
Electronic Structures, and Optical and Magnetic Properties of Quadruple-Decker Phthalocyanines


Department of Materials Science, The University of Shiga Prefecture, 2500 Hassaka, Hikone, Shiga 522-8533, Japan
*
Author to whom correspondence should be addressed.
Magnetochemistry 2017, 3(2), 21; https://doi.org/10.3390/magnetochemistry3020021
Submission received: 16 February 2017
/
Revised: 25 May 2017
/
Accepted: 7 June 2017
/
Published: 9 June 2017
Abstract
:For applications of magnetic devices with operating nuclear-spin-based quantum bits in quantum computing, electronic structures, and magnetic and optical properties of quadruple-decker phthalocyanines with 3d transition metals, such as scandium, yttrium, and lanthanum atoms (M3Pc4: M = Sc, Y, and La), were studied by quantum calculation using density function theory. Electron density distributions at the highest occupied molecular orbital and lowest unoccupied molecular orbital were considerably delocalized on the phthalocyanine ring with considerable bias of the electrostatic potential. The wide energy gaps and the ultraviolet-visible-near infrared spectra of the systems were based on the phthalocyanine ring-ring interactions with overlapping π-orbitals on the phthalocyanine rings. The chemical shift behavior of 13C and 14N-NMR of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4 depended on the deformation of their structures owing to Jahn-Teller splitting of the d-orbital in the metal ligand field, the considerable perturbation of the metal ligand crystal field on the phthalocyanine ring, the electronic structure based on the electron density distribution, and the magnetic interaction of the nuclear quadrupole interaction. The magnetic parameters of the principle g-tensor, the V-tensor of the electronic field gradient, and the asymmetric parameters were influenced by the deformed structures of the complex with the considerable deviation of the charge density distribution. The quadruple-decker metal phthalocyanines using 3d transition metals have an advantage in controlling the electronic structure and magnetic parameters based on the nuclear spin interaction in spin lattice relaxation with respect to applications of single-molecular magnets.
1. Introduction
Electronic structures, and optical and magnetic properties of organometallic transition complexes have been studied for their application in spintronic devices, single-molecule magnetism, and quantum computers. For instance, single-molecule magnets using copper phthalocyanine [1], nitrogen, and phosphorous atom in fullerene cages, such as N@C60 [2], and P@C60, endohedral metallofullerene, such as Sc3N@C80 [3], Sc3-xYN@C80(CF3)n [4,5], GdxSc3-xN@C80 [6], HoSc2N@C80 [7], LnSc2N@C80 [8], (Sc3N@C80)2 dimmer [9], scandium oxide endohedral metallofullerenes Sc4O2@C80(CF3)n, Sc4O2@C80 [10,11], and endohedral metallofullerene encapsulated within single-walled-carbon nanotube (SWCNTs) as peapods [12,13,14], were applied for controlling the magnetic interaction with entanglement of quantum spin in the spin-lattice relaxation process. Experimental verification of spintronics, and nuclear magnetic resonance (NMR) quantum computers using single nitrogen vacancy defects in the center of diamonds [15], perfluorobutadienyl iron complex [16], nitrogen endohedral fullerenes, vanadium phthalocyanines [17], and single-molecule magnets [18,19] were performed for analyzing spin dynamics, Rabi oscillation, and entanglement of nuclear spin as quantum bits in quantum algorithm calculation. The Rabi oscillation at room temperatures was detected in a solid state molecular system using vanadium phthalocyanine mixed with the isostructural diamagnetic host complex using titanyl-phthalocyanine in a molar ratio of 1:1000 [17,18,19].
Multi-decker phthalocyanine complexes with 3d and 4f transition metals [19] have been applied for development of spintronic devices, single-molecule magnets, and quantum computers. The spin dynamics and quantum spin entanglement depend on magnetic interaction with degeneracies of 3d and 4f orbitals in multiple states [20,21]. The magnetic properties of hetero-nuclear quadruple-decker phthalocyanine using lutetium, dysprosium, cadmium, and terbium as rare metals with 4f-electrons have been studied for clarifying the magnetic interaction and the long-range f-f interaction between metal ions separated by a diamagnetic ion. [22]. In the standard case using the phthalocyanine complexes with a 3d transition metal, experimental evidence of magnetic properties using quantum calculations with perturbation theory clarified the electronic structure, magnetic interaction of nuclear spin, and quadrupole interaction based on the electron field gradient (EFG) and asymmetry parameter (η) around the central metal and ligands [23,24]. For complexes with 4f-electrons, experiments of magnetic measurements should be conducted at extremely low temperature owing to a short relaxation time, making it difficult to detect and manipulate the entanglement of nuclear spin as a quantum bit in quantum computing. Quantitative investigation of the nuclear magnetic interaction on the hetero-nuclear quadruple-decker phthalocyanine using rare earth metals with 4f-electrons need to be performed by cryogenic measurements of the magnetic properties with incorporation of high-level sections on 4f spin orbits as the peripheral orbital.
Molecular design of the multi-decker phthalocyanines in multiple states has been fabricated for developing magnetic devices and quantum computers at multi-quantum spin gates with decoupling pulses. Experimental realization with theoretical considerations of the electronic structure and the magnetic properties of the multi-decker phthalocyanine was performed for making clear the magnetic interaction, entanglement of quantum spin with multiplicity, and spin dynamics in spin-lattice relaxation. The magnetic parameters, such as chemical shifts of 13C-NMR, g-tensor, and the A-tensor of the hyperfine structure (hfs) in electron spin resonance (ESR) will depend on the degree of perturbation of the 3d spin orbital of the complex in multiple states. Especially, perturbation of the metal-ligand field of the 3d transition metal in multiple states was controlled for operating the electron-nuclear spin interaction and spin dynamics in the spin lattice relaxation process. Rabi oscillations and coherent single nuclear spin manipulation using all-electrical control of a nuclear spin qubit transistor based on a single molecular magnet of double-decker terbium phthalocyanine through the use of the hyperfine Stark effect were demonstrated as experimental results. For improving the magnetic properties in quantum spin devices, experimental results with theoretical calculation on the electronic structure and magnetic properties of multi-decker phthalocyanine was clarified by scanning tunnelling microscopy and spectroscopy (STM/STS) using density functional theory [25]. The quantum calculation was used for computing the STM images and STS. The experimental results with theoretical calculations in the comparison between YPc2 and Y2(Pc)3 indicated the correlation between the appearance of the Kondo effect and unpaired electrons in the π-orbital.
The electronic structure and the magnetic properties of multi-decker phthalocyanines using rare earth actinide metals in multiple states have been studied for developing single-molecular magnets and electron magnetic devices [26,27,28,29,30]. The experimental results of a strong magnetic coupling between a metal ion and a radical spin in the bis(phtalocyaninato)terbium(III) complex (TbPc2) using rare earth metal with a 4f orbital were reported. The spin dynamic relaxation processes of the triple-decker terbium (III) phthalocyaninato were influenced by the magnetic interaction based on f-f spin-spin interactions, dipole-dipole interaction, electron density distribution with an overlapping π-orbital between the phthalocyanine rings. Alternating current magnetic studies on single molecular magnets using a terbium(III)-phthalocyaninato quintuple-decker complex suggested correlation between the magnetic interaction and the strength of the terbium-terbium interactions. Single-molecular magnetic behaviour for the terbium(III)-phthalocyaninato quintuple-decker complex depended on the magnitude of the terbium-terbium magnetic interaction, and hybrization of d and f-π-orbitals on the extended conjugation system [29,30]. The quantum chemical calculation based on perturbation theory of the multi-decker phthalocyanine using rare earths with 4f-electrons in multiple states will be difficult to make clear the paramagnetic shifts of multinuclear NMR, the g-tensor of ESR, and the long-range f-f magnetic interaction in short relaxation time. The multi-decker phthalocyanines using 3d transition metals have an advantage to control the electronic structure and magnetic parameters, and multi-switching spin dynamics for quantum algorithm calculation with coherent nuclear spin manipulation using electric and magnetic field pulses during the relaxation time.
The purpose of this research is to focus on the electronic structures, the magnetic and optical properties of quadruple-decker phthalocyanine using 3d transition metals for operating the magnetic parameters, and spin dynamics of nuclear spin as a quantum bit in quantum algorithm computing. Especially, the magnetic parameters of the chemical shift of 13C-NMR, 14N-NMR, g-tensor of ESR, and UV-VIS-NIR spectra of the quadruple-decker phthalocyanine using 3d transition metals such as scandium, yttrium, and lanthanum atoms will be investigated by quantum calculation. The magnetic mechanism will be explained on the basis of nuclear spin interaction, spin-local interaction, nuclear-spin interaction, and nuclear quadrupole interaction based on EFG and η related with charge density distribution with the overlapping π-orbital on the phthalocyanine ring.
