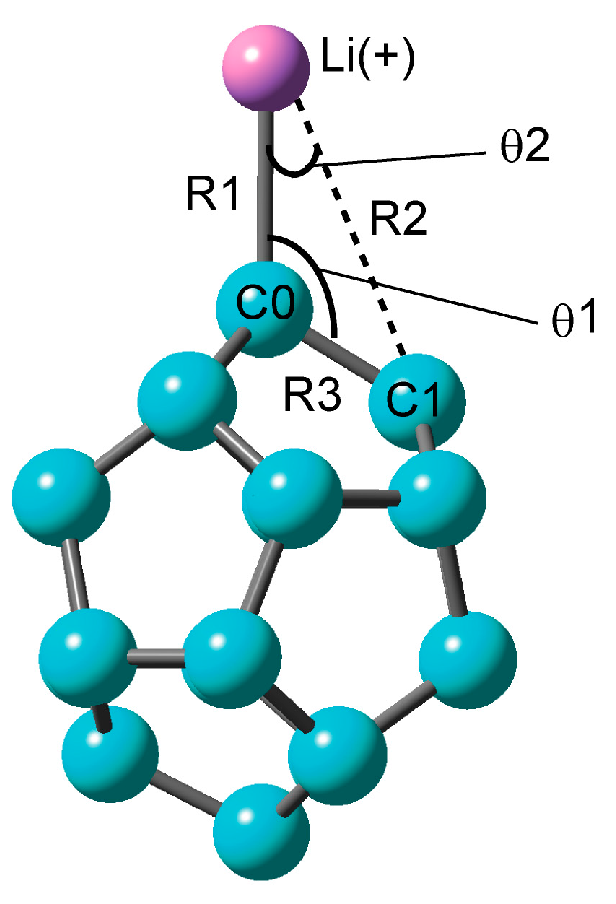
The structures of C20 and C20–Li+ were fully optimized at the CAM-B3LYP level. The structure obtained is illustrated Figure 1. The only stable structure of C20–Li+ was the on-top structure with a Li+ ion on C0. In the pentagonal structure where a Li+ ion is on the carbon five-membered ring, there was a vibration mode of the imaginary part. Thus, it was not a stationary point in this system. The distance R1 between C0 and Li of the on-top structure was 2.055 Å, and the angle θ1 of <C1–C0–Li was 121 degrees (Table 1). The Li–C0 stretching mode (νC0–Li+) was calculated to be 399 cm−1. In our previous research, the hexagonal structure and pentagonal structure were more stable than the on-top structure in C60–Li+ [16]. In addition, it is known that the Li+ ion on graphene has a hexagonal structure. The on-top structure is a structure unique to C20–Li+, which seems to be related to the curvature of C20.
The natural population analysis (NPA) atomic charges on the Li+ ion and C0 were +0.955 and −0.621, respectively. These results imply that the Li+ ion and C0 atom have a positive and a negative charge, respectively. The C0–Li bond is polarized. The binding energy was calculated to be 42 kcal·mol−1. This energy is significantly larger than the binding energy for the C60–Li+ system [16]. The binding energy of the on-top structure of C60–Li+ was about 30 kcal·mol−1, and it was found that C20–Li+ produces a stronger bond than C60–Li+.
2.1.2. Electronic Structures of Pure C20 and C20–Li+
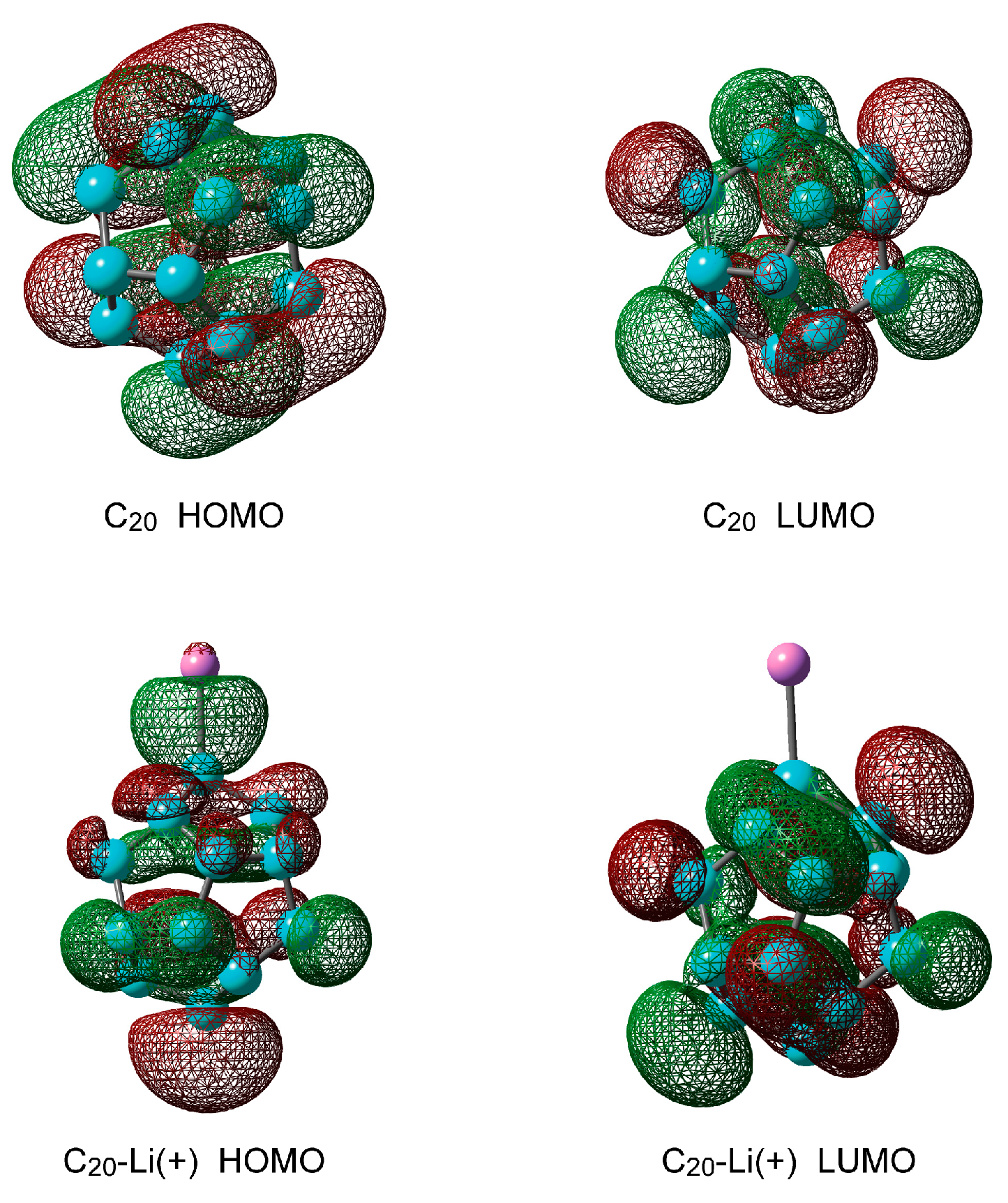
Figure 2 shows the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of C20 and C20–Li+. When doping with Li+ ions, the electronic state of C20 changes drastically. The molecular orbital is spread between C0 and Li+ at HOMO. The energy difference between the HOMO and LUMO orbitals of C20 is 4.2 eV, that is, C20 acts as an insulator. Using the optimized structure of C20 and C20–Li+, the vertical excitation energies were calculated by TD-CAMB3LYP/6-311+G(d). The first excitation energies (S1) of C20 and C20–Li+ were 3.01 eV and 1.48 eV, respectively. These results mean that a charge transfer occurs between C20 and Li+ at HOMO.
