
Fibroblast Fatty-Acid Oxidation Flux Assays Stratify Risk in Newborns with Presumptive-Positive Results on Screening for Very-Long Chain Acyl-CoA Dehydrogenase Deficiency

 ,
,
Abstract
:1. Introduction
2. Patients
3. Methods
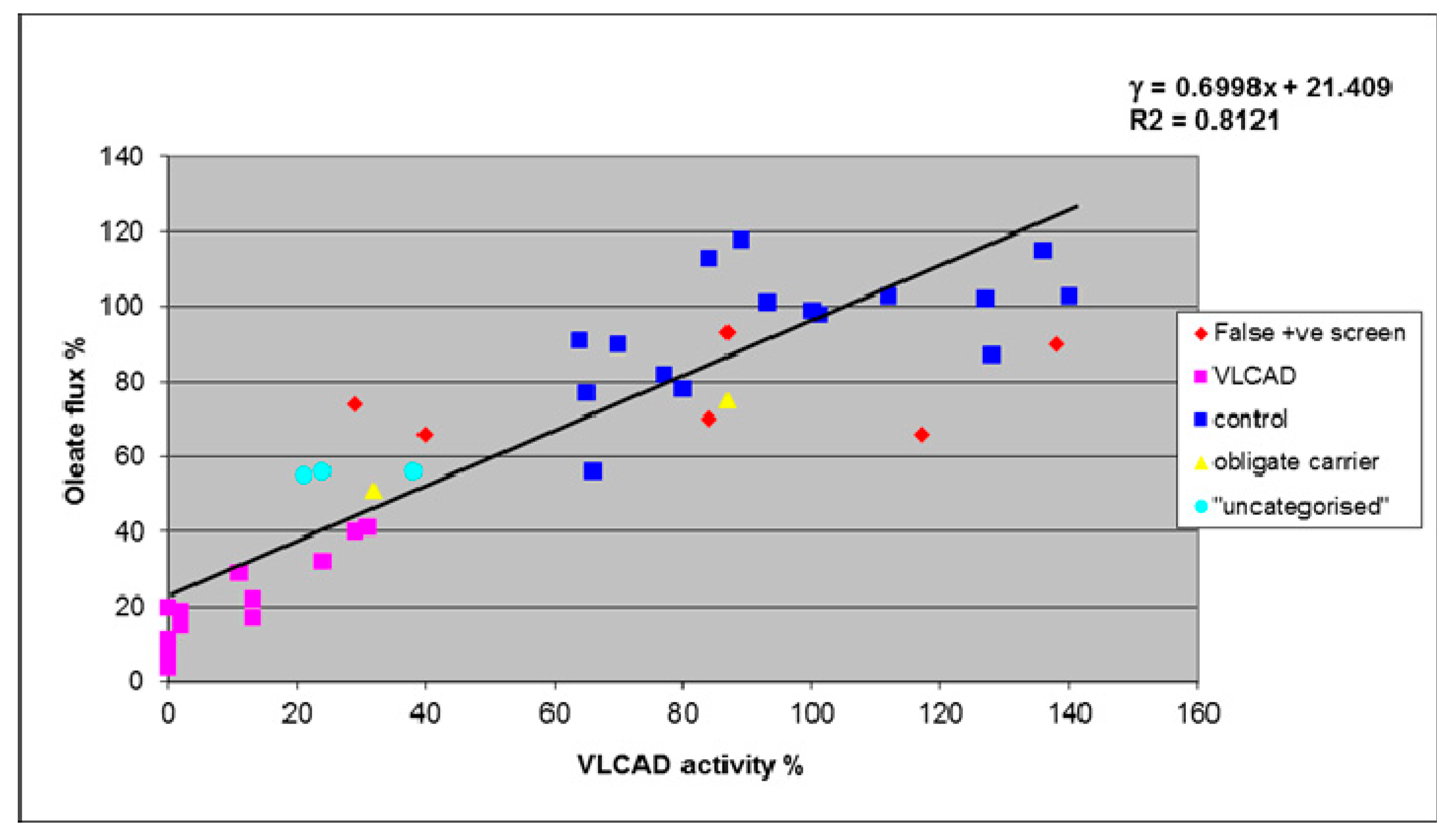
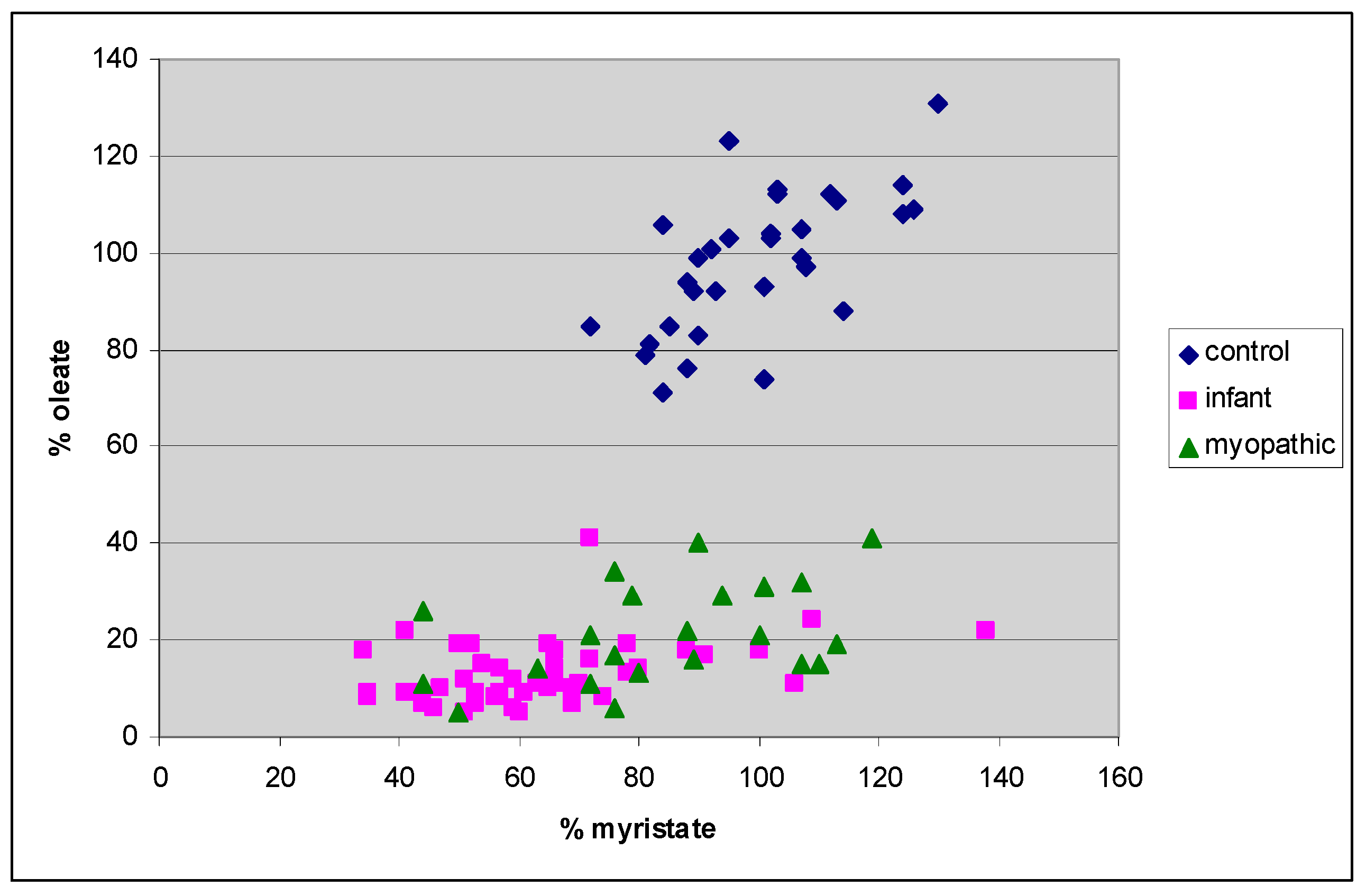
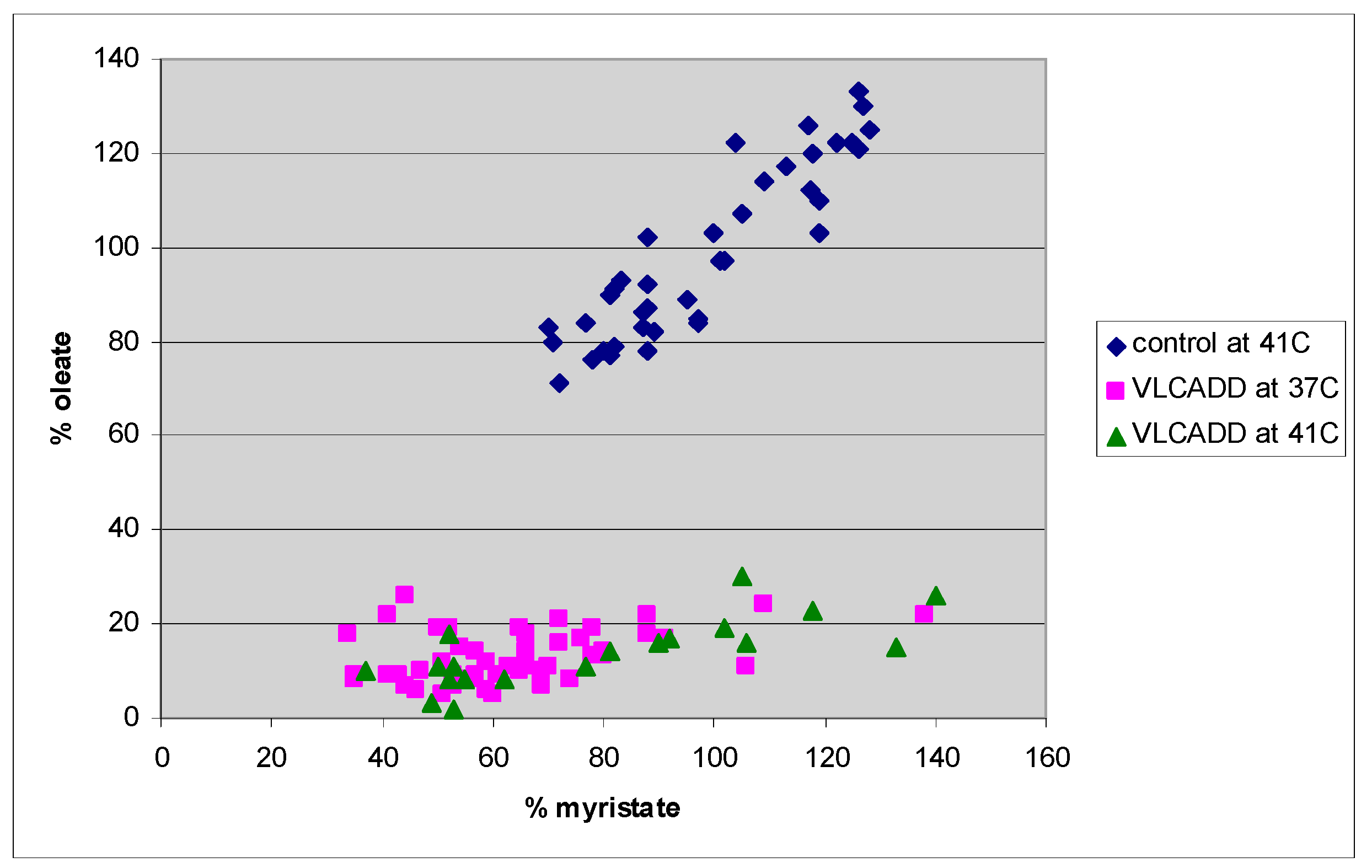
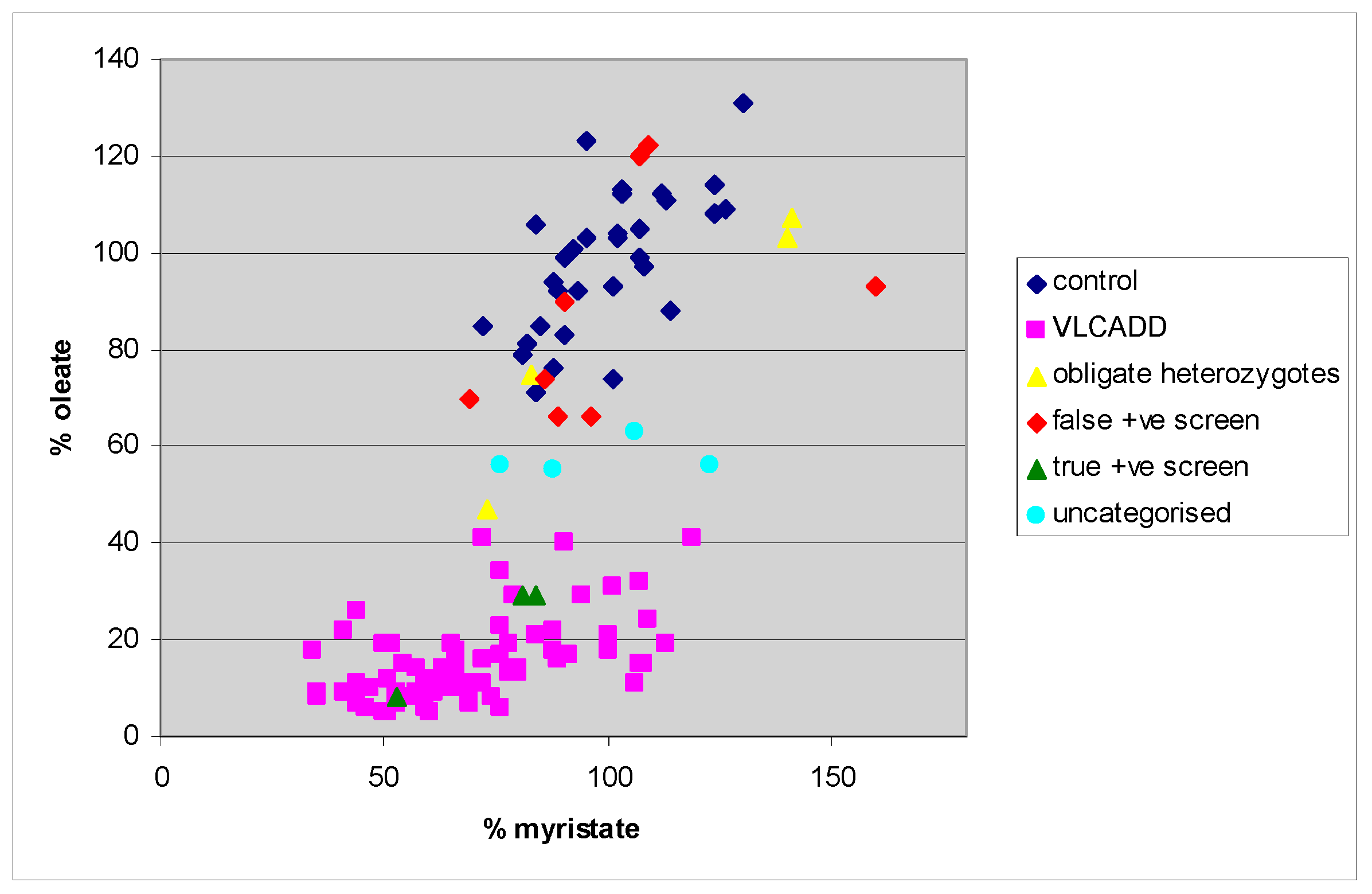
4. Results
5. Discussion
6. Conclusion
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Bennett, M.J. Newborn screening for metabolic diseases: Saving children’s lives and improving outcomes. Clin. Biochem. 2014, 47, 693–694. [Google Scholar] [CrossRef] [PubMed]
- Pollak, A.; Kasper, D.C. Austrian Newborn Screening Program: A perspective of five decades. J. Perinat. Med. 2014, 42, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Coman, D.; Bhattacharya, K. Extended newborn screening: An update for the general paediatrician. J. Paediatr. Child Health 2012, 48, E68–E72. [Google Scholar] [CrossRef] [PubMed]
- Scaturro, G.; Sanfilippo, C.; Piccione, M.; Piro, E.; Giuffrè, M.; Corsello, G. Newborn screening of inherited metabolic disorders by tandem mass spectrometry: Past, present and future. Pediatr. Med. Chir. 2013, 35, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Ryder, B.; Knoll, D.; Love, D.R.; Shepherd, P.; Love, J.M.; Reed, P.W.; de Hora, M.; Webster, D.; Glamuzina, E.; Wilson, C. The natural history of elevated tetradecenoyl-l-carnitine detected by newborn screening in New Zealand: Implications for very long chain acyl-CoA dehydrogenase deficiency screening and treatment. J. Inherit. Metab. Dis. 2016, 39, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.; Wordsworth, S.; Oerton, J.; Khalid, J.M.; Dezateux, C. Synthesis framework estimating prevalence of MCADD and sensitivity of newborn screening programme in the absence of direct evidence. J. Clin. Epidemiol. 