
Mini-Review: Challenges in Newborn Screening for Urea Cycle Disorders

{kind=link}
Abstract
: Urea cycle disorders (UCDs) comprise a group of recessive and one X-linked inherited errors of protein metabolism that, due to insufficient detoxification of excess nitrogen, can lead to severe neurological disease. The key feature, but at the same time only a surrogate marker of UCDs, is the resulting mild to severe hyperammonemia. Biochemical analysis is needed to strengthen the suspicion of any underlying UCD but remains for the majority of cases rather indicative than diagnostic due to the lack of definite markers. Thus, in order to confirm a specific UCD, mutation analysis or enzyme assays are the methods of choice. Because of the drastic clinical complications of severe hyperammonemia, an early diagnosis before onset of symptoms would be desirable. The best way to achieve this would be to implement a general newborn screening for these disorders. However, there are several challenges that need to be overcome before newborn screening for UCDs can be introduced. This review will briefly describe the technical and clinical challenges involved in newborn screening for UCDs and will then discuss current experiences with this approach.1. Introduction
The great success of newborn screening (NBS) for a growing list of mainly but not only metabolic diseases renders the inclusion of further target disorders attractive. A natural choice would be the group of urea cycle disorders (UCDs) as they fulfill most of the criteria set for neonatal mass screening [1,2,3]. However, at the same time controversy remains regarding several important aspects including the choice and availability of biochemical markers for NBS, the question whether early detection of patients will benefit their outcome, and the situation of UCD patients with mild disease [4,5,6,7,8,9,10,11,12].
This mini-review will present an overview of the current situation of NBS for UCDs in different countries. In addition, we will discuss some of the main challenges of NBS for UCDs and hereby focus on the technical and clinical challenges. A major technical challenge is still the sensitivity of NBS for the proximal UCDs. Major clinical challenges lie in the implications of mass screening, identifying severely affected patients with very early onset of their disease possibly before NBS results are available, as well as in the group of patients with rather mild disease in whom any benefit from pre-symptomatic detection of disease remains controversial.
2. Technical Challenges
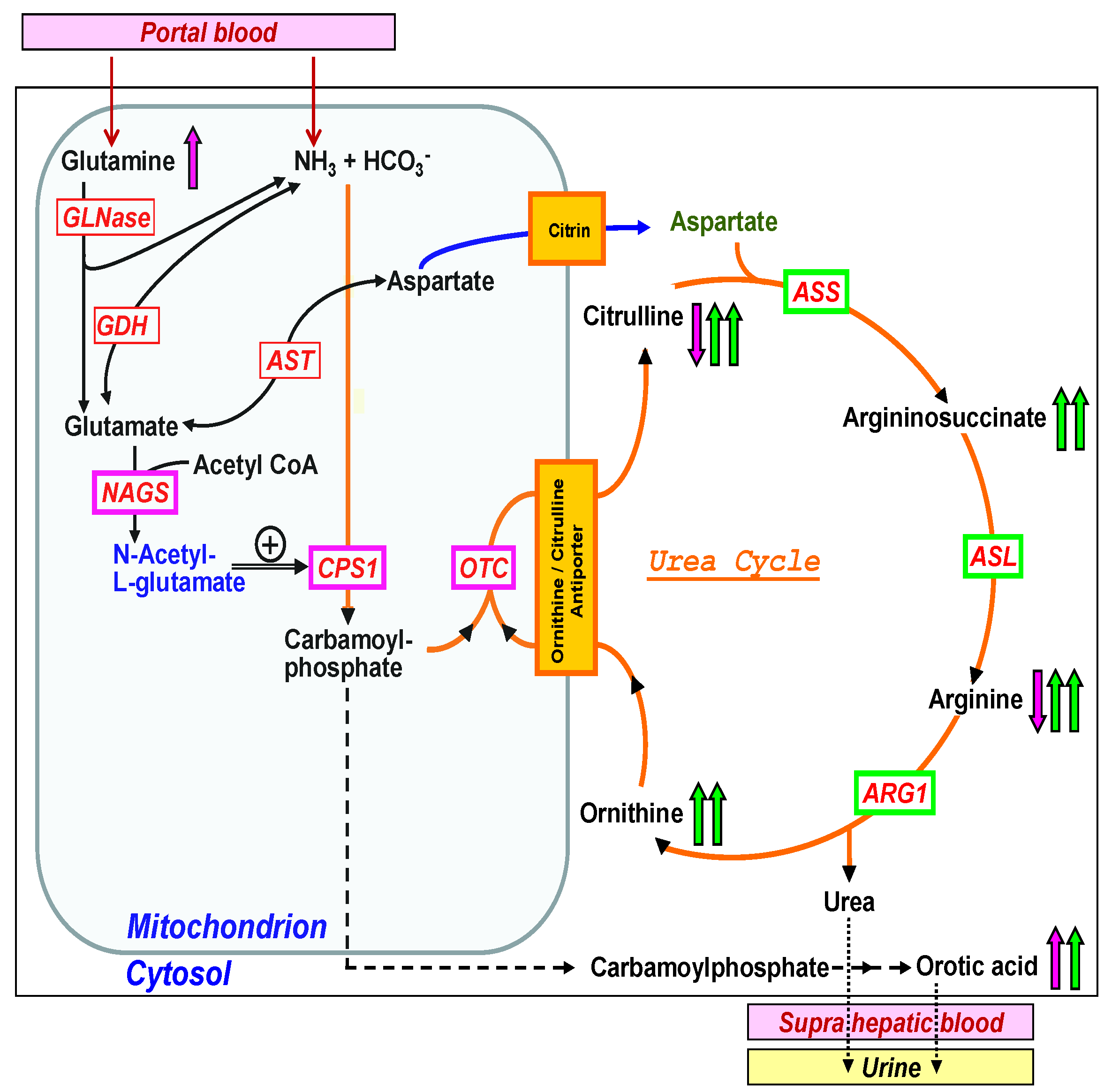