2. Results and Discussion
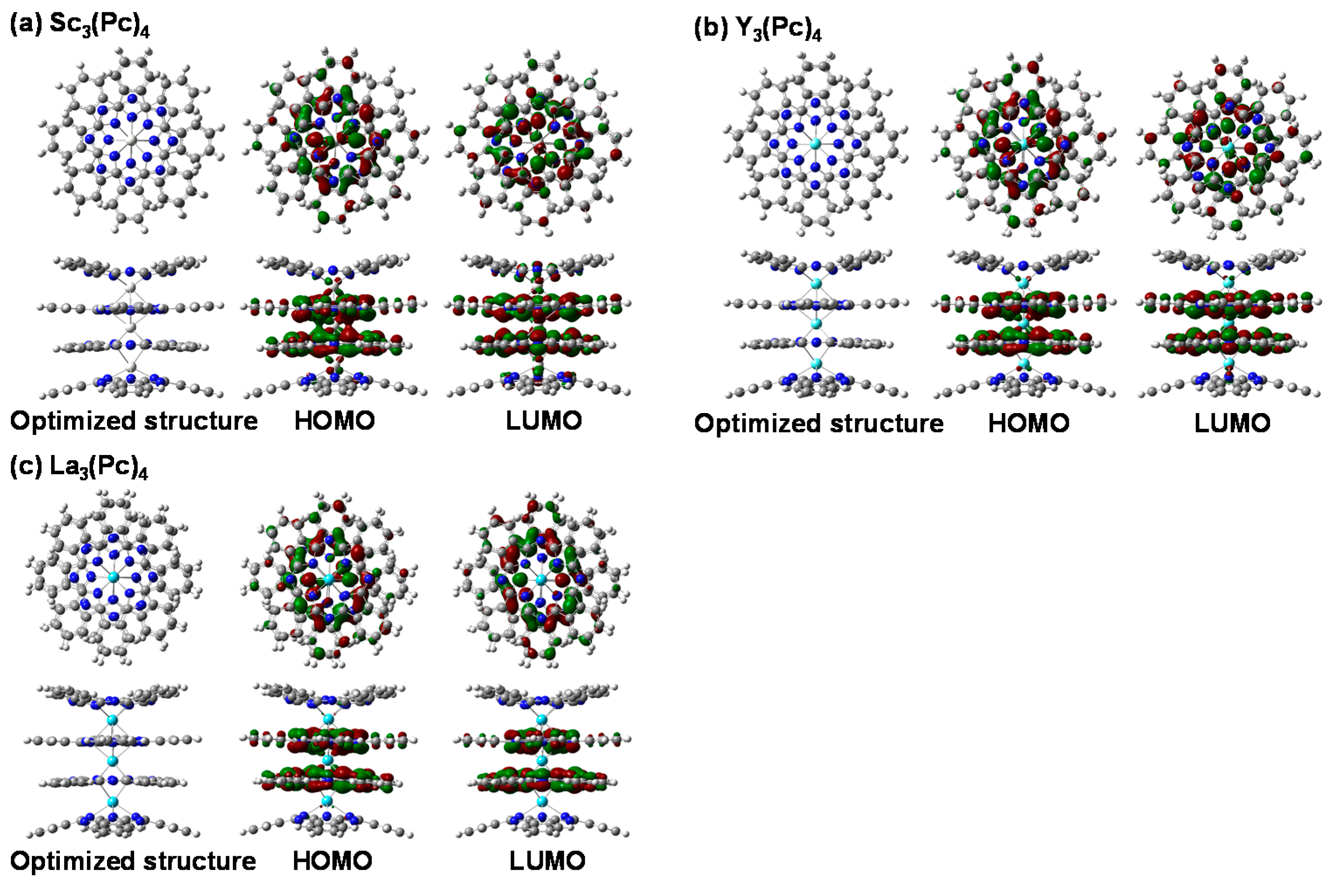
The electronic structures of quadruple-decker metal phthalocyanines at the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4 were calculated by density functional theory (DFT) with hybrid functional calculation with Becke’s three-parameter hybrid functional unrestricted B3LYP (UB3LYP) using LANL2MB as the basis set. The optimized structures, electron density distributions, and the energy levels at the HOMO and LUMO of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4 are shown in Figure 1. The optimized structures of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4 consisted of a double sandwich structure with a degeneration of the energy levels around the frontier orbital. The optimized structures were considerably perturbed by the chemical modification with the central metal diameter varied. The electronic structures were influenced by the deformed structure with the phthalocyanine ring-ring interaction. The electron density distributions were consistent with hybrization between 3d spin orbitals in the metal atom and the nitrogen ligand, and overlapping π-orbitals on the phthalocyanine ring. There was considerable bias of hybrization between the 3d orbital in the metal and nitrogen ligand. Chemical bonds and angles between the metal and ligand are listed in Table 1. The relationship between the lengths of the chemical bond between the metal and nitrogen, and the angles of the N–M–N bonds for M3(Pc)4 was investigated. The bond lengths of Sc-N were slightly decreased, as compared with those for Y3(Pc)4 and La3(Pc)4. The bond angles of N–M–N for Sc3(Pc)4 and Y3(Pc)4 became wider than those for La3(Pc)4. The lengths of the chemical bonds of N–M–N in M3(Pc)4 were increased with increasing metal atomic radii. The chemical bonds and angles depended on the degree of distortion of the molecular structure and intramolecular interaction. The lengths of the chemical bonds of N–M–N in the central part of internal metal were slightly increased, as compared with those in the external metal. The intramolecular interaction between the phthalocyanine rings would influence the electronic structure, the energy gap, and the energy levels at HOMO and LUMO. The deformed structures would resolve the degeneracy of energy levels near the frontier orbital with a narrow band gap between the HOMO and LUMO. The deformed molecular structures will influence the magnetic interaction based on the electron density distribution with the energy levels near the frontier orbital and the band gap.
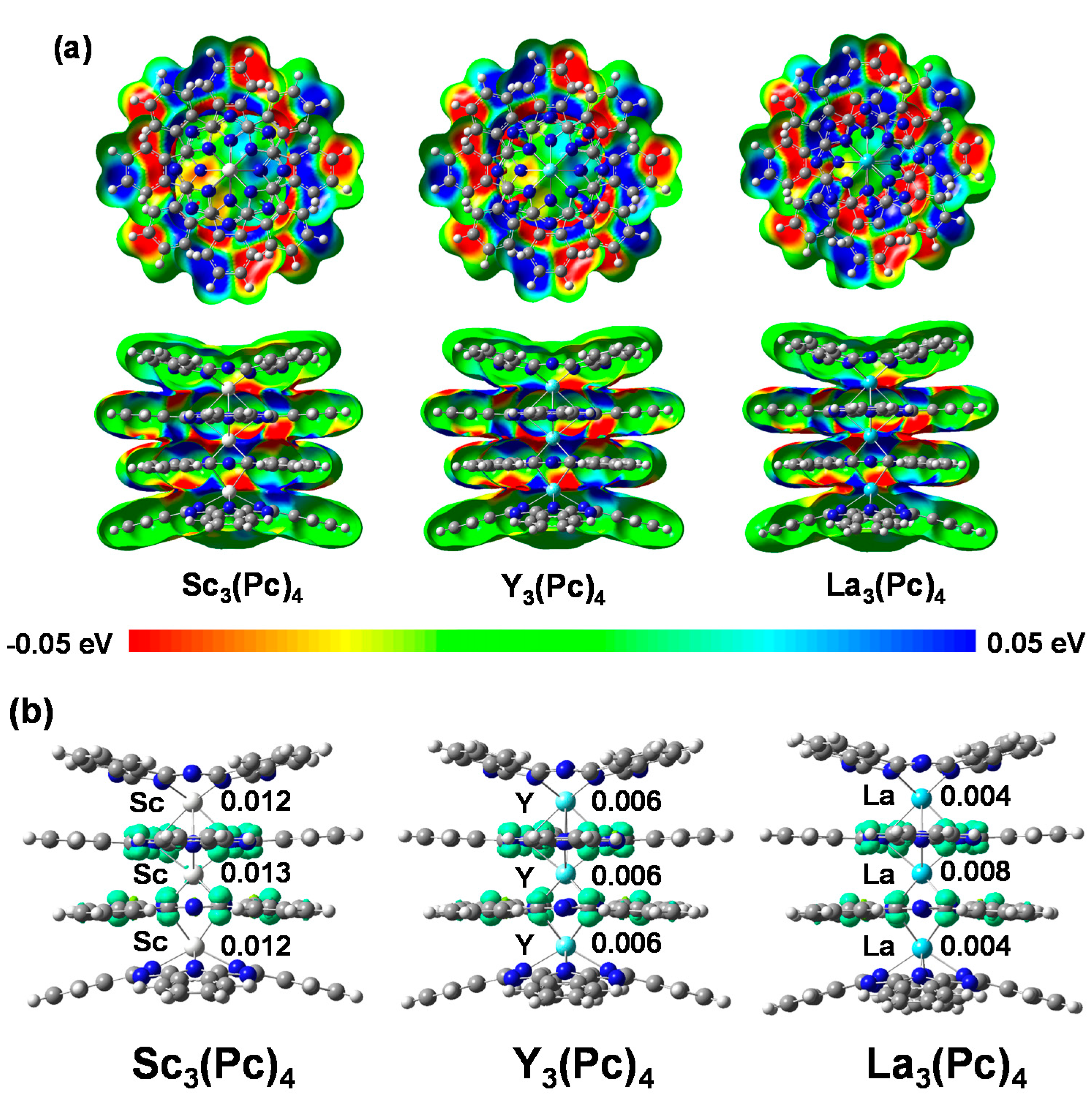
The electrostatic potential (ESP) mappings of M3(Pc)4 (M = Sc, Y, and La) complex are shown in Figure 2a. In the case of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4, the electrostatic potentials showed a slight difference in the distribution of red and blue color contrast near the pyrrole ring on the phthalocyanine ring. This behavior indicates polarization and electronegativity on the phthalocyanine ring, which yields a bias of the ESP phase with a lack of balance in charge near the metal ligand field. The lack of balance in ESP will cause a slight move in the chemical shifts of 13C and 14N-NMR in a low magnetic field. The chemical shifts of 14N-NMR on the phthalocyanine ring will originate from the nuclear spin interactions of nuclear quadrupole interactions based on EFG and η near the metal and nitrogen ligand. The magnetic interaction will depend on the extent of the charge and spin density distribution of the 3d orbital in the metal and the nitrogen ligand, and the hydrization of the 3d-sp2 orbital with an overlapping π-orbital on the phthalocyanine rings. The spin density distributions for M3(Pc)4 are shown in Figure 2b. The spin density distributions in the M atom for M3(Pc)4 were estimated to be 0.012, 0.013 in the outer and central Sc atoms for Sc3(Pc)4, 0.006 in all Y atoms for Y3(Pc)4, and 0.004 and 0.008 in outer and central La atoms for La3(Pc)4, respectively. The spin density distributions were delocalized by a considerable bias of the hybrization between the 3d orbital and sp2 orbital of the metal and the nitrogen ligand, overlapping the π-orbital on the phthalocyanine ring with considerable bias of charge distribution.