2014, 67, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Olpin, S.E. Pathophysiology of fatty acid oxidation disorders and resultant phenotypic variability. J. Inherit. Metab. Dis. 2013, 36, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Andresen, B.S.; Olpin, S.; Poorthuis, B.J.H.M.; Scholte, H.R.; Vianey-Saban, C.; Wanders, R.; Ijlst, L.; Morris, A.; Pourfarzam, M.; Bartlett, K.; et al. Clear correlation of genotype with disease phenotype in very long-chain Acyl-CoA dehydrogenase deficiency. Am. J. Hum. Genet. 1999, 64, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Andresen, B.S.; Nation, J.; Boneh, A. VLCAD deficiency: Follow-up and outcome of patients diagnosed through newborn screening in Victoria. Mol. Genet. Metab. 2016, 118, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Merritt, J.L.; Vedal, S.; Abdenur, J.E.; Au, S.M.; Barshop, B.A.; Feuchtbaum, L.; Harding, C.O.; Hermerath, C.; Lorey, F.; Sesser, D.E.; et al. Infants suspected to have very-long chain acyl-CoA dehydrogenase deficiency from newborn screening. Mol. Genet. Metab. 2014, 111, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, L.; Haussmann, U.; Mueller, M.; Spiekerkoetter, U. VLCAD enzyme activity determinations in newborns identified by screening: A valuable tool for risk assessment. J. Inherit. Metab. Dis. 2012, 35, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, U.; Mueller, M.; Sturm, M.; Hofmann, M.; Schneider, D.T. Lethal undiagnosed very long-chain Acyl-CoA dehydrogenase deficiency with mild c14-acylcarnitine abnormalities on newborn screening. JIMD Rep. 2012, 6, 113–115. [Google Scholar] [PubMed]