Developing a mass screening test requires the availability of a parameter that indicates the presence of a disease. Commonly, metabolites are used as screening parameters, but this still poses a challenge, as already described 34 years ago [13]. As the direct measurement of ammonia is not feasible in NBS, glutamine would be another metabolite that is generally elevated in UCDs [14]. However, glutamine is unfortunately highly unstable and thus spontaneous glutamate and pyroglutamate formation lead to false low glutamine levels [12], rendering glutamine currently not a suitable screening parameter. Likewise, orotic acid, though often elevated in the urine of patients with ornithine transcarbamylase (OTC) deficiency, cannot be used as a marker in blood.
Therefore, in the absence of a universal screening parameter for all UCDs, specific parameters or the combination and ratios of specific parameters may be considered (Figure 1). Despite the fact that all UCDs affect the function of the urea cycle and therefore lead to hyperammonemia, their biochemical profile is very different [15]. The three mitochondrial enzymes, summarized if defective as ‘proximal’ UCDs [16], are the following: Carbamoyl phosphate synthetase 1 (CPS1, if defective leading to CPS1 deficiency, CPS1D, #237300), OTC (if defective leading to OTCD, #311250), and N-acetylglutamate synthase (NAGS, NAGSD, #237310), the latter being responsible for the production of the allosteric activator of CPS1, N-acetylglutamate (NAG). The proximal UCDs result in low or decreased plasma levels of citrulline and arginine, increased ornithine in OTCD and urine orotic acid is often elevated in OTCD (Figure 1). The ‘distal’ UCDs comprise the deficiencies of argininosuccinate synthetase (ASS, ASSD, #215700), argininosuccinate lyase (ASL, ASLD, #207900), and arginase 1 (ARG1, ARG1D, #207800). In contrast to the proximal UCDs with decreased citrulline and arginine, there are elevations of specific metabolites in each distal UCD, namely citrulline in ASSD, argininosuccinate in ASLD, and arginine in ARG1D and these metabolites can be easily used as screening parameters in NBS [13] (Figure 1) as it is current practice in US newborn screening programs to detect these disorders. Recently, postanalytical interpretive tools such as ratios of metabolites have been suggested as an opportunity to improve the sensitivity and specificity of NBS for several disorders including the proximal UCDs but this approach is currently under investigation [17]. In addition, the two transporter defects belonging to the group of UCDs, namely ORNT1 deficiency (hyperammonemia-hyperornithinemia-homocitrullinuria (HHH) syndrome, #238970) and citrin deficiency (citrullinemia type II (CTLN2) #605814, #603471) result in specific elevation of ornithine in ORNT1 and citrulline in citrin deficiency. However, plasma ornithine in HHH syndrome is not a reliable marker as it can still be normal in newborns, while homocitrulline in urine is specific but not feasible for standard NBS using blood.

