Switchable Interfaces: Redox Monolayers on Si(100) by Electrochemical Trapping of Alcohol Nucleophiles

 , and
, and 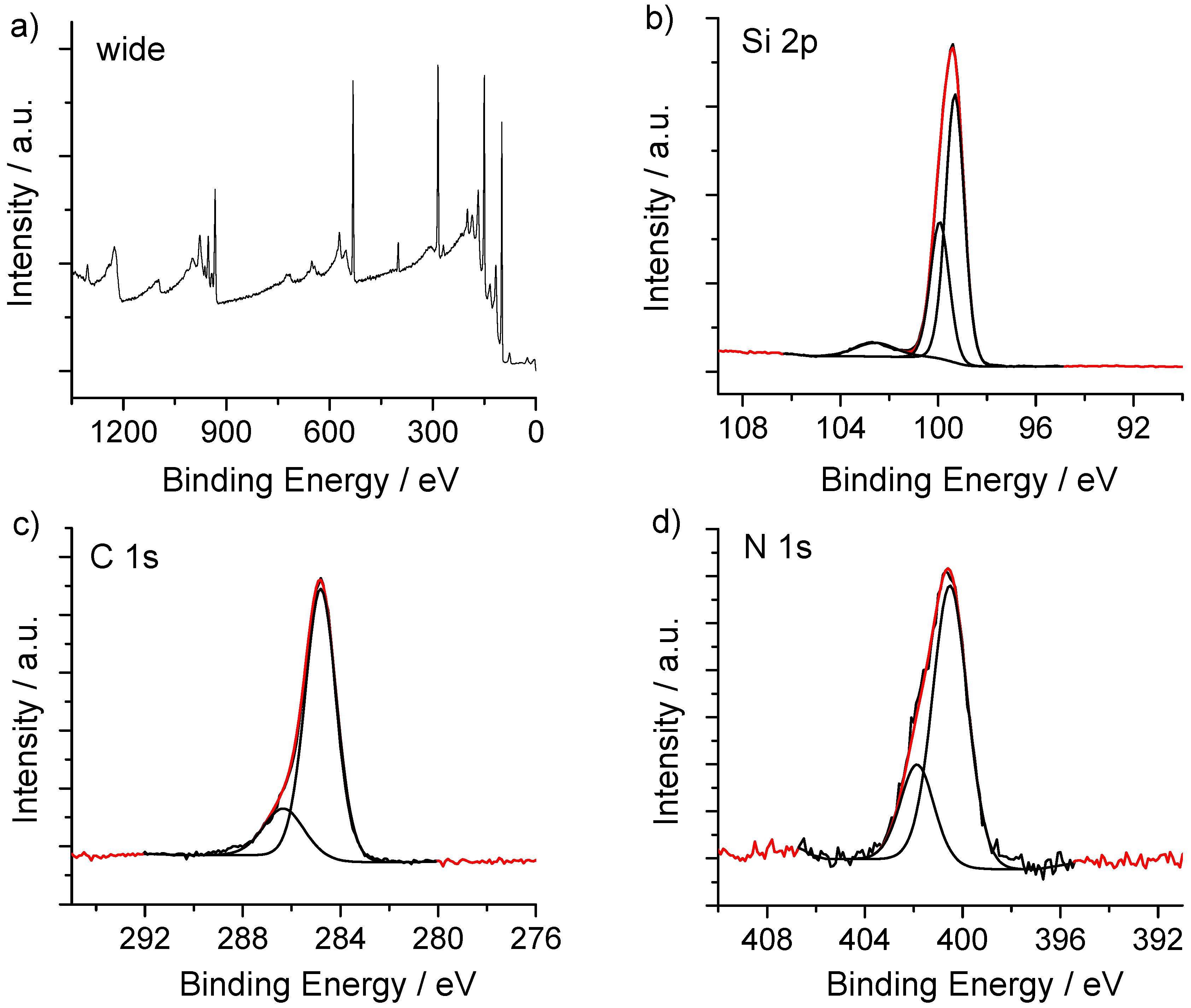{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
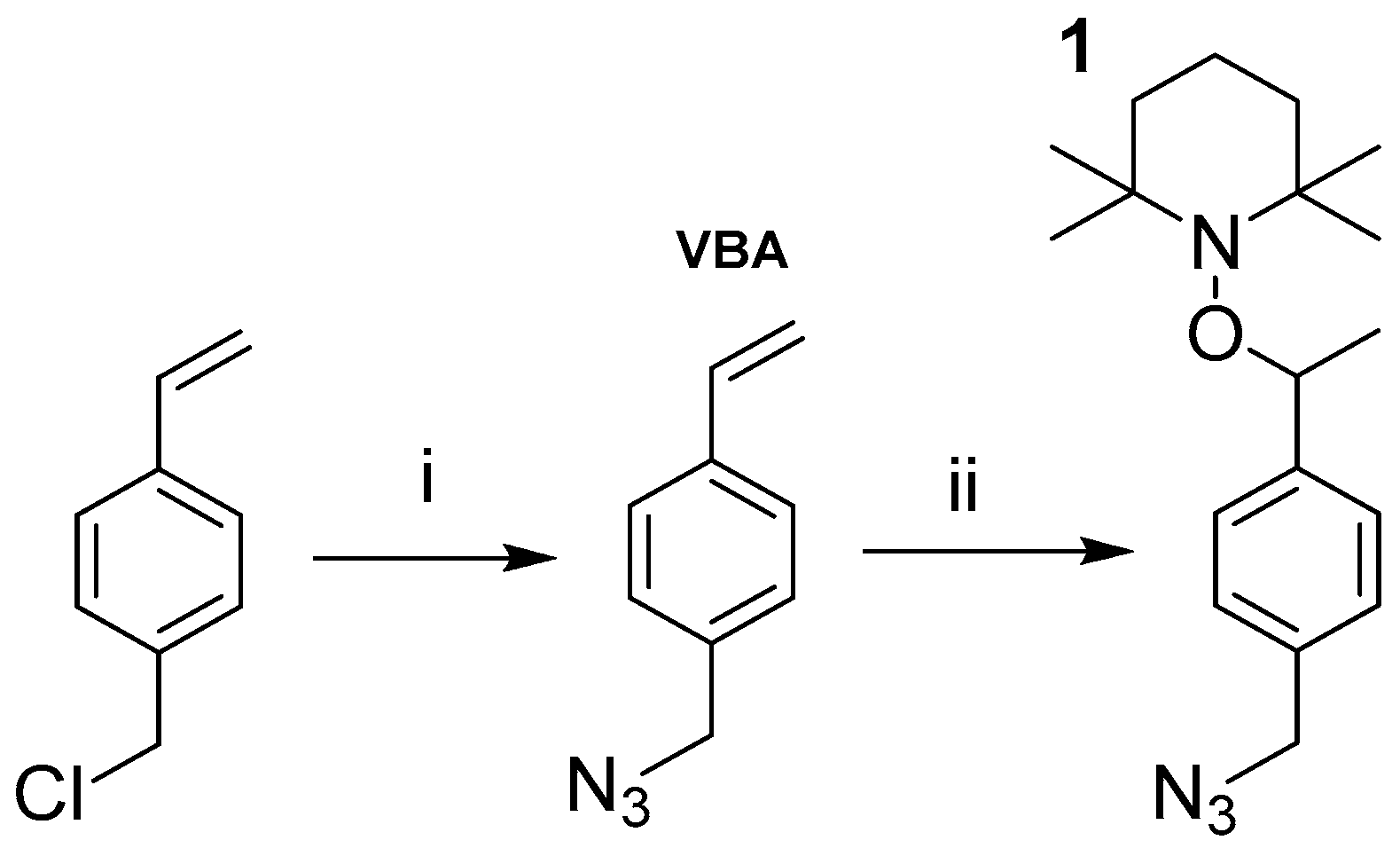
2.2. Synthetic Methods
2.3. Surface Modification
2.3.1. Light-Assisted Hydrosilylation of 1,8-Nonadiyne on Si(100) (S-1)
2.3.2. Click Immobilization of Alkoxyamine 1 (S-2)
2.4. Surface Characterization
2.4.1. X-ray Photoelectron Spectroscopy
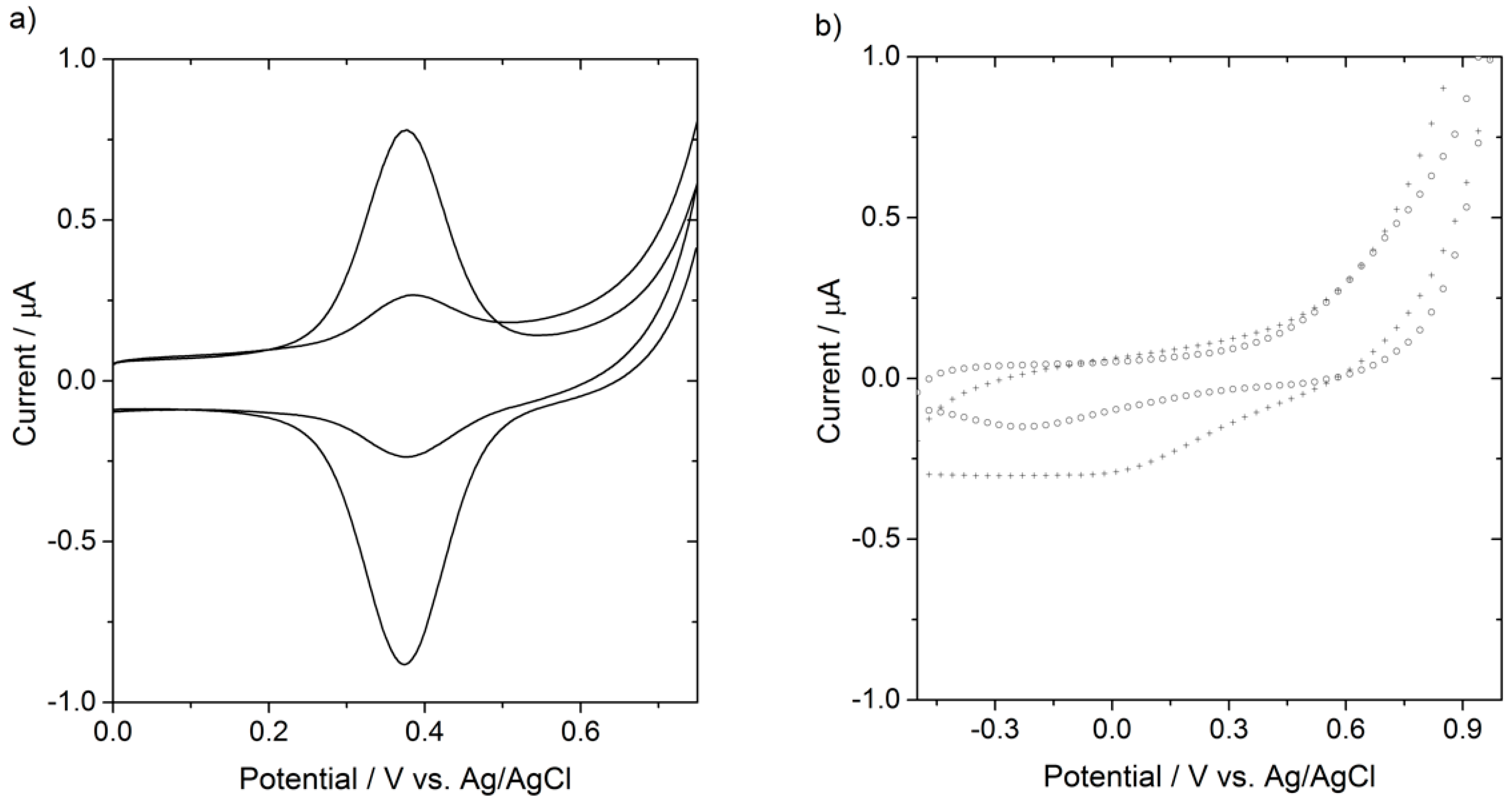
2.4.2. Electrochemical Measurements
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Horn, E.J.; Rosen, B.R.; Baran, P.S. Synthetic organic electrochemistry: An enabling and innately sustainable method. ACS Cent. Sci. 2016, 2, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic organic electrochemistry: Calling all engineers. Angew. Chem. Int. Ed. 2018, 57, 4149–4155. [Google Scholar] [CrossRef] [PubMed]
- Frontana-Uribe, B.A.; Little, R.D.; Ibanez, J.G.; Palma, A.; Vasquez-Medrano, R. Organic electrosynthesis: A promising green methodology in organic chemistry. Green Chem. 2010, 12, 2099–2119. [Google Scholar] [CrossRef]
- Gütz, C.; Selt, M.; Bänziger, M.; Bucher, C.; Römelt, C.; Hecken, N.; Gallou, F.; Galvão, T.R.; Waldvogel, S.R. A novel cathode material for cathodic dehalogenation of 1, 1-dibromo cyclopropane derivatives. Chem. Eur. J. 2015, 21, 13878–13882. [Google Scholar] [CrossRef] [PubMed]
- Gütz, C.; Bänziger, M.; Bucher, C.; Galvão, T.S.R.; Waldvogel, S.R. Development and scale-up of the electrochemical dehalogenation for the synthesis of a key intermediate for NS5A inhibitors. Org. Process Res. Dev. 2015, 19, 1428–1433. [Google Scholar] [CrossRef]