The electronic states of C20 will be changed by the doping of the Li+ ion. To elucidate the electronic states of the binding site, natural bond orbital (NBO) analysis was carried out for the C20–Li+ system. The NBO of the C0–C1 bond of pure C20 was expressed by:
where, C0 and C1 indicate the carbon atom in the addition center and the neighbor-positioned carbon atom, respectively.
σC0–C1 = 0.701 (sp2.03)C0 + 0.713 (sp2.03)C1
After the Li+ ion doping to C20, the NBO was changed to be:
σC0–C1 = 0.690 (sp2.72)C0 + 0.724 (sp2.05)C1
Before the doping of Li+ ion, the carbon atom in the doping site (C0) takes an electronic state of sp2.03. After the doping, the state was changed to sp2.72. This result indicates that the carbon atom (C0) is changed from sp2 to sp3.
2.1.3. Transition State of C20–Li+ and the Movement Path of the Li Ion on the Surface of C20
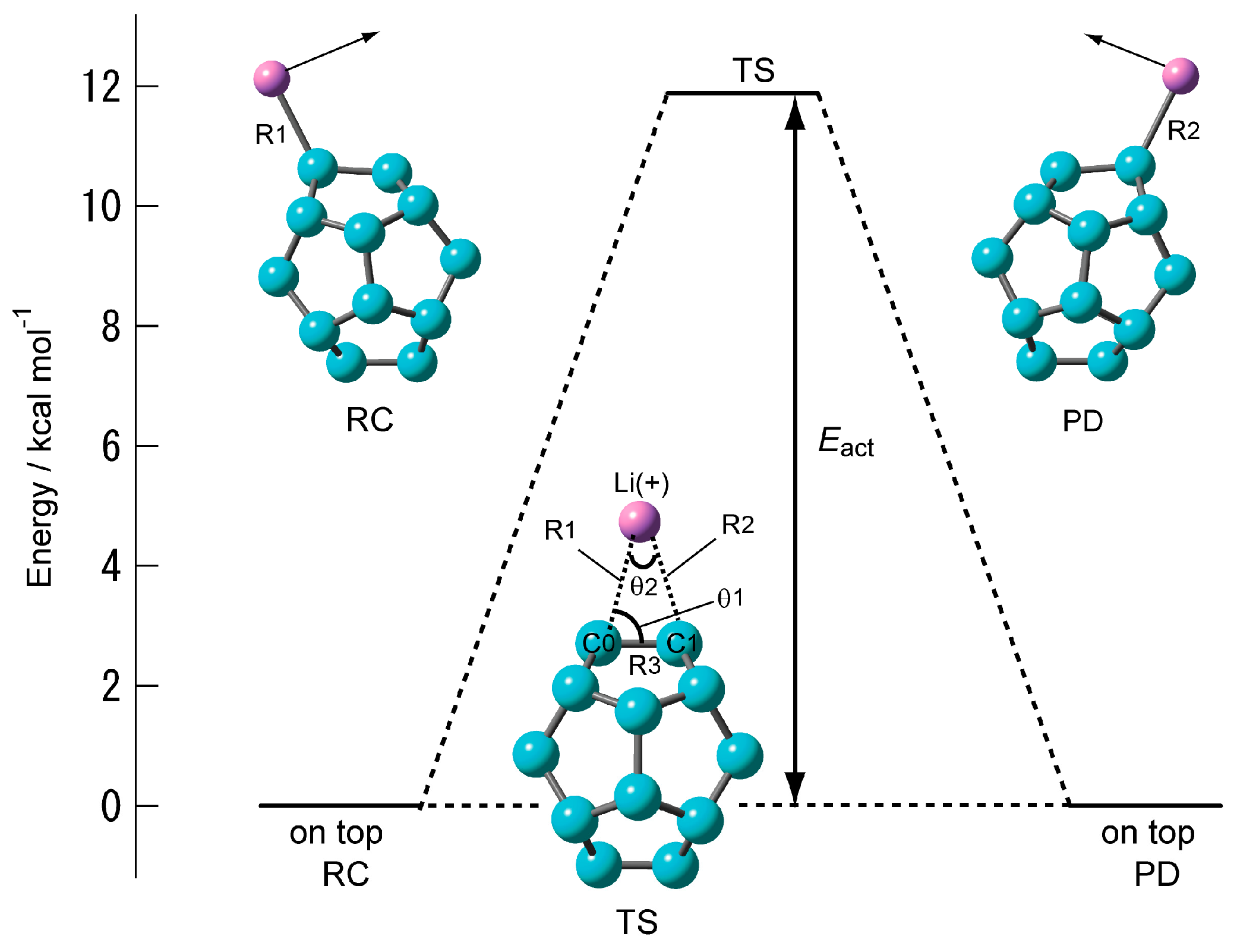
Figure 3 shows the structure of the transition state of C20–Li+ and its energy diagram. The structure of the transition state was found to be a triangular structure with the Li+ ion on the C–C bond (Table 2). Intrinsic reaction coordinate (IRC) analysis was performed, and it was confirmed that the Li+ ion migrated from C0 (RC, reactant) onto the adjacent carbon (C1) (PD, product) via the triangular structure as the transition state. Its activation energy (Eact) is as high as about 12 kcal·mol-1. The structure of the transition state in which the Li+ ion diffuses onto C60 is also a triangular structure. However, its activation energy is less than 4 kcal·mol−1. In other words, the movement of the Li+ ion on the surface of C20 is like a bond alternation. It cannot be said that the Li+ ion diffuses by heat.