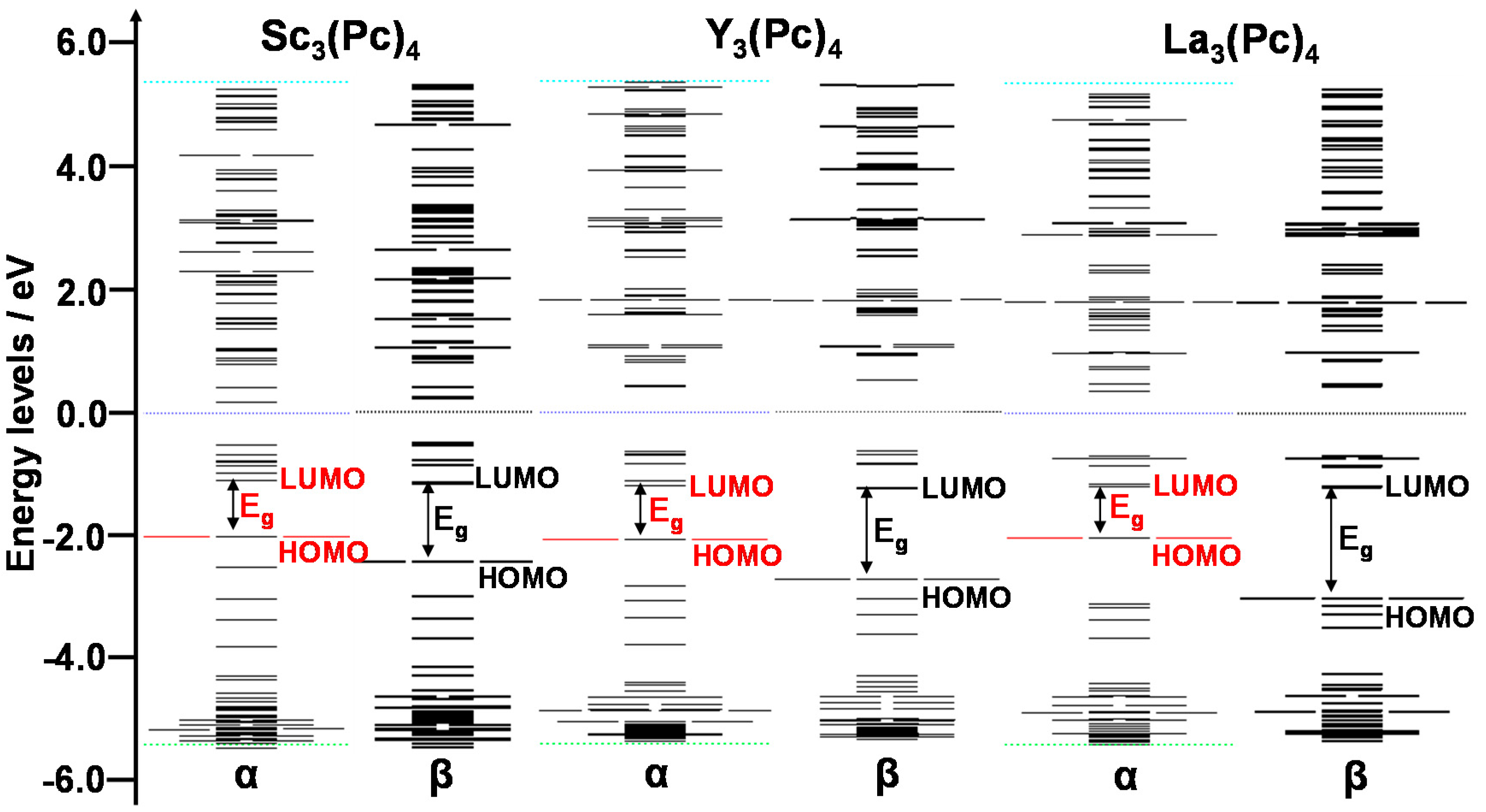
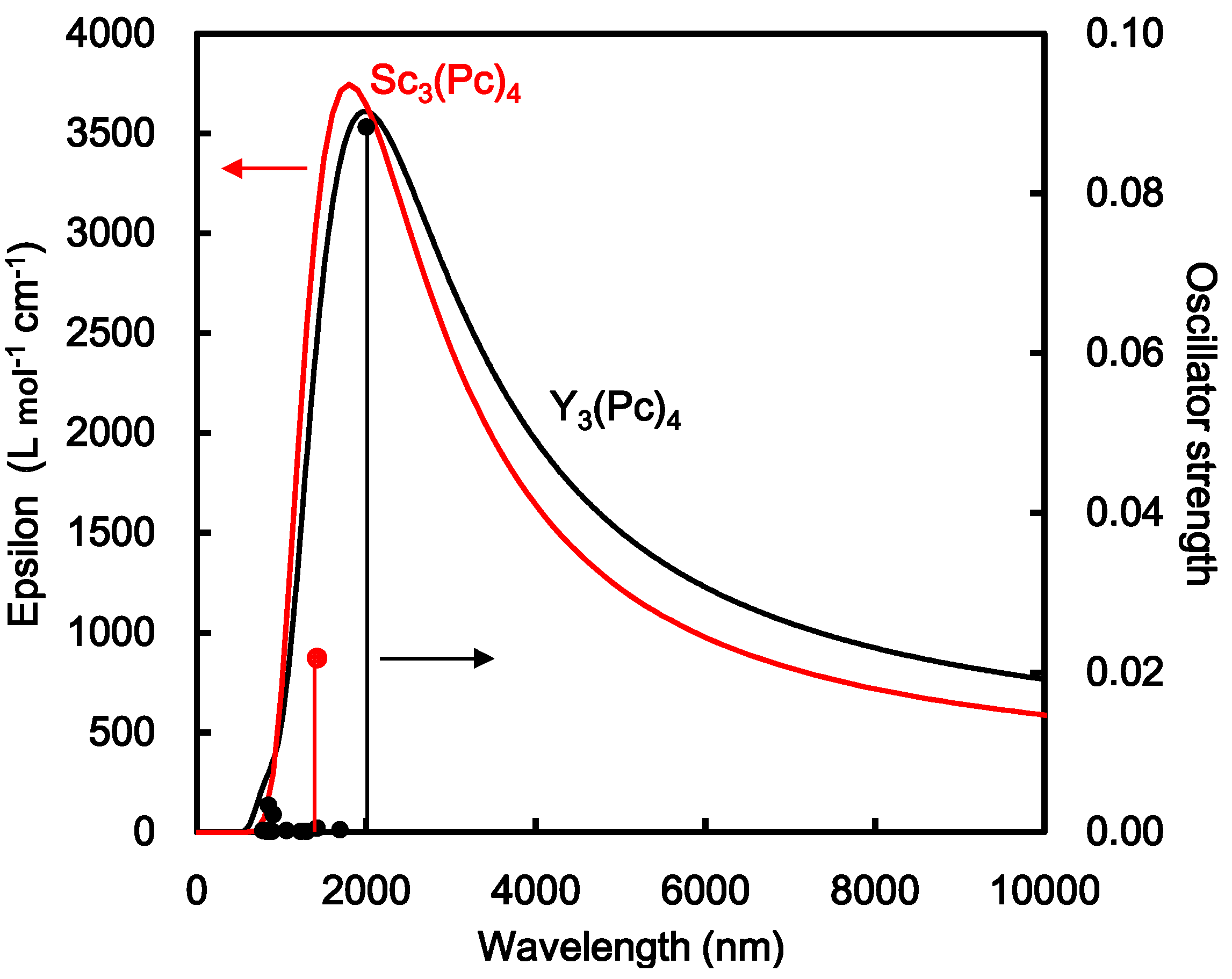
The energy levels and energy gaps between HOMO and LUMO were calculated by DFT. The energy levels near the frontier orbital of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4 are shown in Figure 3. The degeneracy of the energy levels near the frontier orbit was influenced by the deformed structures. In the case of Sc3(Pc)4, the degeneracy of energy levels was separated near the frontier orbital levels. In the case of Y3(Pc)4 and La3(Pc)4, the degenerated molecular orbitals with separated energy levels and wide energy gaps arose from a slightly deformed structure by the Jahan-Teller effect. The energy levels and energy gap near the frontier orbital also depended on the extent of the overlapping π-orbital on the phthalocyanine ring with decreasing metal radii. Excited states, the first excited state, absorption wavelength, and oscillation strength were calculated by time-dependent DFT (TD-DFT). Calculated UV-VIS-NIR spectra of Sc3(Pc)4 and Y3(Pc)4 are shown in Figure 4 and Table 2. In all cases, there was a wide absorption in the UV-VIS-NIR spectra and the excited process in the range of 1000–10,000 nm. In the case of Sc3(Pc)4 and Y3(Pc)4, the first excited state was estimated to be 1417 nm and 1997 nm, which was identified to be the Q absorption band of π-π* excited states. The excited process and energy gaps depended on the extent of overlapping π-orbitals on the phthalocyanine ring and the π-π interaction between the phthalocyanine rings with decreasing metal radii. The tendency of the narrowing energy gaps and long wavelength in UV-VIS-NIR spectra was conspicuous with the multi-decker phthalocyanine ring. The deformation of the molecular structure with the overlapping π-orbital on the phthalocyanine ring will influence the magnetic interaction and parameters of the chemical shift of 13C and 14N-NMR, isotropic g-tensor, V-tensor of EFG, and η, respectively.
