Multiparametric Approach to Arrhythmogenic Cardiomyopathy: Clinical, Instrumental, and Lifestyle Indications


Clinical and Experimental Medicine Department, School of Sports Medicine, Sports Medicine and Exercise Center, 50141 Florence, Italy
*
Author to whom correspondence should be addressed.
J. Funct. Morphol. Kinesiol. 2018, 3(2), 35; https://doi.org/10.3390/jfmk3020035
Submission received: 11 April 2018
/
Revised: 16 May 2018
/
Accepted: 8 June 2018
/
Published: 13 June 2018
(This article belongs to the Special Issue Arrhythmic Events in Sports Medicine and Kinesiology)
Abstract
:Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a genetic disorder with an autosomal dominant inheritance and incomplete penetrance. It predominantly affects the right ventricle (RV), predisposing to the origin of ventricular arrhythmias and sudden death (SD). The structural basis of the disease consists of the progressive loss of myocardium with fibro-adipose replacement. ARVC is a “desmosomes” disease involving mutations of proteins such as placoglobin, desmoplachine, placophylline, desmoglein, and desmocollin. In the “classical” form, the disease mainly implicates the RV chamber, while the left ventricle (LV) is involved in advanced stages. Genotype-phenotype correlation studies have identified some phenotypic variants characterized by an early participation of the LV, which can proceed in parallel toward the two ventricles (“biventricular” variant) or prevails over the RV (variant to “left dominance”). These data led to the evolution of the initial definition of ARVC, which is currently considered a genetic disease of both ventricles and, therefore, deserves the denomination “arrhythmogenic cardiomyopathy”. Many aspects of diagnosis, treatment, and indications for a correct lifestyle are important in sports medicine. This paper will discuss the clinical management of ARVC, with particular reference to diagnosis, risk stratification, therapy, and indications for physical activity.
1. Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiomyopathy that is characterized by ventricular arrhythmias and an increased risk of sudden cardiac death [1]. The literature reports that the rates of sudden death (SD) by cardiovascular diseases were 2.1 in 100,000 athletes per year, compared with 0.7 in 100,000 non-athletes per year. Among the causes of SD, the ARVC has an estimated prevalence of 1/1000, up to 1/5000, in the population [2]. The mean age at presentation is around the third/fourth decade (Table 1). Presentation before the age of 12 is rare. Prevalence of ARVC and related sudden cardiac death (SCD) risk from the UKRR (United Kingdom Regional Registry)
The United States National Registry reports that, with respect to incidence, cardiovascular mortality in African-Americans and other minorities exceeded that in whites by a factor of 4.8. Despite these data, hypertrophic cardiomyopathy persists as the single most common cause of SCD. It was more common among African-Americans and other minorities than among white athletes. On the contrary the ARVC resulted was more common in white athletes, with an incidence of SCD in African-Americans of less than 1% [3]. Less is known about the prevalence of SCD in non-Caucasian Asian probands: the prevalence of desmosomal mutations in non-Caucasian Japanese ARVC probands seems to be identical to that in Caucasians; however, the most prevalent gene mutation was DSG2 [4].
1.1. Italian Profile
ARVC has been studied particularly in Veneto, a region of Italy.
The study was conducted as a special experience to verify the effects of the prevention strategy of the sudden death (SD). The annual incidence of SD found in athletes was 2.3 per 100,000. It was higher if compared to the others reports. The investigation was carried out from 1975 to 1996. It showed an elevated prevalence of ARVC as the cause of sudden death in athletes. Some authors have compared these data with other contexts [5,6] demonstrating that no substantial differences were found. In any case ARVC has been counted among the most common pathologic substrates underlying SD in young athletes [7,8,9]. Attention to the disease started with the importance of risk stratification considering that it is a common cause of SD in young people.
SD can be the first manifestation of ARVC.
From the Register of the Region of Veneto it emerged clearly that some subjects with evidence of ARVC at autopsy showed no alterations or significant morphological findings of the RV, especially in diagnostic tests carried out before the death. This fact highlights the danger of the “hidden phase” ARVC, where fatal alterations, such as heart arrhythmias, can occur in the absence of specific diagnostic markers [1].
1.2. Natural History
The natural history of ARVC is characterized by an instability of the electric ventricular function. This condition is responsible for ventricular tachycardia (VT) and ventricular fibrillation (FV) that can cause SD, or in any case addresses arrhythmic events, especially in young people and athletes. In advanced stages of the disease, the progression of RV dysfunction and the subsequent involvement of the LV can lead to cardiac congestion and failure [10]. Traditionally, the natural history of ARVC is articulated in four phases: (1) “occult”, distinguished by the absence of symptoms and the presence of minor structural changes; (2) “manifest”, characterized by arrhythmic symptoms and abnormalities with cardiac morpho-functional manifestations; (3) “ RV failure”, with severe systolic dysfunction of the RV, but only initial or absent alterations of the LV; and (4) “biventricular heart failure”, with severe systolic dysfunction of both the ventricles that can simulate a dilated cardiomyopathy [11]. A minority of patients presents phenotypic variants characterized by early involvement, sometimes prevalent in the LV [12] chamber.
2. Diagnosis
The diagnostic criteria for arrhythmogenic cardiomyopathy were elaborated by an international task force for the first time in 1994 and subsequently updated in 2010 [13]. The criteria initially aimed to guarantee a high level diagnostic specificity in probands with evident clinical manifestations, to ensure an appropriate differential diagnosis with other diseases, such as dilated cardiomyopathy and idiopathic VT originating from the outflow tract of the RV. On the contrary, the original criteria showed a low level diagnostic specificity for the identification of patients with the “minor” phenotype, particularly family members of patients with ARVC. It also did not provide quantitative values for the evaluation of morphological anomalies and functional features of the RV. The 2010 criteria were then introduced in order to increase the diagnostic sensitivity, without over-compromising its specificity. These criteria belong to six categories which reflect the main clinical-pathological manifestations of the disease. Since no test is diagnostic in itself, the diagnosis of the disease requires the combination of multiple criteria (at least two major criteria, or one major criterion and three minor criteria, or four minor criteria) belonging to different categories. If the number of criteria is too low for reaching a definitive diagnosis, the disease is considered “borderline” (one major and one minor criterion, or three minor criteria) or “possible” (one major criterion or two minor criteria) [14].
