Muscular Dystrophies and Cancer Cachexia: Similarities in Chronic Skeletal Muscle Degeneration


Exercise Physiology Research Group, Department of Movement Sciences, Tervuursevest 101, 3001 KU Leuven, Belgium
J. Funct. Morphol. Kinesiol. 2017, 2(4), 39; https://doi.org/10.3390/jfmk2040039
Submission received: 2 October 2017
/
Revised: 31 October 2017
/
Accepted: 4 November 2017
/
Published: 7 November 2017
(This article belongs to the Special Issue Muscular Dystrophy)
Abstract
:Muscular dystrophies (MDs) are inheritable diseases caused by mutations in specific genes encoding muscle proteins and characterized by progressive muscle wasting and weakness. MDs are classified into nine main categories, encompassing approximately 30 different diseases. Duchenne muscular dystrophy and Becker muscular dystrophy (DMD and BMD) are the most commonly diagnosed neuromuscular disorders, affecting respectively ~1/3500 and 3–6/100,000 male infants worldwide. DMD is characterized by absence of dystrophin function due to complete loss of expression or to the presence of a non-functional dystrophin protein in muscle tissue, while a semi-functional dystrophin isoform is typically present in BMD. Lack of dystrophin function in MDs induces myofiber damage, causing persistent cycles of degeneration/regeneration, exhaustion of regenerative potential and fibrosis, and resulting in muscle degeneration. Cancer cachexia is a multifactorial syndrome characterized by severe body weight loss and muscle degenerative atrophy. Up to 80% of cancer patients show cachexia, and the syndrome has been estimated to cause ~2 million of deaths per year. This cancer-related muscle wasting is mediated by pro-inflammatory cytokines and tumor-released mediators able to activate specific catabolic pathways in muscle tissue. MDs and cancer-associated cachexia (CAC) share some peculiar features, including systemic and muscle-specific inflammation, alteration of myogenic potential, ultrastructural abnormalities, and dysfunction of the dystrophin glycoprotein complex (DCG). This review focuses on the pathophysiological comparison between these two chronic muscle degenerative conditions and discusses the most relevant and promising therapeutic strategies considered so far.
{kind=link}
{kind=link}
1. Introduction
Muscular dystrophies (MDs) and cancer-associated cachexia (CAC) are debilitating degenerative conditions causing loss of both muscle mass and function [1]. The most common muscular dystrophies, Duchenne muscular dystrophy (DMD) and the Becker muscular dystrophy (BMD), are caused by mutations in the DMD gene encoding the 427 kD structural protein called dystrophin [2]. Globally, DMD and BMD affect respectively ~1/3500 and 3–6/100,000 male infants [3]. The main DMD symptom occurring in early childhood is progressive muscle wasting, which starts around three years of age and continues leading to respiratory deficiency, cardiac failure, and premature death, typically by 20 years of age. BMD is less common than DMD and its onset is usually in late childhood or adolescence. BMD is characterized by a slower, less predictable progression and a relatively increased overall survival compared with DMD [2].
Similarly to MDs, CAC is a clinically relevant muscle degenerative condition. CAC is a multifactorial syndrome characterized by severe loss of body weight and skeletal muscle mass (that can be observed with or without loss of fat mass) and occurs in up to 80% of all cancer cases [4]. It has been reported that CAC directly causes 2 million of deaths per year, accounting for 20% of total cancer deaths [4,5]. Similarly to MDs, cachexia syndrome induces progressive muscle degeneration, impairs muscle function, and increases patients’ susceptibility to fatigue [1], while it can also counteract the efficacy of standard anticancer therapies.
Because of the different aetiology between MDs and CAC (i.e., genetic vs. cancer-associated muscle degenerative condition, respectively), such diseases are diagnosed using different criteria to evaluate the extent of muscle degeneration. Specifically, a MD diagnosis is generally based on the results of medical examinations performed to evaluate muscle function and family medical history, together with blood tests for creatine kinase levels determination, a known marker of fiber muscle damage. The measure of dystrophin content by immunohistological analysis of muscular biopsies is a common diagnostic procedure, while sequencing analyses (e.g., RNAseq and whole exome sequencing) are able to identify specific gene mutations [3,6].
The criteria to diagnose cachexia in cancer patients are primarily based on the evaluation of their body mass index (BMI). Specifically, cancer patients are considered cachectic when they lose (i) > 5% of their body weight within six months or they show (ii) a BMI < 20 and any degree of weight loss > 2% or (iii) muscle depletion and any degree of weight loss > 2% [4].
Muscle wasting mediated by both MDs and CAC is associated with high medical costs and economic burden. A study performed in 770 DMD patients from Germany, Italy, the United Kingdom, and the United States showed an annual average of cost per-patient between $23,920 and $54,270, which is up to 16 times higher than the health expenditure per-capita in that countries. Furthermore, the total societal burden has been calculated to be between $80,120 and $120,910/patient per year, while the household burden estimates are in the range $58,440–71,900 [7]. Arthur and colleagues analysed the medical costs associated with CAC treatment for the year 2009 in a cohort of cancer patients in the US [8]. The study revealed that cachexia increased both hospitalization costs and length of inpatient stay for several cancers, including those of the lung, pancreas, esophagus and Kaposi’s sarcomas. The average of inpatients costs for all cancers were $10,633 and $917 for cachectic and not cachectic patients, respectively [8].
2. Alterations in Dystrophin Glycoprotein Complex (DGC) in MDs (Muscular Dystrophies) and CAC (Cancer-Associated Cachexia)
DMD may be caused by recessive frameshift deletions, nonsense mutations and duplications of the DMD gene, which can result in a complete absence of dystrophin or in a nonfunctional protein. However, in BMD myofibers produce a semi-functional form of dystrophin [2,3].