2.2. C20–Li
2.2.1. Optimized Structure of C20–Li
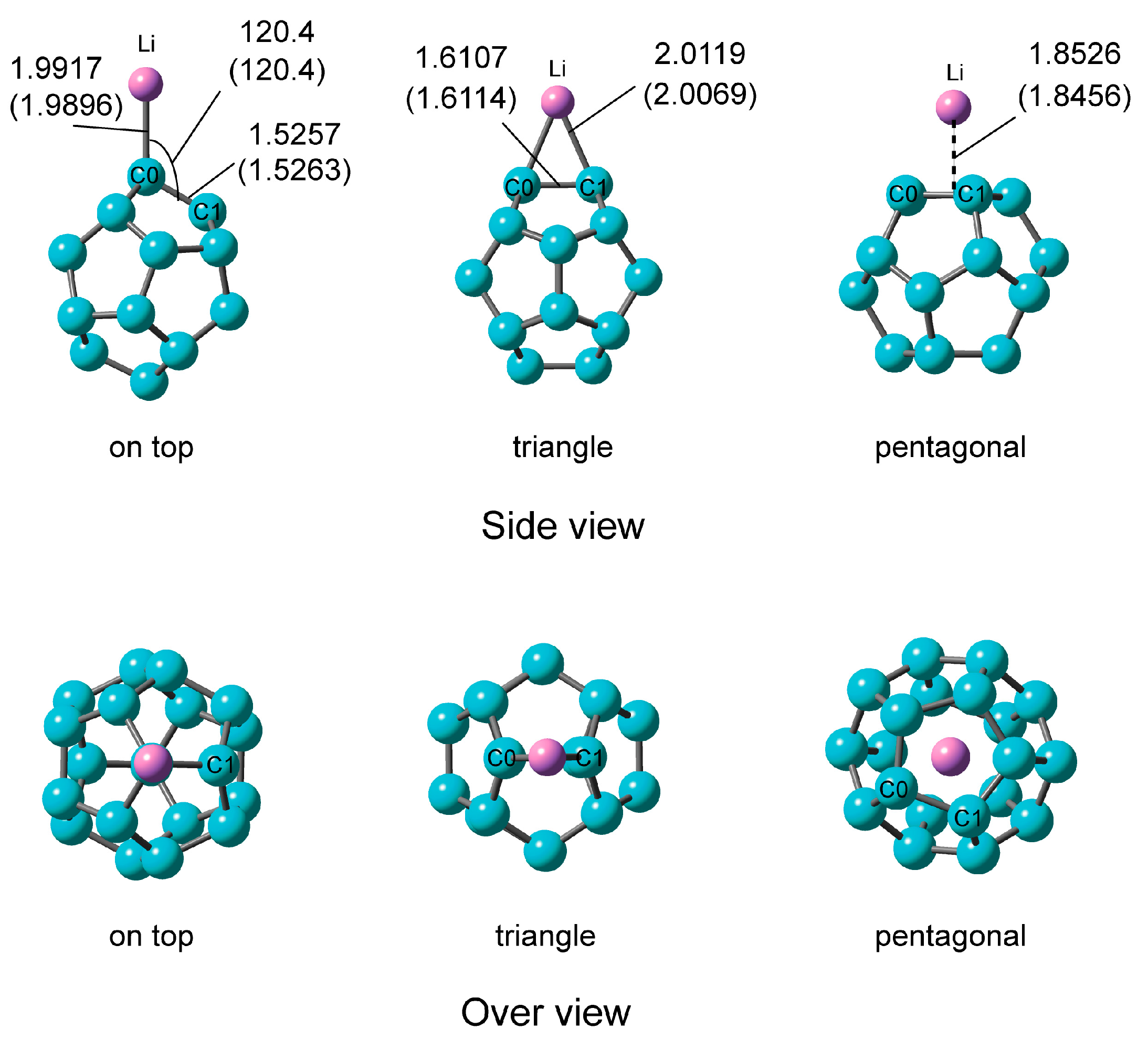
The structures of C20–Li optimized at the CAM-B3LYP/6-311+G(d) level are illustrated in Figure 4. Three structures were found in C20–Li, an on-top structure where the Li atom is located on the carbon atom, a triangular structure where the Li atom binds to the center of the C–C bond (triangle), and a pentagonal structure where the Li atom is located in the center of the five-membered ring (pentagon). There was no vibration mode of the imaginary part. That is, the structures are stationary points, respectively. Very interestingly, the triangular structure, which was in the transition state of C20–Li+, was the most stable in C20–Li. This means that the movement path of the Li+ ion and Li atom on the surface of C20 can be controlled by an electric charge.
The C0–Li distance of C20–Li of the on-top structure is about 0.1 Å shorter than that of C20–Li+. In the triangular structure, the C0–Li distance is about 0.3 Å shorter than that of C20–Li+. In other words, C20–Li formed in the on-top structure and triangular structure is thought to have larger binding energy and charge transfer than C20–Li+.
The binding energy and NPA atomic charge of each atom of C20–Li calculated at the CAM-B3LYP/6-311+G(d) level are provided in Table 3. The NPA atomic charge of the Li atom in the on-top, the triangular, and the pentagonal sites were +0.934, +0.869, and +0.823, respectively.
In all structures, it can be seen that the charge of C20 is changed to negative due to the addition of the Li atom, indicating that charge transfer occurs from Li to C20. The magnitude of the dipole moment is 11.3 D, 7.2 D, and 6.7 D in each of the on-top, triangular, and pentagonal structures, which is related to the charge transfer amount.
The binding energy between C20 and the Li atom was 49.42 kcal mol−1 for on-top structure, 52.96 kcal mol−1 for triangular structure, and 46.89 kcal mol−1 for pentagonal structure. These binding energies are 4.5–10.6 kcal mol−1 greater than that of C20–Li+. This is due to the fact C20–Li produces a strong charge transfer to C20 rather than C20–Li+.
The electronic states of C20 were changed by the doping of Li atoms as well as that of C20–Li+. To elucidate the electronic states of the interaction site, natural bond orbital (NBO) analysis was carried out for the C20–Li system. The NBO of the C0–C1 bond of C20–Li was expressed by:
where, C0 and C1 are carbon atoms of C20 (see Figure 4). After the doping, the electronic states of the on-top and triangular structures were drastically changed. These results indicate that the electronic state in the carbon atom (C0) is changed from sp2 to sp3. In contrast, the hybridization of C0 was hardly changed in the pentagonal structure. It seems that, in this case, the interaction of the Li–C bond was spread into five carbon atoms.