3. Classic Shape
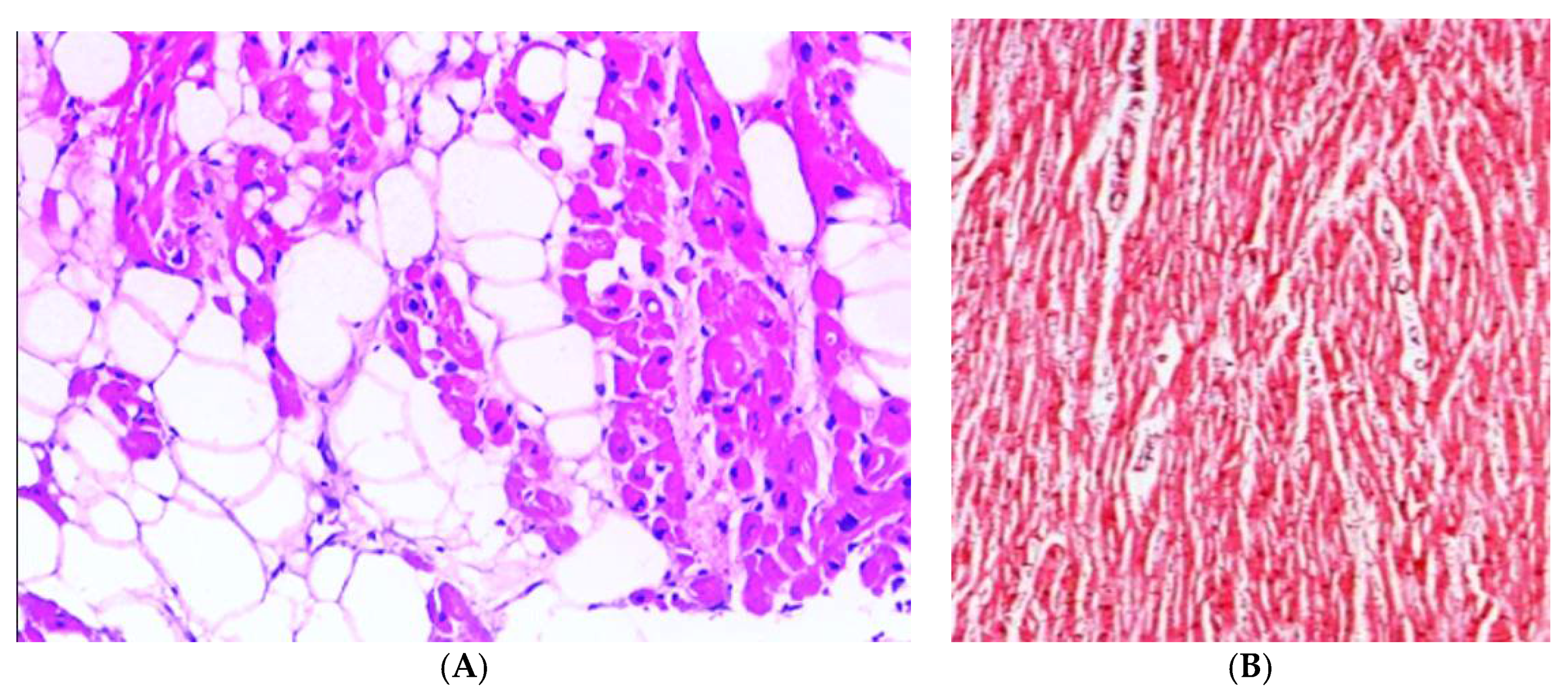
In the classic form of ARVC, there is a prevalent involvement of RV with progressive fibro-adipose replacement of the ventricular myocardium, which is particularly evident at the level of the so-called “triangle of dysplasia” (constituted from the infundibular, apical and diaphragmatic regions). The large fibrous or fibro-adipose substitution of the RV myocardium in endomyocardial biopsy, evident at the histological examination, represents the most specific diagnostic criterion (Figure 1) [15]. The pathological process of progressive fibroadipose cicatrization causes gradual dilatation and systolic dysfunction of the RV, initially of a regional character and subsequently to global extension [16]. The regionality of alterations in RV parietal kinetics (in the form of “bulging”, akinesia or dyskinesia) is highly specific for the diagnosis of ARVC, and allows a differential diagnosis versus other RV diseases (e.g., pulmonary heart, congenital heart disease, and primitive pulmonary hypertension primitive), in which the systolic dilatation/dysfunction uniformly and globally affects the ventricular chamber [17]. Diagnostic criteria based on morpho-functional alterations, studied by imaging techniques such as echocardiography, magnetic resonance, and ventriculography, provide concurrent signs of systolic dilatation/dysfunction of global RV pump and RV regional kinetic parietal alterations.
4. ARVC Variant with Left Dominance
The classical form of ARVC mainly affects RV only affecting the LV in an advanced stage, also resulting in systolic biventricular dysfunction that simulates dilated cardiomyopathy [2].
However, some variants of interest have been described that predominantly affect the LV early, often as a result of a specific genetic predisposition (e.g., gene mutations coding for desmoplachine) [18]. With respect to classical forms the phenotype is mirrored and is characterized by inversion of the T wave in the left precordial derivations (V4–V6) and ventricular arrhythmic events with “QRS wave” morphology of the right bundle branch block (RBB) type.
Unlike the classical forms, the diagnostic power of echocardiography is limited because the alterations of contractility parietal of to both the segmental and global VS, are found in a minority of patients [12]. This particular limit is due to the fact that the process of fibro-adipose substitution of the myocardium initially involves only the epicardial layers of the left ventricular wall left, which contributes only to the development of contractile strength and does not result in evident alterations of regional kinetics. This is a clinically insidious disease, the real incidence of which is likely underestimated. Cardiac MRI with contrast agents increases diagnostic sensitivity because it allows the identification of LV non-transmural scars.
5. From Myocardial Cell Mechanisms to the ECG Pattern
It has been recognized that in the initial phase of the disease a cellular mechanism is responsible for causes ventricular arrhythmias. Arrhythmias are often induced by exacerbations of myocarditis. This is an acute phase, characterized by chest pain, ECG changes of the ventricular repolarization, and an increase in myocardial enzymes. Despite the fact that ARVC is predominantly a genetic disease, a frequent link with an inflammatory process has been demonstrated and myocardial inflammatory infiltrates are frequently detected in patients with ARVC [19]. An etiological role of myocarditis has been supposed, especially in the initial phase of ARVC. In parallel, the reactive nature of myocarditis in ARVC has been supported by the massive inflammatory cell infiltrates found after acute myocardial necrosis in early stages of disease in a desmoglein-2 transgenic animal model [20]. The initial scenario of inflammation could play a pathogenic role in tissue injury and arrhythmogenesis. However, this aspect remains largely unexplored in the literature, especially for the consequent different prognosis and approach that includes major clinical intervention in the case of ARVC, otherwise not considered in myocarditis.
In the most advanced phase, ventricular arrhythmias predominate parallel to the fibro-adipose scar and ventricular tachicardia (VT) from macro re-entry is often found [7]. Experimental studies have recently shown that the genetic alteration of desmosomial proteins can interfere with the function of the intercalated discs of sodium channels, causing electrical instability through the reduction of the intensity of the sodium current. This mechanism appears early and independently from the myocardial structural changes [11].