- Andrieux, C.P.; Gonzalez, F.; Savéant, J.-M. Derivatization of carbon surfaces by anodic oxidation of arylacetates. Electrochemical manipulation of the grafted films. J. Am. Chem. Soc. 1997, 119, 4292–4300. [Google Scholar] [CrossRef]
- Robins, E.; Stewart, M.; Buriak, J. Anodic and cathodic electrografting of alkynes on porous silicon. Chem. Commun. 1999, 2479–2480. [Google Scholar] [CrossRef]
- Hurley, P.T.; Ribbe, A.E.; Buriak, J.M. Nanopatterning of alkynes on hydrogen-terminated silicon surfaces by scanning probe-induced cathodic electrografting. J. Am. Chem. Soc. 2003, 125, 11334–11339. [Google Scholar] [CrossRef] [PubMed]
- Belanger, D.; Pinson, J. Electrografting: A powerful method for surface modification. Chem. Soc. Rev. 2011, 40, 3995–4048. [Google Scholar] [CrossRef] [PubMed]
- Gooding, J.J.; Ciampi, S. The molecular level modification of surfaces: From self-assembled monolayers to complex molecular assemblies. Chem. Soc. Rev. 2011, 40, 2704–2718. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, A.C.; Darwish, N.; Ciampi, S.; Sanz, F.; Gooding, J.J.; Díez-Pérez, I. Single-molecule electrical contacts on silicon electrodes under ambient conditions. Nat. Commun. 2017, 8, 15056–15063. [Google Scholar] [CrossRef] [PubMed]
- Scheres, L.; ter Maat, J.; Giesbers, M.; Zuilhof, H. Microcontact printing onto oxide-free silicon via highly reactive acid fluoride-functionalized monolayers. Small 2010, 6, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Bard, A.J.; Bocarsly, A.B.; Fan, F.R.F.; Walton, E.G.; Wrighton, M.S. The concept of Fermi level pinning at semiconductor/liquid junctions. Consequences for energy conversion efficiency and selection of useful solution redox couples in solar devices. J. Am. Chem. Soc. 1980, 102, 3671–3677. [Google Scholar] [CrossRef]