- Spiekerkoetter, U.; Sun, B.; Zytkovicz, T.; Wanders, R.; Strauss, A.W.; Wendel, U. MS/MS-based newborn and family screening detects asymptomatic patients with very-long-chain acyl-CoA dehydrogenase deficiency. J. Pediatr. 2003, 143, 335–342. [Google Scholar] [CrossRef]
- Ventura, F.V.; Costa, C.G.; Struys, E.A.; Ruiter, J.; Allers, P.; Ijlst, L.; Tavares de Almeida, I.; Duran, M.; Jakobs, C.; Wanders, R. Quantitative acylcarnitine profiling in fibroblasts using [U-13C] palmitic acid: An improved tool for the diagnosis of fatty acid oxidation defects. Clin. Chim. Acta 1999, 281, 1–17. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Ruiter, J.P.N.; Ijlst, L.; Waterham, H.R.; Houten, S.M. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, U.; Haussmann, U.; Mueller, M.; ter Veld, F.; Stehn, M.; Santer, R.; Lukacs, Z. Tandem mass spectrometry screening for very long-chain acyl-CoA dehydrogenase deficiency: The value of second-tier enzyme testing. J. Pediatr. 2010, 157, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Schymik, I.; Liebig, M.; Mueller, M.; Wendel, U.; Mayatepek, E.; Strauss, A.W.; Wanders, R.J.A.; Spiekerkoetter, U. Pitfalls of neonatal screening for very-long-chain acyl-CoA dehydrogenase deficiency using tandem mass spectrometry. J. Pediatr. 2006, 149, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Olpin, S.E.; Manning, N.J.; Pollitt, R.J.; Clarke, S. Improved detection of long-chain fatty acid oxidation defects in intact cells using [9,10-3H]oleic acid. J. Inherit. Metab. Dis. 1997, 20, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Olpin, S.E.; Manning, N.J.; Pollitt, R.J.; Bonham, J.R.; Downing, M.; Clark, S. The use of [9,10-3H]myristate, [9,10-3H]palmitate and [9,10-3H]oleate for the detection and diagnosis of medium and long-chain fatty acid oxidation disorders in intact cultured fibroblasts. Adv. Exp. Med. Biol. 1999, 466, 321–325. [Google Scholar] [PubMed]
