Advanced Fluorescence Microscopy Techniques—FRAP, FLIP, FLAP, FRET and FLIM


Abstract
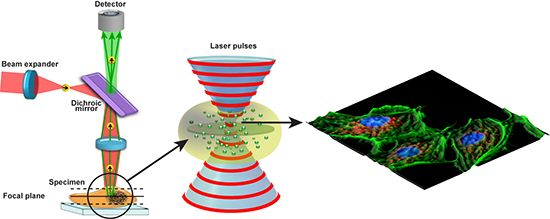
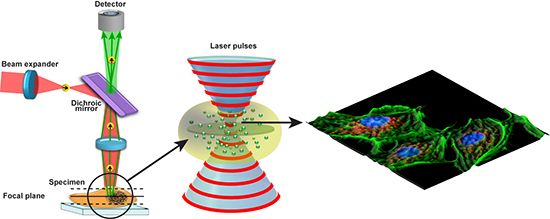
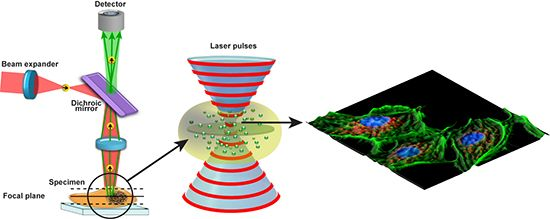
:
1. Introduction
