MCAD-Deficiency with Severe Neonatal Onset, Fatal Outcome and Normal Acylcarnitine Profile

 , ,
, ,
Abstract
:1. Introduction
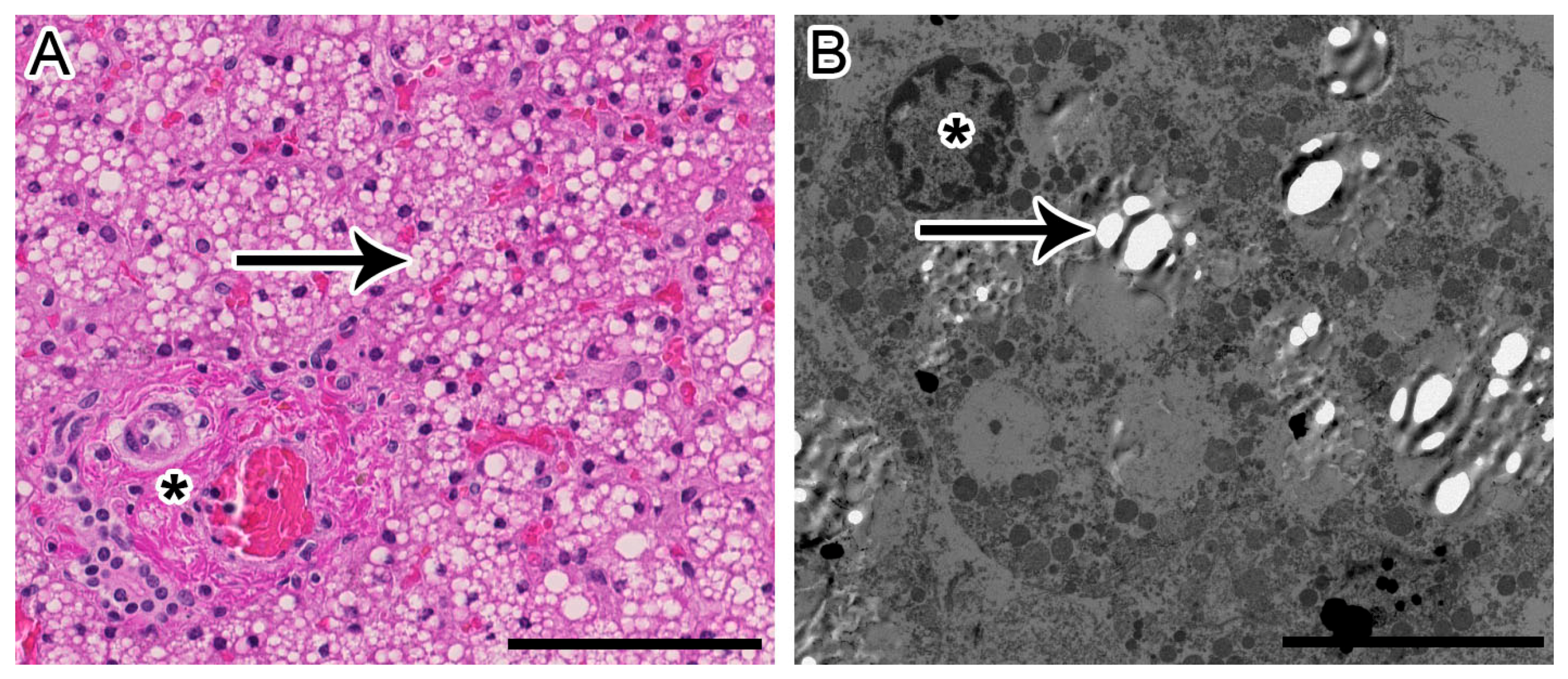
2. Case Report
3. Methods
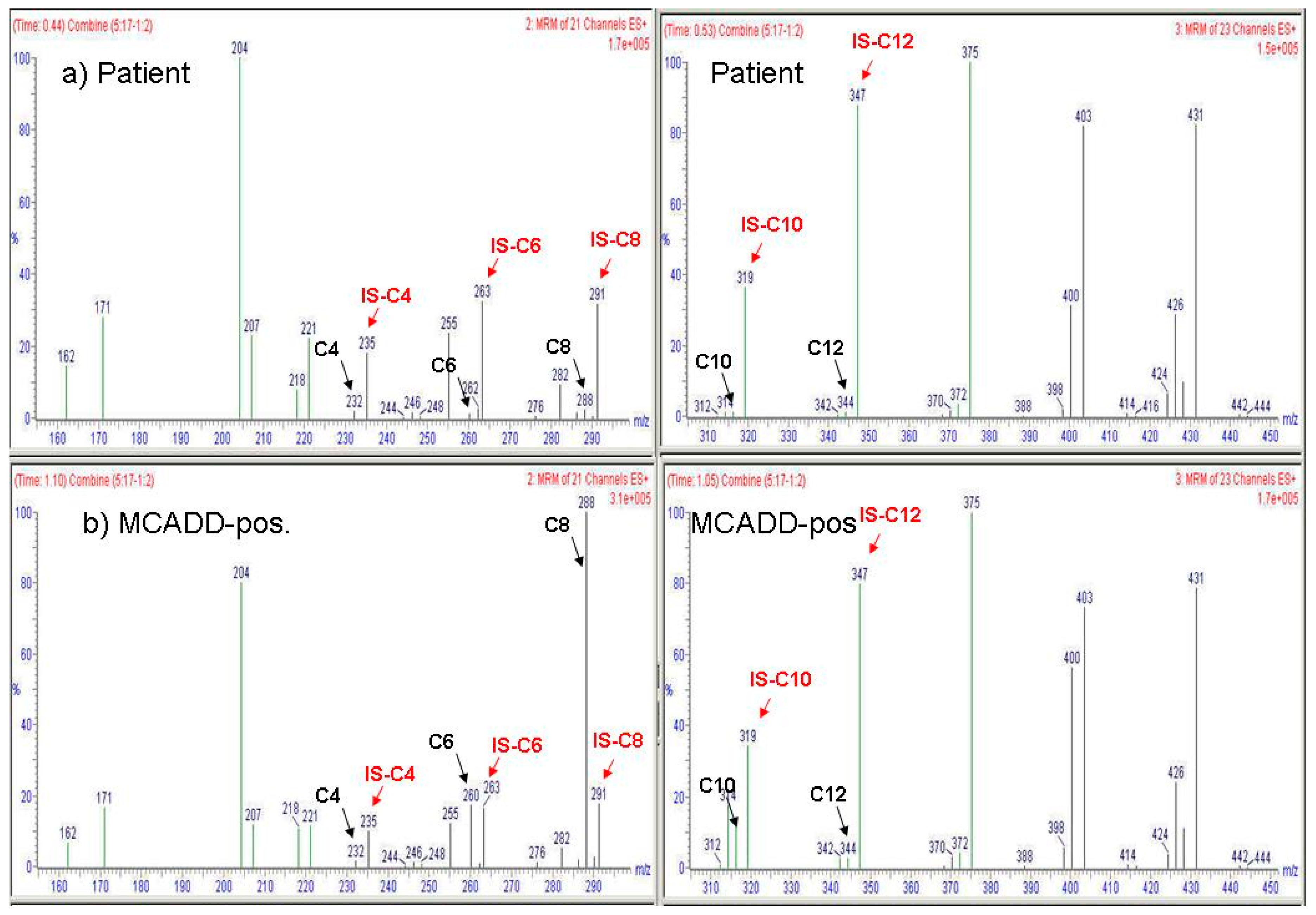
4. Results
5. Discussion
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maier, E.M. Neonatal screening for medium-chain Acyl-CoA deficiency—Insights and unexpected challenges. Int. J. Neonatal Screen. 2015, 1, 79–88. [Google Scholar] [CrossRef]
- Wilson, C.J.; Champion, M.P.; Collins, J.E.; Clayton, P.T.; Leonard, J.V. Outcome of medium chain acyl-CoA dehydrogenase deficiency after diagnosis. Arch. Dis. Child. 1999, 80, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Maier, E.M.; Gersting, S.W.; Kemter, K.F.; Jank, J.M.; Reindl, M.; Messing, D.D.; Truger, M.S.; Sommerhoff, C.P.; Muntau, A.C. Protein Misfolding is the molecular mechanism underlying MCADD identified in newborn screening. Hum. Mol. Genet. 2009, 18, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Jank, J.M.; Maier, E.M.; Reiß, D.D.; Haslbeck, M.; Kemter, K.F.; Truger, M.S.; Sommerhoff, C.P.; Ferdinandusse, S.; Wanders, R.J.; Gersting, S.W.; et al. The Domain-Specific and Temperature-Dependent Protein Misfolding Phenotype of Variant Medium-Chain acyl-CoA Dehydrogenase. PLoS ONE 2014, 9, e93852. [Google Scholar] [CrossRef] [PubMed]
- Maier, E.M.; Liebl, B.; Röschinger, W.; Nennstiehl-Ratzel, U.; Fingerhut, R.; Olgemöller, B.; Busch, U.; Krone, N.; Kries, V.R.; Roscher, A.A. Population Spectrum of ACADM Genotypes Correlated to Biochemical Phenotypes in Newborn Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency. Hum. Mutat. 