3. Clinical Challenges
There are two main clinical issues that should be considered when discussing NBS for UCDs. The first issue is related to the stormy clinical course at the time of presentation in many neonatal-onset UCD patients. Quite often, and especially in severely manifesting proximal UCDs, first symptoms occur soon after birth between 12 and 72 h of age [4,7,18]. Patients may then rapidly deteriorate as a result of the overwhelming hyperammonemic crisis with mainly neurological symptoms including somnolence, vomiting, or coma. In most countries, sampling for NBS is done on day 3 or 4 of life. Even with perfect organization of neonatal mass screening, it will take about three days until the results become available. By that time, patients with a complete loss of enzyme or transporter function within the urea cycle will most likely be in deep hyperammonemic coma, if not already diagnosed and treated on the basis of their clinical presentation [19,20,21]. In other words, the diagnosis from NBS in these patients comes too late unless we assume a situation in which the diagnosis has not yet been made despite the severe clinical deterioration of the patient. The impact on infants presenting clinically before the results of NBS are available has recently been reviewed [4]. The authors stressed that although results were late, some patients still benefited from the availability of results shortly after presentation. Likewise, minimizing total process times from sampling until report of the results can probably reduce the number of infants with a clinical presentation before the results of NBS [4,7].
Besides this scenario, there is another clinical situation that challenges the introduction of mass neonatal screening for UCDs. At the mild end of the spectrum, there are patients described with a late-onset of disease with only a single, few or even absence of symptom(s) and only a biochemical phenotype. These patients were, for instance in the case of ASSD, described as suffering from mild citrullinemia type 1 [22], a condition allelic to classical citrullinemia type 1 but nevertheless much milder and with less, if any need for medical intervention. Such patients were often identified in neonatal screening programs and it has been discussed whether their mild phenotype would result from early detection and initiation of treatment or from a relevant residual enzyme or transporter function [5,6,9,11]. It has been suggested that metabolites and/or mutation analysis may help to identify attenuated patients in an attempt to avoid “medicalization of non-diseases, potentially unnecessary treatment and unnecessary anxiety to parents” [11] but this approach would need further studies.
Based on the fact that in several of the patients with attenuated UCDs mutations have been identified that retain some residual enzyme function [23,24], it is likely that mild ASSD or ASLD result from relevant residual enzyme function. Therefore, the question remains whether such patients should be identified at all by neonatal screening, or whether finding of these patients is unwanted since it results in unnecessary medical intervention and stress for the family. These concerns are of course relevant for most target diseases of neonatal screening, although the proportion of mild cases may be especially high in the case of ASSD and ASLD. It is, however, the combination of the two challenges mentioned here, the possible failure to report classical UCDs early enough together with the high number of mild cases, which provides the particular challenge. To discuss this becomes even more difficult with knowledge about some patients with mild disease who presented with late-onset but still suffered from fatal hyperammonemia [25,26]. In those cases, knowledge about the disease would have most likely prevented the fatal outcome, which can be considered as a strong reason for the introduction of newborn screening.