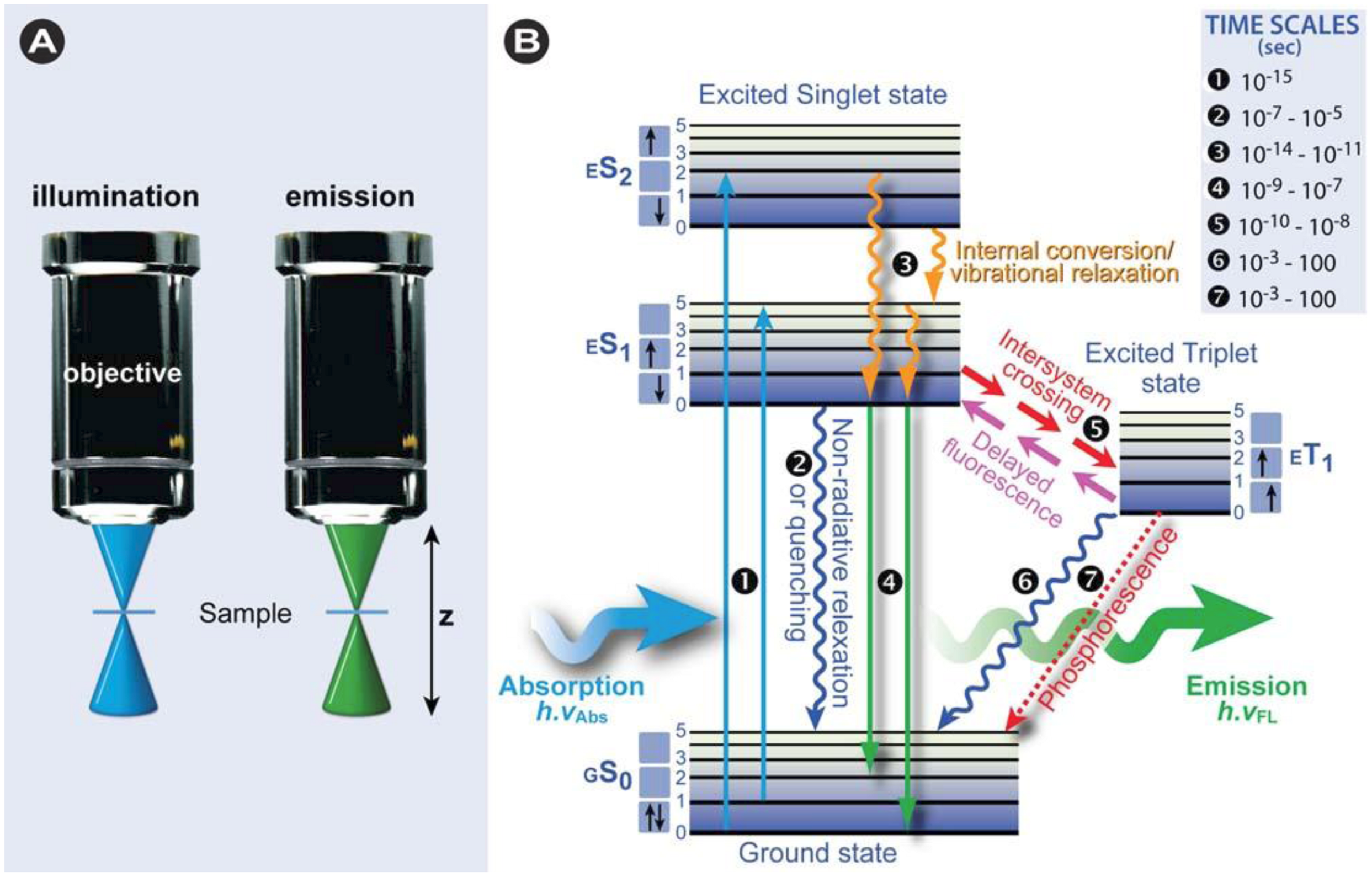
1.1. Introduction to Fluorescence
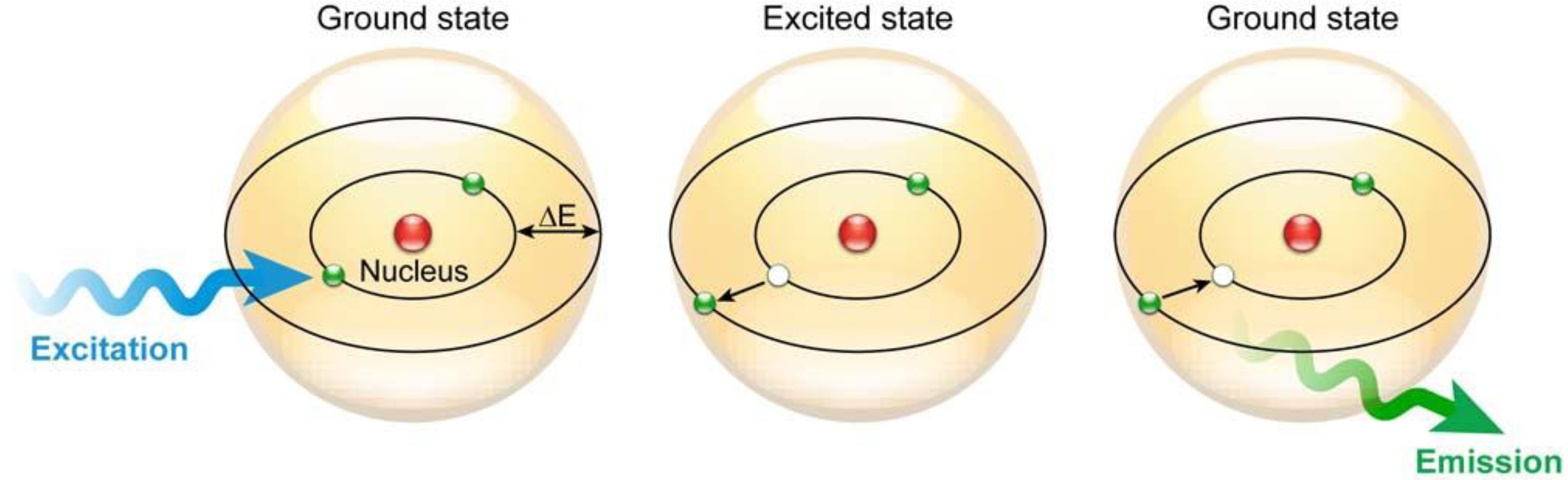
1.1.1. The Physical Phenomenon of Fluorescence

 (1)
(1)

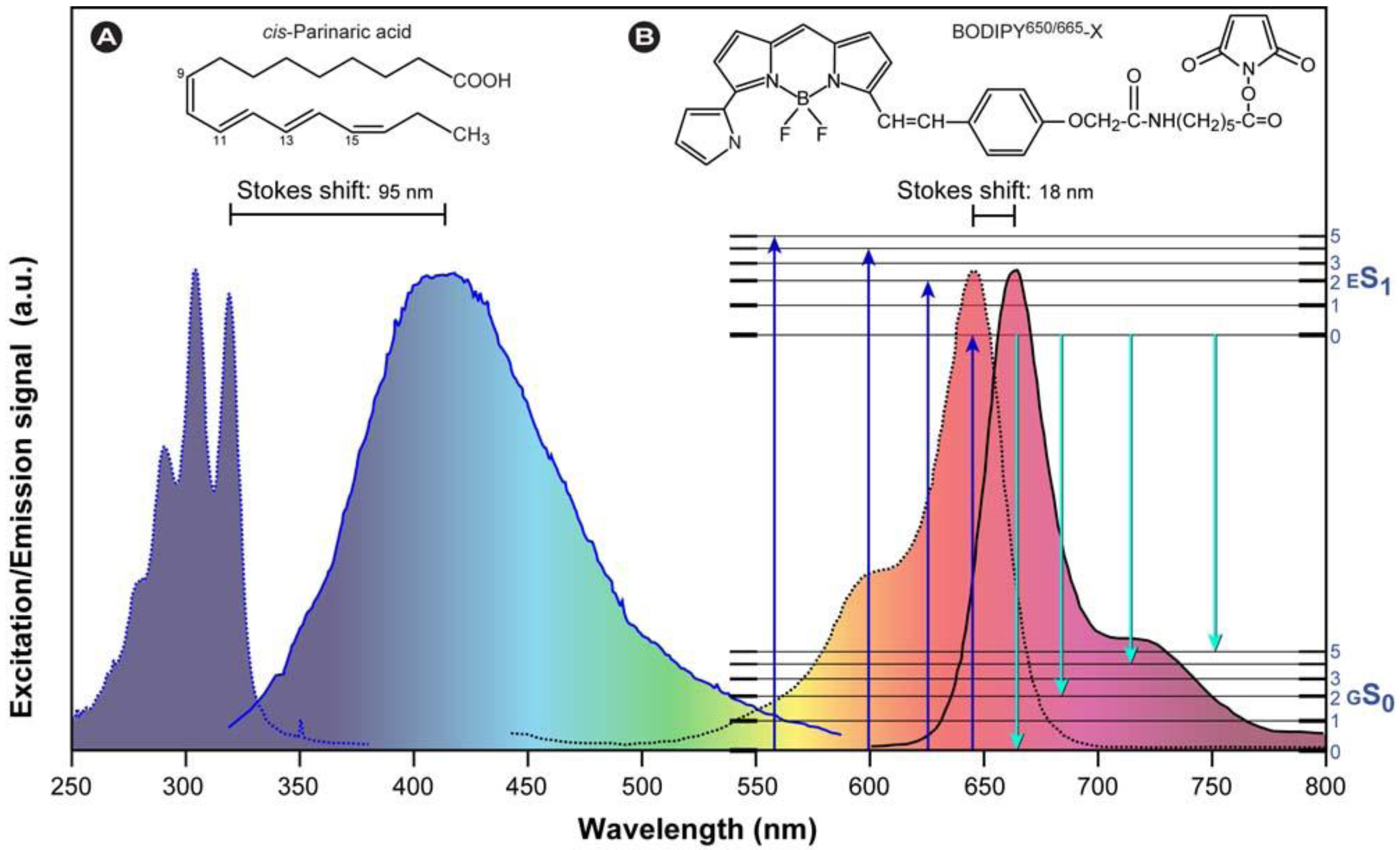
1.1.2. Overview of Fluorescence Characteristics