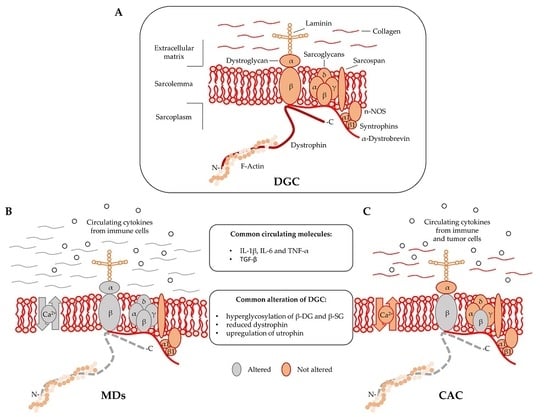
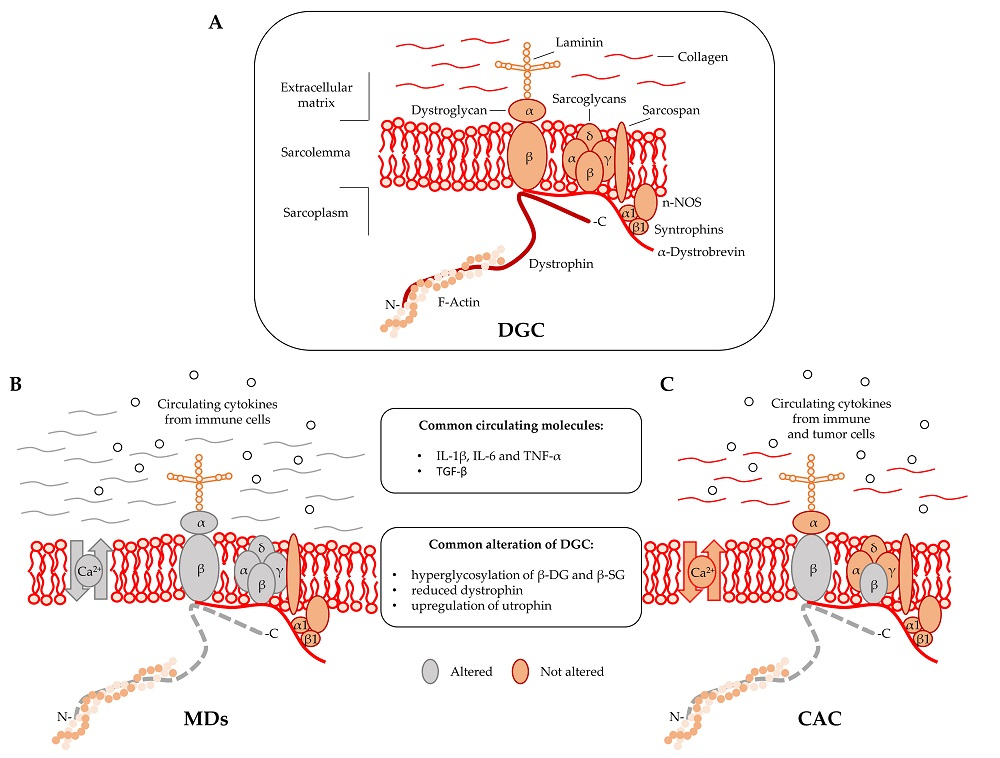
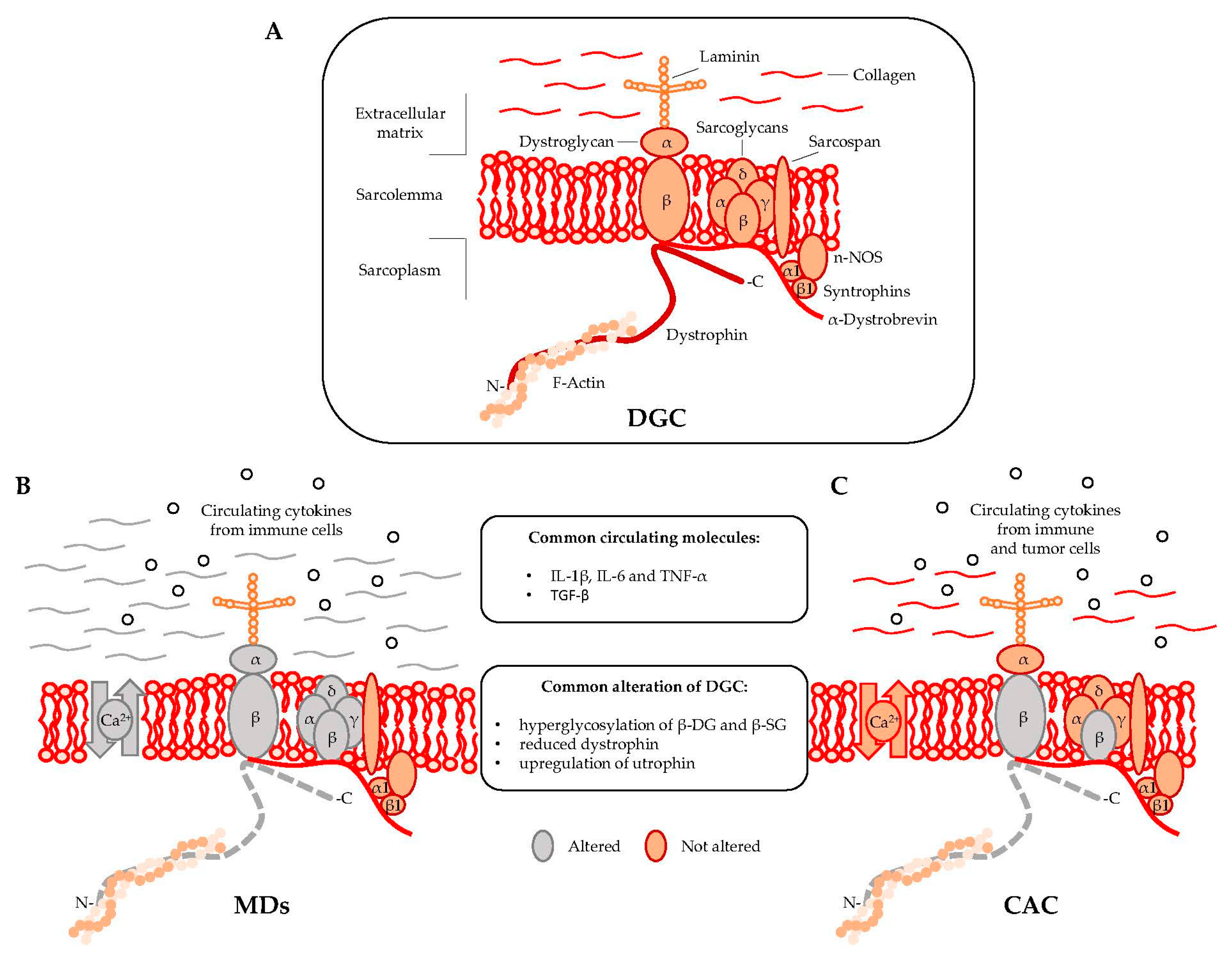
Dystrophin is localized at the inner surface of the sarcolemma. It interacts with the cytoskeletal filamentous actin (F-actin) and with multiple proteins assembling the dystrophyn glycoprotein complex (DGC), a multimeric protein complex connecting the cytoskeleton with the extracellular matrix (ECM). DGC contains intracellular (α1- and β1-syntrophin, α-dystrobrevin, and nNOS), transmembrane (β-dystroglycan, α-, β-, γ-, δ-sarcoglycan, and sarcospan), and extracellular proteins (α-dystroglycan and laminin-2) (Figure 1A) [2,9].
DGC acts as the structural link between the cytoskeleton and ECM [10] and also regulates cellular signaling by interacting with the growth factor receptor bound protein 2 (Grb2) and the neuronal nitric oxide synthase (nNOS) [11]. In particular, the functional interaction of nNOS with syntrophins and dystrobrevin (Figure 1) ensures vasomotility control in response to changes in the energy demand of actively contracting muscle [12]. DGC structural integrity is of pivotal importance for myofiber contraction and for preventing contraction-induced sarcolemma injuries [13]. Mutations in genes encoding specific proteins of the DGC or their posttranslational defects might alter the function of the complex, resulting in sarcolemma damage. Specific sarcoglycanopathies, a subtype of limb-girdle muscular dystrophies, are caused by mutations in the α, β, γ, and δ subunit of the sarcoglycan complex [2]. Moreover, aberrant posttranslational modifications resulting in sarcolemma destabilization include hypoglycosylation of α-dystroglycan (α-DG), which is observed in many MDs and causes a reduction of DGC binding capacity to the ECM proteins [2,14].
A regulatory link between MDs and CAC in DGC dysfunctions has been proposed in 2005 by Acharyya and colleagues [15]. The study showed structural abnormalities in both the sarcolemma and the associated basal lamina of muscles isolated from colon carcinoma C26-bearing mice and cancer patients [15,16]. The results revealed that the sarcolemma abnormalities observed in cachectic muscles were associated with a myofiber damage that was similar to the one observed in DMD patients, although less severe [17]. The muscle analysis of C26-bearing mice also identified common DGC-alterations between MDs and CAC