2005, 25, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Maier, E.M.; Pongratz, J.; Muntau, A.C.; Liebl, B.; Nennstiehl-Ratzel, U.; Busch, U.; Fingerhut, R.; Olgemöller, B.; Roscher, A.A.; Röschinger, W. Validation of MCADD newborn Screening. Clin. Genet. 2009, 76, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Gramer, G.; Haege, G.; Fang-Hoffmann, J.; Hoffmann, G.F.; Bartram, C.R.; Hinderhofer, K.; Burgard, P.; Lindner, M. Medium-Chain Acyl-CoA Dehydrogenase Deficiency: Evaluation of Genotype-Phenotype Correlation in Patients Detected by Newborn Screening. JIMD Rep. 2015, 23, 101–112. [Google Scholar] [PubMed]
- Pollitt, R.J. Neonatal Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency—Alternative Approaches. Int. J. Neonatal Screen. 2016, 2, 79–88. [Google Scholar] [CrossRef]
- Touw, C.M.L.; Smit, G.P.A.; de Vries, M.; de Klerk, J.B.C.; Bosch, A.M.; Visser, G.; Mulder, M.F.; Rubio-Gozalbo, M.E.; Elvers, B.; Niezen-Koning, K.E.; et al. Risk stratification by residual enzyme activity after newborn screening for medium-chain acyl-CoA dehydrogenase deficiency: data from a cohort study. Orphanet J. Rare Dis. 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Catarzi, S.; Caciotti, A.; Thusberg, J.; Tonin, R.; Malvagia, S.; la Marca, G.; Pasquini, E.; Cavicci, C.; Ferri, L.; Donati, M.A.; et al. Medium-Chain Acyl-CoA Deficiency: Outline from Newborn Screening, In Silico Predictions, and Molecular Studies. Sci. World J. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Grünert, S.C.; Wehrle, A.; Villavicencio-Lorini, P.; Lausch, E.; Vetter, B.; Schwab, K.O.; Tucci, S.; Spiekerkötter, U. Medium-chain acyl-CoA dehydrogenase deficiency associated with a novel splice mutation in the ACADM gene missed by newborn screening. BMC Med. Genet. 