- Olpin, S.E.; Clark, S.; Scott, C.; Manning, N.J.; Bonham, J.R.; Hind, H.; Andresen, B.S.; Khan, A.; Sharma, R.; Kirk, R.; et al. Diagnosing very long-chain acyl-CoA dehydrogenase deficiency (VLCAD). J. Inherit. Metab. Dis. 2012, 35 O-043 (Suppl 1), S15. [Google Scholar]
- Diekman, E.F.; Ferdinandusse, S.; van der Pol, L.; Waterham, H.R.; Ruiter, J.P.N.; Ijlst, L.; Wanders, R.J.; Houten, S.M.; Wijburg, F.A.; Blank, A.C.; et al. Fatty acid oxidation flux predicts the clinical severity of VLCAD deficiency. Genet. Med. 2015, 17, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Manning, N.J.J.; Olpin, S.E.E.; Pollitt, R.J.J.; Webley, J. A comparison of [9,10-3H]palmitic and [9,10-3H]myristic acids for the detection of defects of fatty acid oxidation in intact cultured fibroblasts. J. Inherit. Metab. Dis. 1990, 13, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Olpin, S.E.; Manning, N.J.; Carpenter, K.; Middleton, B.; Pollitt, R.J. Differential diagnosis of hydroxydicarboxylic aciduria based on release of 3H2O from [9,10-3H]myristic and [9,10-3H]palmitic acids by intact cultured fibroblasts. J. Inherit. Metab. Dis. 1992, 15, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fukuda, S.; Hasegawa, Y.; Kobayashi, H.; Purevsuren, J.; Mushimoto, Y.; Yamaguchi, S. Effect of heat stress and bezafibrate on mitochondrial β-oxidation: Comparison between cultured cells from normal and mitochondrial fatty acid oxidation disorder children using in vitro probe acylcarnitine profiling assay. Brain Dev. 2010, 32, 362–370. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, L.; Bross, P.; Corydon, T.J.; Olpin, S.E.; Hansen, J.; Kenney, J.M.; McCandless, S.E.; Frazier, D.M.; Winter, V.; Gregersen, N.; et al. The Y42H mutation in medium-chain acyl-CoA dehydrogenase, which is prevalent in babies identified by MS/MS-based newborn screening, is temperature sensitive. Eur. J. Biochem. 2004, 271, 4053–4063. [Google Scholar] [CrossRef] [PubMed]
- Liebig, M.; Schymik, I.; Mueller, M.; Wendel, U.; Mayatepek, E.; Ruiter, J.; Strauss, A.W.; Wanders, R.J.A.; Spiekerkoetter, U. Neonatal screening for very long-chain acyl-CoA dehydrogenase deficiency: Enzymatic and molecular evaluation of neonates with elevated C14:1-carnitine levels. Pediatrics 2006, 118, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Andresen, B.S.; Corydon, M.J.; Corydon, T.J.; Olsen, R.K.J.; Bolund, L.; Bross, P. Mutation analysis in mitochondrial fatty acid oxidation defects: Exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum. Mutat. 2001, 18, 169–189. [Google Scholar] [CrossRef] [PubMed]
- Andresen, B.S.; Vianey-Saban, C.; Bross, P.; Divry, P.; Roe, C.R.; Nada, M.A.; Knudsen, I.; Gregersen, N. The mutational spectrum in very long-chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 1996, 19, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Sims, H.F.; Gopalakrishnan, D.; Gibson, B.; Rinaldo, P.; Vockley, J.; Hug, G.; Strauss, A.W. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation 1999, 99, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Bonham, J.R. Impact of new screening technologies: Should we screen and does phenotype influence this decision? J. Inherit. Metab. Dis. 2013, 36, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Rhead, W.J. Newborn screening for medium-chain acyl-CoA dehydrogenase deficiency: A global perspective. J. Inherit. Metab. Dis. 2006, 29, 370–377. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Age at Time of Investigation/Diagnosis | Detail/Presumed Diagnosis | NBS Cut off and Day of Screen C14:1 µmol/L < 0.6 Day 1 | Mutation | 37 °C Oleate % | 41 °C Oleate % | HPLC % | Current Age | Follow Up |