including, (i) reduced dystrophin; (ii) a compensatory upregulation of utrophin, and (iii) hyperglycosylation of β-dystroglycan (β-DG) and β-sarcoglycan (β-SG) (Figure 1B,C). The hyperglycosylation of β-DG and β-SG was also observed in muscles from mice bearing Lewis lung carcinoma (LLC), another model of cancer cachexia, while the expression and the post-transcriptional modifications of other components of the DGC—such as α-DG, α-SG, δ-SG, dysferlin, syntrophin, and Grb2—were unchanged [15]. DGC dysfunctions appeared together with Atrogin-1/MAFbx and MuRF1 expression, which are known E3 ligases involved in muscle proteolysis during tumor progression. Furthermore, β-DG and β-SG hyperglycosylation in cancer cachexia was also confirmed in muscle biopsies from patients with gastrointestinal tumor and gastroesophageal adenocarcinoma. Overall, this study showed that besides being hallmark of MDs, DGC dysfunction is also an important pathological characteristic of cancer cachexia [15].
3. Other Pathophysiological Features of Muscle Wasting in MDs and CAC
The absence of dystrophin and the aberrant glycosylation of α-dystroglycan (α-DG) induce a chronic influx of calcium into the myofibers, causing the elevated inflammation and necrosis observed in dystrophic muscles [2,3,18]. These conditions stimulate the migration of inflammatory cells, which produce pro-inflammatory cytokines such as IL-1β and IL-6 and TNF-α that play an important role in the pathogenesis of MDs. Moreover, transforming growth factor beta (TGF-β), a fibrogenic cytokine produced by muscle resident fibroblasts, induces ECM deposition and inhibits satellite cells differentiation [19]. These events impair muscle tissue’s homeostasis and results in a chronic degeneration sustained by altered and persistent cycles of regeneration/degeneration. In this picture, satellite cells, located between the basal lamina and the sarcolemma [20], and other muscle progenitors [21] are constantly activated to regenerate new fibers that should replace the damaged ones. The muscle repair process also includes over-synthesis and accumulation of ECM, which progressively replaces necrotic fibers, while the continue activation of satellite cells impairs their myogenic potential and ultimately induce their exhaustion. Thus, impairment of muscle regeneration, together with the accumulation of ECM, contributes to the massive fibrosis observed in MDs [3].