σC0–C1 (on top) = 0.695 (sp2.72)C0 + 0.719 (sp2.06)C1
σC0–C1 (triangle) = 0.707 (sp3.13)C0 + 0.707 (sp3.13)C1
σC0–C1 (pentagonal) = 0.707 (sp2.14)C0 + 0.708 (sp2.15)C1
2.2.2. Transition State of C20–Li and the Movement Path of the Li atom on the Surface of C20
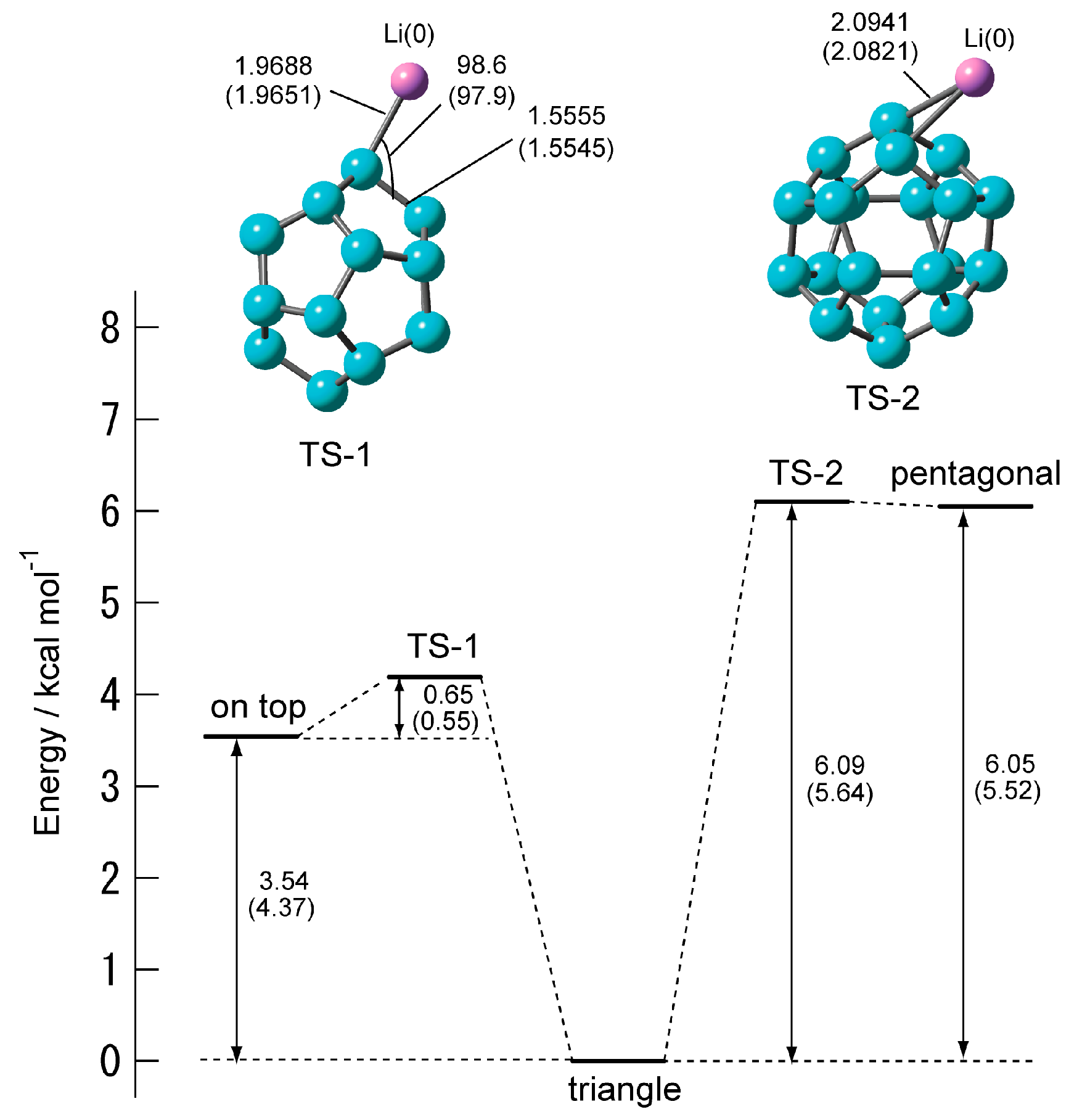
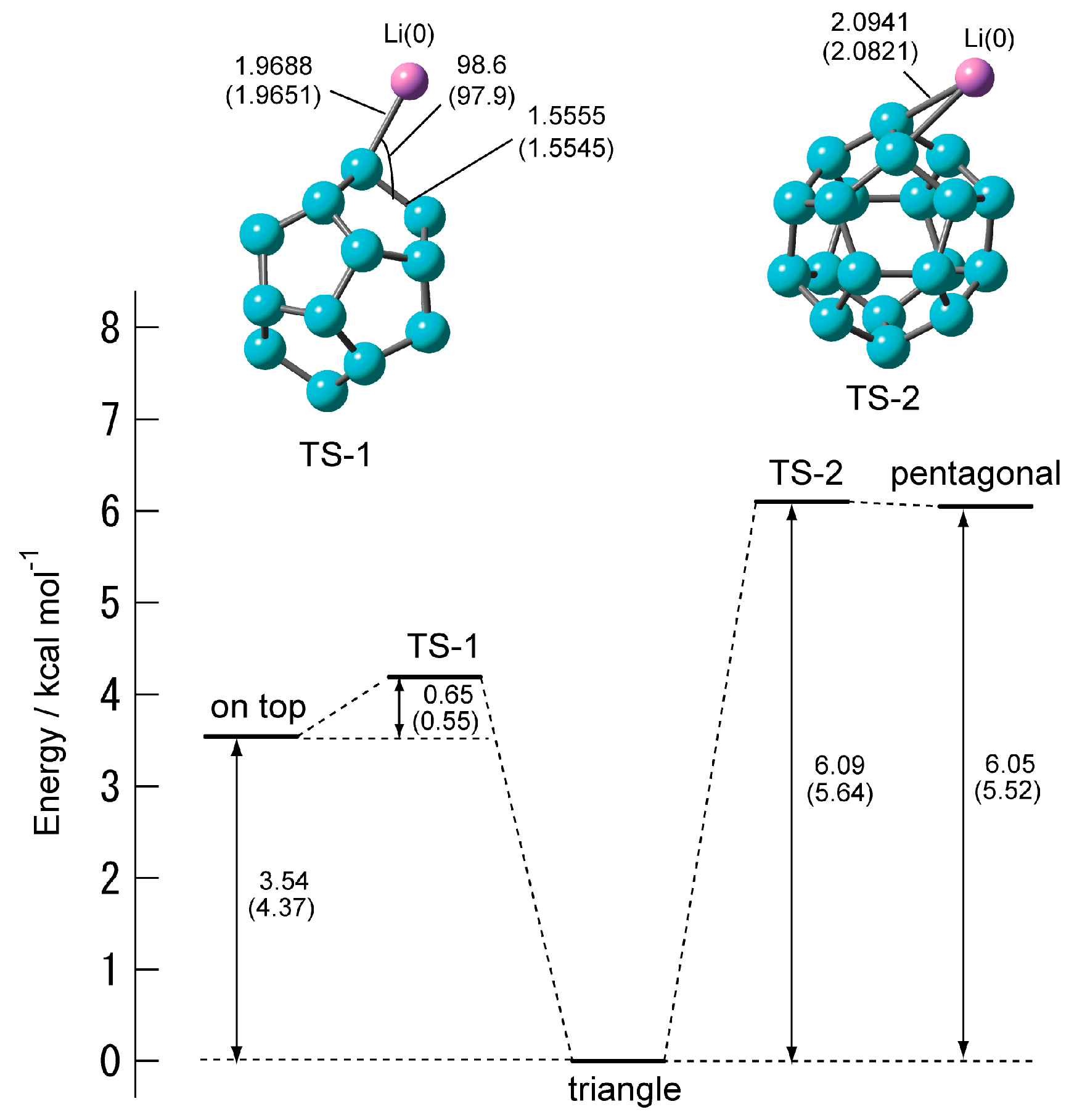
Figure 5 shows the structures of the transition state and energy diagram of C20–Li. When the position of the Li atom changes from the on-top site and pentagonal site to the triangular site, it turns out that there is one transition state in each. The transition state when changing from the on-top structure to the triangular structure is TS-1, and the transition state when changing from the pentagonal structure to the triangular structure is TS-2. TS-1 and TS-2 each has a vibrational mode of one imaginary part. IRC confirmed that the on-top structure and the pentagonal structure are triangular structures via TS-1 and TS-2, respectively.