4. Current Experience with Neonatal Mass Screening for Urea Cycle Disorders
First neonatal mass screening programs in some states in the US, in Canada, and Australia in the 1970s and 1980s have used urine chromatography (as nicely reviewed in [13]). In parallel, several screening labs in Austria, different states in the US, and New Zealand have used an enzyme-multiple auxotroph screening test in dried blood spots for detection of ASLD, but also for ASSD and ARG1D [27]. This test was however soon abandoned in most places apart from Austria where screening for ASLD was continued until the year 2000 but then as well stopped because of a high rate of newborns with only mild disease [6].
Introduction of tandem mass spectrometry has brought a new technology allowing the analysis of urea cycle metabolites as part of the amino acids profile in parallel to screening for other amino acid disorders [8,28,29]; technical aspects are reviewed in [30]. Probably the largest experience in screening for UCDs with this new technique was reported from Australia, where part of the population was screened leading to identification of seven patients (4 OTCD, 2 ASLD, 1 CPS1D) between 1998 and 2002 [31]. While numbers in this study were too low to assess the impact of NBS on outcome of the patients, the feasibility of the approach was proven even when OTCD and CPS1D were included. At the same time, this study underlined previous concerns regarding the detection of mild cases, described as “extra diagnoses in the screened group” [31]. Using arginine and citrulline as markers, ASSD and ASLD are included in the expanded newborn screening program in some states in the US (California, Massachusetts, Michigan, New York, Newark, Wisconsin) since 2001. Using citrulline as a marker, ASSD and ASLD have been screened for as part of the ‘Recommended Uniform Screening Panel’ in all of the United States since 2006. Published data from 6,077,736 births (covering years from 2001 to 2012 for different states) resulted in a cumulative incidence of 1 in 117,000 newborns for the two disorders [32].
Highlighting the difficulty to use low citrulline as a marker for the detection of proximal UCDs, a study in Tuscany applying LC-MS/MS was performed between 2001 and 2008 [10]. The authors concluded that “hypocitrullinemia is not a reliable marker for OTCD newborn screening, especially for late-onset forms that may exhibit normal citrulline levels”. It will be interesting to learn from the experience in those states in the US (California, Connecticut, Massachusetts) that screen for OTC deficiency by use of glutamine/citrulline (and other) ratios. Low citrulline concentrations may also be found in other metabolic disorders further challenging its use as screening marker [33,34]. In a recent UCD guideline, it was therefore concluded that NBS for proximal disorders cannot currently be recommended, but it may be considered for the distal UCDs [35].
5. Summary
It is mainly the clinical challenges that has let some authors to state that “population screening for UCD is … not indicated at present” [12]. Nevertheless, outcome of UCDs remains unsatisfactory for many of the severely affected patients. Importantly, also late-onset UCD patients may face a bleak prognosis if not identified and treated very fast [19,36]. Therefore, it seems to be indicated to follow all strategies that possibly result in an improvement of the prognosis. NBS would clearly be one option to contribute to that aim and should thus be included in the package of measures [17]. The authors therefore welcome the currently running pilot studies and also advocate a broad discussion including healthcare professionals and patient advocate group to come to a balanced agreement on the still controversial aspects of NBS for UCDs.
Until UCDs are widely included in NBS programs, it remains essential to identify patients as early as possible. This requires a high index of suspicion within healthcare professionals but may also be achieved by automatic ‘red flags’ for an improved surveillance as recently suggested [37].
Five decades after start of mass screening, we are still challenged on several levels but should accept this as motivation to work for an improved outcome of patients with UCDs.
Acknowledgment
The authors are grateful to the Swiss National Science Foundation for support of their studies of urea cycle disorders (grant 310030_153196 to JH).