 (2)
(2)
 (3)
(3) (4)
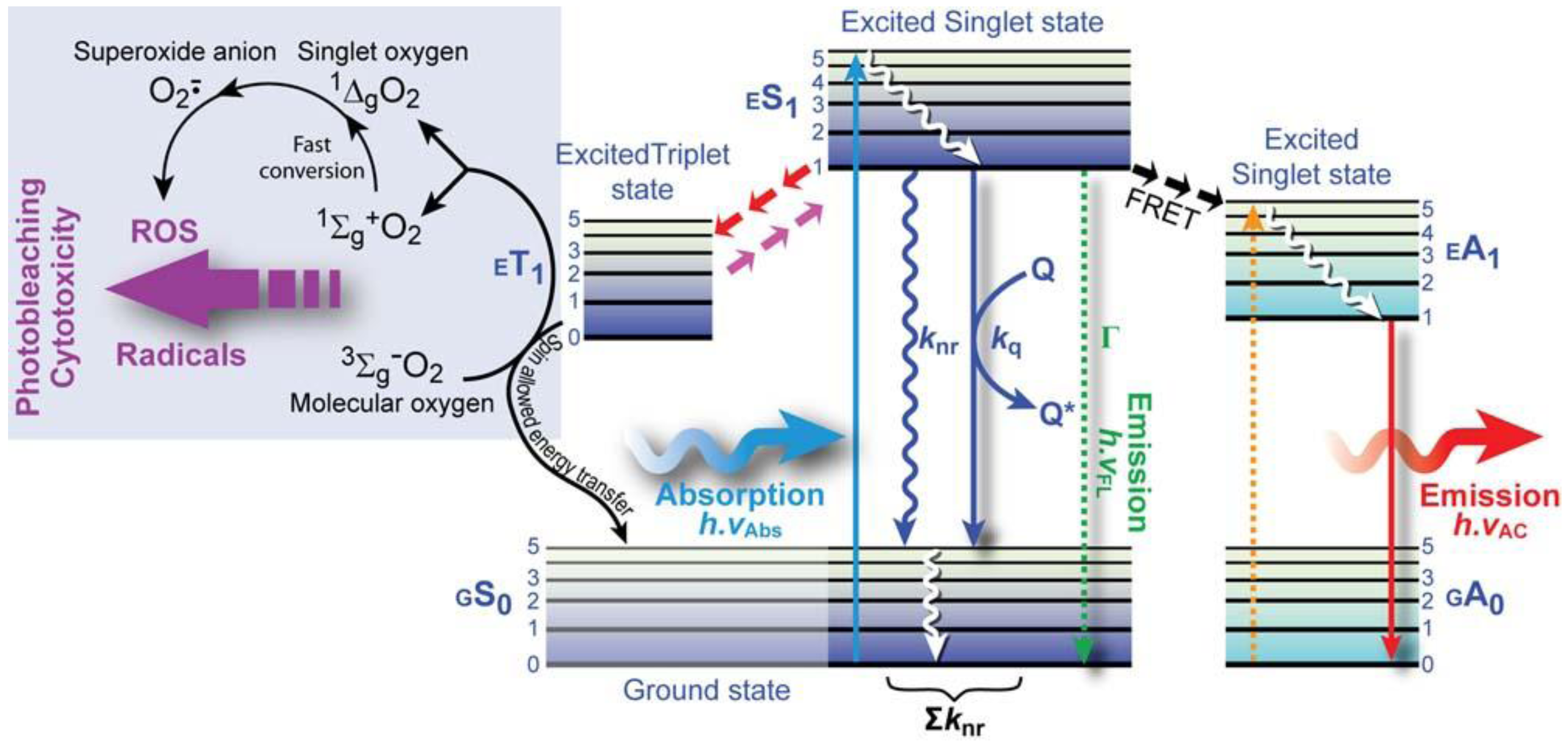
(4)- ➲ Dynamic quenching occurs through collision of the quencher and the excited state fluorochrome, which leads to a decrease in the lifetime and emission intensity.
