Molecular Analysis of PKU-Associated PAH Mutations: A Fast and Simple Genotyping Test

Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Single-Nucleotide Polymorphism Loci and Assay Design
2.2. Samples
2.3. Bioinformatic Tools: Probe Design
2.4. Polymerase Chain Reaction Multiplex Amplification
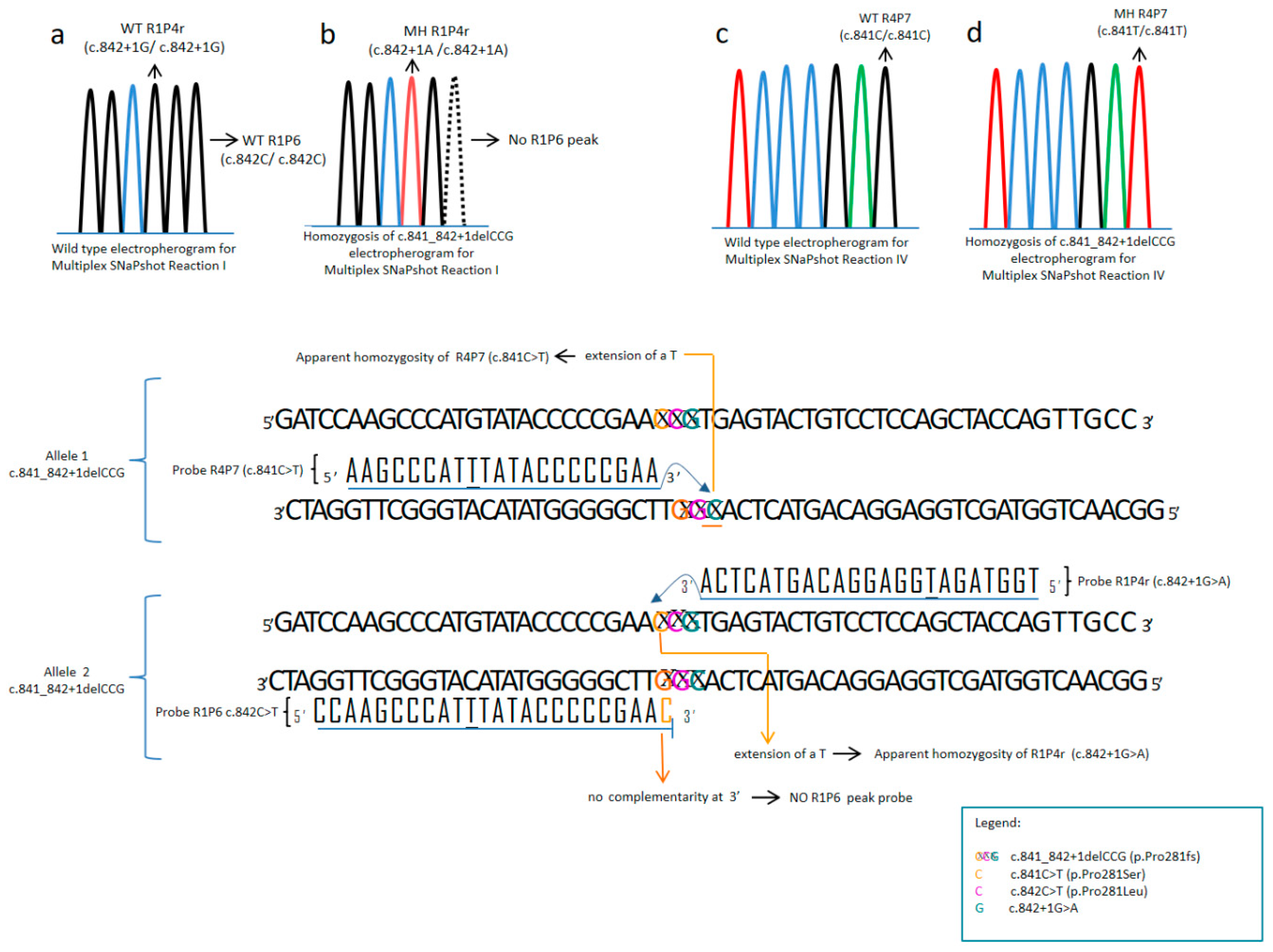
2.5. Multiplex SNaPshot Reactions
2.6. Next Generation Sequencing and Sanger Sequencing
3. Results
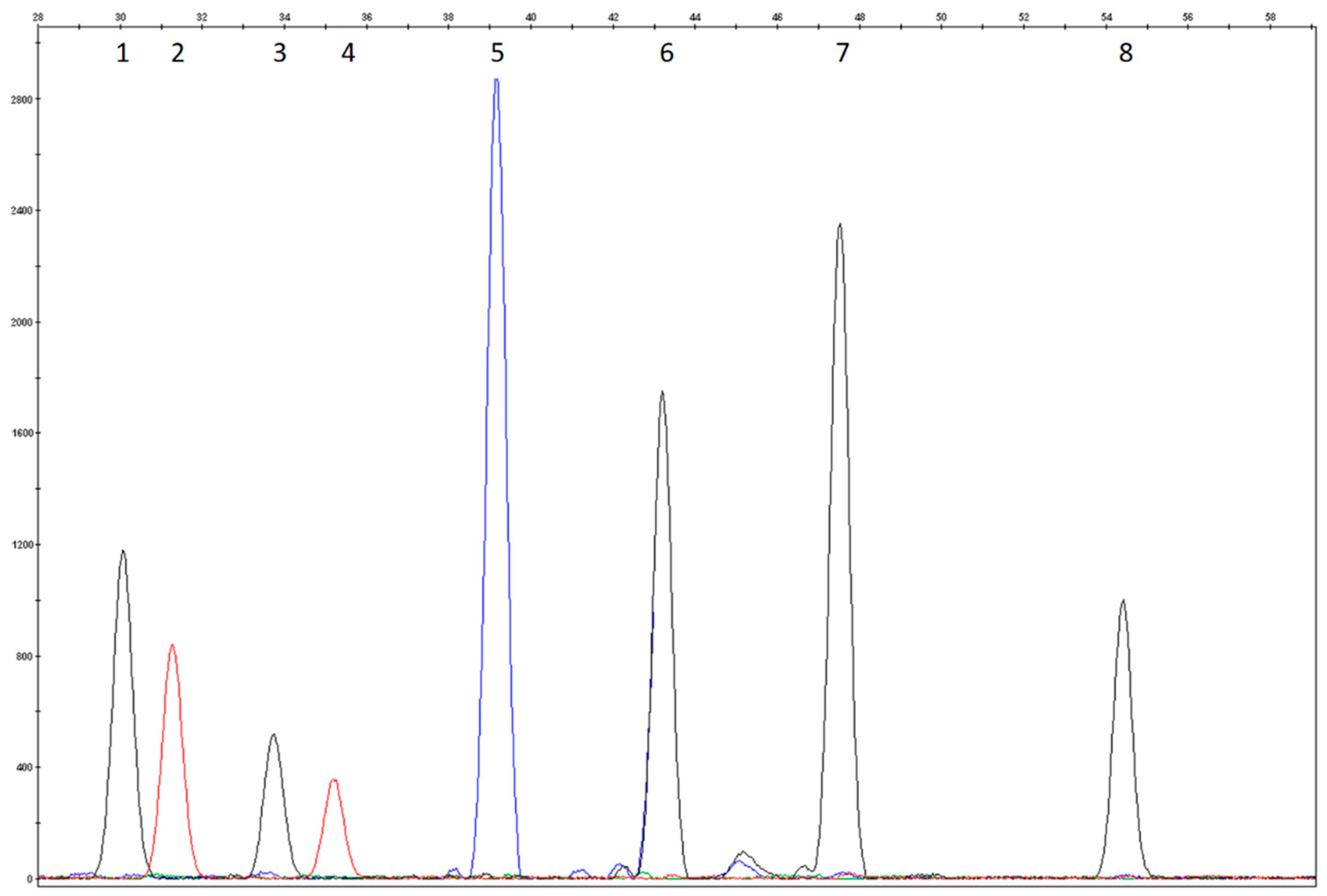
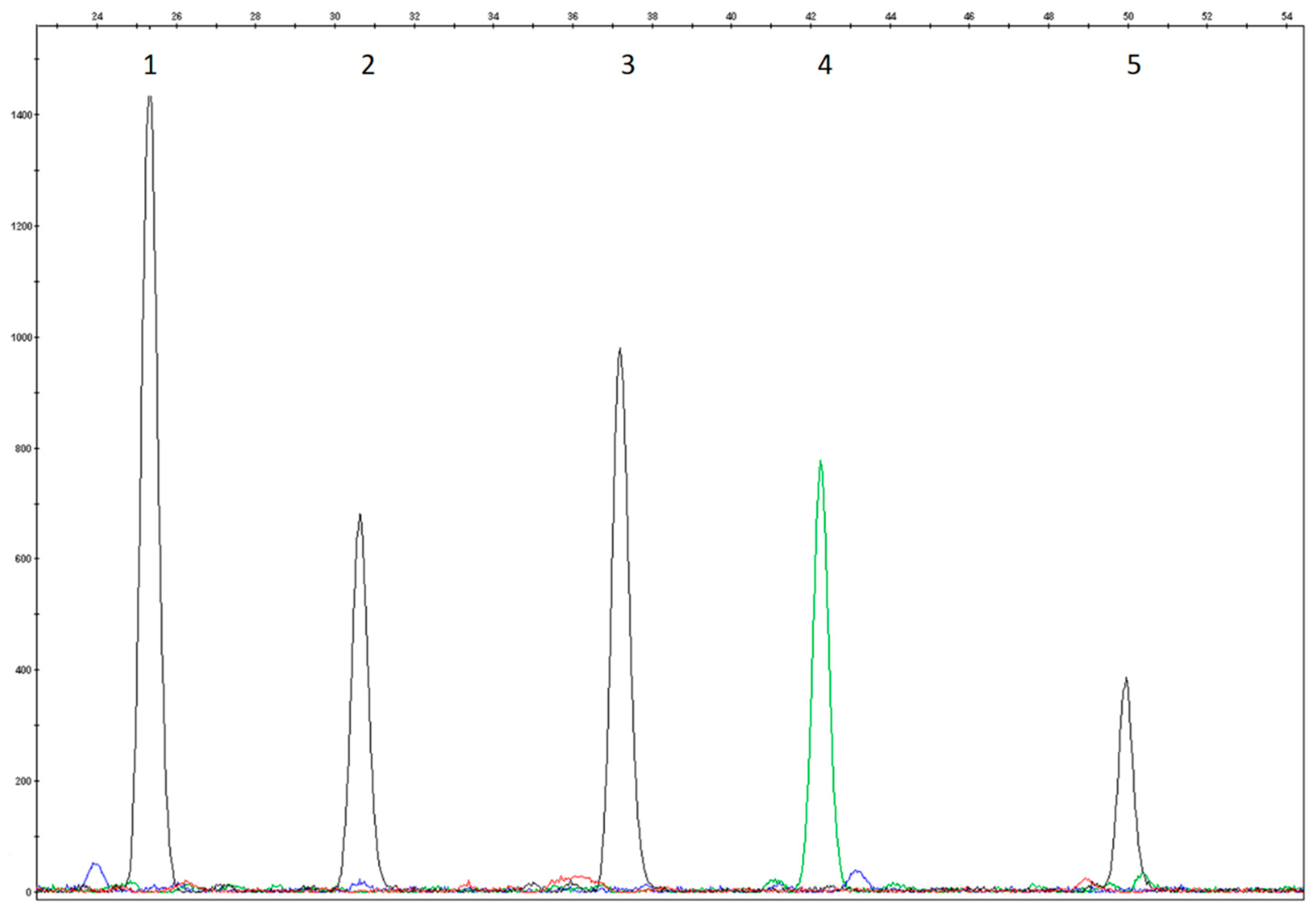
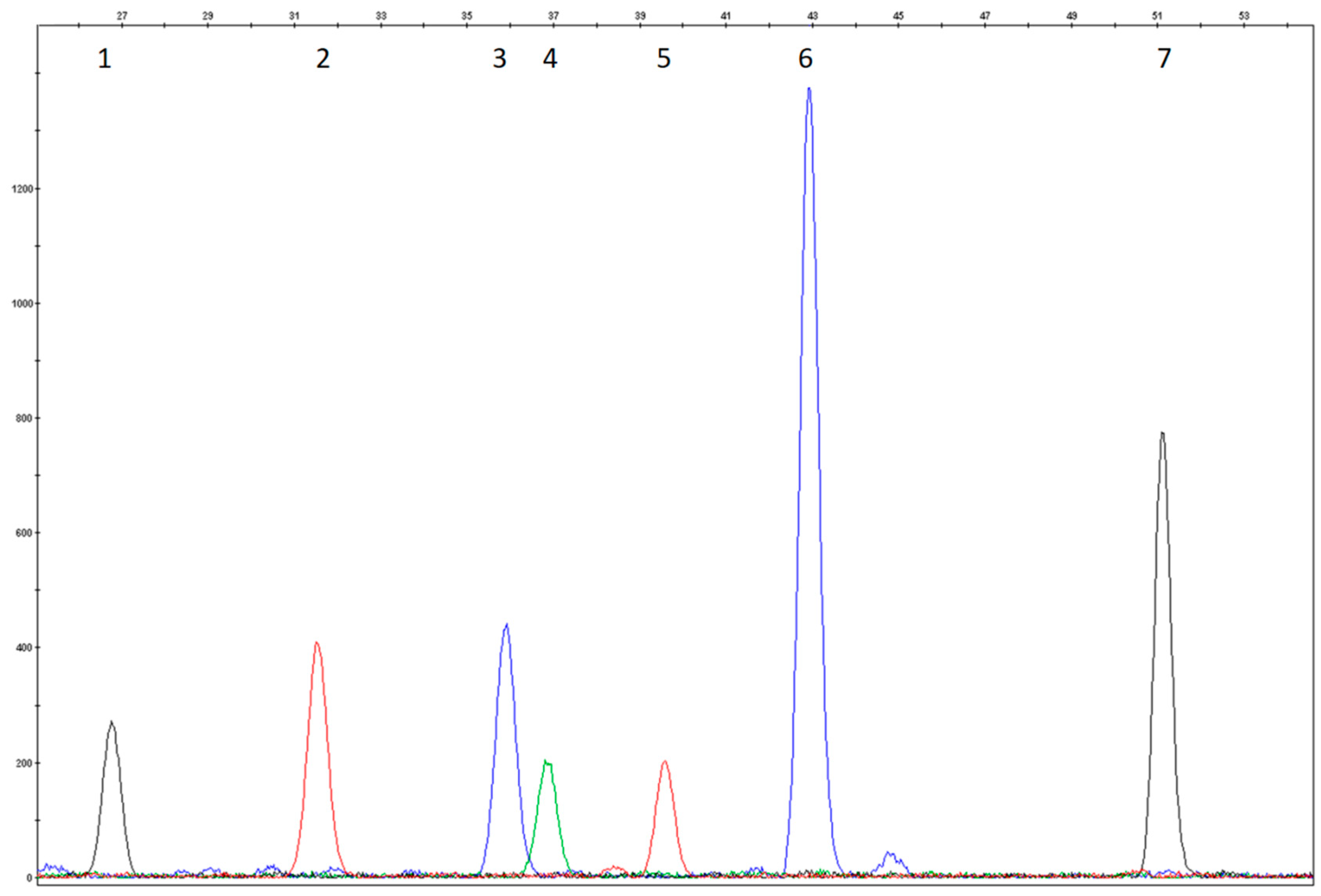
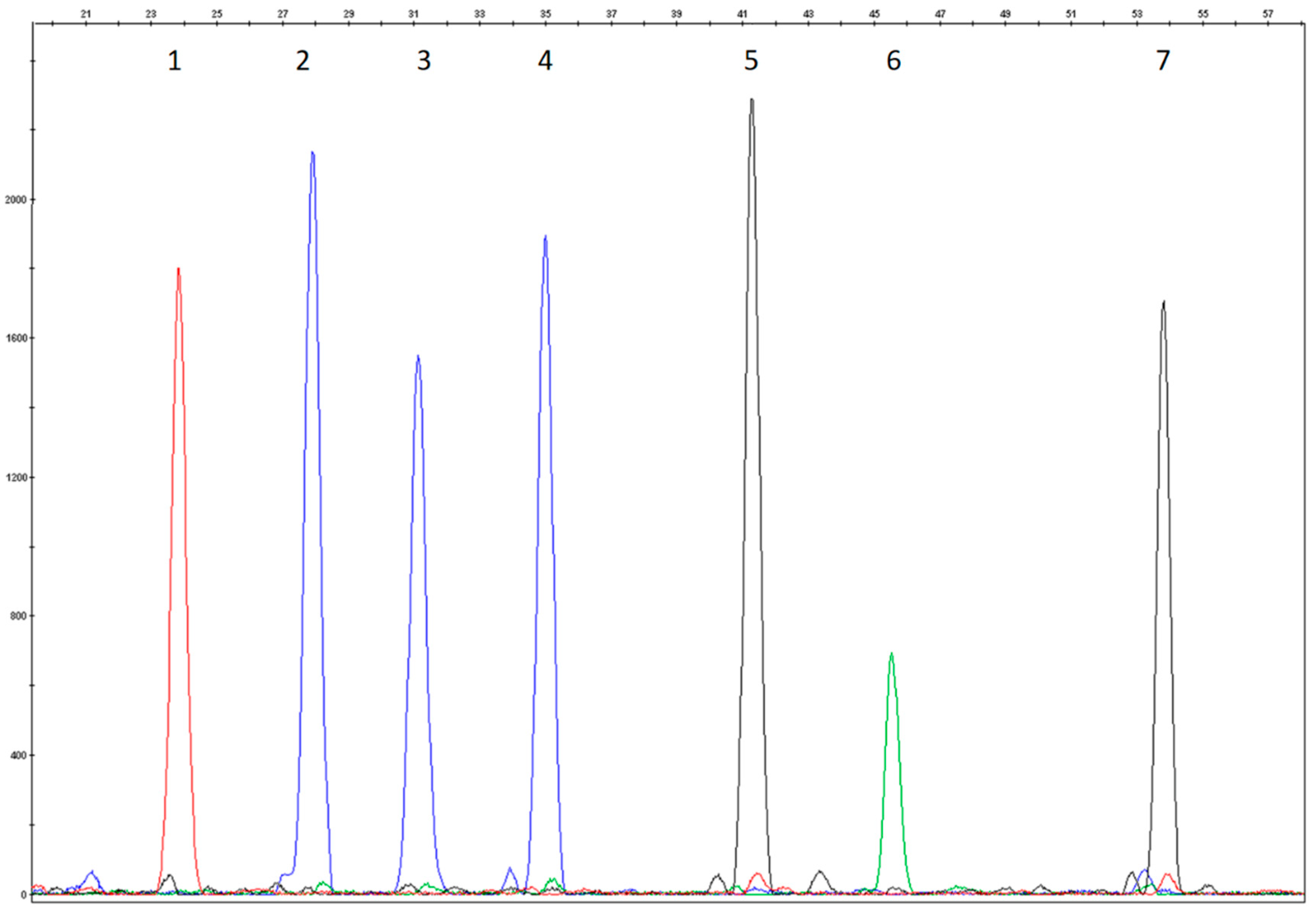
3.1. Study Design and Assay Optimization
3.2. Analytical Validation
3.3. Clinical Validation