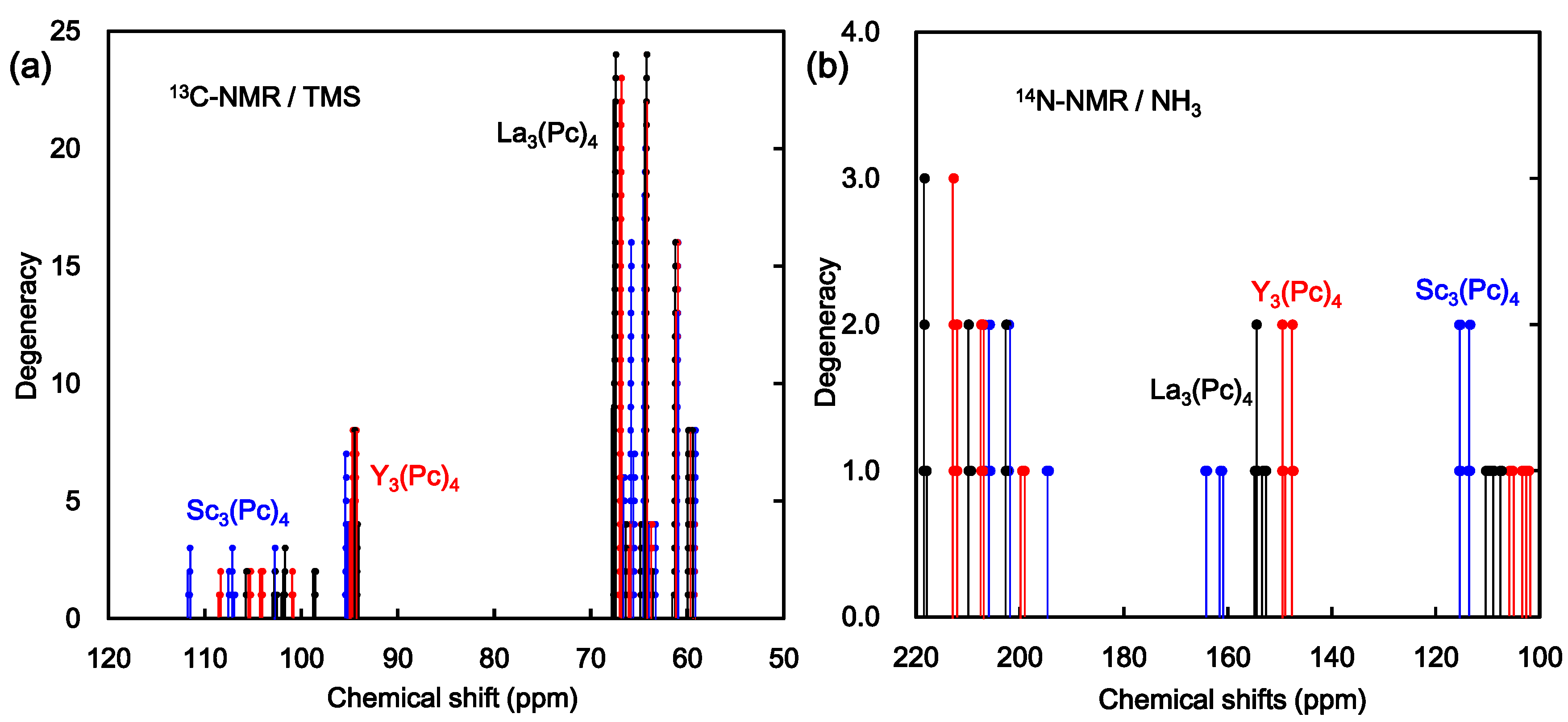
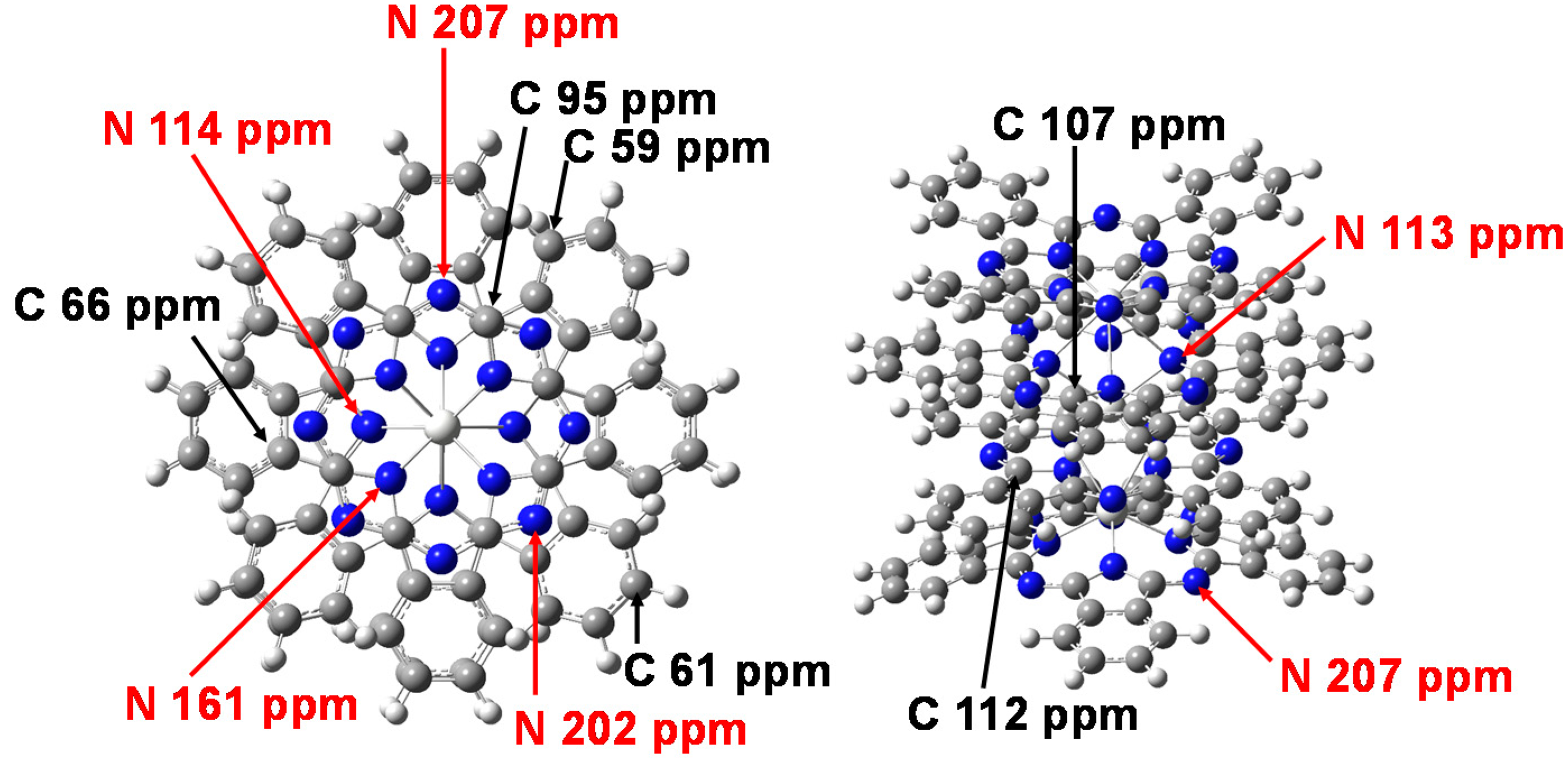
The chemical shifts of 13C and 14N-NMR were calculated by DFT using gauge-independent atomic orbitals (GIAO). The chemical shifts of 13C and 14N-NMR of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4 are shown in Figure 5. The chemical shifts of 13C-NMR, as referred by tetramethylsilane (TMS), were considerably separated around 100–110 ppm, 98 ppm, and 60–70 ppm. The chemical shifts of 14N-NMR, as referred by NH3, were considerably separated around 100–120 ppm, 150–160 ppm, and 190–220 ppm. Assignments of the chemical shifts of 13C and 14N-NMR of Sc3(Pc)4 are shown in Figure 6. The separation of chemical shifts of 13C and 14N-NMR were mainly caused by the deformed structure with considerable bias of the electron density distribution between the central metal and ligand. The chemical shift was based on nuclear spin magnetic interaction with a paramagnetic term of spin-local interaction, and the excited process from the ground state. The chemical shifts were explained by the theoretical study of the NMR spectra [32,33,34].
The quantum chemical calculation based on perturbation theory expects the electronic structure and magnetic interaction of nuclear quadrupole interaction based on EFG, η, and the excited process. Generally, a chemical shift is a sum of diamagnetic and paramagnetic terms. According to the Karplus-Pople equation, the chemical shift is mainly dominated by paramagnetic terms with spin-local interaction, hybridization of the molecular orbital, and nuclear quadrupole interaction based on EFG and η. The magnetic parameters of the chemical shift are based on the spin-local interaction, and the excited process from the ground state to excited states. The excited process depends on the extent of the overlapping π-orbital between the phthalocyanine rings with decreasing metal radii. The magnetic parameters, chemical shift, principle g-tensor, V-tensor of EFG, and η are considerably influenced by Jahn-Teller distortion of the 3d orbital in the metal ligand field on the phthalocyanine rings with considerable bias of the charge density distribution.
Mulliken atomic charge, spin density distribution, principle g-tensor, V-tensor of EFG, and η for Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4 are listed in Table 3. Especially, the Sc3(Pc)4 system had a Mulliken atomic charge of 0.32 e, 0.49 e, spin density of 0.0123, and 0.0133 for the scandium atom. The magnetic parameters of the principle g-tensor, V-tensor of EFG, and η for Sc1 in Sc3(Pc)4 were calculated to be gxx = 1.96695, gyy = 2.00180, gzz = 2.00300, Vxx = −0.0247, Vyy = −0.0356, Vzz = 0.0603, and η = 0.18, respectively. In other cases of M3(Pc)4 (M = Y and La), anisotropic magnetic parameters of the g-tensor was estimated to be gxx = 1.85325, gyy = 1.99824, gzz = 2.00298 for Y3(Pc)4, gxx = 1.54111, gyy = 1.98794, and gzz = 2.00117 for La3(Pc)4, respectively. The symmetry parameters of η for Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4 were decreased by reducing the bias of the V-tensor of EFG at the top and bottom of the M atom. The anisotropic parameters caused a wide separation and splitting of chemical shifts in a low magnetic field. The system had a polarization of charge distribution around the metal-ligand field in Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4. The magnetic parameters were based on the deformed phthalocyanine ring with considerable polarization of the charge distribution.