Author Contributions
Both authors contributed equally to all aspects of writing this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wilson, J.M.G.; Jungner, G. Principles and Practice of Screening for Disease; World Health Organization: Geneva, Switzerland, 1968; pp. 26–39. [Google Scholar]
- Bickel, H.; Bachmann, C.; Beckers, R.; Brandt, N.J.; Clayton, B.E.; Corrado, G.; Feingold, H.J.; Giardini, O.; Hammersen, G.; Schonberg, D. Neonatal mass-screening for metabolic disorders—Summaryof recent sessions of the committee of experts to study inborn metabolic diseases, public-health committee, council-of-europe—Review. Eur. J. Pediatr. 1981, 137, 133–139. [Google Scholar]
- Bearn, A.G.; Cusworth, D.C.; Dean, G.; Frézal, J.P.; Neifakh, S.A.; Scheinberg, I.H.; Scriver, C.R.; Szeinberg, A. Screening for Inborn Errors of Metabolism; World Health Organization: Geneva, Switzerland, 1968; pp. 1–57. [Google Scholar]
- Tal, G.; Pitt, J.; Morrisy, S.; Tzanakos, N.; Boneh, A. An audit of newborn screening procedure: Impact on infants presenting clinically before results are available. Mol. Genet. Metab. 2015, 114, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.; Janzen, N.; Sander, S.; Steuerwald, U.; Das, A.M.; Scholl, S.; Trefz, F.K.; Koch, H.G.; Häberle, J.; Korall, H.; et al. Neonatal screening for citrullinaemia. Eur. J. Pediatr. 2003, 162, 417–420. [Google Scholar] [PubMed]
- Mercimek-Mahmutoglu, S.; Moeslinger, D.; Häberle, J.; Engel, K.; Strobl, M.W.; Scheibenreiter, S.; Herle, M.; Muehl, A.; Stockler-Ipsiroglu, S. Long-term outcome of patients with argininosuccinate lyase deficiency diagnosed by newborn screening in austria. Mol. Genet. Metab. 2010, 100, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Gramer, G.; Haege, G.; Fang-Hoffmann, J.; Schwab, K.O.; Tacke, U.; Trefz, F.K.; Mengel, E.; Wendel, U.; Leichsenring, M.; et al. Efficacy and outcome of expanded newborn screening for metabolic diseases--report of 10 years from south-west germany. Orphanet J. Rare Dis. 2011, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Frazier, D.M.; Millington, D.S.; McCandless, S.E.; Koeberl, D.D.; Weavil, S.D.; Chaing, S.H.; Muenzer, J. The tandem mass spectrometry newborn screening experience in north carolina: 1997–2005. J. Inherit. Metab. Dis. 2006, 29, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Ficicioglu, C.; Mandell, R.; Shih, V.E. Argininosuccinate lyase deficiency: Longterm outcome of 13 patients detected by newborn screening. Mol. Genet. Metab. 2009, 98, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Cavicchi, C.; Malvagia, S.; la Marca, G.; Gasperini, S.; Donati, M.A.; Zammarchi, E.; Guerrini, R.; Morrone, A.; Pasquini, E. Hypocitrullinemia in expanded newborn screening by lc-ms/ms is not a reliable marker for ornithine transcarbamylase deficiency. J. Pharm. Biomed. Anal. 2009, 49, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Barends, M.; Pitt, J.; Morrissy, S.; Tzanakos, N.; Boneh, A. Biochemical and molecular characteristics of patients with organic acidaemias and urea cycle disorders identified through newborn screening. Mol. Genet. Metab. 2014, 113, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, C. Long-term outcome of patients with urea cycle disorders and the question of neonatal screening. Eur. J. Pediatr. 2003, 162 (Suppl. 1), S29–S33. [Google Scholar] [CrossRef] [PubMed]