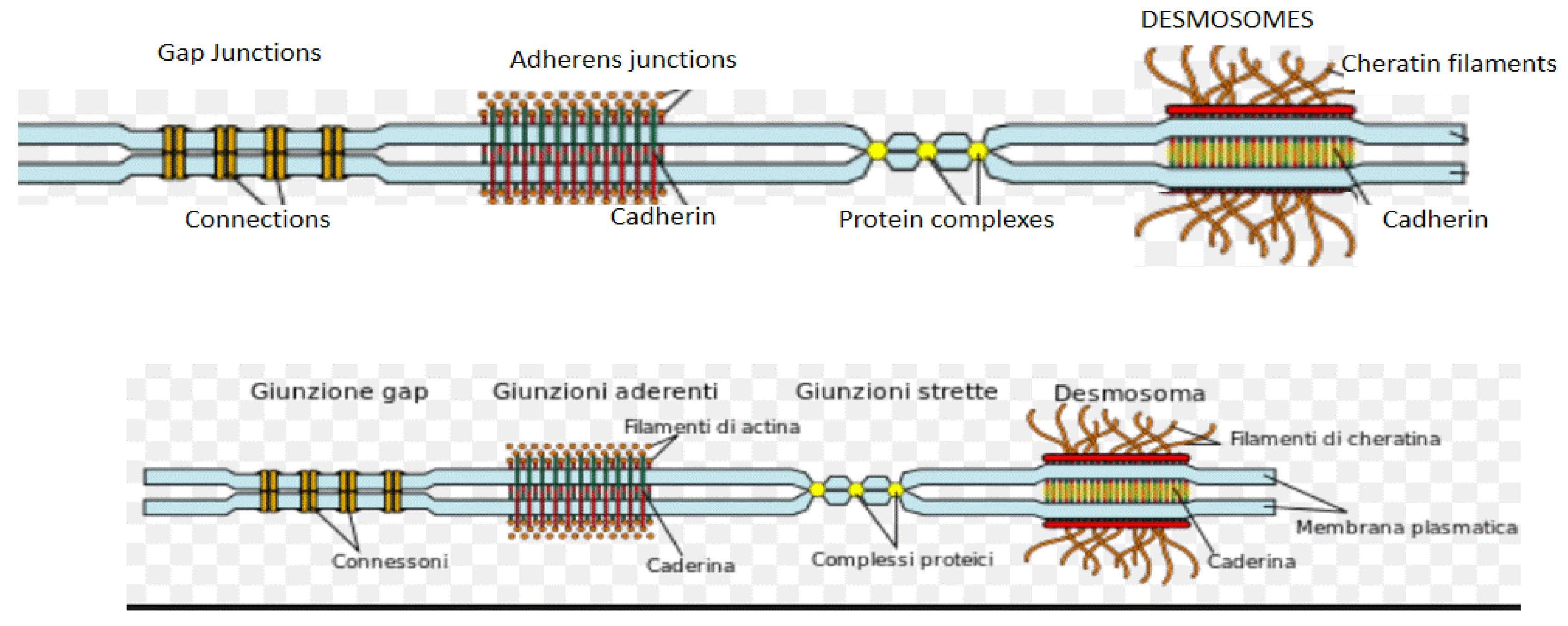
Desmosomes are specialized structures, functionally related to the sodium channels, with the task of maintaining mechanical adhesion between cardiomyocytes. There are three principal components in the desmosomes: desmoplakin, which binds cytoskeleton intermediate filaments, transmembrane proteins (desmocollin 2 and desmoglein 2), and armadillo proteins (plakoglobin and plakophilin 2), which mediate the interactions between transmembrane proteins and desmoplakin (Figure 2)
Desmosomes are essential for myocardial mechanical continuity and the E-C coupling. A deficiency of these structures can induce electrical instability.
Causative mutations in ARVC have been detected in cardiac desmosomal genes encoding the constituent proteins: desmoplakin (DSP), desmoglein-2 (DSG2), plakophilin-2 (PKP2), desmocollin-2 (DSC2), and plakoglobin (JUP) [21,22]
ARVC can result from mutations in at least 13 desmosomal genes. The most common mutation found in 60% of the population is PKP2, a desmosomal gene mutation.
A prevalent genetic pattern is summarized in Table 2.
Secondly ARVC involves a reduction of the sodium current and predisposes to malignant arrhythmia with a mechanism similar to Brugada syndrome.
This explains the existence of some “mixed” phenotypes, particularly in those patients with SD, presenting some structural alterations typical of arrhythmogenic right ventricular cardiomyopathy at the autopsy, and ECG compatible with Brugada Type 1. The recent identification of functional inter-cellular interactions could explain the close relationship between the two diseases.
6. ARVC General ECG Criteria
ECG is often the first clinical examination carried out, the results of which may arouse suspicion of ARVC. In ARVC the ECG pattern offers some specific aspects summarized in Table 3 as major and minor criteria.
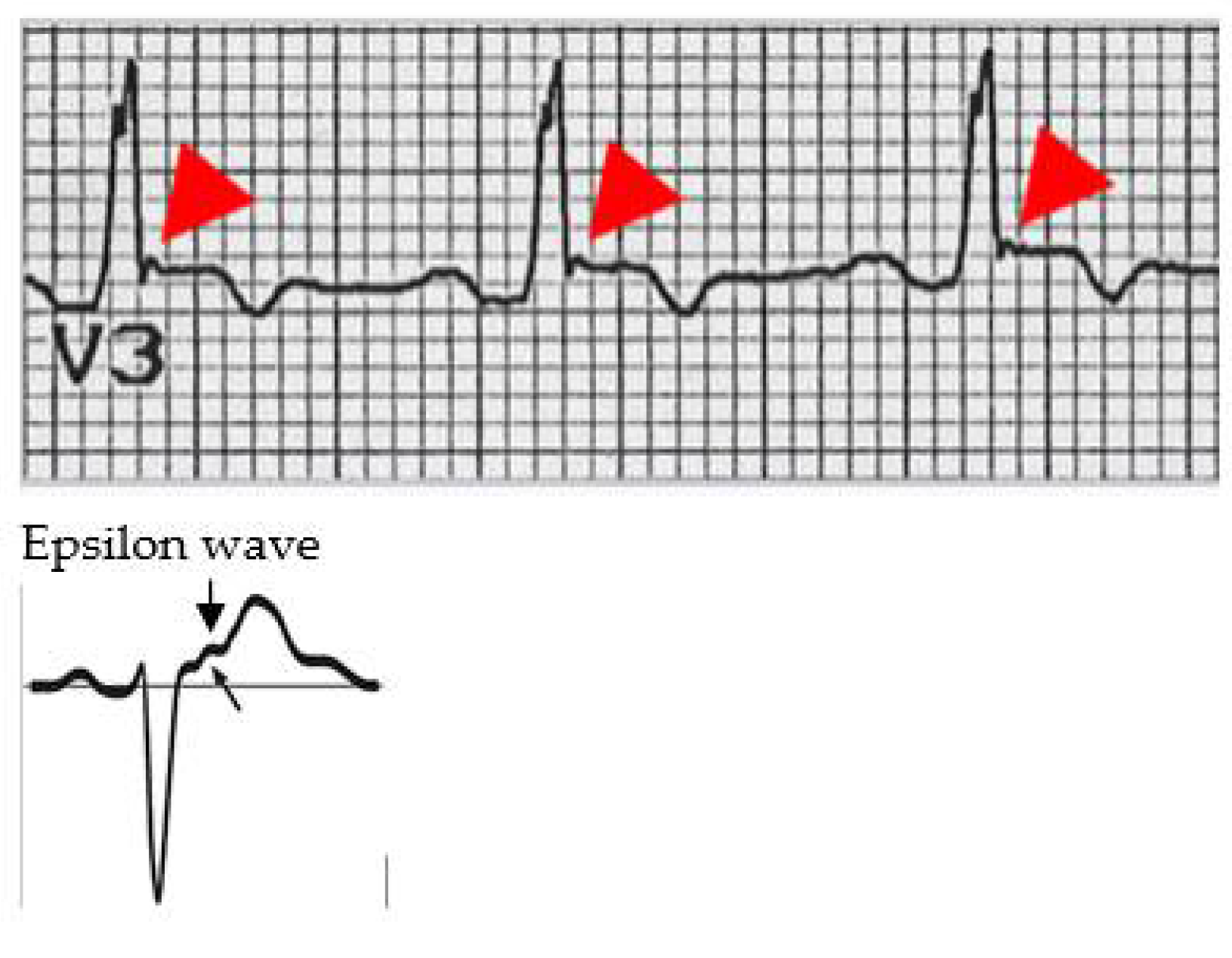
The specific ECG characteristics of ARVC are shown in the Figure 3.