2015, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Lovera, C.; Porta, F.; Caciotta, A.; Catarzi, S.; Cassanallo, M.; Caruso, U.; Gallina, M.R.; Morrone, A.; Spada, M. Sudden unexpected infant death (SUDI) in a newborn due to medium chain acyl CoA dehydrogenase (MCAD) deficiency with an unusual severe genotype. Ital. J. Pediatr. 2012, 38, 59. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.M.; Laing, I.A.; Smith, N.; Uttley, W.S. Neonatal presentation of medium-chain acyl-CoA dehydrogenase deficiency in two families. J. Inherit. Metab. Dis. 1996, 19, 370. [Google Scholar] [CrossRef] [PubMed]
- Brackett, J.C.; Sims, H.F.; Steiner, R.D.; Nunge, M.; Zimmerman, E.M.; deMartinville, B.; Rinaldo, P.; Slaugh, R.; Strauss, A.W. A novel mutation in medium chain acyl-CoA dehydrogenase causes sudden neonatal death. J. Clin. Investig. 1994, 94, 1477. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Carpenter, K.H.; Hammond, J. Neonatal symptoms in medium chain acyl coenzyme A dehydrogenase deficiency. Arch. Dis. Child. 1993, 69, 292. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compound | Patient * | Reference Range # | Median # | % of Direct Measurement |
|---|---|---|---|---|
| Free carnitine | 25 | 5–60 | 15 | 145 |
| Acetylcarnitine (C2) | 3.2 | 6–60 | 18 | 55 |
| Propionylcarnitine (C3) | 0.24 | <6.0 | 1.5 | 40 |
| Butyrylcarnitine (C4) | 0.07 | <1.0 | 0.25 | 63 |
| Isovalerylcarnitine (C5) | 0.04 | <0.5 | 0.11 | 94 |
| Hexanoylcarnitine (C6) | 0.04 | <0.2 | 0.04 | 13 |
| Octanoylcarnitine (C8) | 0.16 | <0.6 | 0.04 | 29 |