- Naylor, E.W. Newborn screening of urea cycle disorders. Pediatrics 1981, 68, 453–457. [Google Scholar] [PubMed]
- Maestri, N.E.; McGowan, K.D.; Brusilow, S.W. Plasma glutamine concentration: A guide in the management of urea cycle disorders. J. Pediatr. 1992, 121, 259–261. [Google Scholar] [CrossRef]
- Brusilow, S.W.; Maestri, N.E. Urea cycle disorders: Diagnosis, pathophysiology, and therapy. Adv.Pediatr. 1996, 43, 127–170. [Google Scholar] [PubMed]
- Ah Mew, N.A.; Krivitzky, L.; McCarter, R.; Batshaw, M.; Tuchman, M.; Consortium, U.C.D. Clinical outcomes of neonatal onset proximal versus distal urea cycle disorders do not differ. J. Pediatr. 2013, 162, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.L.; Marquardt, G.; McHugh, D.M.; Currier, R.J.; Tang, H.; Stoway, S.D.; Rinaldo, P. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet. Med. 2014, 16, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.V.; Morris, A.A.M. Diagnosis and early management of inborn errors of metabolism presenting around the time of birth. Acta Paediatr. 2006, 95, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Nassogne, M.C.; Heron, B.; Touati, G.; Rabier, D.; Saudubray, J.M. Urea cycle defects: Management and outcome. J. Inherit. Metab. Dis. 2005, 28, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Uchino, T.; Endo, F.; Matsuda, I. Neurodevelopmental outcome of long-term therapy of urea cycle disorders in japan. J. Inherit. Metab. Dis. 1998, 21 (Suppl. 1), 151–159. [Google Scholar] [CrossRef] [PubMed]
- Kido, J.; Nakamura, K.; Mitsubuchi, H.; Ohura, T.; Takayanagi, M.; Matsuo, M.; Yoshino, M.; Shigematsu, Y.; Yorifuji, T.; Kasahara, M.; et al. Long-term outcome and intervention of urea cycle disorders in japan. J. Inherit. Metab. Dis. 2012, 35, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Häberle, J.; Pauli, S.; Schmidt, E.; Schulze-Eilfing, B.; Berning, C.; Koch, H.G. Mild citrullinemia in caucasians is an allelic variant of argininosuccinate synthetase deficiency (citrullinemia type 1). Mol. Genet. Metab. 2003, 80, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Berning, C.; Bieger, I.; Pauli, S.; Vermeulen, T.; Vogl, T.; Rummel, T.; Hohne, W.; Koch, H.G.; Rolinski, B.; Gempel, K.; et al. Investigation of citrullinemia type i variants by in vitro expression studies. Hum. Mutat. 2008, 29, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Pandey, A.V.; Balmer, C.; Eggimann, S.; Rüfenacht, V.; Nuoffer, J.M.; Häberle, J. Unstable argininosuccinate lyase in variant forms of the urea cycle disorder arginino-succinic aciduria. J. Inherit. Metab. Dis. in press.