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scriver, C.R.; Kaufman, S. The hyperphenylalaninemias. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; McGraw-Hill: New York, NY, USA, 2001; pp. 1667–1724. [Google Scholar]
- Guldberg, P.; Rey, F.; Zschocke, J.; Romano, V.; François, B.; Michiels, L.; Ullrich, K.; Hoffmann, G.F.; Burgard, P.; Schmidt, H.; et al. A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am. J. Hum. Genet. 1998, 63, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Bénit, P.; Rey, F.; Blandin-Savoja, F.; Munnich, A.; Abadie, V.; Rey, J. The mutant genotype is the main determinant of the metabolic phenotype in phenylalanine hydroxylase deficiency. Mol. Genet. Metab. 1999, 68, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Blau, N.; Shen, N.; Carducci, C. Molecular genetics and diagnosis of phenylketonuria: State of the art. Expert Rev. Mol. Diagn. 2014, 6, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Brosco, J.P.; Jeffrey, P.; Diane, B.P. The political history of PKU: reflections on 50 years of newborn screening. Pediatrics 2013, 132, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Blau, N.; Erlandsen, H. The metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Mol. Genet. Metab. 2004, 82, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Lidsky, A.S.; Güttler, F.; Woo, S.L. Prenatal diagnosis of classic phenylketonuria by DNA analysis. Lancet 1985, 1, 549–551. [Google Scholar] [CrossRef]
- Romano, V.; Dianzani, I.; Ponzone, A.; Zammarchi, E.; Eisensmith, R.; Ceratto, N.; Bosco, P.; Indelicato, A. Prenatal diagnosis by minisatellite analysis in Italian families with phenylketonuria. Prenat. Diagn. 