Table 4 shows the activation energies (Eact) and the vibrational frequencies for the imaginary modes. TS-1 and TS-2 have one vibration mode of the imaginary part, respectively. Both Eact of TS-1 and TS-2 were less than 1 kcal·mol−1. This was significantly smaller than that of the Eact of the C20–Li+. This means that the on-top structure and the pentagonal structures are easily changed to the triangular structure by thermal activation. However, the triangular structure is 6.1 kcal mol−1 more stable than the pentagonal structure. Therefore, it is unlikely that the Li atom will diffuse the surface of C20 via TS-2.
On the other hand, the difference in heat of formation between the triangular structure and the on-top structure is 3.5 kcal mol−1. The activation energy for Li+ ions to diffuse onto the surface of C60 is ca. 4 kcal mol−1 [16]. Thus, there is a possibility that the Li atom diffuses its surface along the C–C bond of C20.
3. Method of Calculation
All hybrid DFT calculations were carried out using the Gaussian 09 program package [19]. Clusters of C20 and Li+ ions or Li atoms were examined. All calculations performed in this study are only Γ points. The structures and electronic states of C20, C20–Li+ (denoted by C20–Li+), and C20–Li atom (denoted by C20–Li) were calculated using the CAM-B3LYP functional, which produces accurate results for long-range interactions, and these results were combined with several basis sets [20,21,22,23]. Calculations on C20 and C20–Li+ were performed using the RCAM-B3LYP functional. Since C20–Li is an open shell, the calculation was made using the UCAM-B3LYP functional. The geometries of all systems were fully optimized at the CAM-B3LYP/6-311G(d) and CAM-B3LYP/6-311+G(d) levels of theory. The convergence criterion of SCF (self-consistent field) was set to 10−7. These levels of theory have given reasonable electronic structures of C20–Li+ and C20–Li systems [13,14,15,24,25]. All of the geometries studied here were characterized as stationary or transition states by analytically vibrational frequency calculation.
Furthermore, each transition state was checked to be connected to the initial state and the final state by IRC analysis.
4. Conclusions
In this study, the interaction between C20 and Li+ ions and Li atoms were investigated by density functional theory. The only stable structure of C20–Li+ is the on-top structure, where the Li+ ion is on top of the carbon atom of C20. The Li+ ion migrates to an adjacent carbon atom through a triangular structure consisting of C–C and Li+. Its activation energy is 12 kcal·mol−1, and the migration of the Li+ ion is due to coupling substitution. On the other hand, C20–Li has three structures: the on-top structure, triangular structure, and pentagonal structure. The most stable is the triangular structure, while the other two are metastable. The activation energy from on-top structure and pentagonal structure to the triangular structure is smaller than 1 kcal·mol−1. From the magnitude of the relative energy of each structure, it was revealed that the Li atom diffuses with heat along the C–C bond of C20.
Acknowledgments
The author acknowledges partial support from JSPS KAKENHI Grant Number 15K05371 and MEXT KAKENHI Grant Number 25108004.