- Häberle, J.; Vilaseca, M.A.; Meli, C.; Rigoldi, M.; Jara, F.; Vecchio, I.; Capra, C.; Parini, R. First manifestation of citrullinemia type i as differential diagnosis to postpartum psychosis in the puerperal period. Eur.J.Obstet.Gynecol.Reprod. Biol. 2010, 149, 228–229. [Google Scholar] [CrossRef]
- Wong, L.J.; Craigen, W.J.; O’Brien, W.E. Postpartum coma and death due to carbamoyl-phosphate synthetase i deficiency. Ann. Intern. Med. 1994, 120, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Talbot, H.W.; Sumlin, A.B.; Naylor, E.W.; Guthrie, R. A neonatal screening-test for argininosuccinic acid lyase deficiency and other urea cycle disorders. Pediatrics 1982, 70, 526–531. [Google Scholar] [PubMed]
- Naylor, E.W.; Chace, D.H. Automated tandem mass spectrometry for mass newborn screening for disorders in fatty acid, organic acid, and amino acid metabolism. J. Child Neurol. 1999, 14, S4–S8. [Google Scholar] [PubMed]
- Huang, H.P.; Chu, K.L.; Chien, Y.H.; Wei, M.L.; Wu, S.T.; Wang, S.F.; Hwu, W.L. Tandem mass neonatal screening in taiwan—Report from one center. J. Formos. Med. Assoc. 2006, 105, 882–886. [Google Scholar] [CrossRef]
- Rashed, M.S. Clinical applications of tandem mass spectrometry: Ten years of diagnosis and screening for inherited metabolic diseases. J. Chromatogr. B 2001, 758, 27–48. [Google Scholar] [CrossRef]
- Wilcken, B.; Haas, M.; Joy, P.; Wiley, V.; Bowling, F.; Carpenter, K.; Christodoulou, J.; Cowley, D.; Ellaway, C.; Fletcher, J.; et al. Expanded newborn screening: Outcome in screened and unscreened patients at age 6 years. Pediatrics 2009, 124, e241–e248. [Google Scholar] [CrossRef] [PubMed]
- Summar, M.L.; Koelker, S.; Freedenberg, D.; le Mons, C.; Häberle, J.; Lee, H.S.; Kirmse, B. The incidence of urea cycle disorders. Mol. Genet. Metab. 2013, 110, 179–180. [Google Scholar] [CrossRef] [PubMed]
- Atkuri, K.R.; Cowan, T.M.; Kwan, T.; Ng, A.; Herzenberg, L.A.; Enns, G.M. Inherited disorders affecting mitochondrial function are associated with glutathione deficiency and hypocitrullinemia. Proc. Natl. Acad. Sci. USA 2009, 106, 3941–3945. [Google Scholar] [CrossRef] [PubMed]
- De Sain-van der Velden, M.G.; Rinaldo, P.; Elvers, B.; Henderson, M.; Walter, J.H.; Prinsen, B.H.; Verhoeven-Duif, N.M.; de Koning, T.J.; van Hasselt, P. The proline/citrulline ratio as a biomarker for oat deficiency in early infancy. JIMD Rep. 2012, 6, 95–99. [Google Scholar] [PubMed]
- Häberle, J.; Boddaert, N.; Burlina, A.; Chakrapani, A.; Dixon, M.; Huemer, M.; Karall, D.; Martinelli, D.; Sanjurjo Crespo, P.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J. Rare Dis. 2012, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Rüegger, C.M.; Lindner, M.; Ballhausen, D.; Baumgartner, M.R.; Beblo, S.; Das, A.; Gautschi, M.; Glahn, E.M.; Grunert, S.C.; Hennermann, J.; et al. Cross-sectional observational study of 208 patients with non-classical urea cycle disorders. J. Inherit. Metab. Dis. 2014, 37, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Vergano, S.A.; Crossette, J.M.; Cusick, F.C.; Desai, B.R.; Deardorff, M.A.; Sondheimer, N. Improving surveillance for hyperammonemia in the newborn. Mol. Genet. Metab. 2013, 110, 102–105. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rüfenacht, V.; Häberle, J. Mini-Review: Challenges in Newborn Screening for Urea Cycle Disorders. Int. J. Neonatal Screen. 2015, 1, 27-35. https://doi.org/10.3390/ijns1010027
Rüfenacht V, Häberle J. Mini-Review: Challenges in Newborn Screening for Urea Cycle Disorders. International Journal of Neonatal Screening. 2015; 1(1):27-35. https://doi.org/10.3390/ijns1010027
Chicago/Turabian StyleRüfenacht, Véronique, and Johannes Häberle. 2015. "Mini-Review: Challenges in Newborn Screening for Urea Cycle Disorders" International Journal of Neonatal Screening 1, no. 1: 27-35. https://doi.org/10.3390/ijns1010027