- Ciampi, S.; James, M.; Le Saux, G.; Gaus, K.; Justin Gooding, J. Electrochemical “switching” of Si(100) modular assemblies. J. Am. Chem. Soc. 2012, 134, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Ciampi, S.; Harper, J.B.; Gooding, J.J. Wet chemical routes to the assembly of organic monolayers on silicon surfaces via the formation of Si−C bonds: Surface preparation, passivation and functionalization. Chem. Soc. Rev. 2010, 39, 2158–2183. [Google Scholar] [CrossRef] [PubMed]
- Ciampi, S.; Eggers, P.K.; Le Saux, G.; James, M.; Harper, J.B.; Gooding, J.J. Silicon (100) Electrodes resistant to oxidation in aqueous solutions: An unexpected benefit of surface acetylene moieties. Langmuir 2009, 25, 2530–2539. [Google Scholar] [CrossRef] [PubMed]
- Noufi, R.; Frank, A.J.; Nozik, A.J. Stabilization of n-type silicon photoelectrodes to surface oxidation in aqueous electrolyte solution and mediation of oxidation reaction by surface-attached organic conducting polymer. J. Am. Chem. Soc. 1981, 103, 1849–1850. [Google Scholar] [CrossRef]
- Nemanick, E.J.; Hurley, P.T.; Webb, L.J.; Knapp, D.W.; Michalak, D.J.; Brunschwig, B.S.; Lewis, N.S. Chemical and electrical passivation of single-crystal silicon(100) surfaces through a two-step chlorination/alkylation process. J. Phys. Chem. B 2006, 110, 14770–14778. [Google Scholar] [CrossRef] [PubMed]
- Linford, M.R.; Chidsey, C.E.D. Alkyl monolayers covalently bonded to silicon surfaces. J. Am. Chem. Soc. 1993, 115, 12631–12632. [Google Scholar] [CrossRef]