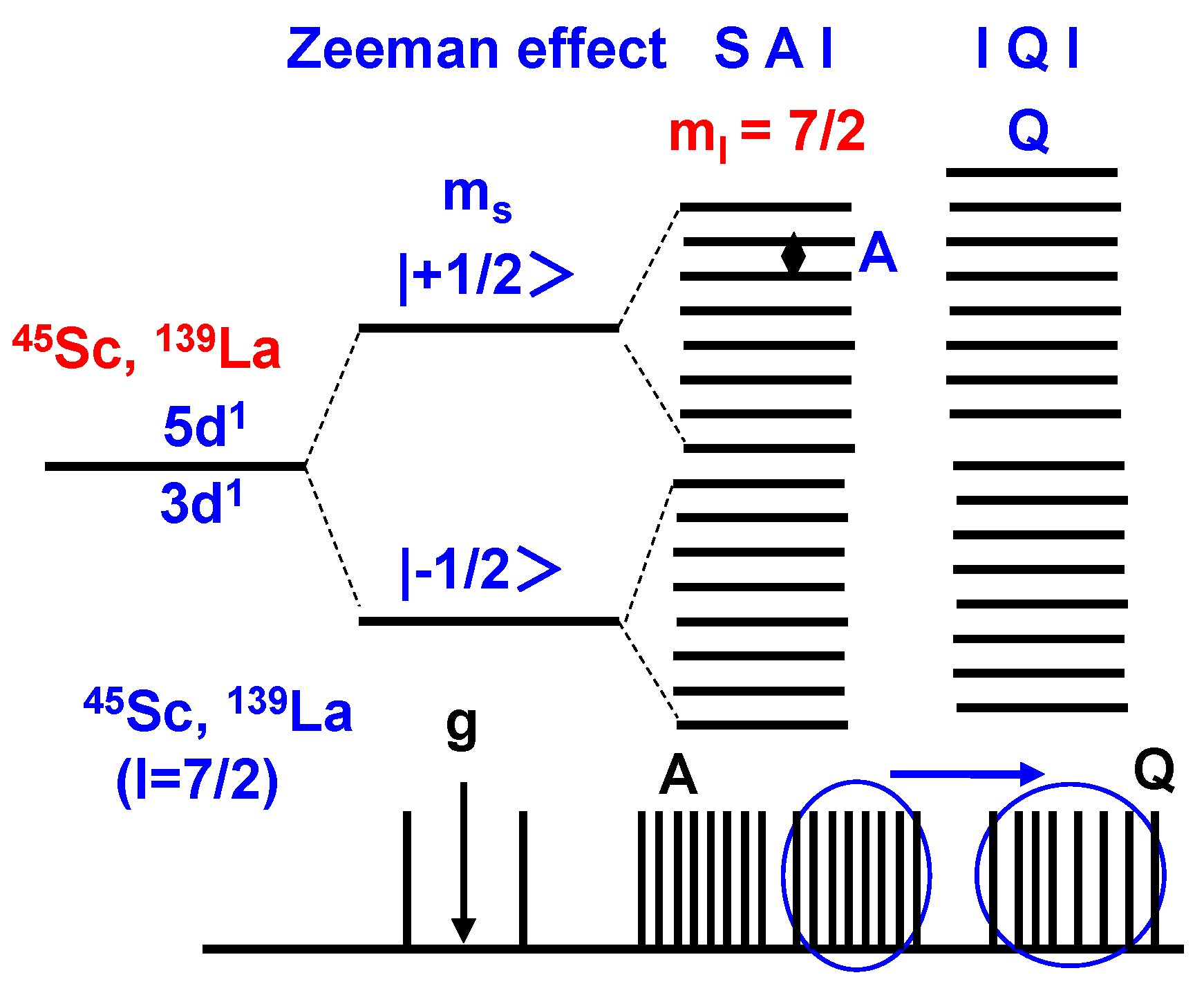
The allowed ESR and NMR transitions will be explained by an energy level diagram of 45Sc and 139La atoms, as shown in Figure 7. The degeneracy of the doublet splits each ground state into eight different quantum states in the magnetic field. The separated states are considerably shifted by the nuclear quadrupole interaction based on EFG and η of 45Sc and 139La at I = 7/2. The allowed ESR and NMR transitions are obeyed by the separated energy level. Spin Hamiltonian and g-tensor are written by following Equations (1) and (2). Spin Hamiltonian (HQ) regarding with nuclear quadrupole interaction is formulated by Equations (3) and (4). The magnetic parameters of ge and gN represent a g factor of free electrons and nuclear spin. β and βN means the Bohr magneton and nuclear magneton. A-tensor means the spin coupling constant in the hfs as electron-nuclear spin interaction. S and I mean the quantum numbers of electrons and nuclear spin. H represents the magnetic field. The parameters of φ, L, εm, and εp represent the wave function, operator of the orbital angular momentum, energy levels of each orbital, and the energy level at the ground state, respectively. eQ is the quadrupole moment, and eqzz is the maximum principal value of the Vzz along the z axis.
As noted by Equation (1), the spin Hamiltonian is a sum of Zeeman effects of electrons and nuclear spin under the magnetic field in first and second terms, electron-nuclear spin interaction (SAI) as a dipole-dipole interaction in the third term, and nuclear quadrupole interaction (IQI) in the fourth term based on EFG and η generated by the charge distribution around nucleus. As noted by Equation (2), the magnetic parameter of the g-tensor depends on spin local interaction, electron density distribution, and energy transition process from the ground state to the excited states. The excited transition is based on the resolved degeneracy of the 3d orbital with the overlapping π-orbital on the phthalocyanine ring.
In the case of Sc3(Pc)4 and La3(Pc)4 with nuclear spin at I = 7/2 of 45Sc, 139La, and I = 1 of 14N atoms, the influence of nuclear quadrupole interaction for 45Sc, 139La, and 14N atoms on the magnetic parameters of chemical shift needs to be considered in accordance with the first and second perturbation. As noted by Equations (3) and (4), the nuclear quadrupole interaction is related to the nuclear quadrupole moment based on the Q-tensor, EFG, and η generated by the charge distribution around the nucleus. The chemical shift of 13C and 14N-NMR would be remarkably shifted by nuclear quadrupole interaction in proportion to the extent of the V-tensor of EFG and η with a lack of balance in the charge density distribution. The chemical shifts of 45Sc, 139La, and 14N-NMR depends on multi-separate transition states, and spin dynamics based on the nuclear spin interaction in the spin lattice relaxation process. Multi-separate transition states are based on the hyperfine coupling between the electron and nuclear spin in 45Sc and 139La atoms at I = 7/2 with nuclear quadrupole interaction. The nuclear magnetic interaction, multi-separation, and spin oscillation are important factors to control the spin dynamics of the quantum spin bit in the spin lattice relaxation time.
The spin-lattice relaxation time of T1 of 45Sc, 139La, and 14N atoms in quadruple-decker phthalocyanines is important to control the entanglement of multi-spin gates and spin dynamics for quantum computers. The relaxation time of T1 of 45Sc, 139La, and 14N atoms for isotropic orientation motion with correlation time using η is given by Equation (5) [35,36]:
where I is the number of nuclear spin, e is electronic charge, h is Planck constant, Vzz is electronic field gradient, and τ is correlation time. The correlation time of τ = 2a2/9D related with sphere radius of a by Stokes law and the self-diffusion coefficient of D will be detected on the basis of the experimental data. Using the parameters of τ, η, Vzz, and eQ/h, the calculated rate of T1−1 will be compared with the measured rate by 45Sc, 139La, and 14N-NMR. From comparisons between the DFT quantum calculations and the experimental results using 45Sc, 139La, and 14N-NMR, the damping nuclear spin-oscillation of the nuclear spin for 45Sc, 139La, and 14N atoms of Mc3Pc4 will be generated by energy exchange through nuclear quadrupole interaction.
Electron and nuclear spin of the 3d transition metal in Mc3Pc4 in multiple states will work the magnetic interaction with entanglement and spin dynamics based on the nuclear spin interaction in the multi-parallel process. The resonance frequency of the nuclear spin 3d transition metal for M3Pc4 will be controlled independently. The nuclear quadrupole interaction is an important factor for controlling the spin gate with damping spin-oscillation in the long relaxation time. Considerable distorted electron and charge density distribution of M3Pc4 plays an important role in affecting the magnetic interaction of chemical shifts with splitting and separation in the relaxation time. The relaxation mechanism will be explained by the spin-lattice vibration modes (phonon) on the phthalocyanine ring as spin-lattice relaxation. The spin dynamics in the spin lattice relaxation depends on the molecular modification. The quadruple-decker metal phthalocyanines with the 3d transition metal in multiple states have an advantage to control the electronic structure and the magnetic parameters based on nuclear spin interaction in spin-lattice relaxation for applications in single-molecular magnets and quantum computers.
3. Calculation Method
The isolated molecular structures of quadruple-decker metal phthalocyanines with 3d transition metals, such as scandium, yttrium, and lanthanum atoms (M3(Pc)4: M = Sc, Y, and La) in the doublet state were optimized by ab initio quantum chemical calculation using unrestricted Hartree–Fock as the unrestricted open-shell self-consistent field calculation and DFT calculation using the Gaussian 03 program (Gaussian, Inc.) with Becke’s three-parameter hybrid functional UB3LYP [37,38] method in order of STO-3G*, 3-31G*, 6-31G*, and LANL2MB as the basis set. The HOMO, HOMO − 1, LUMO, LUMO + 1, and the HOMO-LUMO band gap (Eg) were calculated. Mulliken atomic charges, electron density distributions, and the ESP image on the phthalocyanine ring were estimated by Mulliken population analysis. After calculating the optimized structures and the electronic structures near the frontier molecular orbital, the isotropic chemical shifts of 13C and 14N-NMR, principle g-tensors, V-tensors of EFG, and the η of Sc, Y, and La atoms in the quadruple-decker phthalocyanines were calculated by DFT using the gauge-independent atomic orbital (GIAO) [39] with hybrid functional UB3LYP method, and LANL2MB as the basis set. UV-VIS-NIR spectra and excited processes of the optimized structures were calculated by TD-DFT with UB3LYP with LANL2MB as the basis set [40]. The TD-DFT intense excitation energies, molar absorption coefficients (Epsilon), oscillator strengths, and transition process expressed in terms of the frontier molecular orbitals were also computed.