Author Contributions
Hiroshi Kawabata and Hiroto Tachikawa designed this study and wrote the paper; All of the computer experiments and analyses were conducted by Hiroshi Kawabata.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Qin, L.-C.; Zhao, X.; Hirahara, K.; Miyamoto, Y.; Ando, Y.; Iijima, S. The smallest carbon nanotube. Nature 2000, 408, 50. [Google Scholar] [CrossRef] [PubMed]
- Parasuk, V.; Almof, J. C20: The smallest fullerene? Chem. Phys. Lett. 1991, 184, 187–190. [Google Scholar] [CrossRef]
- Prinzbach, H.; Weiler, A.; Landenberger, P.; Wahl, F.; Wörth, J.; Scott, L.T.; Gelmont, M.; Olevano, D.; Issendorff, B.V. Gas-phase production and photoelectron spectroscopy of the smallest fullerene, C20. Nature 2000, 407, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, S.; Yamamoto, D.; Hirashige, K.; Sakai, A. Possible formation of one-dimensional chains of C20 fullerenes observed by scanning tunneling microscopy. Appl. Phys. Express 2016, 9, 045102. [Google Scholar] [CrossRef]
- Lüssem, B.; Riede, M.; Leo, K. Doping of organic semiconductor. Phys. Status. Solidi. A 2013, 210, 9–43. [Google Scholar]
- Salzmann, I.; Heimel, G. Toward a comprehensive understanding of molecular doping organic semiconductors. J. Electron Spectrosc. Relat. Phenom. 2015, 204, 208–222. [Google Scholar] [CrossRef]
- Holczer, K.; Whetten, R.L. Superconducting and normal state properties of the A3C60 compounds. Carbon 1992, 30, 1261–1276. [Google Scholar] [CrossRef]
- Moriyama, H.; Kobayashi, H.; Kobayashi, A.; Watanabe, T. ESR spectra on single crystals of alkali metal fulleride complexes by means of wet-chemical synthesis. Chem. Phys. Lett. 1995, 238, 116–121. [Google Scholar] [CrossRef]
- Dalchiele, E.A. Rosolen. J.M.; Decker, F. Electrochemically intercalated MxC60 thin films in a solid state cell (M = Li, K): Optical and photoelectrochemical characterization. Appl. Phys. A Mater. Sci. 1996, 63, 487–494. [Google Scholar]
- Zhang, Z.; Chen, C.C.; Kelty, S.P.; Dai, H.J.; Lieber, C.M. The superconducting energy gap of Rb3C60. Nature 1991, 353, 333–335. [Google Scholar] [CrossRef]
- Cazorla, C.; Shevlin, S.A.; Guo, Z.X. Calcium-based functionalization of carbon materials for CO2 capture: A first-principles computational study. J. Phys. Chem. C 2011, 115, 10990–10995. [Google Scholar] [CrossRef]
- Cazorla, C. Ab initio study of the binding of collagen amino acids to graphene and A-doped (A = H, Ca) graphene Original Research Article. Thin Solid Films 2010, 518, 6951–6961. [Google Scholar] [CrossRef]
- Abe, S.; Kawano, S.; Toida, Y.; Nakamura, M.; Inoue, S.; Sano, H.; Yoshida, Y.; Kawabata, H.; Tachikawa, H. Electronic states of alkyl-radical-functionalized C20 fullerene using density functional theory. Jpn. J. Appl. Phys. 2016, 55, 03DD03. [Google Scholar] [CrossRef]
- Iyama, T.; Abe, S.; Tachikawa, H. Density functional theory (DFT) study on the addition of hydroxyl radical (OH) to C20. Mol. Cryst. Liq. Cryst. 2012, 567, 200–206. [Google Scholar] [CrossRef]
- Fukuzumi, T.; Tachikawa, H.; Azumi, K. DFT Study on the interaction of carbon nano-materials with sodium ion and atom. Mol. Cryst. Liq. Cryst. 2011, 538, 61–66. [Google Scholar] [CrossRef]
- Tachikawa, H. Diffusion of the Li+ Ion on C60: A DFT and Molecular Dynamics Study. J. Phys. Chem. C 2011, 115, 20406–20411. [Google Scholar] [CrossRef]
- Tachikawa, H. Diffusion dynamics of the Li ion on C60: A direct molecular orbital−molecular dynamics study. J. Phys. Chem. C 2007, 111, 13087–13091. [Google Scholar] [CrossRef]
- Tachikawa, H.; Shimizu, A. Diffusion Dynamics of the Li+ Ion on a Model Surface of Amorphous Carbon: A direct molecular orbital dynamics study. J. Phys. Chem. B 2005, 109, 13255–13262. [Google Scholar] [CrossRef] [PubMed]
- Gaussian 09, Revision D.01, Gaussian, Inc.: Wallingford CT, 2013.
- Kobayashi, R.; Amos, R.D. The application of CAM-B3LYP to the charge-transfer band problem of the zincbacteriochlorin–bacteriochlorin complex. Chem. Phys. Lett. 2006, 420, 106–109. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange—Correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Cazorla, C.; Shevlin, S.A. Accuracy of density functional theory in the prediction of carbon dioxide adsorbent materials. Dalton Trans. 2013, 42, 4670–4676. [Google Scholar] [CrossRef] [PubMed]
- Cazorla, C. The role of density functional theory methods in the prediction of nanostructured gas-adsorbent materials. Coordination Chem. Rev. 2015, 300, 142–163. [Google Scholar] [CrossRef]
- Tachikawa, H.; Kawabata, H. Electronic states of alkali metal-NTCDA complexes: A DFT study. Solid State Sci. 2015, 48, 141–146. [Google Scholar] [CrossRef]
- Iyama, T.; Kawabata, H.; Tachikawa, H. Origin of Spectrum Shifts of Benzophenone-Water Clusters: DFT Study J. Solution Chem. 2014, 43, 1676–1686. [Google Scholar] [CrossRef]
Figure 1.
Optimized structure of C20–Li+ calculated at the CAM-B3LYP/6-311+G(d) level.
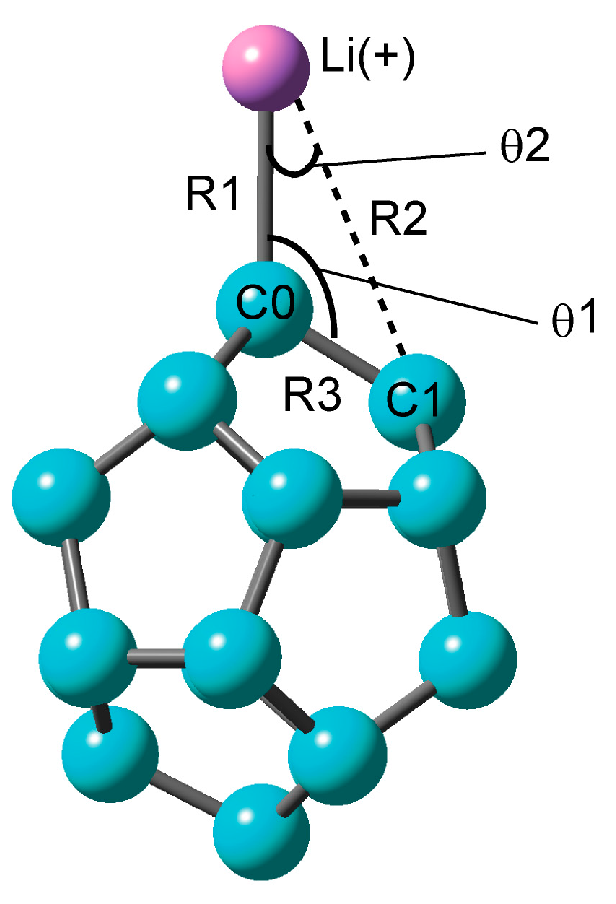
Figure 2.
Molecular orbitals of C20 and C20–Li+ calculated at the CAM-B3LYP/6-311+G(d) level.