- Sun, Q.-Y.; de Smet, L.C.P.M.; van Lagen, B.; Wright, A.; Zuilhof, H.; Sudhöelter, E.J.R. Covalently attached monolayers on hydrogen-terminated Si(100): Extremely mild attachment by visible light. Angew. Chem. Int. Ed. 2004, 43, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Vogel, Y.B.; Noble, B.B.; Gonçales, V.R.; Darwish, N.; Brun, A.L.; Gooding, J.J.; Wallace, G.G.; Coote, M.L.; Ciampi, S. TEMPO monolayers on Si(100) electrodes: Electrostatic effects by the electrolyte and semiconductor space-charge on the electroactivity of a persistent radical. J. Am. Chem. Soc. 2016, 138, 9611–9619. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.-I.; Kataoka, K.; Horcajada, R.; Nagaki, A. Modern strategies in electroorganic synthesis. Chem. Rev. 2008, 108, 2265–2299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Laborda, E.; Darwish, N.; Noble, B.B.; Tyrell, J.H.; Pluczyk, S.; Le Brun, A.P.; Wallace, G.G.; Gonzalez, J.; Coote, M.L. Electrochemical and electrostatic cleavage of alkoxyamines. J. Am. Chem. Soc. 2018, 140, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Danko, M.; Szabo, E.; Hrdlovic, P. Synthesis and spectral characteristics of fluorescent dyes based on coumarin fluorophore and hindered amine stabilizer in solution and polymer matrices. Dyes Pigments 2011, 90, 129–138. [Google Scholar] [CrossRef]
- Vogel, Y.B.; Zhang, L.; Darwish, N.; Gonçales, V.R.; Le Brun, A.; Gooding, J.J.; Molina, A.; Wallace, G.G.; Coote, M.L.; Gonzalez, J. Reproducible flaws unveil electrostatic aspects of semiconductor electrochemistry. Nat. Commun. 