4. Conclusions
The electronic structures, and optical and magnetic properties of quadruple-decker phthalocyanines with 3d transition metals in multiple states were studied for applications in magnetic devices that manipulate nuclear-spin-based quantum bits in quantum computing. The electronic structures, chemical shifts of 13C and 14N-NMR, the principle g-tensors and V-tensors of EFG, η, and UV-VIS-NIR spectra were calculated by density functional theory. The electron density distributions at HOMO and LUMO of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4 were delocalized on the π-orbital of the phthalocyanine rings with hybrization of the 3d orbital near metal ligand field. The narrow Eg of Sc3(Pc)4 and Y3(Pc)4 was based on the strong phthalocyanine ring-ring interactions with decreasing metal radii, suggesting a broad absorption of UV-VIS-NIR spectra for the π-π* transition process of the phthalocyanine rings. Considerable solvable perturbation of degeneracy of 3d orbital for M3(Pc)4 influenced the magnetic parameters of chemical shifts in 13C and 14N-NMR, the isotropic g-tensor, the V-tensor of EFG, and η. The considerable splitting and separations on 13C and 14N-NMR chemical shifts of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4 were obeyed by Jahn-Teller splitting of the 3d orbital, the perturbation of the metal ligand field on the phthalocyanine ring, the electronic structure, and magnetic interaction of the nuclear quadrupole interaction. The magnetic parameters of the principle g-tensor, the V-tensor of EFG, and η depended on the deformation of the molecular structures of the complex with the considerable deviation of the electron and charge density distribution. The magnetic properties will be controlled by the molecular modification with a lack of balance in electron and charge density distribution on the conjugated system of the complex. The considerable solvable perturbation of the metal ligand field on the multi-decker of the phthalocyanine ring will influence the magnetic resonance frequency, splitting, separation of the chemical shift, and spin dynamics in the relaxation time. The quadruple-decker metal phthalocyanines with 3d transition metals have an advantage to control the electronic structure and the magnetic parameters based on the nuclear spin interaction in the spin-lattice relaxation for applications in single-molecular magnets and quantum computer.
Author Contributions
A. Suzuki and T. Oku conceived and designed the quantum calculations; A. Suzuki performed the quantum calculations; A. Suzuki analyzed the data; A. Suzuki and T. Oku contributed reagents/materials/analysis tools; A. Suzuki wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Warner, M.; Din, S.; Tupitsyn, I.S.; Morley, G.W.; Stoneham, A.M.; Gardener, J.A.; Wu, Z.; Fisher, A.J.; Heutz, S.; Kay, C.W.M.; et al. Potential for spin-based information processing in a thin-film molecular semiconductor. Nature 2013, 503, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.J.L.; Tyryshkin, A.M.; Ardavan, A.; Benjamin, S.C.; Porfyrakis, K.; Lyon, S.A.; Briggs, G.A.D. The N@C60 nuclear spin qubit: Bang-bang decoupling and ultrafast phase gates. Phys. Status Solidi B 2006, 243, 3028–3031. [Google Scholar] [CrossRef]
- Popov, A.A.; Shustova, N.B.; Svitova, A.L.; Mackey, M.A.; Coumbe, C.E.; Phillips, J.P.; Stevenson, S.; Strauss, S.H.; Boltalina, O.V.; Dunsch, L. Redox-tuning endohedral fullerene spin states: From the dication to the trianion radical of Sc3N@C80(CF3)2 in five reversible single-electron steps. Chem. Eur. J. 2010, 16, 4721–4724. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Oku, T. Electronic structure and magnetic properties of endohedral metallofullerenes based on mixed metal cluster within fullerene cage with trifluoromethyl groups. J. Phys. Conf. Ser. 2013, 433, 012004. [Google Scholar] [CrossRef]
- Suzuki, A.; Oku, T. Influence of chemical substitution in ScxY3-xN@C80(CF3)n endohedral fullerenes on magnetic properties. Physica B 2013, 428, 18–26. [Google Scholar] [CrossRef]
- Svitova, A.L.; Krupskaya, Y.; Samoylova, N.; Kraus, R.; Geck, J.; Dunsch, L.; Popov, A.A. Magnetic moments and exchange coupling in nitride cluster fullerenes GdxSc3–xN@C80 (x = 1–3). Dalton Trans. 2014, 43, 7387–7390. [Google Scholar] [CrossRef] [PubMed]
- Dreiser, J.; Westerström, R.; Zhang, Y.; Popov, A.A.; Dunsch, L.; Krämer, K.; Liu, S.X.; Decurtins, S.; Greber, T. The metallofullerene field-induced single-ion magnet HoSc2N@C80. Chem. A Eur. J. 2014, 20, 13536–13540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Krylov, D.; Rosenkranz, M.; Schiemenz, S.; Popov, A.A. Magnetic anisotropy of endohedral lanthanide ions: Paramagnetic NMR study of MSc2N@C80-Ih with M running through the whole 4f row. Chem. Sci. 2015, 6, 2328–2341. [Google Scholar] [CrossRef]
- Suzuki, A.; Oku, T. Electronic structures and magnetic properties of Sc2YN@C80(CF3)2 dimer. Jpn. J. Appl. Phys. 2014, 53, 05FN01. [Google Scholar] [CrossRef]
- Abe, Y.; Suzuki, A.; Oku, T. Electronic structures and magnetic properties of Sc4O2@C80(CF3)n (n = 2 and 4). Jpn. J. Appl. Phys. 2014, 53, 05FN02. [Google Scholar] [CrossRef]
- Valencia, R.; Fortea, A.R.; Stevenson, S.; Balch, A.L.; Poblet, J.M. Electronic structures of scandium oxide endohedral metallofullerenes, Sc4(μ3-O)n@Ih-C80 (n = 2, 3). Inorg. Chem. 2009, 48, 5957–5961. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Oku, T. Geometrical effects of (14N@C60)2, 14N@C60 and C59N endohedral fullerenes within single-walled carbon nanotube as peapods on electronic structure and magnetic properties. Physica B 2011, 406, 3274–3278. [Google Scholar] [CrossRef]
- Suzuki, A.; Oku, T. Electronic structure and magnetic properties of 31P@C60-SWCNT as peapods. J. Phys. Conf. Ser. 2012, 352, 012012. [Google Scholar] [CrossRef]
- Warner, J.H.; Watt, A.A.R.; Ge, L.; Porfyrakis, K.; Akachi, T.; Okimoto, H.; Ito, Y.; Ardavan, A.; Montanari, B.; Jefferson, J.H.; et al. Dynamics of paramagnetic metallofullerenes in carbon nanotube peapods. Nano Lett. 2008, 8, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Mizuochi, N.; Neumann, P.; Rempp, F.; Beck, J.; Jacques, V.; Siyushev, P.; Nakamura, K.; Twitchen, D.J.; Watanabe, H.; Yamasaki, S.; et al. Coherence of single spins coupled to a nuclear spin bath of varying density. Phys. Rev. B 2009, 80, 041201. [Google Scholar] [CrossRef]
- Vandersypen, L.M.K.; Steffen, M.; Breyta, G.; Yannoni, C.S.; Sherwood, M.H.; Chuang, I.L. Experimental realization of Shor’s quantum factoring algorithm using nuclear magnetic resonance. Nature 2001, 414, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.; Tesi, L.; Morra, E.; Chiesa, M.; Sorace, L.; Sessoli, R. Room-temperature quantum coherence and Rabi oscillations in vanadyl phthalocyanine: Toward multifunctional molecular spin qubits. J. Am. Chem. Soc. 2016, 138, 2154–2157. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.S.; Deng, Y.F.; Zheng, Y.Z. The rise of single-ion magnets as spin qubits. Magnetochemistry 2016, 2, 40. [Google Scholar] [CrossRef]
- Thiele, S.; Balestro, F.; Ballou, R.; Klyatskaya, S.; Ruben, M.; Wernsdorfer, W. Electrically driven nuclear spin resonance in single-molecule magnets. Science 2014, 344, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Komeda, T.; Isshiki, H.; Liu, J.; Zhang, Y.F.; Lorente, N.; Katoh, K.; Breedlove, B.K.; Yamashita, M. Observation and electric current control of a local spin in a single-molecule magnet. Nat. Commun. 2011, 2, 217. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, H.; Liu, J.; Katoh, K.; Yamashita, M.; Miyasaka, H.; Breedlove, B.K.; Takaishi, S.; Komeda, T. Scanning tunneling microscopy investigation of tris(phthalocyaninato)yttrium triple-decker molecules deposited on Au(111). J. Phys. Chem. C 2010, 114, 12202–12206. [Google Scholar] [CrossRef]
- Shang, H.; Zeng, S.; Wang, H.; Dou, J.; Jiang, J. Peripheral substitution: An easy way to tuning the magnetic behavior of tetrakis(phthalocyaninato)dysprosium(III) SMMs. Sci. Rep. 2015, 5, 8838. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Oku, T. Effects of central metal on electronic structure, magnetic properties, infrared and Raman spectra of double-decker phthalocyanine. Appl. Surf. Sci. 2016, 380, 127–134. [Google Scholar] [CrossRef]
- Suzuki, A.; Oku, T. Theoretical study of NMR, infrared and Raman spectra on triple-decker phthalocyanines. AIP Conf. Proc. 2016, 1709, 020013. [Google Scholar] [CrossRef]
- Katoh, K.; Komeda, T.; Yamashita, M. Surface morphologies, electronic structures, and Kondo effect of lanthanide(III)-phthalocyanine molecules on Au(111) by using STM, STS and FET properties for next generation devices. Dalton Trans. 2010, 39, 4708–4723. [Google Scholar] [CrossRef] [PubMed]
- Urdampilleta, M.; Klayatskaya, S.; Ruben, M.; Wernsdorfer, W. Magnetic interaction between a radical spin and a single-molecule magnet in a molecular spin-valve. ACS Nano 2015, 9, 4458–4464. [Google Scholar] [CrossRef] [PubMed]
- Horii, Y.; Katoh, K.; Yasuda, N.; Breedlove, B.K.; Yamashita, M. Effects of f-f interactions on the single-molecule magnet properties of terbium(III)–phthalocyaninato quintuple-decker complexes. Inorg. Chem. 2015, 54, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Horii, Y.; Katoh, K.; Cosquer, G.; Breedlove, B.K.; Yamashita, M. Weak DyIII–DyIII interactions in DyIII–phthalocyaninato multiple-decker single-molecule magnets effectively suppress magnetic relaxation. Inorg. Chem. 2016, 55, 11782–11790. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Katoh, K.; Breedlove, B.K.; Yamashita, M. Controlling the dipole-dipole interactions between terbium(III) phthalocyaninato triple-decker moieties through spatial control using a fused phthalocyaninato ligand. Inorg. Chem. 2013, 52, 13555–13561. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Matsumura, K.; Ishikawa, N. Influence of intramolecular f-f Interactions on nuclear spin driven quantum tunneling of magnetizations in quadruple-decker phthalocyanine complexes containing two terbium or dysprosium magnetic centers. J. Phys. Chem. A 2013, 117, 10447–10454. [Google Scholar] [CrossRef] [PubMed]
- Slater, J.C. Atomic Radii in Crystals. J. Chem. Phys. 2016, 41, 3199. [Google Scholar] [CrossRef]
- Sumimoto, M.; Kawashima, Y.; Horia, K.; Fujimoto, H. Theoretical study on the stability of double-decker type metal phthalocyanines, M(Pc)2 and M(Pc)2+ (M = Ti, Sn and Sc): A critical assessment on the performance of density functionals. Phys. Chem. Chem. Phys. 2015, 17, 6478–6483. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Iino, T.; Kaizu, Y. Study of 1H-NMR spectra of dinuclear complexes of heavy lanthanides with phthalocyanines based on separation of the effects of two paramagnetic centers. J. Phys. Chem. A 2003, 107, 7879–7884. [Google Scholar] [CrossRef]
- Polovkova, M.A.; Martynov, A.G.; Birin, K.P.; Nefedov, S.E.; Gorbunova, Y.G.; Tsivadze, A.Y. Determination of the structural parameters of heteronuclear(phthalocyaninato)bis(crownphthalo cyaninato)lanthanide(III)triple-deckers in solution by simultaneous analysis of NMR and single-crystal X-ray data. Inorg. Chem. 2016, 55, 9258–9269. [Google Scholar] [CrossRef] [PubMed]
- Abragam, A. Principles of Nuclear Magnetism; Oxford Clarendon Press: Oxford, UK, 1961. [Google Scholar]
- Suzuki, A.; Oku, T. Role of scandium atom in ScxY3-xN@C80(CF3)n (n = 0, 2) on nuclear quadrupole interaction, electric field gradient and asymmetric parameters. Microelectron. Eng. 2014, 126, 113–117. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1998, 98, 1372–1377. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Liu, J.; Liang, W. Analytical approach for the excited-state Hessian in time-dependent density functional theory: Formalism, implementation, and performance. J. Chem. Phys. 2011, 135, 184111. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Optimized structures and electron density distribution (isovalue 0.02 a.u.) at HOMO and LUMO of (a) Sc3(Pc)4; (b) Y3(Pc)4; and (c) La3(Pc)4, calculated by DFT using hybrid functional calculation UB3LYP and LANL2BM as the basis set. Dark blue atoms are nitrogen, dark gray atoms are carbon, and light white atoms are hydrogen.