Epsilon wave is a typical ECG expression in the ARVC of late potentials. It is found at the end of the QRS complex and at the beginning of the ST stretch. The epsilon wave is present in 33% of patients affected by ARVC and is often found in V1–V3 leads.
The ultimate diagnosis of illness requires the combination of multiple criteria (at least two major criteria, or one major criterion and three minor criteria, or four minor criteria) belonging to different categories.
7. Echocardiographic Assessment
The echocaradiographic manifestations are related to the RV morphology and functioning.
RV dilatation is frequently found in individuals with ARVC. In particular, the enlargement of the RVOT (right ventricle outflow tract) is present. It can be studied in the parasternal long-axis view and in the short axis view, where the main measurements of PLAX >32 mm and PSAX >36 mm are diagnostic.
Increase of trabecular derangement is also evident in RV, are localized aneurysms in the free wall of the same chamber.
Major criteria are the presence of RV dilatation and acinetic or discinetik areas, associated with:
- RV outflow tract diameter (long axis) >32 mm, or
- RV outflow tract diameter (short axis) >36 mm, or
- RV shortening fraction <33%
Recommendations for the echocardiographic assessment of RV size is one of the most important elements for the identification of ARVC. The morphological manifestations of ARVC can be illustrated in the echocardiogram. The echo examination often shows the presence of RV involvement in the late phase of the disease, particularly when clinical manifestations are already evident.
In the first case, as the ASE guidelines indicate, the RV linear dimensions are dependent on probe rotation and different RV views; in order to permit inter-study comparison, the echocardiographic report should state the window from which measurement was performed.
In the second case the measurements are easier.
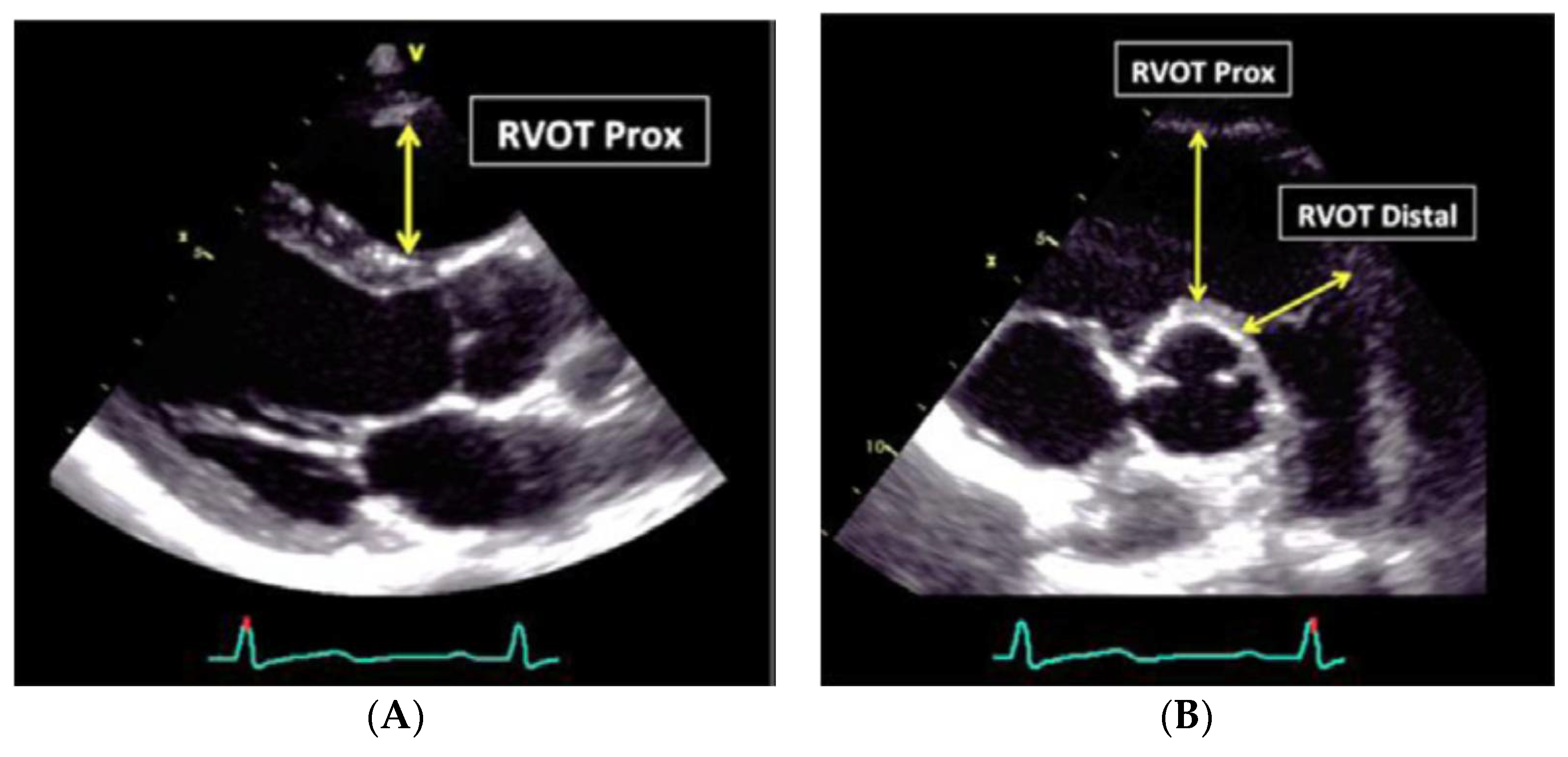
The outflow measurement needs to also include the distal RV outflow diameter (RVOT distal) in two sessions: just proximal to the pulmonary valve at end-diastole (Figure 5, Table 4).