1994, 14, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Zschocke, J.; Haverkamp, T.; Møller, L.B. Clinical utility gene card for: Phenylketonuria. Eur. J. Hum. Genet. 2012, 20, e1–e3. [Google Scholar] [CrossRef] [PubMed]
- D’Apice, M.R.; Novelli, G.; Sangiuolo, F. Diagnostic CFTR mutation analysis. Expert Opin. Med. Diagn. 2008, 2, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Aulehla-Scholz, C.; Heilbronner, H. Mutational spectrum in German patients with phenylalanine hydroxylase deficiency. Hum. Mutat. 2003, 21, 399–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zschocke, J. Phenylketonuria mutations in Europe. Hum. Mutat. 2003, 21, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaruzelska, J.; Matuszak, R.; Lyonnet, S.; Rey, F.; Rey, J.; Filipowicz, J.; Borski, K.; Munnich, A. Genetic background of clinical homogeneity of phenylketonuria in Poland. J. Med. Genet. 1993, 30, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Van Spronsen, F.J.; van Wegberg, A.M.J.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017, 5, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Giannattasio, S.; Dianzani, I.; Lattanzio, P.; Spada, M.; Romano, V.; Calì, F.; Andria, G.; Ponzone, A.; Marra, E.; Piazza, A. Genetic heterogeneity in five Italian regions: analysis of PAH mutations and minihaplotypes. Hum. Hered. 2001, 52, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Ensembl Home Page. Available online: http://www.ensembl.org (accessed on 28 June 2018).
- BIOPKU Home Page. Available online: http://www.biopku.org (accessed on 28 June 2018).