2017, 8, 2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciampi, S.; Böcking, T.; Kilian, K.A.; James, M.; Harper, J.B.; Gooding, J.J. Functionalization of acetylene-terminated monolayers on Si(100) surfaces: A click chemistry approach. Langmuir 2007, 23, 9320–9329. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.H.; Ciampi, S.; Yang, Y.; Tavallaie, R.; Zhu, Y.; Zarei, L.; Gonçales, V.R.; Gooding, J.J. Connecting electrodes with light: One wire, many electrodes. Chem. Sci. 2015, 6, 6769–6776. [Google Scholar] [CrossRef] [PubMed]
- Vogel, Y.B.; Gonçales, V.R.; Gooding, J.J.; Ciampi, S. Electrochemical microscopy based on spatial light modulators: A projection system to spatially address electrochemical reactions at semiconductors. J. Electrochem. Soc. 2018, 165, H3085–H3092. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Espíndola, R.B.D.; Noble, B.B.; Gonçales, V.R.; Wallace, G.G.; Darwish, N.; Coote, M.L.; Ciampi, S. Switchable Interfaces: Redox Monolayers on Si(100) by Electrochemical Trapping of Alcohol Nucleophiles. Surfaces 2018, 1, 3-11. https://doi.org/10.3390/surfaces1010002
Zhang L, Espíndola RBD, Noble BB, Gonçales VR, Wallace GG, Darwish N, Coote ML, Ciampi S. Switchable Interfaces: Redox Monolayers on Si(100) by Electrochemical Trapping of Alcohol Nucleophiles. Surfaces. 2018; 1(1):3-11. https://doi.org/10.3390/surfaces1010002
Chicago/Turabian StyleZhang, Long, Ruth Belinda Domínguez Espíndola, Benjamin B. Noble, Vinicius R. Gonçales, Gordon G. Wallace, Nadim Darwish, Michelle L. Coote, and Simone Ciampi. 2018. "Switchable Interfaces: Redox Monolayers on Si(100) by Electrochemical Trapping of Alcohol Nucleophiles" Surfaces 1, no. 1: 3-11. https://doi.org/10.3390/surfaces1010002