Muscle biopsies from cancer patients as well as skeletal muscle tissue from C26-bearing mice do not reveal signs of necrotic fibers, massive infiltration of inflammatory cells or an extensive presence of regenerating fibers (e.g., centrally nucleated fibers), if compared with those from MDs patients or mdx mice, used as a murine model of DMD [15]. However, similarly to MDs, also in CAC the dysregulation of muscle precursors impairs muscle regenerative program [16]. Specifically, the upregulation of Pax7, a marker of satellite cells, and the increase of Pax7 positive cells not followed by their proper myogenic differentiation have been observed in muscles from C26 [16,22] and LLC-bearing mice [16], as well as in six-months-old mdx mice [23]. This highlights a common involvement of satellite cells dysfunction in the muscle wasting observed in both MDs and CAC.
Circulating pro-inflammatory cytokines, such as IL-1β, IL-6, tumor necrosis factor-α (TNF-α), TWEAK (an emerging cytokine and member of the TNF-α superfamily), and TGF-β are abundant in CAC (Figure 1). Interferon-γ (INF-γ) and other tumor mediators such as the proteolysis-inducing factor (PIF) have been established as pivotal activators of specific catabolic pathways, including ubiquitin-proteasome pathway (UPP), proteases-mediated degradation, and autophagocytosis. The perturbation of such catabolic processes leads to an imbalance in the rates of muscle protein synthesis and degradation [24,25,26]. Specifically, pro-inflammatory cytokines derived from either immune or tumor cells (e.g., IL-1β, IL-6, and TNF-α) have been shown to trigger muscle wasting through activation of nuclear factor-κB (NF-κB) and the JAK/STAT pathways, respectively. The involvement of NF-κB signaling pathway has been linked to the loss of skeletal muscle mass in many chronic degenerative conditions, including MDs and CAC [27]. NF-κB is activated by the binding of TNF-α with its receptor, TNFR1 which can induce apoptosis via the TRADD/FADD/Caspase 8 pathway, or protein degradation via NF-κB. TWEAK also activates NF-kB by binding the fibroblast growth factor-inducible 14 (Fn14) receptor [28]. The pathophysiological role of TWEAK/Fn14 in CAC has been demonstrated in both LLC and C26-bearing mice [29], while the expression of Fn14 correlates with the severity of muscle degeneration in myotonic dystrophy type 1 (DM1) [30]. Overall, the activation of NF-κB during muscle degeneration induces the overexpression of muscle ring finger protein 1 (MuRF-1), an ubiquitin E3 ligase, and subsequent protein degradation through the UUP [28]. The UPP is also regulated by atrophy gene 1/muscle atrophy F-box (Atrogin-1/MAFbx) that together with MuRF-1 act as the two main regulators of muscle protein breakdown [31]. Perturbation of the UPP system can also affects MDs [32]. In particular, mutations in Trim32, another E3 ubiquitin ligases, have been described in limb girdle muscular dystrophy type 2H [33], a mild form of muscular dystrophy, as well as in sarcotubular myopathy [34].
4. Therapeutic Approaches in MDs and CAC
4.1. MDs
No effective therapies against the chronic muscle degeneration observed in both MDs and CAC are available so far. Because of the high membrane susceptibility to damage in dystrophic muscles, many treatments trying to stabilize the sarcolemma integrity by attenuating the inflammation have been proposed [3]. In particular, promising results have been obtained for the treatment of MDs with non-steroidal anti-inflammatory drugs (NSAIDs), such as nabumetone, ibuprofen, and isosorbide dinitrate that counteract the systemic burden of TGF-β and the excessive ECM deposition [1,35].
Other pharmacological approaches have been proposed to treat the inflammation and the necrosis mediated by both mitochondrial dysfunction and the altered metabolism of dystrophic muscles [18]. The use of synthetic glucocorticoids, (e.g., α-methylprednisolone) which normalizes cytosolic calcium in dystrophic muscles, showed positive results in counteracting apoptosis and/or necrosis [36]. Among corticosteroids drugs, prednisone and deflazacort were proposed for their ability to ameliorate muscle function by preventing myofiber damage, weakness and loss of muscle contraction [1,3]. Another therapeutic strategy that has been adopted aims to restore the transcription of utrophin as compensatory mechanism [1,2,3]. Nabumetone is a small anti-inflammatory molecule able to inhibit cyclooxygenase and partially restore the utrophin expression [1,3,37]. However, also other small molecules, such as RTC13, RTC14, ataluren, and PTC124A as well as aminoglycosides antibiotics (e.g., gentamicin) can partially restore full-length dystrophin in patients with stop codon mutations by the ribosomal read-through process [1,3].