- IDT Integrated DNA Technologies Home Page. Available online: http://eu.idtdna.com (accessed on 19 June 2017).
- BLAST Basic Local Alignment Search Tool Home Page. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 8 March 2017).
- Mattocks, C.J.; Morris, M.A.; Matthijs, G.; Swinnen, E.; Corveleyn, A.; Dequeker, E.; Müller, C.R.; Pratt, V.; Wallace, A.; Euro Gentest Validation Group. A standardized framework for the validation and verification of clinical molecular genetic tests. Eur. J. Hum. Genet. 2010, 18, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Association for Molecular Pathology Clinical Practice Committee 2009. Molecular Diagnostic Assay Validation. Available online: https://www.amp.org/AMP/assets/File/resources/AssayValidation_Final.pdf (accessed on 28 March 2018).
- Bik-Multanowski, M.; Kaluzny, L.; Mozrzymas, R.; Oltarzewski, M.; Starostecka, E.; Lange, A.; Didycz, B.; Gizewska, M.; Ulewicz-Filipowicz, J.; Chrobot, A.; et al. Molecular genetics of PKU in Poland and potential impact of mutations on BH4 responsiveness. Acta Biochim. Pol. 2013, 60, 613–616. [Google Scholar] [PubMed]
- Polak, E.; Ficek, A.; Radvanszky, J.; Soltysova, A.; Urge, O.; Cmelova, E.; Kantarska, D.; Kadasi, L. Phenylalanine hydroxylase deficiency in the Slovak population: Genotype–phenotype correlations and genotype-based predictions of BH4-responsiveness. Gene 2013, 526, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Rad, I.A.; Jazani, N.H.; Zarrin, R.; Ghazavi, A. Mutation analysis of the phenylalanine hydroxylase gene in Azerbaijani population, a report from West Azerbaijan province of Iran. Iran J. Basic Med. Sci. 2015, 18, 649. [Google Scholar] [PubMed]
- Ozgüç, M.; Ozalp, I.; Coşkun, T.; Yilmaz, E.; Erdem, H.; Ayter, S. Mutation analysis in Turkish phenylketonuria patients. J. Med. Genet. 1993, 30, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.L.; Castro-Magalhães, M.; Fonseca, C.G.; Starling, A.L.P.; Januário, J.N.; Aguiar, M.J.B.; Carvalho, M.R.S. PKU in minas Gerais state, Brazil: Mutation analysis. Ann. Hum. Genet. 2008, 72, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Eisensmith, R.C.; Martinez, D.R.; Kuzmin, A.I.; Goltsov, A.A.; Brown, A.; Singh, R.; Woo, S.L. Molecular Basis of Phenylketonuna and a Correlation Between Genotype and Phenotype in a Heterogeneous Southeastern US Population. Pediatrics 1996, 97, 512–516. [Google Scholar] [PubMed]
- Biglari, A.; Saffari, F.; Rashvand, Z.; Alizadeh, S.; Najafipour, R.; Sahmani, M. Mutations of the phenylalanine hydroxylase gene in Iranian patients with phenylketonuria. Springerplus 2015, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chen, H.; Zhang, Y.; Li, L.; Mi, H.; Xu, Q.; Zhu, B. Mutations of phenylalanine hydroxylase gene detected in 20 patients with phenylketonuria from Yunnan Province. Chin. J. Med. Genet. 2015, 32, 153–157. [Google Scholar]
- Li, N.; Jia, H.; Liu, Z.; Tao, J.; Chen, S.; Li, X.; Liang, Y. Molecular characterisation of phenylketonuria in a Chinese mainland population using next-generation sequencing. Sci. Rep. 2015, 5, 15769. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Exon | Our Frequency | Frequency by Giannattasio et al. [15] |