|---|---|---|---|---|---|---|---|---|---|
| 77 | Neonate | False positive screen 1 mutation only | 1.11 | Heterozygous c.848T>C (p.Val283Ala) | 70 | 66 | 84 | 6 years 7 months | Asymptomatic; 10 month follow up |
| 78 | Neonate | False positive screen 1 mutation only | 1.21 | Heterozygous c.1316G>A (p.Gly439Asp) | 66 | 70 | 40 | 6 years 8 months | Asymptomatic; 10 month follow up |
| 76 | Neonate | False positive screen 1 mutation only | 0.79 | Heterozygous c.848T>C (p.Val283Ala) | 93 | 134 | 87 | 6 years 9 months | Asymptomatic; 9 month follow up |
| 37 | Neonate | False positive screen 1 mutation only | 1.21 | Heterozygous c.897G>C (p.Lys299Asn) | 74 | 83 | 29 | 6 years 10 months | Remains asymptomatic |
| 91 | Neonate | False positive screen 2 mutations | 1.08 | c.1405C>T (p.Arg469Trp) c.686G>A (p.Arg229Gln) | 66 | 66 | 117 | 5 years 9 months | Asymptomatic; 5 year 2 month follow up |
| 78 | Neonate | False positive screen No mutation | 0.91 | No mutations detected | 90 | 93 | 138 | 6 years 10 months | Asymptomatic; 8 month follow up |
| * NBS cut off < 1.3 Day 2 | |||||||||
| 17 | Neonate | False positive screen | 2.14 | Homozygous c.1226C>T (p.Thr409Met) | 120 | 126 | 70 | 4 years 6 months | Well. No more ER. Discharged |
| 519 | Neonate | False positive screen | 2.98 | Homozygous c.1226C>T (p.Thr409Met) | 122 | 155 | NA | 5 years 2 months | Well. No more ER. Discharged |
| 12 | Neonate | True positive screen. Treated from birth. Previous sib died of VLCAD on day 1 of life | 5.45 | c.942_947delAGTACG (p.Arg316_Val317del) c.343delG (p.(Glu115fs)) | 8 | 8 | NA | 4 years 1 month | Severe infantile: Treated from birth with MCT and ++ calories. High CK despite high calorie and MCT diet and overnight feeding |