The gene replacement strategy with lentiviral vectors and adeno-associated viruses (AAV) delivering micro-dystrophin or mini-utrophin was historically conceived to counteract the lack of dystrophin in DMD and BDM patients, while the use of exon skipping has been used to increase the endogenous expression of dystrophin [1,3,38]. However, clinical studies revealed only limited restoration of dystrophin in small areas of muscles and only poor outcomes in terms of muscle function restoration [38,39,40,41]. A clinically relevant problem that emerged from these studies is due to the strong immune responses caused by the use of viral vectors [3,38]. TALEN (transcription activator-like effector nucleases) and CRISPR (clustered regularly interspaced short palindromic repeats) technologies represent the new frontier of gene editing. Specifically, TALENs are endonucleases containing DNA binding and DNA cleavage domains that induce a double strand break in the DNA following by a homologous recombination of the DNA target [3]. CRISPR is a gene-editing system based on an RNA-guide that is also able to induce a double strand break in any location of DNA by delivering the Cas9 protein and appropriate guide RNAs sequence complementary to the DNA target [3,38]. Such powerful technology can be used to perform gene editing in theoretically any post-natal somatic cells without the use of viral vectors [3]. CRISPR/Cas9-mediated genome editing of dystrophin in germ line of mdx mice was adopted by Long and colleagues in 2014. This approach produced animals with up to 100% dystrophin gene correction [42]. More recently, new studies performed in the mdx model confirmed the feasibility of CRISPR/Cas9 therapy in vivo [43,44,45]. However, despite the encouraging results obtained in these preclinical studies, the technology itself is still in its infant stage. Some issues need to be addressed before gene editing techniques can be effectively translated into clinical trials, including the immune response to the bacterial-derived Cas9 protein [38].
Besides pharmacological and gene therapy approaches adopted in the treatment of MDs [1,3,18,46], in the last years, preclinical studies in murine and canine models investigated the myogenic potential of muscle precursors, such as satellite cells (SC), skeletal myogenic precursors (SMPS), muscle side population (SP), fibro-adipogenic progenitors (FAPs), and mesoangiblasts (MABs) [1,3,21]. These studies revealed new potential therapeutic perspectives for the treatment of MDs based on muscle progenitor cell transplantation as revealed by numerous interventional trials [21].
4.2. CAC
Since the pathogenesis of CAC has a multifactorial origin, several therapeutic approaches have been tested. Treatments included nutritional support alone or in association with anabolic drugs, anti-inflammatory agents, and molecules targeting the molecular pathways involved in cancer-induced muscle wasting [1,3]. However, despite the efforts made in preclinical models and cancer patients, only limited successes have been obtained [47].
The eicosapentaenoic acid (EPA), fish oil-enriched nutritional supplement, known as a source of omega-3 fatty acids, and docosahexaenoic acid (DHA), have been tested in the treatment of cancer patients for their ability to down-regulate the high levels of IL-1β, IL-6, TNF-α, and PIF [47,48,49]. The administration of fish oil in patients with advanced pancreatic cancer showed reduction of body weight loss and improvement of performance status, appetite, and quality of life [48,50,51,52,53].
Ghrelin is an anabolic peptide hormone proposed for the treatment of CAC. Preclinical studies revealed the ability of Ghrelin to counteract muscle atrophy by stimulating the production of IL-10, an anti-inflammatory cytokine able to neutralize the action of several pro-inflammatory cytokines, including as IL-1β, IL-6, and TNF-α [54,55]. Grehlin also suppresses NFκB, which is involved in the ubiquitin-proteasome pathway, resulting in a downregulation of both Atrogin-1/MAFbx and MuRF-1 [55]. Studies in cancer patients showed that Ghrelin can counteract body weight loss, increase energy intake, and improve muscle function [49,56].
Anabolic drugs such as anamorelin [57,58,59,60], an orally active ghrelin receptor agonist, or enobosarm [61,62], an oral nonsteroidal selective androgen receptor modulator, have been tested in phase II and III trials showing promising results [47,61,62]. In particular, cachectic cancer patients after a 12-week-long period of anamorelin administration showed gain of body weight and lean mass [57,58,60]. Minor changes were observed in muscle strength [60,62]. However, patients recruited in the trial testing anamorelin developed several side effects, including hyperglycemia, nausea, and dizziness [60], while cases of pneumonia and febrile neutropenia were observed in patients enrolled in the enobosarm trial [61].
Since circulating pro-inflammatory cytokines mediate a pivotal role in muscle wasting, many efforts have been made to lower their levels in the bloodstream. Monoclonal antibodies against IL-1, IL-6 and TNF-α have been tested in clinical trials [47,63,64]. In particular, monoclonal antibodies anti IL-1 (i.e., MABp1), IL-6 (i.e., siltuximab), or anti IL-6 receptor (i.e., tocilizumab) showed promising results in counteracting the pathophysiological features of CAC in ovarian and prostate cancer patients [65,66]. Improvement of muscle function obtained by rescue of fatigue and muscle mass loss was also reported in patients with non-small cell lung cancer treated with a humanized anti-IL-6 antibody (ALD518) [67]. Moreover, it has been reported that treatment with antibody against Fn14 prevents tumor-mediated cachexia in murine models of CAC by counteracting body weight loss, as well as fat and muscle mass loss [29]. Overall, the use of antibodies targeting of IL-6 and TWEAK/Fn14 showed promising results. However, their therapeutic value in CAC are still limited because their molecular mechanisms of action and the transduction pathways involved are still not completely known. Furthermore, considering the multifactorial origin of CAC, combined therapies aiming to counteract the inflammatory burden and the proteolytic phenomena while increasing protein synthesis need to be further developed.