| Decanoylcarnitine (C10) | 0.00 | <0.3 | 0.05 | 33 |
| Decenoylcarnitine (C10:1) | 0.01 | <0.2 | 0.03 | 80 |
| Dodecanoylcarnitine (C12) | 0.01 | <0.3 | 0.07 | 95 |
| Tetradecanoylcarnitine (C14) | 0.01 | <0.5 | 0.18 | 49 |
| Tetradecenoylcarnitine (C14:1) | 0.01 | <0.3 | 0.07 | 67 |
| Tetradecadienoylnoylcarnitine (C14:2) | 0.01 | <0.05 | 0.01 | 67 |
| Hexadecanoylcarnitine (C16) | 0.25 | <8 | 2.9 | 69 |
| Hexadecenoylcarnitine (C16:1) | 0.01 | <0.7 | 0.22 | 56 |
| Octadecanoylcarnitine (C18) | 0.2 | <2.2 | 0.82 | 81 |
| Octadecenoylcarnitine (C18:1) | 0.06 | <4.0 | 1.5 | 66 |
| Octadecadienoylcarnitine (C18:2) | 0.02 | <0.9 | 0.15 | 63 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fingerhut, R.; Joset, P.; Rupp, N.J.; Girsberger, M.; Sluka, S.H.M.; Herget, T.; Azzarello-Burri, S.M.; Rauch, A.; Baumgartner, M. MCAD-Deficiency with Severe Neonatal Onset, Fatal Outcome and Normal Acylcarnitine Profile. Int. J. Neonatal Screen. 2017, 3, 21. https://doi.org/10.3390/ijns3030021
Fingerhut R, Joset P, Rupp NJ, Girsberger M, Sluka SHM, Herget T, Azzarello-Burri SM, Rauch A, Baumgartner M. MCAD-Deficiency with Severe Neonatal Onset, Fatal Outcome and Normal Acylcarnitine Profile. International Journal of Neonatal Screening. 2017; 3(3):21. https://doi.org/10.3390/ijns3030021
Chicago/Turabian StyleFingerhut, Ralph, Pascal Joset, Niels J. Rupp, Martin Girsberger, Susanna H.M. Sluka, Theresia Herget, Silvia Miranda Azzarello-Burri, Anita Rauch, and Matthias Baumgartner. 2017. "MCAD-Deficiency with Severe Neonatal Onset, Fatal Outcome and Normal Acylcarnitine Profile" International Journal of Neonatal Screening 3, no. 3: 21. https://doi.org/10.3390/ijns3030021