- ➲ Static quenching arises from direct interaction of the fluorochrome and quenching molecules, for instance by forming a non-fluorescent ground state complex. This form of quenching does not necessarily decrease the measured emission lifetime and often occurs simultaneous with dynamic quenching.
- ➲ In self-quenching (concentration quenching), the fluorochrome quenches its own fluorescence because of close proximity of identical molecules at high concentration. Various mechanisms underlie self-quenching, including radiationless energy transfer–this occurs particularly in fluorochromes with small Stokes shifts–or formation of molecular aggregates. Self-quenching occurs in particular in biomembranes, where the lipid bilayer behaves as a two dimensional fluid with different domains of fluidity where fluorochromes can be concentrated or when labeling proteins with multiple labels.
- ➲ Color-quenching is a process in which emitted photons are absorbed by a strongly colored component such as β-carotene. This leads to a decrease in intensity, but not the fluorescence lifetime.
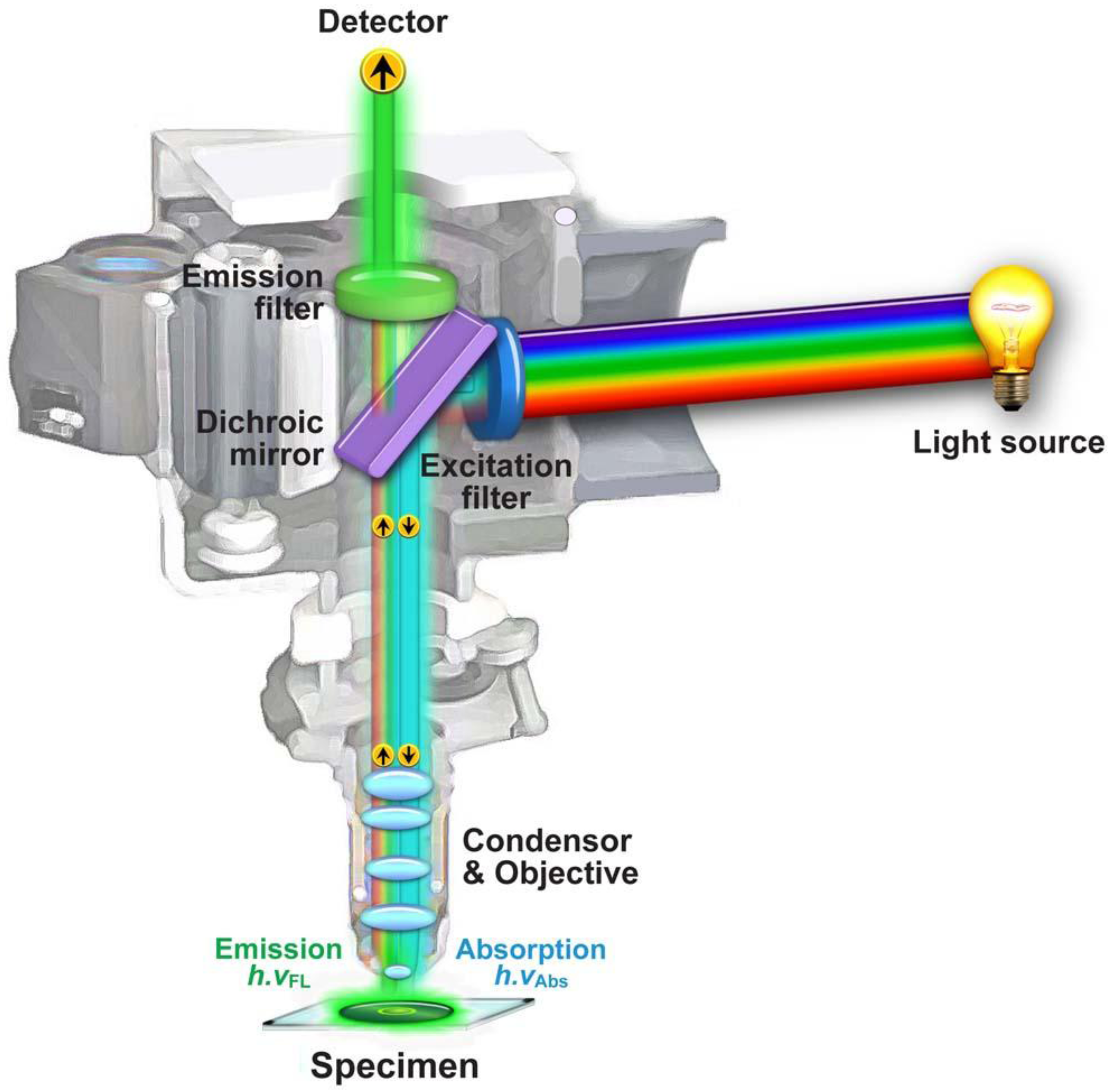
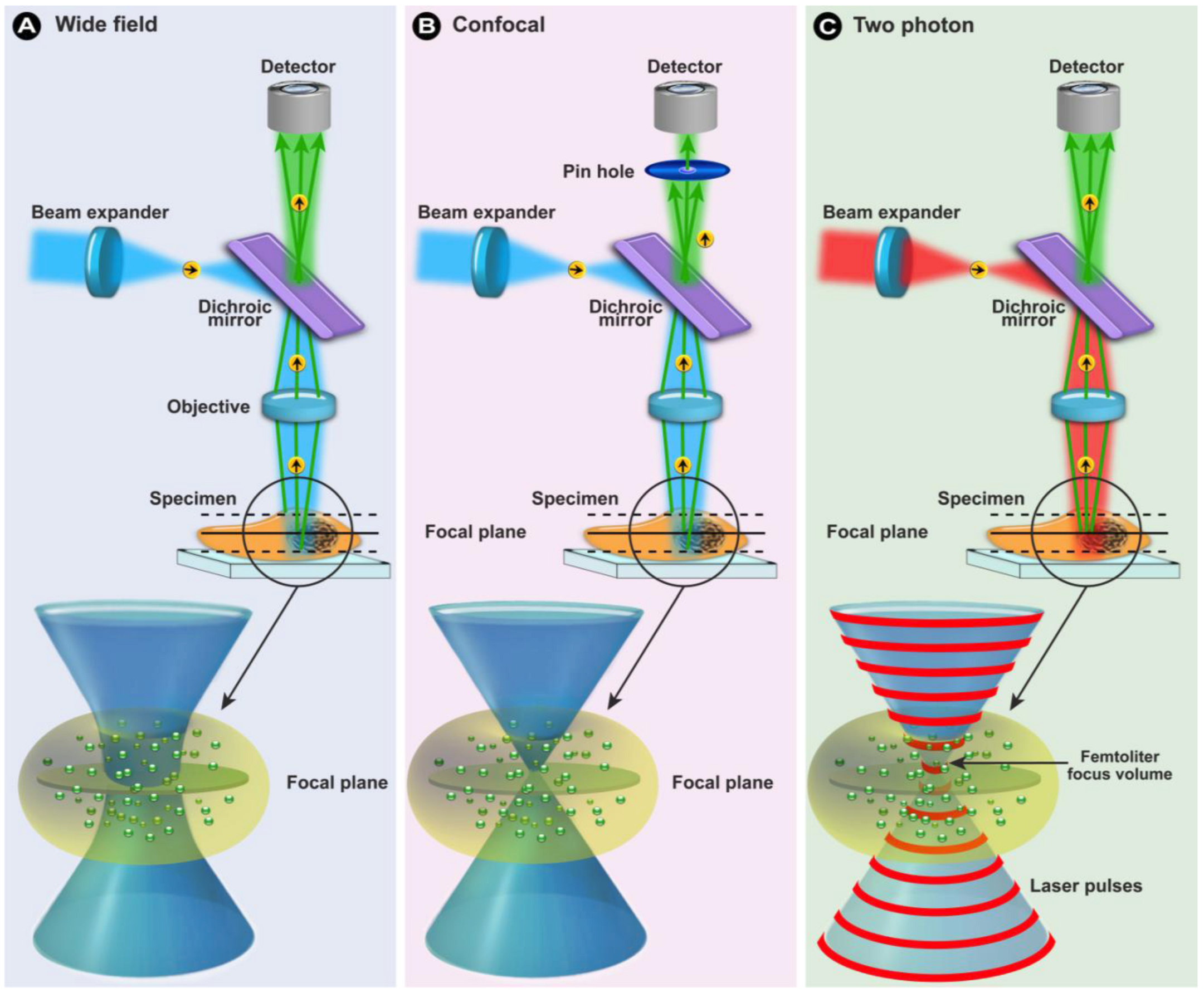
1.2. Fluorescence Microscopy
1.2.1. General Concepts



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
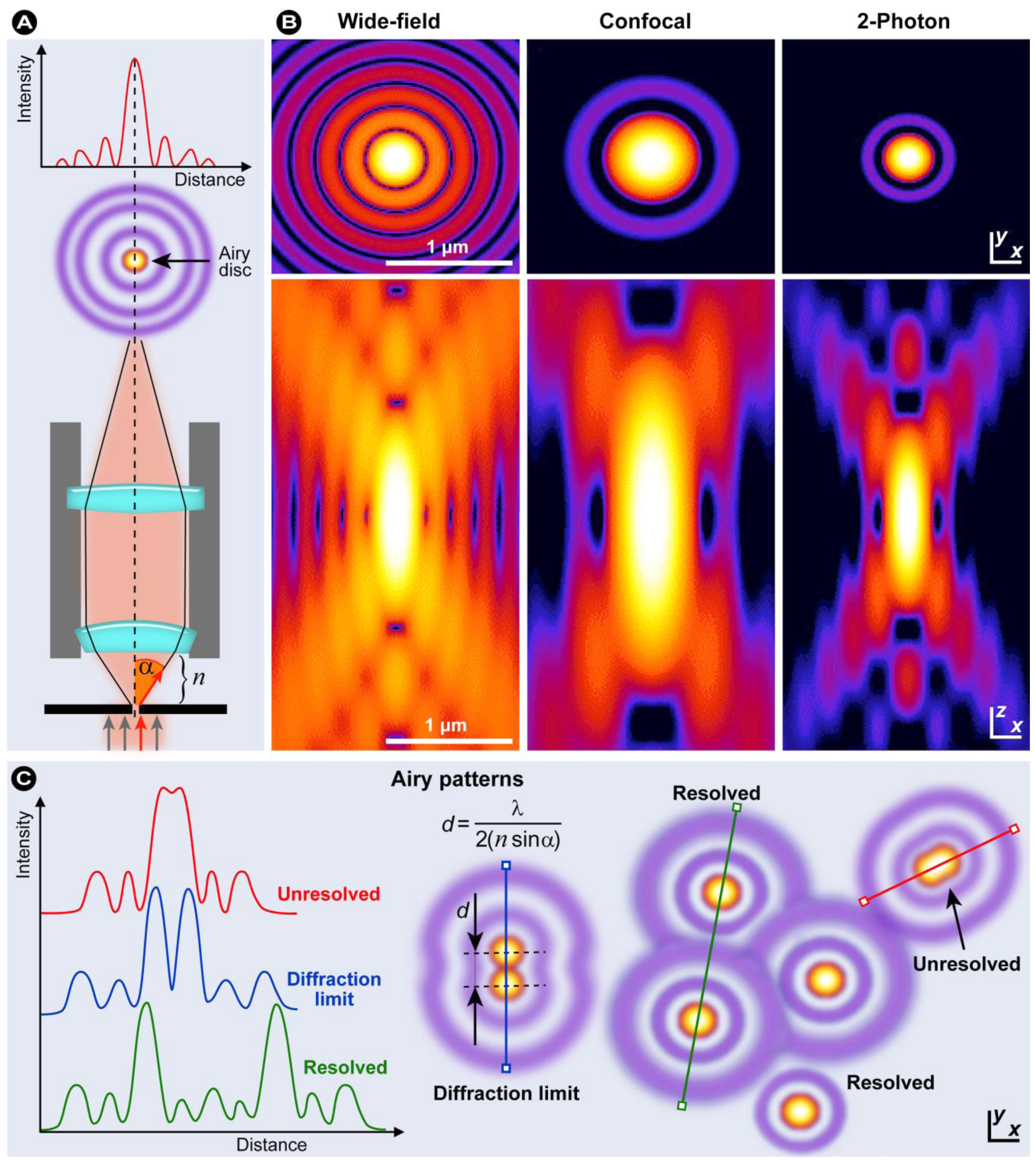
1.2.2. Resolution in Fluorescence Microscopy
 (5)
(5)
 (6)
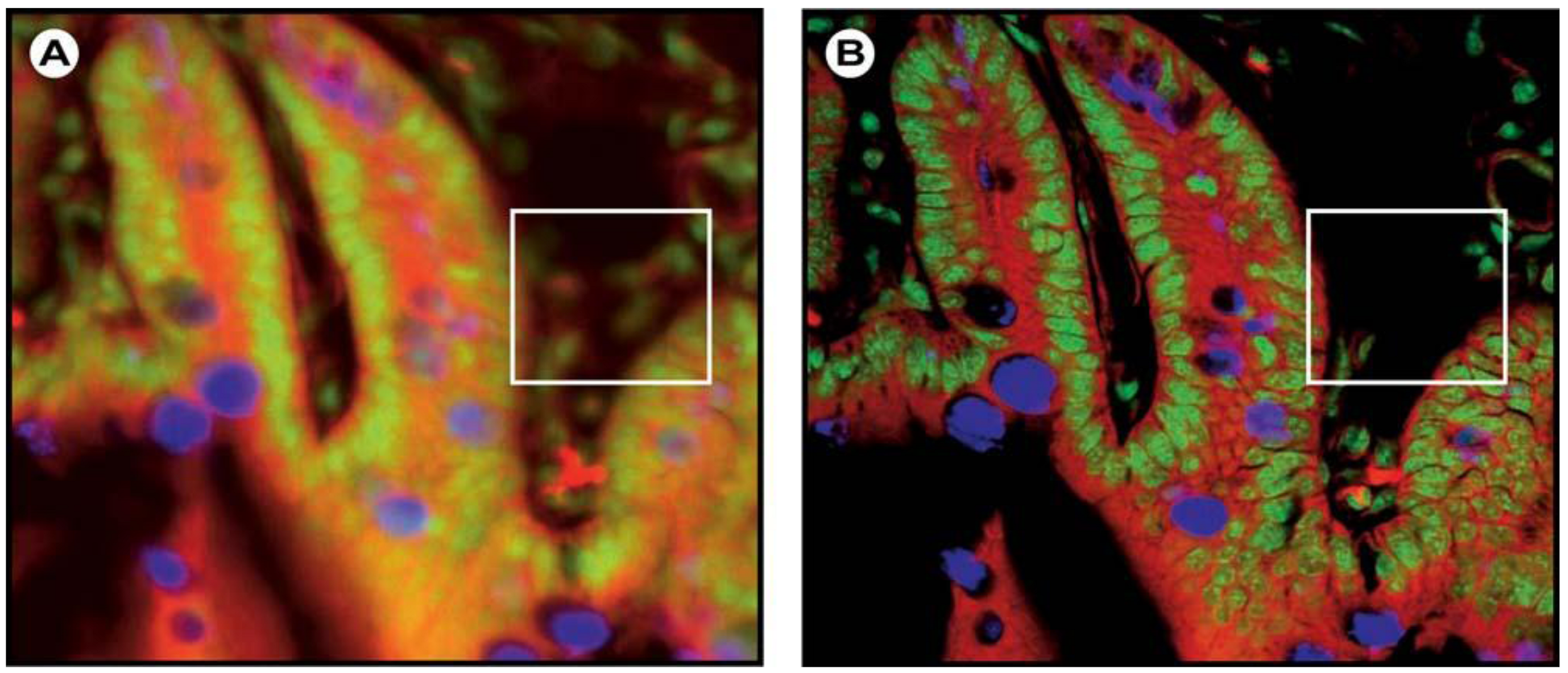