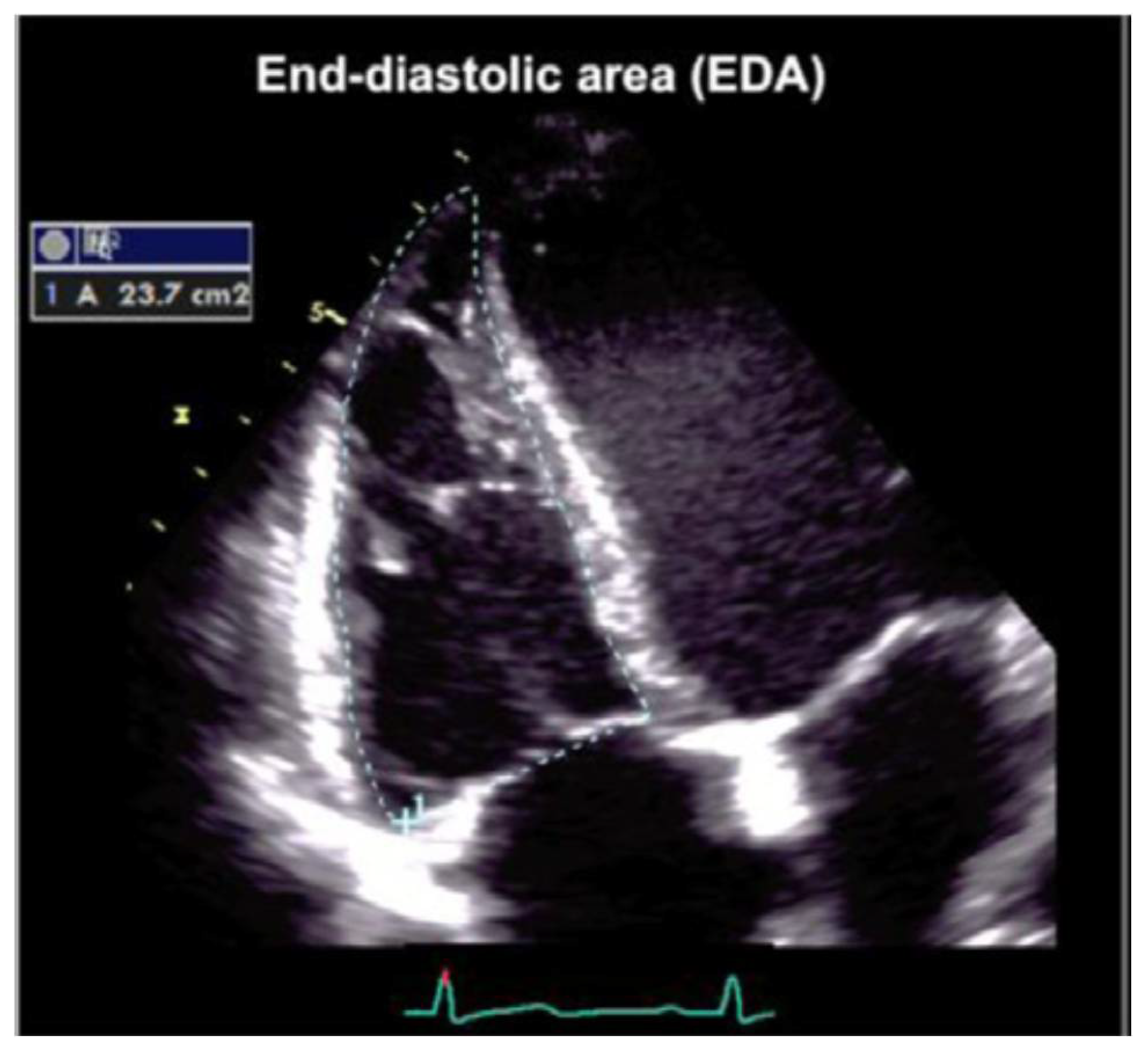
A manual tracing of the RV endocardial border from the lateral tricuspid annulus, along the free wall to the apex and back to the medial tricuspid annulus, and along the interventricular septum at the end-diastole and at the end-systole is also recommended (Figure 6).
8. Prognosis
ARVC was initially considered to have poor short-term prognosis. This was based on studies carried out in third-level centers on patients at high risk or with severe clinical manifestations, requiring advanced therapeutic intervention such as ablation, transcatheter implantation, or cardioverter-defibrillator implantation (ICD). Subsequent studies on populations offered a less pessimistic view of the course of ARVC, more often asymptomatic and with better prognosis, with an annual mortality rate <1%. It should be emphasized, however, that these studies describe the “unnatural” history of the disease, referring to those patients with diagnosed and treated ARVC. They do not include the young SD victims, previously asymptomatic and for whom diagnosis was made only at the autopsy [23]. Cardiac magnetic resonance (CMR) imaging, and endomiocardial biopsy can contribute to predict final diagnosis of ARVC, especially in the presence of ventricular arrhythmias.
9. Risk Stratification
Clinical and pathological studies have identified numerous possible clinical predictors of arrhythmic risk [18]. Symptomatic patients with previous cardiac arrest due to ventricular fibrillation (VF) or sustained ventricular tachicardia (VT) can be considered in the group with the highest risk of recurrent arrhythmics and SD; unexplained syncopes (non-vasovagal) are proved to be a significant risk factor only in some studies.
Numerous markers have been identified in the asymptomatic patient among arrhythmic risk patients whose predictive value is documented from a limited number of observational studies, sometimes with mixed results. Unsustained TV and moderate–severe ventricular dysfunction could be the main indicators of arrhythmic risk among asymptomatic patients. Other prognostically-relevant factors include proband status and the number and complexity of premature ventricular beats to the Holter and the TV/FV inducibility to programmed ventricular stimulation. Considering arrhythmia is a genetic disorder at different penetrance, genetic screening could be extended to other members of a patient’s family. Literature reports the incidence of SD in subjects without phenotypic evidence of the AVRD. This approach needs to be considered in the context of the SD prevention and the family members of victims can be considered a restricted group to investigate.
10. Lifestyle Modification—Exercise Limitations
Studies have linked ARVC onset or progression to exercise. Adolescents and young adults with ARVC who practice competitive sports have a five-fold higher SD risk than the their sedentary counterparts; therefore, early detection (often in the pre-symptomatic phase) of ARVC in the athlete, through medical sports-screening examination (with consequent eventual exclusion from athletics due to medical contraindications) could well turn out to be life-saving [12,18]. Furthermore, experimental studies have shown that exercise promotes the development and the progression of the disease in animal models, with desmosomal mutations [21]. Recent studies have confirmed that endurance sports and intense, continuous exercise increase the chances of a carrier of a desmosomal mutation in early development of the phenotype of the disease, and becomes symptomatic for arrhythmias and/or heart failure. Patients cannot participate in either competitive or most recreational sports [24,25,26]. Authors have demonstrated that endurance exercise accelerates desmosomal dysfunction [27,28].
Increased myocardial stress due to prolonged training is determinant for disclosing hidden disease, especially in a wide sports context, where the RV structure could reveal a remodeling caused by the impact of exercise.
Endurance exercise may facilitate myocyte uncoupling at defective desmosomes leading to inflammation, fibrosis, adipocytosis and can subsequently lead to an impairment in electrical coupling. The interaction between exercise and AVRD is a topic that has long been widely discussed by researchers in the field of the sports medicine discipline with the aim of finding a definition [29]. The mechanical impact of exercise may predispose, in the presence of a genetic pattern, to apoptosis, fibrosis, and arrhythmogenicity, which most commonly involves the right ventricle [30].
Taking into consideration the stamens made on lifestyle by subjects with ARVC, specific attention has been paid to the relationship of dose-response to exercise intensity-duration and myocardial damage [31]. The general conclusion is that exercise might promote progressive heart failure by a continuum of both intensity and duration. In the new “threshold theory” hypothesis the presence of a positive phenotypic expression when training practice is regularly undertaken, could represent evidence of the basic and extreme causes of the disease.
Particularly, in this last context it appears to be very difficult to decide how to proceed with genetic testing in the case of a potential positive phenotype, especially if detected in the echocardiographic findings. Genetic testing or genetic counseling sessions usually follow the review of a patient’s diagnosis, and it should be considered in cases of positive clinical history.
Sports and, therefore, physical sports activity, should be discouraged not only for patients with ascertained diagnosis of AVRC, but also for carriers of healthy mutation or with signs mild illnesses. There is no absolute contraindication for the practice of physical activity with low cardiovascular involvement, especially if the patient is being treated with beta-blockers [31].
11. Therapy: Transcatheter Ablation
Transcatheter ablation is a therapeutic option reserved for patients with VT, which in most cases recognizes a macro-re-entry mechanism related to fibroadipose myocardial scar. More rarely VT in ARVC can have a focal automatic source (“triggered activity”) [32]. The results of acute transcatheter ablation are generally satisfactory, especially in the most recent cases [33]. However, the incidence of arrhythmic recurrences in follow-up is high, above all because the progressive nature of the disease predisposes to the development over time of new arrhythmic outbreaks and circuits multiple return. The fact that the disease mainly affects the epicardial layer of the ventricular wall explains the reason for the disappointing results of the conventional technique of endocardial mapping/ablation. More promising results come from the combined interventional approach: endocardial (for transvenous route) and epicardial (by pericardial percutaneous access), with efficacy of interruption of the re-entry circuit of VT significantly better than the traditional endocardial technique [34].