|---|---|---|---|
| c.1066-11G>A (IVS10-11G>A) | 1 | 10.65 | 19.4 |
| p.Arg261Gln (c.782G>A) | 7 | 9.6 | 13.5 |
| p.Ala403Val (c.899C>T) | 12 | 7.2 | 2.1 |
| p.Arg158Gln (c.473G>A) | 5 | 6 | 4.8 |
| p.Leu48Ser (c.143T>C) | 2 | 4.3 | 9.7 |
| p.Arg261* (c.781C>T) | 7 | 4.2 | 3.8 |
| p.Arg408Trp (c.1222C>T) | 12 | 4 | 1 |
| p.Pro281Leu (c.842C>T) | 7 | 4 | 3.1 |
| p.Ala300Ser (c.898G>T) | 8 | 3.5 | 3.8 |
| p.Tyr414Cys (c.1241A>G) | 12 | 3 | 2.1 |
| c.441+5G>T (IVS4+5G>T | 4 | 2.5 | / |
| p.Glu390Gly (c.1169A>G) | 11 | 2.0 | 1 |
| p.Arg252Trp (c.754C>T) | 7 | 1.8 | 3.8 |
| p.Phe55del (c.163_165delTT) | 2 | 1.7 | 3.1 |
| c.842+1G>A (IVS7+1G>A) | 7 | 1.5 | 2.1 |
| p.Val245Ala (c.734T>C) | 7 | 1.5 | / |
| c.1315+1G>A (IVS12+1G>A) | 12 | 1.3 | 1 |
| p.Arg243* (c.727C>T) | 7 | 1.3 | 0.7 |
| PROBE NAME | PROBE SEQUENCE (5′-3′) | PROBES LENGTH | MUTATION(S) | |
|---|---|---|---|---|
| Multiplex SNaPshot Reaction I | R1P1 | Tail-TTGCACTGGTTTCCGCCTC | 30 bp | p.Arg243* (c.727C>T) |
| R1P2 | Tail-GGGTGGCCTGGCCTTC | 35 bp | p.Arg261* (c.781C>T) | |
| R1P3r | Tail-CAGGACACCCAAGAAATCCC r | 38 bp | p.Arg252Trp (c.754C>T) | |
| R1P4r | Tail-TGGTAGATGGAGGACAGTACTCA R | 44 bp | c.842+1G>A (IVS7+1G>A) | |
| R1P5r | Tail-ATGATGTACTGTGTGCAGTGGAAACT R | 48 bp | p.Arg261Gln (c.782G>A) | |
| R1P6 | Tail-CCAAGCCCATTTATACCCCCGAAC a | 53 bp | p.Pro281Leu (c.842C>T) p.Pro281Arg (c.842C>G) b | |
| Multiplex SNaPshot Reaction II | R2P1r | Tail-CCTTACCTGGGAAAACTGGG r | 21 bp | p.Ala300Ser (c.898G>T) |
| R2P2 | Tail-ACTCAAAGAAGAAGTTGGTGCAT | 29 bp | p.Leu48Ser (c.143T>C) | |
| R2P3r | Tail-GCAATGTCAGCAAACTGCTTC r | 35 bp | p.Arg158Gln (c.473G>A) | |
| R2P4r | Tail-AGATGATTGTAGCACTGACCTCAA r | 39 bp | p.Phe55del(c.163_165delTT) | |
| R2P5 | Tail-A ATC TCA TCC TAC GTG CCA TGG A | 48 bp | c.441+5G>T (IVS4+5G>T) | |
| Multiplex SNaPshot Reaction III | R3P1 | Tail-TGGTATTGGTCTTAGGAACTTTG | 23 bp | p.Ala403Val (c.899C>T) |
| R3P2r | Tail-AATCCTTTGGGTGTATGGGTCG r | 27 bp | p.Tyr414Cys (c.1241A>G) | |
| R3P3r | Tail-GCGAACTGAGAAGGGCC r | 33 bp | p.Arg408Trp (c.1222C>T) | |
| R3P4r | Tail-ACCTTACTTTCTCCTTGGCATCACTAAAACTC r | 36 bp | p.Glu390Gly (c.1169A>G) | |
| R3P5 | Tail-GCAGATTAAGATTTTGGCTGATTCCATTAACA | 41 bp | c.1315+1G>A (IVS12+1G>A) | |
| R3P6r | Tail-CTTCTCTGATAAGCAGTACTGTAG GCCC r | 53 bp | c.1066-11G>A (IVS10-11G>A) | |
| Multiplex SNaPshot Reaction IV | R4P1r | Tail-TTTCCGCCTCCGACCTG a,r | 17 bp | p.Val245Ala (c.734T>C) p.Val245Glu (c.734T>A) b |
| R4P2 | Tail-CCTTACCTGGGAAAACTGG | 25 bp | p.Ala300Val (c.899C>T) | |
| R4P3 | Tail-AATCCTTTGGGTGTATGGGTC | 27 bp | p.Tyr414Tyr (c.1242C>T) | |
| R4P4 | Tail-CTGATTTCCGCCTCCGACCT a | 33 bp | p.Val245Ile (c.733G>A) b p.Val245Leu (c.733G>C) b | |
| R4P5 | Tail-AGCGAACTGAGAAGGGC | 38 bp | p.Arg408Gln (c.1223G>A) | |
| R4P6 | Tail-TGGTAGATGGAGGACAGTACTC | 46 bp | c.842+2T>A (IVS7+2T>A) | |
| R4P7 | Tail-AAGCCCATTTATACCCCCGAA a | 52 bp | p.Pro281Ser (c.841C>T) b p.Pro281Ala (c.841C>G) b |