Figure 1.
Optimized structures and electron density distribution (isovalue 0.02 a.u.) at HOMO and LUMO of (a) Sc3(Pc)4; (b) Y3(Pc)4; and (c) La3(Pc)4, calculated by DFT using hybrid functional calculation UB3LYP and LANL2BM as the basis set. Dark blue atoms are nitrogen, dark gray atoms are carbon, and light white atoms are hydrogen.

Figure 2.
(a) ESP mapping with negative and positive potential values in red and blue; and (b) spin density distribution of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4.
Figure 2.
(a) ESP mapping with negative and positive potential values in red and blue; and (b) spin density distribution of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4.
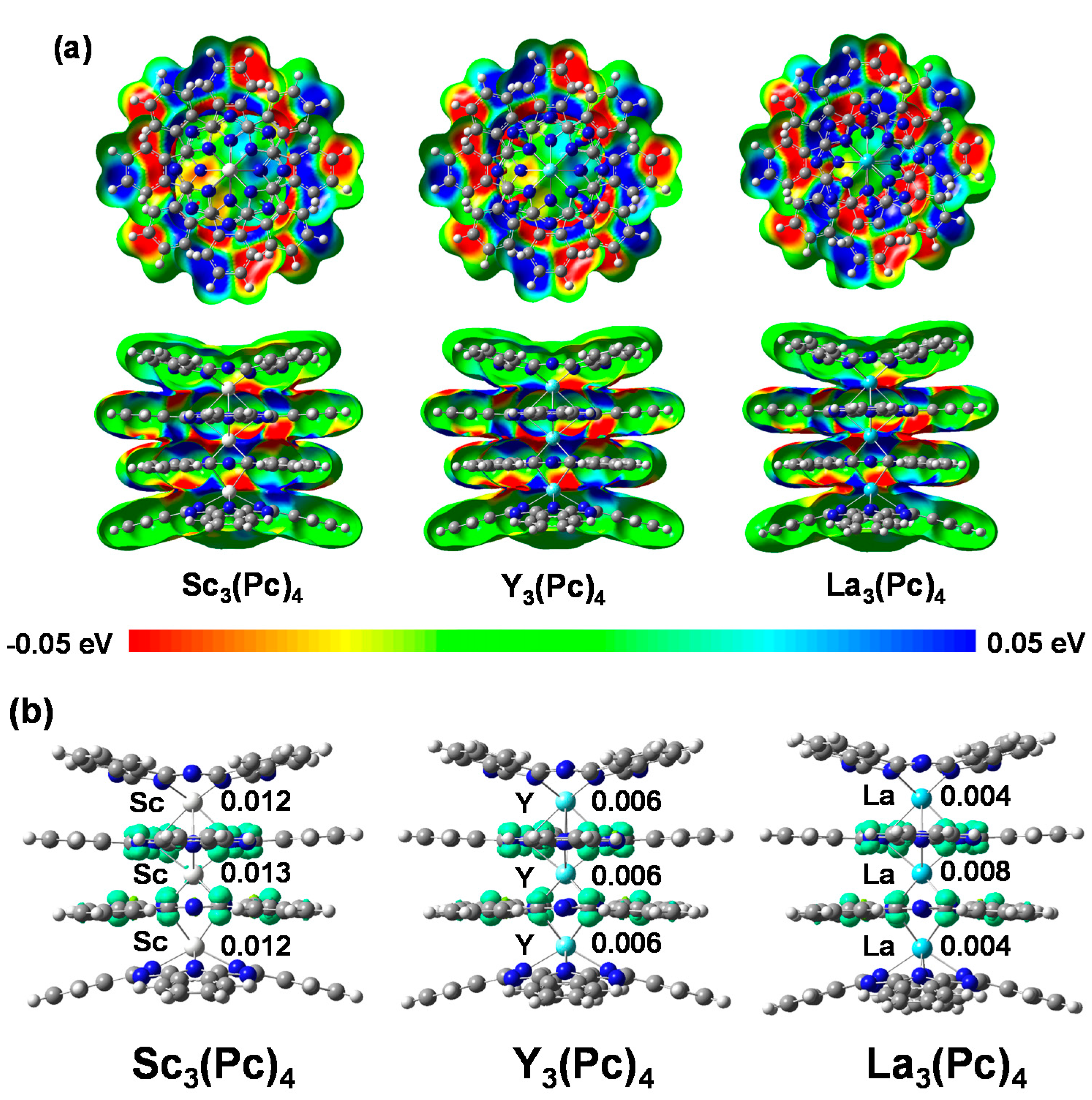
Figure 3.
Energy levels near the frontier orbital and energy gaps (Eg) of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4. α and β correspond to upward and downward spins, respectively.
Figure 3.
Energy levels near the frontier orbital and energy gaps (Eg) of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4. α and β correspond to upward and downward spins, respectively.
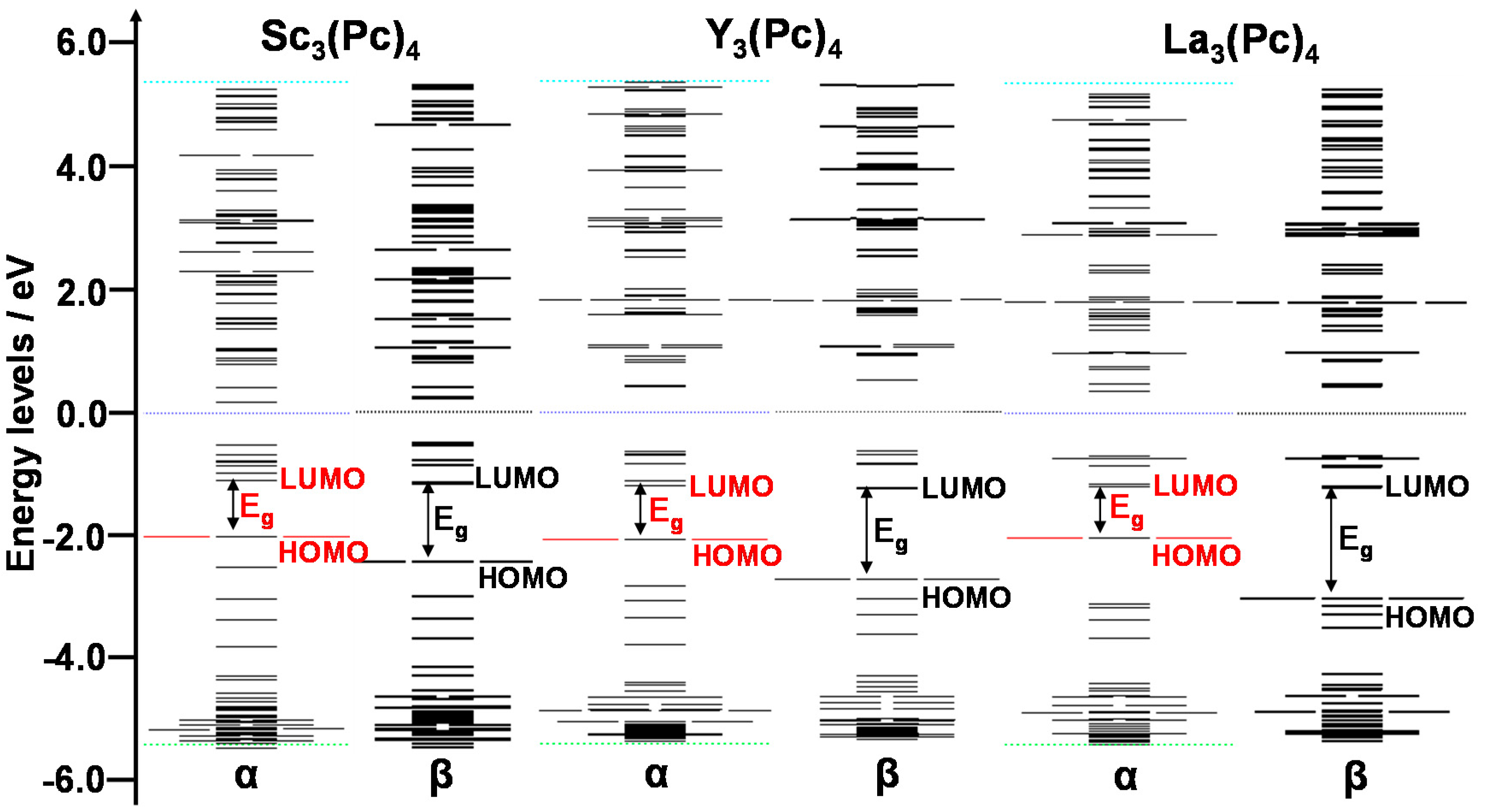
Figure 4.