Finally, it is important to remember that, similarly to how much has already been mentioned for antiarrhythmic drugs, transcatheter ablation of TV does not protect the patient from SD and cannot be considered an alternative to ICD implantation.
Evidence in literature highlights ICD implantation as the most strategic therapy to control the electrical instability of the disease [35]. Some authors support this treatment, suggesting ICD implantation for all subjects affected by AVRC [36]. The indication to use ICD as the primary prevention is, however, actually debated [37].
Despite ICD implantation being a simple procedure, it is, however, not completely free of complications. The prophylactic use of this device needs to take into account the incidence of mechanical and electrical complications of the procedure, such as emothorax, infections, malfunctioning of the electrodes, at a short and long time after implantation. It is important to also consider the potential cost and the significant impact on the quality of life, especially in young patients. Thus, in the case of asymptomaticity (no cardiac arrest of major ventricular arrhythmias) and with a low risk profile, ICD implantation is not indicated.
12. Pharmacological Therapy
Pharmacological treatment options in patients with ARVC includes the use of beta-blockers, antiarrhythmics, and drugs for the treatment of heart failure.
13. Beta Blockers
Ventricular arrhythmias and cardiac arrest in ARVC are often favored by adrenergic stimulation and typically arise during or immediately after a physical effort.
Authors have demonstrated that patients with ARVD have autonomic dysfunction with increased orthosympathic neurovegetative tone and a reduced density of the beta-receptors [38]. The prescription of beta-blockers is recommended in patients with ventricular arrhythmias, but is also suggested for asymptomatic patients with an ascertained diagnosis of ARVC. Instead, there is currently no consensus on the indication for prophylactic beta-blocker treatment in healthy carriers of desmosomal mutations without the disease phenotype.
14. Antiarrhythmic Drugs
Prospective and randomized studies are not available for antiarrhythmic drugs. In ARVC the therapeutic efficacy of a single drug is difficult to evaluate, given the high incidence of arrhythmic episodes that involves frequent therapeutic changes. Therefore, the indication for antiarrhythmic drug treatment and the choice of a specific drug are usually the result of an empirical approach. Current clinical experience indicates that sotalol and amiodarone (alone or in combination with beta-blockers) can represent the most effective and safe drugs for symptomatic treatment of ventricular arrhythmias, but do not guarantee the prevention of SD. By contrast, studies on patients wearing ICD show that most of the appropriate shocks triggered by the device for malignant tachyarrhythmia are not prevented from a concurrent use of antiarrhythmic drugs [36].
For these reasons, the main goal of the antiarrhythmic drug treatment is to alleviate arrhythmic symptoms and contain the number of arrhythmic episodes (such as TV recurrences), keeping in mind that drugs do not constitute an alternative therapeutic strategy to ICD for SD prevention.
15. Conclusions
The diagnosis of ARCV has a multiparametric approach and the main purpose of treatment is to prevent arrhythmic SD. Nevertheless, some controversial aspects are evident. The implantable cardioverter-defibrillator (ICD) is the only effective treatment for preventing SD. Indications for this system represent what is still a controversial topic: the general opinion is that this treatment should be reserved for those patients who have already suffered a cardiac arrest from ventricular fibrillation or sustained ventricular tachycardia (secondary prevention) and those with major risk factors, including unexplained syncope (not vasovagal), unsustained ventricular tachycardia, and moderate ventricular dysfunction. The selection of patients must take into consideration the high incidence of complications: costs and significant psychological impact are aspects of ICD, especially in young patients. Therapeutic measures currently available are palliative. A proven cure for ARVC will require targeted intervention in the future, directly blocking the molecular mechanisms of the disease. Changes in lifestyle are one of the most challenging limits for young people with suspected ARVC. Competitive sports activity has been shown to increase the risk of SD five-fold in adolescents and young adults with ARVC [27]. Early (i.e., presymptomatic) identification of affected athletes by preparticipation screening and their disqualification from competitive sports activity may be ‘life-saving’ [36]. In addition, physical exercise has been implicated as a factor promoting development and progression of the ARVC phenotype [12]. It has been demonstrated that, in heterozygous plakoglobin-deficient mice, endurance training accelerated the development of RV dilatation, dysfunction, and ventricular ectopy, suggesting that chronically-increased ventricular load might contribute to the worsening of the ARVC phenotype. It has been postulated that impairment of myocyte cell-to-cell adhesion may lead to tissue and organ vulnerability, which may promote myocyte death, especially during mechanical stress, which occurs during competitive sports activity [36]. Studies in humans confirmed that endurance sports and frequent exercise increase age-related penetrance, risk of VT/VF, and occurrence of heart failure in ARVC desmosomal-gene carriers [37]. In summary, further research is necessary in order to provide multiple data and to combine ECG and morphological aspects of ARVC to support this important decision, especially for young asymptomatic subjects.
Author Contributions
L.S. conceived and wrote the paper; B.T. contributed to updating the literature’ and G.G. has revised and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Corrado, D.; Marcus, F.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Maron, B.J.; Haas, T.S.; Ahluwalia, A.; Murphy, C.J.; Garberich, R.F. Demographics and Epidemiology of Sudden Deaths in Young Competitive Athletes: From the United States National Registry. Am. J. Med. 2016, 129, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Ohno, S.; Aiba, T.; Horie, M. Unique genetic background and outcome of non-Caucasian Japanese probands with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Mol. Genet. Genom. Med. 2017, 5, 639–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Haas, T.S.; Doerer, J.J.; Thompson, P.D.; Hodges, J.S. Comparison of U.S. and Italian Experiences with Sudden Cardiac Deaths in Young Competitive Athletes and Implications for Preparticipation Screening Strategies. Am. J. Cardiol. 2009, 104, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Papadakis, M.; Robertus, J.L.; Dhutia, H.; Steriotis, A.K.; Tome, M.; Mellor, G.; Merghani, A.; Malhotra, A.; Behr, E.; et al. Etiology of Sudden Death in Sports: Insights from a United Kingdom Regional Registry. J. Am. Coll. Cardiol. 2016, 67, 2108–2115. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Thiene, G.; Nava, A.; Rossi, L.; Pennelli, N. Sudden death in young competitive athletes: Clinico-pathologic correlations in 22 cases. Am. J. Med. 1990, 89, 588–596. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Schiavon, M.; Thiene, G. Screening for hypertrophic cardiomyopathy in young athletes. N. Engl. J. Med. 1998, 339, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Rizzoli, G.; Schiavon, M.; Thiene, G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J. Am. Coll. Cardiol. 2003, 42, 1959–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemola, K.; Brunckhorst, C.; Helfenstein, U.; Oechslin, E.; Jenni, R.; Duru, F. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: Long term experience of a tertiary care centre. Heart 2005, 91, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Migliore, F.; Zorzi, A.; Silvano, M.; Silvano, M.; Rigato, I.; Basso, C.; Thiene, G.; Corrado, D. Clinical management of arrhythmogenic right ventricular cardiomyopathy: An update. Curr. Pharm. Des. 2010, 16, 2918–2928. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Thiene, G.; Nava, A.; Fontaliran, F.; Blomstrom-Lundqvist, C.; Fontaine, G.; Camerini, F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br. Heart J. 1994, 71, 215–218. [Google Scholar] [PubMed]
- Hamid, M.S.; Norman, M.; Quraishi, A.; Firoozi, S.; Thaman, R.; Gimeno, J.R.; Sachdev, B.; Rowland, E.; Elliott, P.M.; McKenna, W.J. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J. Am. Coll. Cardiol. 2002, 40, 1445–1450. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Ronco, F.; Marcus, F.; Abudureheman, A.; Rizzo, S.; Frigo, A.C.; Bauce, B.; Maddalena, F.; Nava, A.; Corrado, D.; et al. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: An in vitro validation of diagnostic criteria. Eur. Heart J. 2008, 29, 2760–2771. [Google Scholar] [CrossRef] [PubMed]
- Yoegher, D.M.; Marcus, F.; Sherrill, D.; Calkins, H.; Towbin, J.A.; Zareba, W.; Picard, M.H. Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: New insights from the multidisciplinary study of right ventricular dysplasia. J. Am. Coll. Cardiol. 2005, 45, 860–865. [Google Scholar]
- Rizzo, S.; Pilichou, K.; Thiene, G.; Basso, C. The changing spectrum of arrhythmogenic (right ventricular) cardiomyopathy. Cell Tissue Res. 2012, 348, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Ponsiglione, A.; Puglia, M.; Morisco, C.; Barbuto, L.; Rapacciuolo, A.; Santoro, M.; Spinelli, L.; Trimarco, B.; Cuocolo, A.; Imbriaco, M. A unique association of arrhythmogenic right ventricular dysplasia and acute myocarditis, as assessed by cardiac MRI: A case report. BMC Cardiovasc. Disord. 2016, 16, 230. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.; de Bakker, J.M.; et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-relatedarrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Pavei, A.; Michieli, P.; Schiavon, M.; Thiene, G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA 2006, 296, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Schmied, C.; Basso, C.; Borjesson, M.; Schiavon, M.; Pelliccia, A.; Vanhees, L.; Thiene, G. Risk of sports: Do we need a pre-participation screening for competit,ive and leisure athletes? Eur. Heart J. 2011, 32, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Fabritz, L.; Zwiener, M.; Witt, H.; Schäfers, M.; Zellerhoff, S.; Paul, M.; Athai, T.; Hiller, K.H.; Baba, H.A.; et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobindeficient mice. Circulation 2006, 114, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/ cardiomyopathy-associated desmosomal mutation carriers. J. Am. Coll. Cardiol. 2013, 62, 1290. [Google Scholar] [CrossRef] [PubMed]
- Saberniak, J.; Hasselberg, N.E.; Borgquist, R.; Platonov, P.G.; Sarvari, S.I.; Smith, H.J.; Ribe, M.; Holst, A.G.; Edvardsen, T.; Haugaa, K.H. Vigorous physical activity impairs myocardialfunction in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur. J. Heart Fail. 2014, 16, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Heidbuchel, H.; Hoogsteen, J.; Fagard, R.; Vanhees, L.; Ector, H.; Willems, R.; Van Lierde, J. High prevalence of right ventricular involvement in endurance athletes with ventricular arrhythmias. Role of an electrophysiologic study in risk stratification. Eur. Heart J. 2003, 24, 1473–1480. [Google Scholar] [CrossRef]
- La Gerche, A. Defining the interaction between exercise and arrhythmogenic right ventricular Cardiomyopathy. Eur. J. Heart Fail. 2015, 17, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Berruezo, A.; Fernandez-Armenta, J.; Mont, L.; Zeljko, H.; Andreu, D.; Herczku, C.; Boussy, T.; Tolosana, J.M.; Arbelo, E.; Brugada, J. Combined endocardial and epicardial catheter ablation in arrhythmogenic right ventricular dysplasia incorporating scar dechanneling technique. Circ. Arrhythm. Electrophysiol. 2012, 5, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Garcia, F.C.; Bazan, V.; Zado, E.S.; Ren, J.F.; Marchlinski, F.E. Epicardial substrate and outcome with epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2009, 120, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Schuler, P.K.; Haegeli, L.M.; Saguner, A.M.; Wolber, T.; Tanner, F.C.; Jenni, R.; Corti, N.; Lüscher, T.F.; Brunckhorst, C.; Duru, F. Predictors of appropriate ICD therapy in patients with arrhythmogenic right ventricular cardiomyopathy: Long term experience of a tertiary care center. PLoS ONE 2012, 7, e39584. [Google Scholar] [CrossRef] [PubMed]
- Bhonsale, A.; James, C.A.; Tichnell, C.; Murray, B.; Gagarin, D.; Philips, B.; Dalal, D.; Tedford, R.; Russell, S.D.; Abraham, T.; et al. Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter-defibrillator implantation for primary prevention. J. Am. Coll. Cardiol. 2011, 58, 1485–1496. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Calkins, H.; Link, M.S.; Leoni, L.; Favale, S.; Bevilacqua, M.; Basso, C.; Ward, D.; Boriani, G.; Ricci, R.; et al. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation 2010, 122, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Wichter, T.; Hindricks, G.; Lerch, H.; Bartenstein, P.; Borggrefe, M.; Schober, O.; Breithardt, G. Regional myocardial sympathetic dysinnervation in arrhythmogenic right ventricular cardiomyopathy. An analysis using 123Imeta- iodobenzylguanidine scintigraphy. Circulation 1994, 89, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Cao, K.; Yang, B.; Chen, M.; Shan, Q.; Chen, C.; Li, W.; Haines, D.E. Dynamic substrate mapping and ablation of ventricular tachycardias in right ventricular dysplasia. J. Int. Card. Electrophysiol. 2004, 11, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Miljoen, H.; State, S.; De Chillou, C.; Magnin-Poull, I.; Dotto, P.; Andronache, M.; Abdelaal, A.; Aliot, E. Electroanatomic mapping characteristics of ventricular tachycardia in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Europace 2005, 7, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Histological characteristics of RV dysplasia vs normal chamber tissue. The presence of fibro-adipose tissue in the RV chamber (A) vs. normal tissue (B).
Figure 1.
Histological characteristics of RV dysplasia vs normal chamber tissue. The presence of fibro-adipose tissue in the RV chamber (A) vs. normal tissue (B).

Figure 2.
Desmosome structures. The principal components of the cardiac contractile compartment and non-cardiac cells.
Figure 2.
Desmosome structures. The principal components of the cardiac contractile compartment and non-cardiac cells.

Figure 3.
ECG epsilon wave.