| Parameters | Sample Size | Results | |
|---|---|---|---|
| Accuracy | TR/TR+FR | N = 19 | 100% |
| Sensitivity | TP/TP+FN | N = 19 | 100% |
| Specificity | TN/TN+FP | N = 10 | 100% |
| Precision (Calculated on size peaks for each probe in each run) | Repeatability and Reproducibility | N = 5 TO 12 Replicates | SD = FROM 0.01 TO 0.3 |
| Apparently Aberrant Patterns | ||
| Peak Absence | Present in Homozygosis | Genotype (in Homozygosis) |
| c.842C > T (p.Pro281Leu) | c.841C > T(p.Pro281Ser) | c.841C > T (p.Pro281Ser) |
| c.899C > T (p.Ala300Val) | c.898G > T (p.Ala300Ser) | c.898G > T (p.Ala300Ser) |
| c.734T > C (p.Val245Ala) | c.733G > A (p.Val245Ile) | c.733G > A (p.Val245Ile) |
| c.733G > C (p.Val245Leu) | c.733G > C (p.Val245Leu) | |
| c.1241A > G (p.Tyr414Cys) | c.1242C > T (p.Tyr414Tyr) | c.1242C > T (p.Tyr414Tyr) |
| c.1222C > T (p.Arg408Trp) | c.1223G > A (p.Arg408Gln) | c.1223G > A (p.Arg408Gln) |
| c.842+1G > A | c.842+2T > A | c.842+2T > A |
| Complex Apparently Aberrant Pattern | Reason | Recommendation |
| No c.842C>T (p.Pro281Leu) peak + c.841C>T (p.Pro281Ser) (MH) + c.842+1G>A (MH) | c.841_842+1delCCG (p.Pro281fs) (MH) | Sequencing exon 7 |
| c.842C>T (p.Pro281Leu) (MH) + c.841C>T (p.Pro281Ser) (HZ) | c.842C>T (p.Pro281Leu)/c.841C>T (p.Pro281Ser) (CHZ) | Sequencing exon 7 |
| c.841C>T (p.Pro281Ser) (HZ) + | c.842C>T (p.Pro281Leu)/c.841_842+1delCCG (p.Pro281fs) (CHZ) | Sequencing exon 7 |
| c.841C>T (p.Pro281Ser) (HZ) + c.842C>T (p.Pro281Leu) (HM)+ c.842+1G>A (HZ) | c.842C>T (p.Pro281Leu)/c.841_842+1delCCG (p.Pro281fs) (CHZ) | Sequencing exon 7 |
| No c.842C>T (p.Pro281Leu) peak + c.841C>T (p.Pro281Ser) (MH) + c.842+1G>A (HZ) | c.841C>T (p.Pro281Ser)/c.841_842+1delCCG (p.Pro281fs) (CHZ) | Sequencing exon 7 |
| c.841C>T (p.Pro281Ser) (HZ) +c.842+1G>A (MH) | c.841_842+1delCCG (p.Pro281fs)/c.842+1G>A (CHZ) | Sequencing exon 7 |
| Reflex Testing | ||
| Apparent Compound Heterozygote | Reason | Recommendation |
| c.841C>T (p.Pro281Ser)/c.842+1G>A | c.841_842+1delCCG (p.Pro281fs) in heterozygosis | Sequencing exon 7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolve, M.; Artiola, C.; Pasquali, A.; Giovanniello, T.; D’Amici, S.; Angeloni, A.; Pizzuti, A.; Carducci, C.; Leuzzi, V.; Carducci, C. Molecular Analysis of PKU-Associated PAH Mutations: A Fast and Simple Genotyping Test. Methods Protoc. 2018, 1, 30. https://doi.org/10.3390/mps1030030
Tolve M, Artiola C, Pasquali A, Giovanniello T, D’Amici S, Angeloni A, Pizzuti A, Carducci C, Leuzzi V, Carducci C. Molecular Analysis of PKU-Associated PAH Mutations: A Fast and Simple Genotyping Test. Methods and Protocols. 2018; 1(3):30. https://doi.org/10.3390/mps1030030
Chicago/Turabian StyleTolve, Manuela, Cristiana Artiola, Amelia Pasquali, Teresa Giovanniello, Sirio D’Amici, Antonio Angeloni, Antonio Pizzuti, Claudia Carducci, Vincenzo Leuzzi, and Carla Carducci. 2018. "Molecular Analysis of PKU-Associated PAH Mutations: A Fast and Simple Genotyping Test" Methods and Protocols 1, no. 3: 30. https://doi.org/10.3390/mps1030030