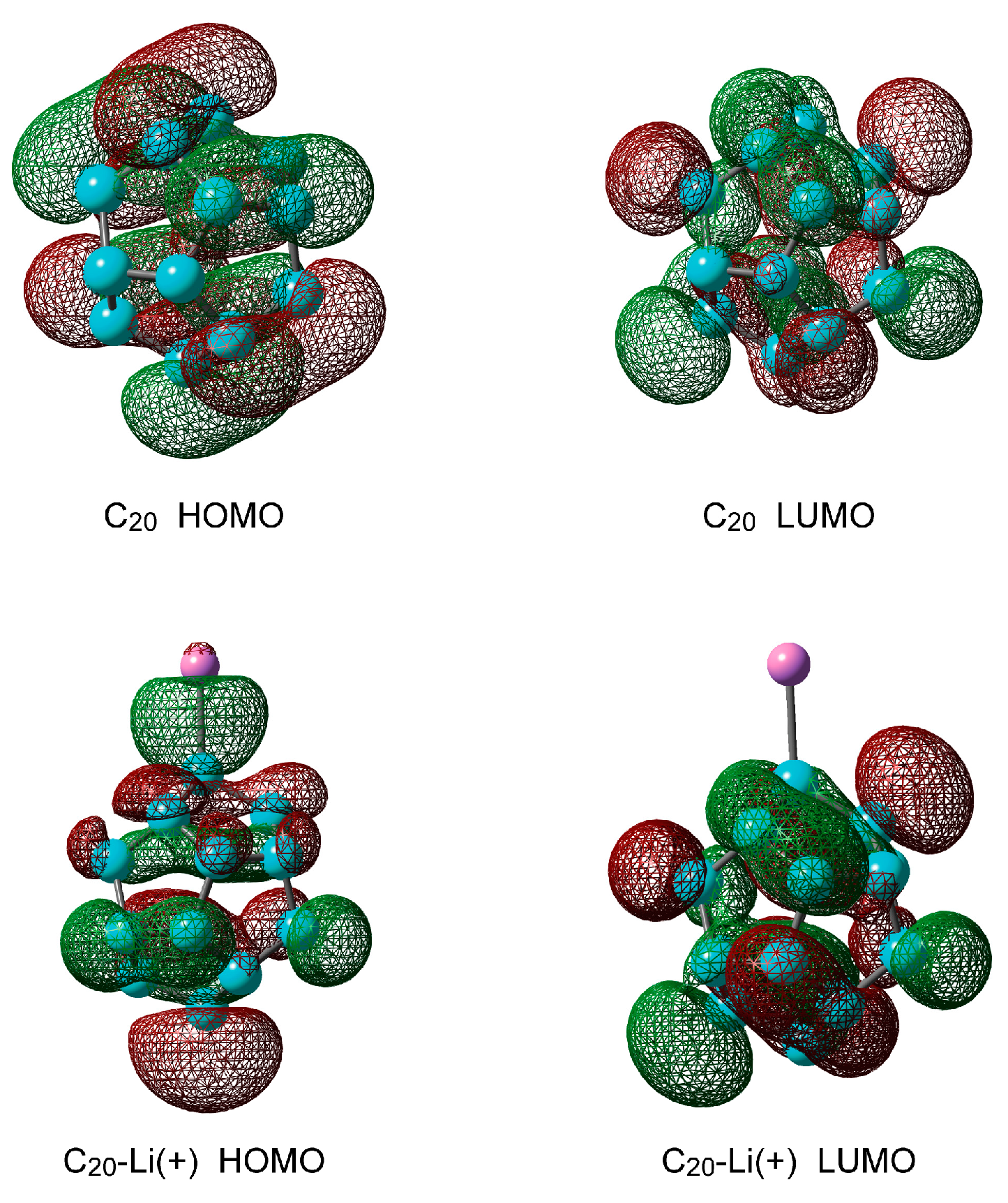
Figure 3.
Energy diagram of Li+ ion migration reaction on the surface of C20 calculated at the CAM-B3LYP/6-311+G(d) level.
Figure 3.
Energy diagram of Li+ ion migration reaction on the surface of C20 calculated at the CAM-B3LYP/6-311+G(d) level.
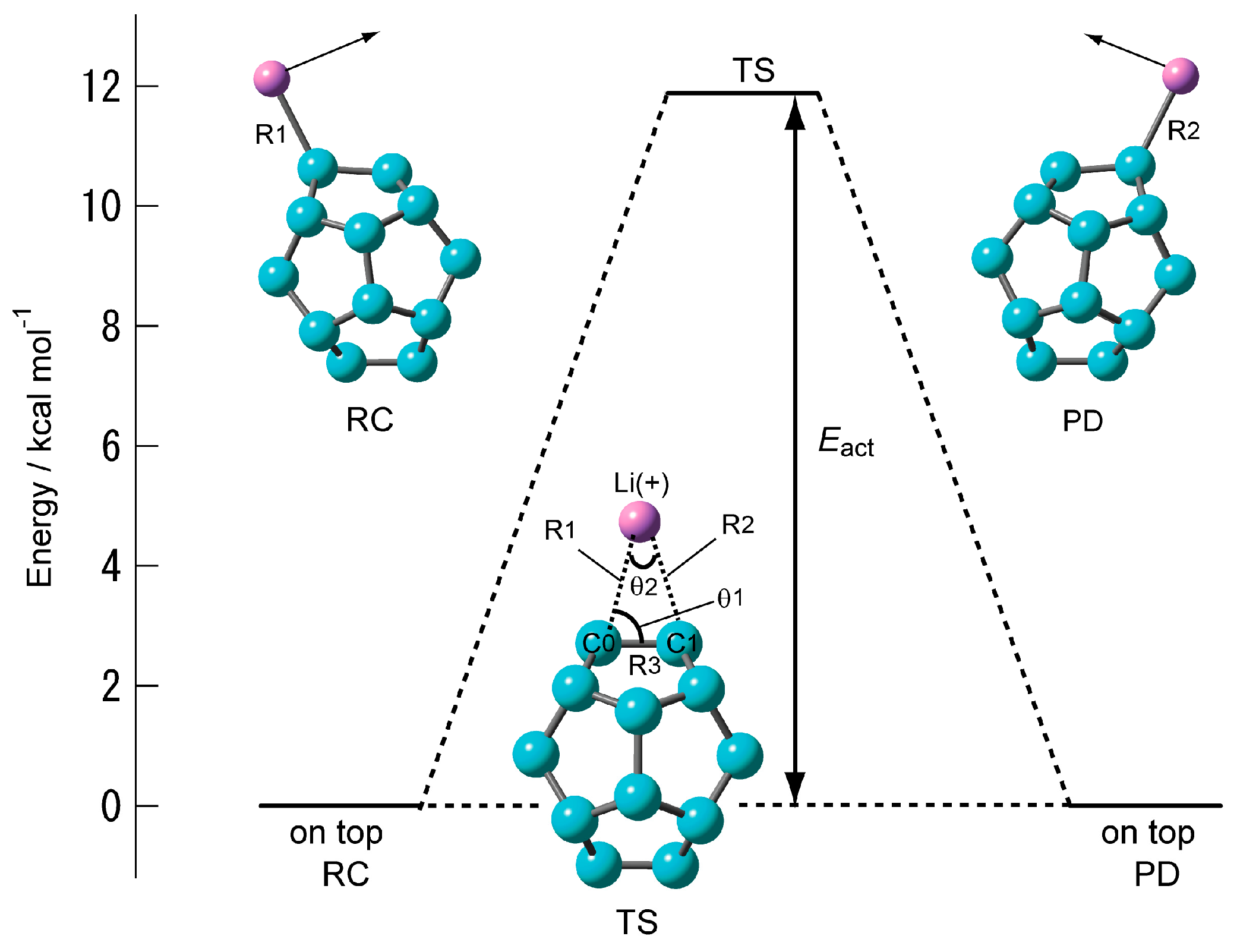
Figure 4.