5. Conclusions
In the last decades, many molecular mechanisms and mediators of chronic muscle degeneration have been discovered. Loss of muscle integrity and contraction force, increase of protein catabolism, decrease of protein synthesis, and alteration of muscle regeneration are common pathophysiological features of muscle wasting mediated by different chronic diseases, including MDs and CAC [1]. Dysfunctions in DGC similar to those observed in MDs have been identified in cancer-related muscle wasting, suggesting that myofiber damage might proceed via common mechanisms shared by these two different muscle degenerative conditions [15]. Thanks to the knowledge acquired so far, several therapeutic approaches have been proposed [1]. Unfortunately, gene therapy produced poor results in terms of dystrophin restoration and safety [1,3,38], while non-steroidal anti-inflammatory drugs and corticosteroids currently in use for dystrophic patients can marginally delay the progression of the disease by counteracting muscle inflammation and necrosis [1,3]. On the other hand, nutritional supplements, anti-inflammatory drugs, anabolic drugs and molecules targeting the signalling pathways involved in cancer-induced muscle wasting showed promising results in counteracting body weight loss, decreasing systemic inflammation and enhancing appetite [47,49]. However, less results were obtained in terms of recovery of muscle function and quality of life improvement [47]. The side effects emerged in the clinical trials testing new agents in both dystrophic [46,68] and cancer patients [47] represent another important issue in the treatment of MDs and CAC. Taken together, these evidences highlight the need for additional studies aimed to further investigate the molecular mechanisms orchestrating muscle wasting in chronic diseases. The presence of common features in the pathophysiology of muscle degeneration between MDs and CAC suggests the possibility that other characteristics can be shared between several chronic diseases such as HIV, chronic obstructive pulmonary disease (COPD), multiple sclerosis, congestive heart failure, sepsis, diabetes, rheumatoid arthritis, and tuberculosis.
Acknowledgments
This study was supported by Research Fund Flanders (FWO) Postdoctoral Fellowship (12D2816N) and grant (1525315N).
Conflicts of Interest
The author declares no conflict of interest.
References
- Berardi, E.; Annibali, D.; Cassano, M.; Crippa, S.; Sampaolesi, M. Molecular and cell-based therapies for muscle degenerations: A road under construction. Front. Physiol. 2014, 5, 119. [Google Scholar] [CrossRef] [PubMed]
- Rahimov, F.; Kunkel, L.M. The cell biology of disease: Cellular and molecular mechanisms underlying muscular dystrophy. J. Cell Biol. 2013, 201, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardi, E.; Sampaolesi, M. Novel Therapeutic Approaches for Skeletal Muscle Dystrophies; Chapter 14; INTECH: Winchester, UK, 2015; pp. 393–412. [Google Scholar]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Von Haehling, S.; Anker, S.D. Cachexia as a major underestimated and unmet medical need: Facts and numbers. J. Cachexia Sarcopenia Muscle 2010, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ghaoui, R.; Cooper, S.T.; Lek, M.; Jones, K.; Corbett, A.; Reddel, S.W.; Needham, M.; Liang, C.; Waddell, L.B.; Nicholson, G.; et al. Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: Outcomes and lessons learned. JAMA Neurol. 2015, 72, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Landfeldt, E.; Lindgren, P.; Bell, C.F.; Schmitt, C.; Guglieri, M.; Straub, V.; Lochmuller, H.; Bushby, K. The burden of duchenne muscular dystrophy: An international, cross-sectional study. Neurology 2014, 83, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Arthur, S.T.; Van Doren, B.A.; Roy, D.; Noone, J.M.; Zacherle, E.; Blanchette, C.M. Cachexia among us cancer patients. J. Med. Econ. 2016, 19, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Rando, T.A. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve 2001, 24, 1575–1594. [Google Scholar] [CrossRef] [PubMed]
- Kobzik, L.; Reid, M.B.; Bredt, D.S.; Stamler, J.S. Nitric oxide in skeletal muscle. Nature 1994, 372, 546–548. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Michele, D.E.; Campbell, K.P. Dystrophin-glycoprotein complex: Post-translational processing and dystroglycan function. J. Biol. Chem. 2003, 278, 15457–15460. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Butchbach, M.E.; Sahenk, Z.; Wang, H.; Saji, M.; Carathers, M.; Ringel, M.D.; Skipworth, R.J.; Fearon, K.C.; Hollingsworth, M.A.; et al. Dystrophin glycoprotein complex dysfunction: A regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005, 8, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. Nf-kappab-mediated pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef] [PubMed]
- Mokri, B.; Engel, A.G. Duchenne dystrophy: Electron microscopic findings pointing to a basic or early abnormality in the plasma membrane of the muscle fiber. Neurology 1975, 25, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Davies, K.E. Pharmacological advances for treatment in duchenne muscular dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, B.; De Bleecker, J.L. Cytokines and chemokines as regulators of skeletal muscle inflammation: Presenting the case of duchenne muscular dystrophy. Mediat. Inflamm. 2013, 2013, 540370. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, D.; Berardi, E.; Ceccarelli, G.; Sampaolesi, M. Adult stem cells and skeletal muscle regeneration. Curr. Gene. Ther. 2015, 15, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Coletti, D.; Aulino, P.; Pigna, E.; Barteri, F.; Moresi, V.; Annibali, D.; Adamo, S.; Berardi, E. Spontaneous physical activity downregulates pax7 in cancer cachexia. Stem Cells Int. 2016, 2016, 6729268. [Google Scholar] [CrossRef] [PubMed]