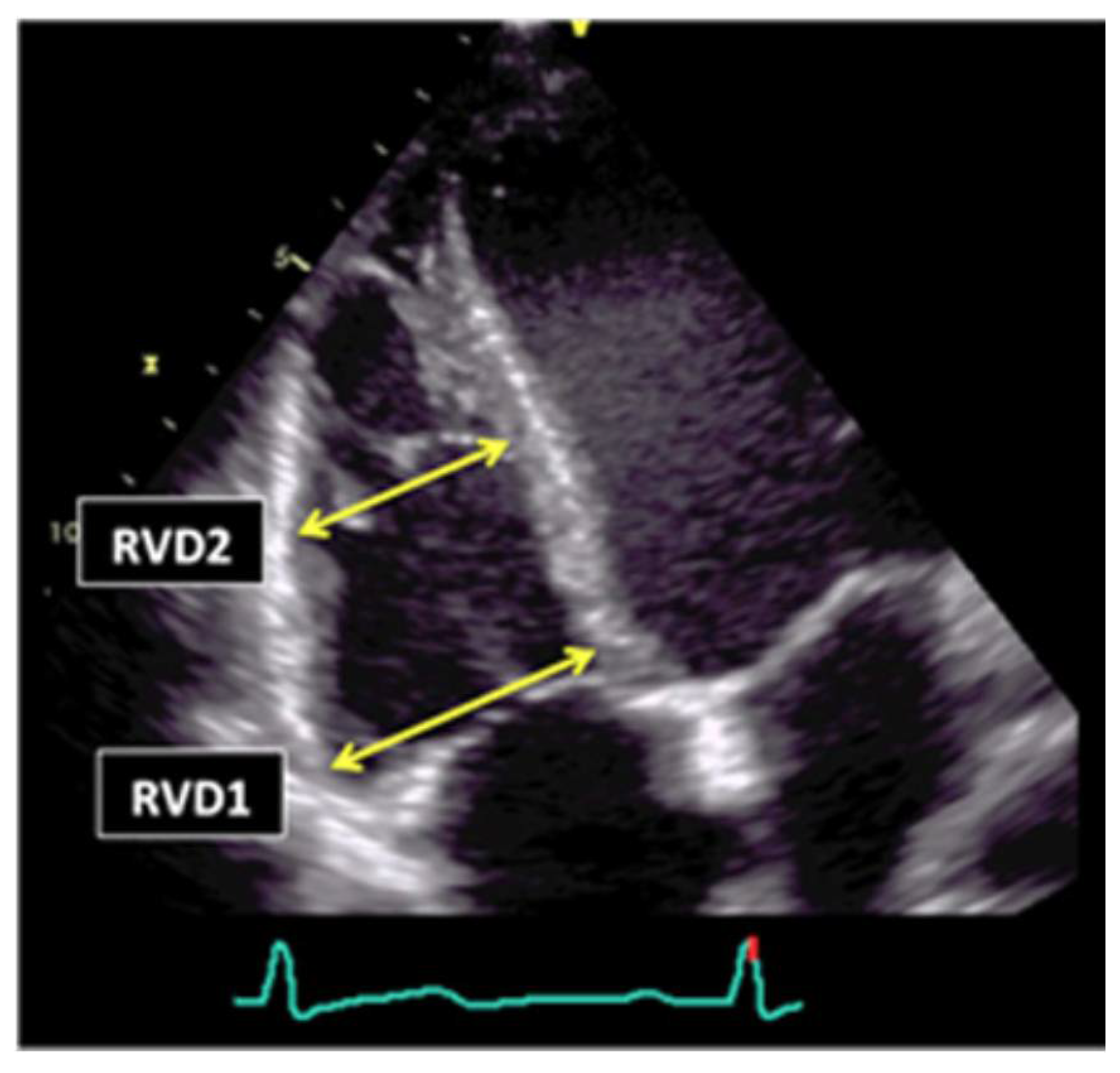
Figure 4.
RV linear dimensions (inflow).

Figure 5.
RV linear dimensions (outflow). (A) Parasternal long axis view from the anterior free wall RV wall up to the septal aortic junction (RVOT Prox); (B) linear transversal dimension measured just proximal to the pulmonary valve at end-diastole, by short axis view.
Figure 5.
RV linear dimensions (outflow). (A) Parasternal long axis view from the anterior free wall RV wall up to the septal aortic junction (RVOT Prox); (B) linear transversal dimension measured just proximal to the pulmonary valve at end-diastole, by short axis view.

Figure 6.
Tracing of the RV endocardial border for RVAC calculation. Manual tracing of the RV endocardial border. Trabeculations, papillary muscles, and moderator band are included in the cavity area.
Figure 6.
Tracing of the RV endocardial border for RVAC calculation. Manual tracing of the RV endocardial border. Trabeculations, papillary muscles, and moderator band are included in the cavity area.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Prevalence by age of ARVC association to sudden death.
| AGE (Years) | <18 | 18–35 | >35 |
|---|---|---|---|
| SD prevalence | <6% | 14% | 18% |
Legend: SD (sudden death).
Table 2.
Pattern of genetic mutations of ARVC.
| Disease Gene | Gene | Locus | Mode of Inheritance | Associated Phenotype |
|---|---|---|---|---|
| Desmosomal genes | ||||
| Plakoglobin | JUP | 17q21.2 | AD/AR | AR form: Naxos disease |
| Desmoplakin | DSP | 6p24.3 | AD/AR | AR form: cardiocutaneous syndrome |
| Plakophilin-2 | PKP-2 | 12p11.21 | AD/AR | |
| Desmoglein-2 | DSG2 | 18q12.1 | AD/AR | |
| Desmocollin-2 | DSC2 | 18q12.1 | AD/AR | |
| Non-desmosomal genes | ||||
| Transforming growth factor-β-3 | TGFB3 | 14q24.3 | AD | |
| Transmembrane protein 43 | TMEM43 | 3p25.1 | AD | |
| Ryanodine receptor | RYR2 | 1q42–q43 | AD? | |
| Desmin | DES | 2q35 | AD | Overlap syndrome (DC HC phenotype, early conduction disease) |
| Phospholamban | PLP | 6q22.31 | AD | |
| Titin | TNT | 2q31.2 | AD | Overlap syndrome (early conduction disease) |
| Lamin A/C | LMNA | 1q22 | AD | Overlap syndrome |
| α-T-catenin | CTNAA3 | 10q21.3 | AD | |
| Filamin-C | FLNC | 7q32.1 | AD | Overlap syndrome (HC and DC phenotype) |
| N-Cadherin | CDH2 | 18q12.1 | AD | |
Table 3.
ECG diagnostic criteria for ARVC.
| Major Criteria | Epsilon Wave/QRS >110 ms | V1–V3 |
|---|---|---|
| Repolarization abnormalities: Inverted T wave >14 years and in absence of RBB | V1–V3 | |
| Minor Criteria | Inverted T wave (>14 years) in presence of RBB | V1–V4 |
| Inverted T wave | V4–V6 | |
| Late activation time (S wave) >50 ms | V1–V3 | |
| Late potential at high amplification at Signal Averaged ECG (SAECG) exam | SAECG |
Table 4.
Pathological values compatible with ARVC and measured by 2D echocardiography.
| Parameter | Pathological Values | Normal Range |
|---|---|---|
| RV Area Change | Fractional Area Change <33 | >40% |
| RVOT PLAX diameter | >33 mm | 20–30 mm |
| RVOT prox diameter | >36 mm | 21–35 mm |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stefani, L.; Tosi, B.; Galanti, G. Multiparametric Approach to Arrhythmogenic Cardiomyopathy: Clinical, Instrumental, and Lifestyle Indications. J. Funct. Morphol. Kinesiol. 2018, 3, 35. https://doi.org/10.3390/jfmk3020035
AMA Style
Stefani L, Tosi B, Galanti G. Multiparametric Approach to Arrhythmogenic Cardiomyopathy: Clinical, Instrumental, and Lifestyle Indications. Journal of Functional Morphology and Kinesiology. 2018; 3(2):35. https://doi.org/10.3390/jfmk3020035
Chicago/Turabian StyleStefani, Laura, Benedetta Tosi, and Giorgio Galanti. 2018. "Multiparametric Approach to Arrhythmogenic Cardiomyopathy: Clinical, Instrumental, and Lifestyle Indications" Journal of Functional Morphology and Kinesiology 3, no. 2: 35. https://doi.org/10.3390/jfmk3020035