Optimized structures of C20–Li system; on top, triangular, and pentagonal structures calculated at the CAM-B3LYP/6-311+G(d). In parentheses, values were calculated at the CAM-B3LYP/6-311G(d) level. The bond distances and angles are in Å and in degrees, respectively.
Figure 4.
Optimized structures of C20–Li system; on top, triangular, and pentagonal structures calculated at the CAM-B3LYP/6-311+G(d). In parentheses, values were calculated at the CAM-B3LYP/6-311G(d) level. The bond distances and angles are in Å and in degrees, respectively.

Figure 5.
Energy diagram and structures of the transition state of C20–Li system calculated at the CAM-B3LYP/6-311+G(d) level. In parentheses are the values calculated at the CAM-B3LYP/6-311G(d) level.
Figure 5.
Energy diagram and structures of the transition state of C20–Li system calculated at the CAM-B3LYP/6-311+G(d) level. In parentheses are the values calculated at the CAM-B3LYP/6-311G(d) level.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimized structural parameters, natural population analysis (NPA) atomic charge on C0 and Li+, and binding energies of Li+ to C20 calculated at several levels of theory. Zero point energy corrected values are in parentheses.
Table 1.
Optimized structural parameters, natural population analysis (NPA) atomic charge on C0 and Li+, and binding energies of Li+ to C20 calculated at several levels of theory. Zero point energy corrected values are in parentheses.
| Method | Structural Parameter | NPA Atomic Charge | Ebind(a)/kcal mol−1 | |||||
|---|---|---|---|---|---|---|---|---|
| R1/Å | R2/Å | θ1/° | θ2/° | νC0–Li+/cm−1 | C0 | Li+ | ||
| CAM-B3LYP/6-311G(d) | 2.0548 | 3.1288 | 121.0 | 24.7 | 399 | −0.630 | 0.957 | 43.24 (41.31) |
| CAM-B3LYP/6-311+G(d) | 2.0547 | 3.1293 | 121.0 | 24.7 | 399 | −0.621 | 0.955 | 42.36 (40.40) |
(a) The energy of the Li+ ion binding to C20 (Ebind) is defined by −Ebind = E(C20–Li+) − [ E(C20) + E(Li+)]. Here, E(C20–Li+) represents the total energy of the Li+ ion added to C20, E(C20) represents that of the free C20 molecule, and E(Li+) represents that of the free Li+ ion, respectively.
Table 2.
Structural parameters of the transition state and activation energies (Eact in kcal·mol–1) calculated at several levels of theory. Zero point energy corrected values are in parentheses.
Table 2.
Structural parameters of the transition state and activation energies (Eact in kcal·mol–1) calculated at several levels of theory. Zero point energy corrected values are in parentheses.
| System | Structural Parameter | Eact/kcal·mol–1 | ||||
|---|---|---|---|---|---|---|
| R1/Å | R3/Å | θ1/° | θ2/° | νC0–Li+/cm–1 | ||
| CAM-B3LYP/6-311G(d) | 2.2856 | 1.4347 | 71.7 | 36.6 | 399 i | 11.94 (11.34) |
| CAM-B3LYP/6-311+G(d) | 2.2884 | 1.4352 | 71.7 | 36.5 | 400 i | 11.88 (11.31) |
Table 3.
Binding energies of Li to C20, and NPA atomic charges of C20, C0, and Li calculated at the CAM-B3LYP/6-311+G(d) level. Zero point energy corrected values are in parentheses.
Table 3.
Binding energies of Li to C20, and NPA atomic charges of C20, C0, and Li calculated at the CAM-B3LYP/6-311+G(d) level. Zero point energy corrected values are in parentheses.
| System | Ebind (b)/kcal·mol−1 | NPA Atomic Charge | ||
|---|---|---|---|---|
| Part of C20 | C(0) | Li | ||
| On-top | 49.42 (49.32) | –0.934 | –0.662 | +0.934 |
| Triangle | 52.96 (52.85) | –0.869 | +0.869 | |
| Pentagonal | 46.89 (47.95) | –0.823 | +0.823 | |
(b) The energy of the Li atom binding to C20 (Ebind) is defined by −Ebind = E(C20–Li) − [E(C20) + E(Li)]. Here, E(C20–Li) represents the total energy of the Li atom added to C20, E(C20) represents that of the free C20 molecule, and E(Li) represents that of the free Li atom, respectively.
Table 4.
The activation energies (Eact in kcal mol−1) and imaginary frequencies (νi in cm−1) of TS-1 and TS-2 in the C20–Li system calculated at the CAM-B3LYP/6-311+G(d) level. In parentheses, values were calculated at the CAM-B3LYP/6-311G(d) level.
Table 4.
The activation energies (Eact in kcal mol−1) and imaginary frequencies (νi in cm−1) of TS-1 and TS-2 in the C20–Li system calculated at the CAM-B3LYP/6-311+G(d) level. In parentheses, values were calculated at the CAM-B3LYP/6-311G(d) level.
| State | Eact/kcal mol−1 | νi/cm−1 |
|---|---|---|
| TS-1 | 0.65 (0.55) | 252 i (182 i) |
| TS-2 | 0.04 (0.12) | 110 i (120 i) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kawabata, H.; Tachikawa, H. DFT Study on the Interaction of the Smallest Fullerene C20 with Lithium Ions and Atoms. C 2017, 3, 15. https://doi.org/10.3390/c3020015
AMA Style
Kawabata H, Tachikawa H. DFT Study on the Interaction of the Smallest Fullerene C20 with Lithium Ions and Atoms. C. 2017; 3(2):15. https://doi.org/10.3390/c3020015
Chicago/Turabian StyleKawabata, Hiroshi, and Hiroto Tachikawa. 2017. "DFT Study on the Interaction of the Smallest Fullerene C20 with Lithium Ions and Atoms" C 3, no. 2: 15. https://doi.org/10.3390/c3020015
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.