- Duddy, W.; Duguez, S.; Johnston, H.; Cohen, T.V.; Phadke, A.; Gordish-Dressman, H.; Nagaraju, K.; Gnocchi, V.; Low, S.; Partridge, T. Muscular dystrophy in the mdx mouse is a severe myopathy compounded by hypotrophy, hypertrophy and hyperplasia. Skelet Muscle 2015, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic exercise and pharmacological treatments counteract cachexia by modulating autophagy in colon cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Malhotra, S.; Kumar, A. Nuclear factor-κb signaling in skeletal muscle atrophy. J. Mol. Med. 2008, 86, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, B.; Liang, C.; Li, Y.; Song, Y.H. Cytokine signaling in skeletal muscle wasting. Trends Endocrinol. Metab. 2016, 27, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.J.; Murphy, K.T.; Jenkinson, L.; Laine, D.; Emmrich, K.; Faou, P.; Weston, R.; Jayatilleke, K.M.; Schloegel, J.; Talbo, G.; et al. Targeting of fn14 prevents cancer-induced cachexia and prolongs survival. Cell 2015, 162, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Yadava, R.S.; Foff, E.P.; Yu, Q.; Gladman, J.T.; Kim, Y.K.; Bhatt, K.S.; Thornton, C.A.; Zheng, T.S.; Mahadevan, M.S. Tweak/fn14, a pathway and novel therapeutic target in myotonic dystrophy. Hum. Mol. Genet. 2015, 24, 2035–2048. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Hanna, D.L.; Zhang, W.; Baba, H.; Lenz, H.J. Molecular pathways: Cachexia signaling—A targeted approach to cancer treatment. Clin. Cancer Res. 2016, 22, 3999–4004. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the e3 ubiquitin ligases murf1 and mafbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, E.; Kudryashov, D.; Kramerova, I.; Spencer, M.J. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2h that binds to skeletal muscle myosin and ubiquitinates actin. J. Mol. Biol. 2005, 354, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Schoser, B.G.; Frosk, P.; Engel, A.G.; Klutzny, U.; Lochmuller, H.; Wrogemann, K. Commonality of trim32 mutation in causing sarcotubular myopathy and lgmd2h. Ann. Neurol. 2005, 57, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Cossu, M.V.; Cattaneo, D.; Fucile, E.; Pellegrino, P.; Baldelli, S.; Cozzi, V.; Capetti, A.; Clementi, E. Combined isosorbide dinitrate and ibuprofen as a novel therapy for muscular dystrophies: Evidence from phase i studies in healthy volunteers. Drug Des. Dev. Ther. 2014, 8, 411–419. [Google Scholar]
- Ruegg, U.T.; Nicolas-Metral, V.; Challet, C.; Bernard-Helary, K.; Dorchies, O.M.; Wagner, S.; Buetler, T.M. Pharmacological control of cellular calcium handling in dystrophic skeletal muscle. Neuromuscul. Disord. 2002, 12, S155–S161. [Google Scholar] [CrossRef]
- Moorwood, C.; Lozynska, O.; Suri, N.; Napper, A.D.; Diamond, S.L.; Khurana, T.S. Drug discovery for duchenne muscular dystrophy via utrophin promoter activation screening. PLoS ONE 2011, 6, e26169. [Google Scholar] [CrossRef] [PubMed]
- Duan, D. Dystrophin gene replacement and gene repair therapy for duchenne muscular dystrophy in 2016: An interview. Hum. Gene. Ther. Clin. Dev. 2016, 27, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in duchenne's muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.B.; Braun, S.; Benveniste, O.; Leturcq, F.; Hogrel, J.Y.; Morris, G.E.; Barois, A.; Eymard, B.; Payan, C.; Ortega, V.; et al. Phase I study of dystrophin plasmid-based gene therapy in duchenne/becker muscular dystrophy. Hum. Gene Ther. 2004, 15, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide pro051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by crispr/cas9-mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Long, C.Z.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Rivera, R.M.C.; Madhavan, S.; Pan, X.F.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.X.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, J.M.; Kunkel, L.M. Pharmacological therapeutics targeting the secondary defects and downstream pathology of duchenne muscular dystrophy. Expert Opin. Orphan Drugs 2016, 4, 1179–1194. [Google Scholar] [CrossRef] [PubMed]
- Molfino, A.; Amabile, M.I.; Rossi Fanelli, F.; Muscaritoli, M. Novel therapeutic options for cachexia and sarcopenia. Expert Opin. Biol. Ther. 2016, 16, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Giacosa, A.; Rondanelli, M. Fish oil and treatment of cancer cachexia. Genes Nutr. 2008, 3, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Penet, M.F.; Bhujwalla, Z.M. Cancer cachexia, recent advances, and future directions. Cancer J. 2015, 21, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.D.; Ross, J.A.; Voss, A.C.; Tisdale, M.J.; Fearon, K.C.H. The effect of an oral nutritional supplement enriched with fish oil on weight loss in patients with pancreatic cancer. Br. J. Cancer 1999, 81, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Colomer, R.; Moreno-Nogueira, J.M.; Garcia-Luna, P.P.; Garcia-Peris, P.; Garcia-de-Lorenzo, A.; Zarazaga, A.; Quecedo, L.; del Llano, J.; Usan, L.; Casimiro, C. N-3 fatty acids, cancer and cachexia: A systematic review of the literature. Br. J. Nutr. 2007, 97, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.H.; von Meyenfeldt, M.F.; Moses, A.G.W.; van Geenen, R.; Roy, A.; Gouma, D.J.; Giacosa, A.; Van Gossum, A.; Bauer, J.; Barber, M.D.; et al. Effect of a protein and energy dense n-3 fatty acid enriched oral supplement on loss of weight and lean tissue in cancer cachexia: A randomised double blind trial. Gut 2003, 52, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Wigmore, S.J.; Ross, J.A.; Falconer, J.S.; Plester, C.E.; Tisdale, M.J.; Carter, D.C.; Fearon, K.C.H. The effect of polyunsaturated fatty acids on the progress of cachexia in patients with pancreatic cancer. Nutrition 1996, 12, S27–S30. [Google Scholar] [CrossRef]