| 233 | Neonate | True positive screen. Avoidance of fasting, emergency regimen | 6.9 | c.848T>C (p.Val283Ala) c.1616C>A (p.Ala539Asp) | 29 | 16 | NA | 3 years 10 months | On normal diet, max fasting time of 12 h + ER. Yearly CK, Liver USS, echo. Asymptomatic |
| 18 | Neonate | True positive screen | 8.2 | c.622G>C (p.Gly208Arg) last nucleotide in exon—likely to be splicing mutation. c.1838G>A (p.Arg613Gln) | 29 | 17 | 11 | 4 years 2 months | Loose low fat, high MCT diet. Short fasting time. Strict ER. At 4 years, resting CK increased, corn starch introduced |
| 16 | Neonate | Uncategorised positive screen | 3.81 | c.1226C>T (p.Thr409Met) c.1616C>A (p.Ala539Asp) | 63 | 64 | NA | 4 years 5 months | Well. No medical problems. No diet. Self-discharged |
| 22 | Neonate | Uncategorised positive screen. Avoidance of fasting, emergency regimen | 2.73 | c.1226C>T (p.Thr409Met) c.1322G>A (p.Gly441Asp) | 56 | 21 | 24 | 4 years 9 months | Well. Discharged |
| 52 | # Symptomatic adult | Uncategorised. Single episode of hyperthermia and rhabdomyolysis following unaccustomed extended aerobic exercise, became encephalopathic, hospitalised and developed multi-organ failure. No biochemical markers suggestive of VLCAD. CPT2 deficiency excluded | Within normal reference range | Heterozygous c.1837C>T (p.Arg613Trp) | 56 | 46 | 38 | ||
| 19 | # Symptomatic adult | Uncategorised. Atypical neurological symptoms not suggestive of VLCAD. No history of hypoglycaemia or rhabdomyolysis. CPT2, LCHAD, TFP also excluded | Within normal reference range | Heterozygous c.1019G>T (p.Gly340Val) + c.753-27C>T Possible double mutant allele, identical genotype detected in an unrelated newborn (unpublished data) | 55 | 46 | 21 | ||