Calculated UV-VIS-NIR spectra of Sc3(Pc)4 and Y3(Pc)4.

Figure 5.
Calculated chemical shifts of (a) 13C-NMR; and (b) 14N-NMR for Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4.
Figure 5.
Calculated chemical shifts of (a) 13C-NMR; and (b) 14N-NMR for Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4.

Figure 6.
Assignments of the chemical shifts of 13C-NMR and 14N-NMR for Sc3(Pc)4.
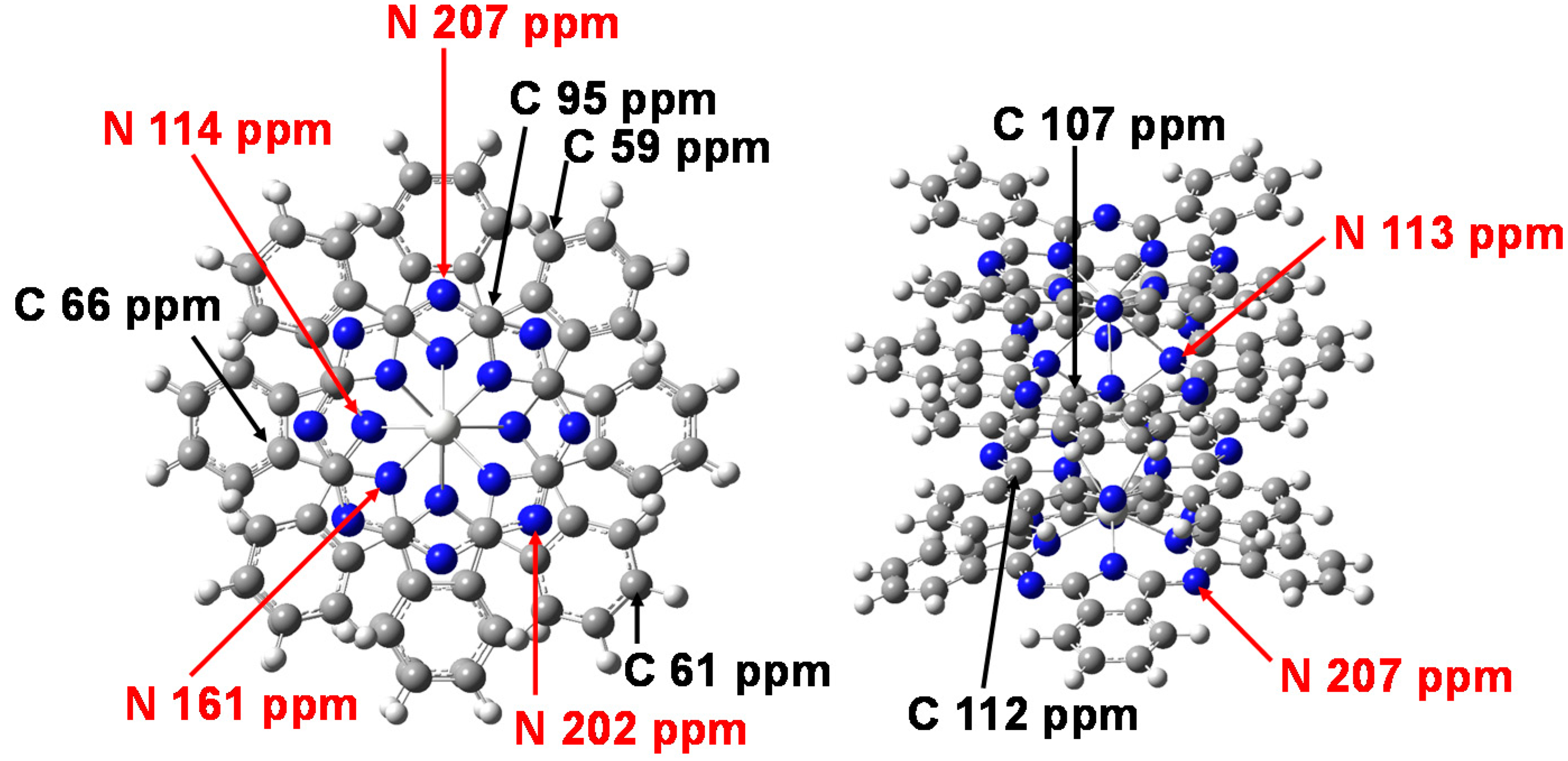
Figure 7.
Schematic illustration of the energy level diagram for 45Sc and 139La in a magnetic field, which shows the allowed ESR and NMR transitions.
Figure 7.
Schematic illustration of the energy level diagram for 45Sc and 139La in a magnetic field, which shows the allowed ESR and NMR transitions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Atomic radii of the central metal, bond lengths, and angles between the metal atom and nitrogen ligand in the optimized structure of M3(Pc)4.
Table 1.
Atomic radii of the central metal, bond lengths, and angles between the metal atom and nitrogen ligand in the optimized structure of M3(Pc)4.
| M3(Pc)4 | Atomic Radii (pm) [31] | M–N (Å) | Angle N–M–N (°) |
|---|---|---|---|
| Sc3(Pc)4 | 160 | 2.21 | 126 |
| 2.39 | 105 | ||
| 2.21 | 126 | ||
| Y3(Pc)4 | 180 | 2.33 | 119 |
| 2.48 | 104 | ||
| 2.33 | 119 | ||
| La3(Pc)4 | 195 | 2.46 | 113 |
| 2.60 | 99 | ||
| 2.46 | 113 |
Table 2.
Energy levels, wavelengths, and oscillator strengths in the excitation processes of Sc3(Pc)4 and Y3(Pc)4.
Table 2.
Energy levels, wavelengths, and oscillator strengths in the excitation processes of Sc3(Pc)4 and Y3(Pc)4.
| Sc3(Pc)4 | No. | Energy (eV) | Wavelength (nm) | Oscillator Strength |
|---|---|---|---|---|
| Excited state | 1 | 0.8747 | 1417 | 0.0218 |
| Y3(Pc)4 | ||||
| Excited state | 1 | 0.6209 | 1997 | 0.0883 |
| 2 | 1.3731 | 903 | 0.0022 | |
| 3 | 1.4644 | 847 | 0.0034 |
Table 3.
Magnetic parameters, principle g-tensors, V-tensors of EFG, and η of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4.
Table 3.
Magnetic parameters, principle g-tensors, V-tensors of EFG, and η of Sc3(Pc)4, Y3(Pc)4, and La3(Pc)4.
| Sc3(Pc)4 | Mulliken Atomic Charge | Spin Density | g-tensor | EFG | η | ||||
|---|---|---|---|---|---|---|---|---|---|
| gxx | gyy | gzz | Vxx | Vyy | Vzz | ||||
| Sc1 | 0.32 | 0.0123 | 1.96695 | 2.00180 | 2.00300 | −0.0247 | −0.0356 | 0.0603 | 0.18 |
| Sc2 | 0.49 | 0.0133 | −0.0153 | −0.0370 | 0.0523 | 0.42 | |||
| Sc3 | 0.32 | 0.0122 | −0.0246 | −0.0356 | 0.0602 | 0.18 | |||
| Y3(Pc)4 | |||||||||
| Y1 | 0.31 | 0.0064 | 1.85325 | 1.99824 | 2.00298 | −0.0057 | −0.0079 | 0.0136 | 0.16 |
| Y2 | 0.53 | 0.0059 | −0.0063 | −0.0092 | 0.0155 | 0.19 | |||
| Y3 | 0.31 | 0.0064 | −0.0057 | −0.0079 | 0.0136 | 0.16 | |||
| La3(Pc)4 | |||||||||
| La1 | 0.41 | 0.0039 | 1.54111 | 1.98794 | 2.00117 | −0.0073 | −0.0087 | 0.0160 | 0.09 |
| La2 | 0.66 | 0.0078 | −0.0071 | −0.0086 | 0.0157 | 0.10 | |||
| La3 | 0.41 | 0.0039 | −0.0073 | −0.0087 | 0.0160 | 0.09 | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Suzuki, A.; Oku, T. Electronic Structures, and Optical and Magnetic Properties of Quadruple-Decker Phthalocyanines. Magnetochemistry 2017, 3, 21. https://doi.org/10.3390/magnetochemistry3020021
AMA Style
Suzuki A, Oku T. Electronic Structures, and Optical and Magnetic Properties of Quadruple-Decker Phthalocyanines. Magnetochemistry. 2017; 3(2):21. https://doi.org/10.3390/magnetochemistry3020021
Chicago/Turabian StyleSuzuki, Atsushi, and Takeo Oku. 2017. "Electronic Structures, and Optical and Magnetic Properties of Quadruple-Decker Phthalocyanines" Magnetochemistry 3, no. 2: 21. https://doi.org/10.3390/magnetochemistry3020021
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.