- Akamizu, T.; Kangawa, K. Ghrelin for cachexia. J. Cachexia Sarcopenia Muscle 2010, 1, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Reano, S.; Ferrara, M.; Angelino, E.; Gnocchi, V.F.; Prodam, F.; Ronchi, G.; Fagoonee, S.; Fornaro, M.; et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J. Clin. Investig. 2013, 123, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Molfino, A.; Formiconi, A.; Rossi Fanelli, F.; Muscaritoli, M. Ghrelin: From discovery to cancer cachexia therapy. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.M.; Boccia, R.V.; Graham, C.D.; Yan, Y.; Duus, E.M.; Allen, S.; Friend, J. Anamorelin for patients with cancer cachexia: An integrated analysis of two phase 2, randomised, placebo-controlled, double-blind trials. Lancet Oncol. 2015, 16, 108–116. [Google Scholar] [CrossRef]
- Garcia, J.M.; Friend, J.; Allen, S. Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: A multicenter, randomized, double-blind, crossover, pilot study. Support Care Cancer 2013, 21, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Pietra, C.; Takeda, Y.; Tazawa-Ogata, N.; Minami, M.; Xia, Y.F.; Duus, E.M.; Northrup, R. Anamorelin hcl (ono-7643), a novel ghrelin receptor agonist, for the treatment of cancer anorexia-cachexia syndrome: Preclinical profile. J. Cachexia Sarcopenia 2014, 5, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Temel, J.S.; Abernethy, A.P.; Currow, D.C.; Friend, J.; Duus, E.M.; Yan, Y.; Fearon, K.C. Anamorelin in patients with non-small-cell lung cancer and cachexia (romana 1 and romana 2): Results from two randomised, double-blind, phase 3 trials. Lancet Oncol. 2016, 17, 519–531. [Google Scholar] [CrossRef]
- Dobs, A.S.; Boccia, R.V.; Croot, C.C.; Gabrail, N.Y.; Dalton, J.T.; Hancock, M.L.; Johnston, M.A.; Steiner, M.S. Effects of enobosarm on muscle wasting and physical function in patients with cancer: A double-blind, randomised controlled phase 2 trial. Lancet Oncol. 2013, 14, 335–345. [Google Scholar] [CrossRef]
- Srinath, R.; Dobs, A. Enobosarm (gtx-024, s-22): A potential treatment for cachexia. Future Oncol. 2014, 10, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Gueta, I.; Altman, A.; Shoenfeld, Y. The effect of blocking tnf-alpha in patients with cancer-related cachexia and anorexia. Harefuah 2010, 149, 512–514, 550, 551. [Google Scholar] [PubMed]
- Wu, C.; Fernandez, S.A.; Criswell, T.; Chidiac, T.A.; Guttridge, D.; Villalona-Calero, M.; Bekaii-Saab, T.S. Disrupting cytokine signaling in pancreatic cancer: A phase i/ii study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas 2013, 42, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Hui, D.; Bruera, E.; Janku, F.; Naing, A.; Falchook, G.S.; Piha-Paul, S.; Wheler, J.J.; Fu, S.; Tsimberidou, A.M.; et al. Mabp1, a first-in-class true human antibody targeting interleukin-1alpha in refractory cancers: An open-label, phase 1 dose-escalation and expansion study. Lancet Oncol. 2014, 15, 656–666. [Google Scholar] [CrossRef]
- Yao, X.; Huang, J.; Zhong, H.; Shen, N.; Faggioni, R.; Fung, M.; Yao, Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 2014, 141, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, T.J.; Smith, J.T.; Schuster, M.; Dragnev, K.H.; Rigas, J.R. A humanized anti-il-6 antibody (ald518) in non-small cell lung cancer. Expert Opin. Biol. Ther. 2011, 11, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef]
Figure 1.
Muscular dystrophies (MDs) and cancer-associated cachexia (CAC) share peculiar abnormalities of the dystrophin glycoprotein complex (DCG). (A) DGC Model; (B) DGC in MDs is characterized by specific mutations (grey color) in α, β, γ, and δ subunit of the sarcoglycan complex (in sarcoglycanopathies), hyperglycosylation of α-dystroglycan (α-DG), β-dystroglycan (β-DG) and β-sarcoglycan (β-SG), reduced expression of dystrophin, altered intracellular influx of calcium, and abundant deposition of collagen in the extracellular matrix; (C) DGC in CAC is characterized by hyperglycosylation of β-dystroglycan (β-DG) and β-sarcoglycan (β-SG) and reduced content of dystrophin.
Figure 1.
Muscular dystrophies (MDs) and cancer-associated cachexia (CAC) share peculiar abnormalities of the dystrophin glycoprotein complex (DCG). (A) DGC Model; (B) DGC in MDs is characterized by specific mutations (grey color) in α, β, γ, and δ subunit of the sarcoglycan complex (in sarcoglycanopathies), hyperglycosylation of α-dystroglycan (α-DG), β-dystroglycan (β-DG) and β-sarcoglycan (β-SG), reduced expression of dystrophin, altered intracellular influx of calcium, and abundant deposition of collagen in the extracellular matrix; (C) DGC in CAC is characterized by hyperglycosylation of β-dystroglycan (β-DG) and β-sarcoglycan (β-SG) and reduced content of dystrophin.
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Berardi, E. Muscular Dystrophies and Cancer Cachexia: Similarities in Chronic Skeletal Muscle Degeneration. J. Funct. Morphol. Kinesiol. 2017, 2, 39. https://doi.org/10.3390/jfmk2040039
AMA Style
Berardi E. Muscular Dystrophies and Cancer Cachexia: Similarities in Chronic Skeletal Muscle Degeneration. Journal of Functional Morphology and Kinesiology. 2017; 2(4):39. https://doi.org/10.3390/jfmk2040039
Chicago/Turabian StyleBerardi, Emanuele. 2017. "Muscular Dystrophies and Cancer Cachexia: Similarities in Chronic Skeletal Muscle Degeneration" Journal of Functional Morphology and Kinesiology 2, no. 4: 39. https://doi.org/10.3390/jfmk2040039