| 32 | Adult | obligate heterozygote | NA | Heterozygous c.1360G>A (p.Asp454Asn) | 107 | NA | NA | Asymptomatic | |
| 33 | Adult | obligate heterozygote | NA | Heterozygous c.1375C>T (p.Arg459Trp) | 103 | NA | NA | Asymptomatic | |
| 34 | Adult | obligate heterozygote | NA | Heterozygous c.848T>C (p.Val283Ala) | 51 | 52 | 32 | Asymptomatic | |
| 35 | Adult | obligate heterozygote | NA | None detected | 71 | 63 | 87 | Asymptomatic | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olpin, S.E.; Clark, S.; Dalley, J.; Andresen, B.S.; Croft, J.; Scott, C.A.; Khan, A.; Kirk, R.J.; Sparkes, R.; Chard, M.; et al. Fibroblast Fatty-Acid Oxidation Flux Assays Stratify Risk in Newborns with Presumptive-Positive Results on Screening for Very-Long Chain Acyl-CoA Dehydrogenase Deficiency. Int. J. Neonatal Screen. 2017, 3, 2. https://doi.org/10.3390/ijns3010002
Olpin SE, Clark S, Dalley J, Andresen BS, Croft J, Scott CA, Khan A, Kirk RJ, Sparkes R, Chard M, et al. Fibroblast Fatty-Acid Oxidation Flux Assays Stratify Risk in Newborns with Presumptive-Positive Results on Screening for Very-Long Chain Acyl-CoA Dehydrogenase Deficiency. International Journal of Neonatal Screening. 2017; 3(1):2. https://doi.org/10.3390/ijns3010002
Chicago/Turabian StyleOlpin, Simon E., Shirley Clark, Jane Dalley, Brage S. Andresen, Joanne Croft, Camilla A. Scott, Aneal Khan, Richard J. Kirk, Rebecca Sparkes, Marisa Chard, and et al. 2017. "Fibroblast Fatty-Acid Oxidation Flux Assays Stratify Risk in Newborns with Presumptive-Positive Results on Screening for Very-Long Chain Acyl-CoA Dehydrogenase Deficiency" International Journal of Neonatal Screening 3, no. 1: 2. https://doi.org/10.3390/ijns3010002