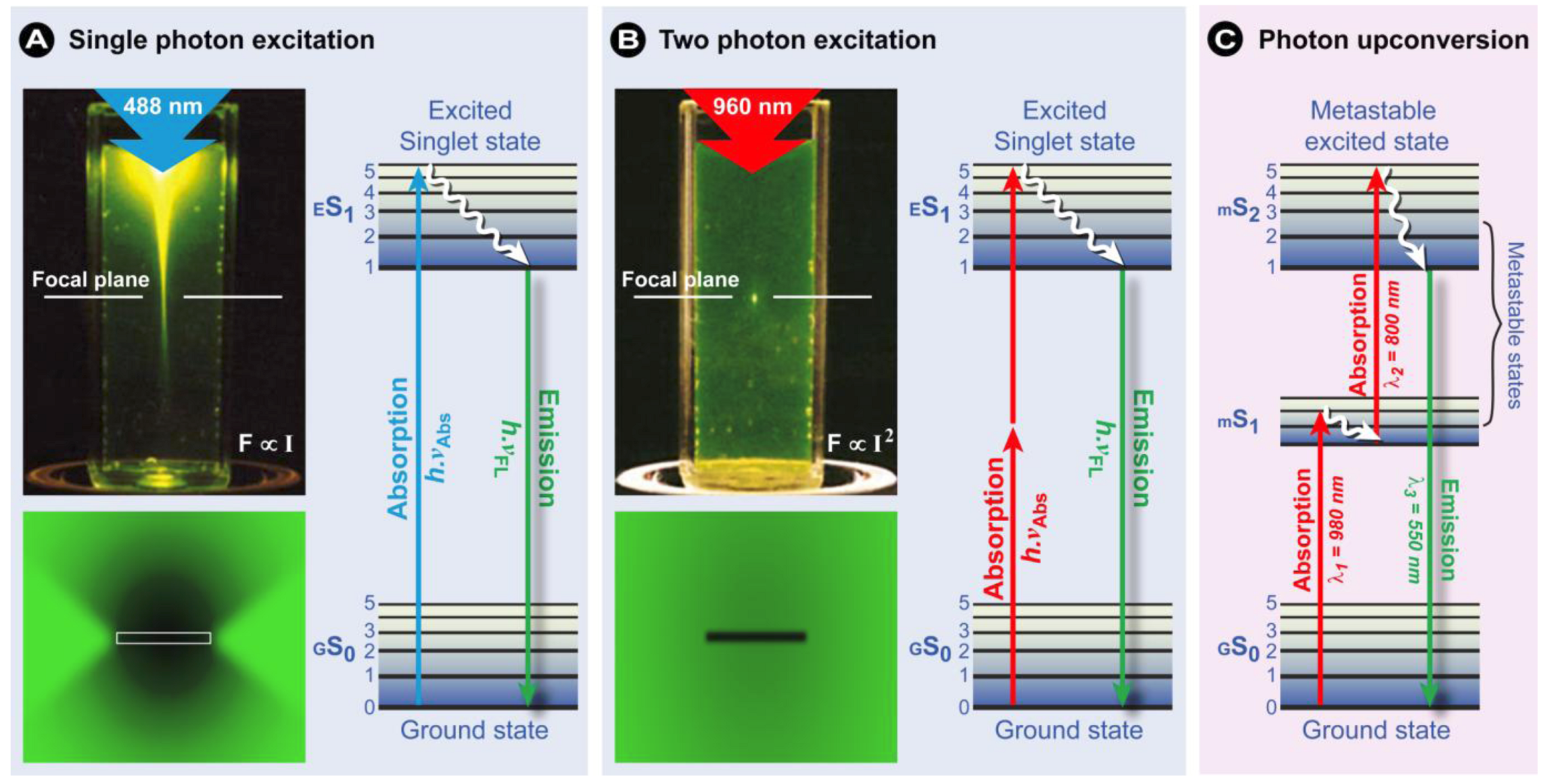
(6)1.2.3. Confocal Laser Scanning Microscopy (CLSM)


1.2.4. Multiphoton Fluorescence Microscopy

2. Photobleaching-based Techniques for Assessing Cellular Dynamics
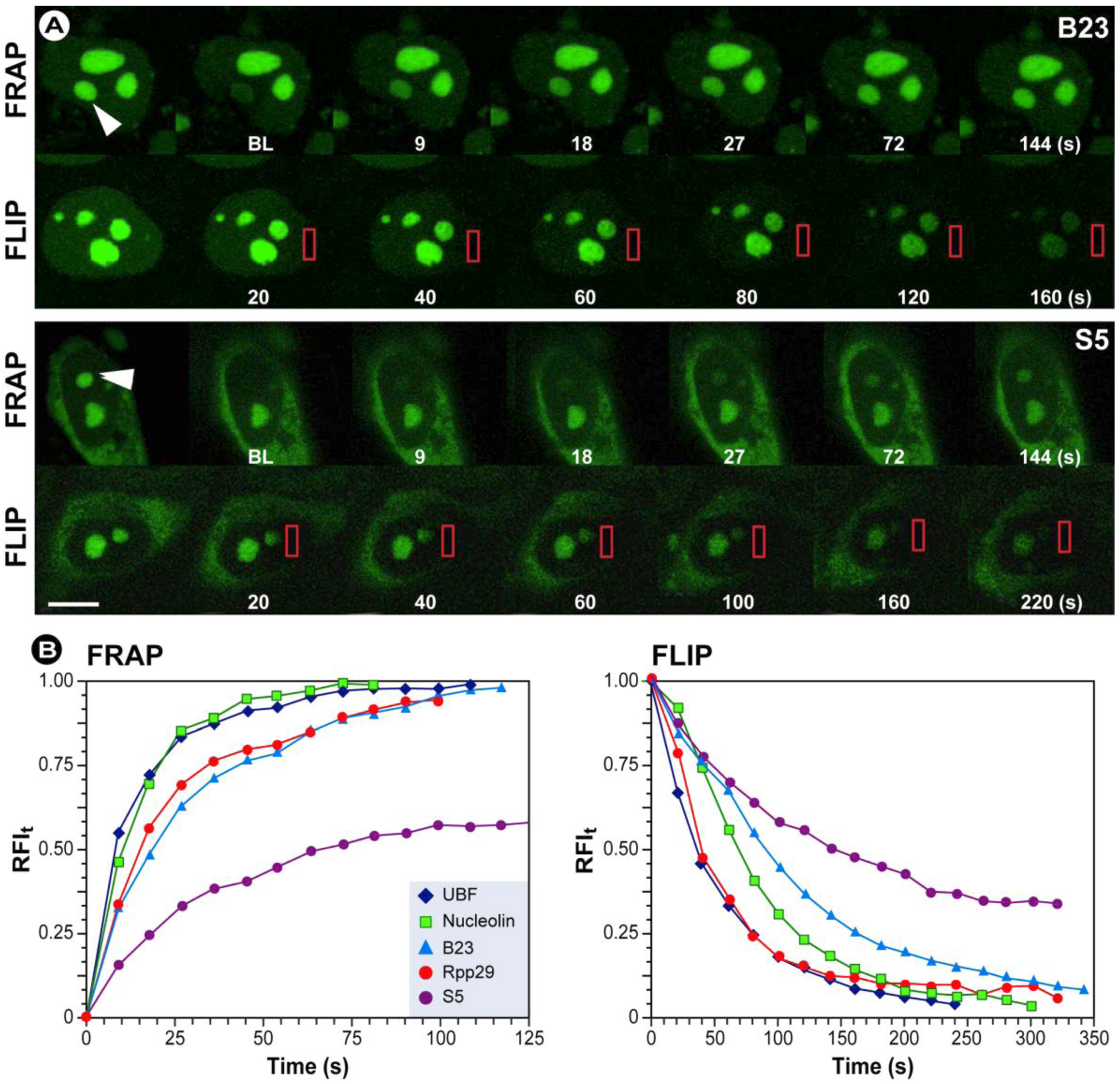
2.1. Fluorescence Recovery after Photobleaching (FRAP)
- ➲ Protein/molecule movement and diffusion (diffusional speed).
- ➲ Compartmentalization and connections between intracellular compartments.
- ➲ The speed of protein/molecule exchange between compartments (exchange speed).
- ➲ Binding characteristics between proteins. Additionally, the effect of mutations that alter individual amino acids on protein association, and the effect of small molecules, such as drugs or inhibitors, on protein pairs can effectively be studies using FRAP.
- ➲ Immobilization of proteins that bind to large structures, e.g., DNA, nuclear envelope, membranes, cytoskeletal elements, etc.
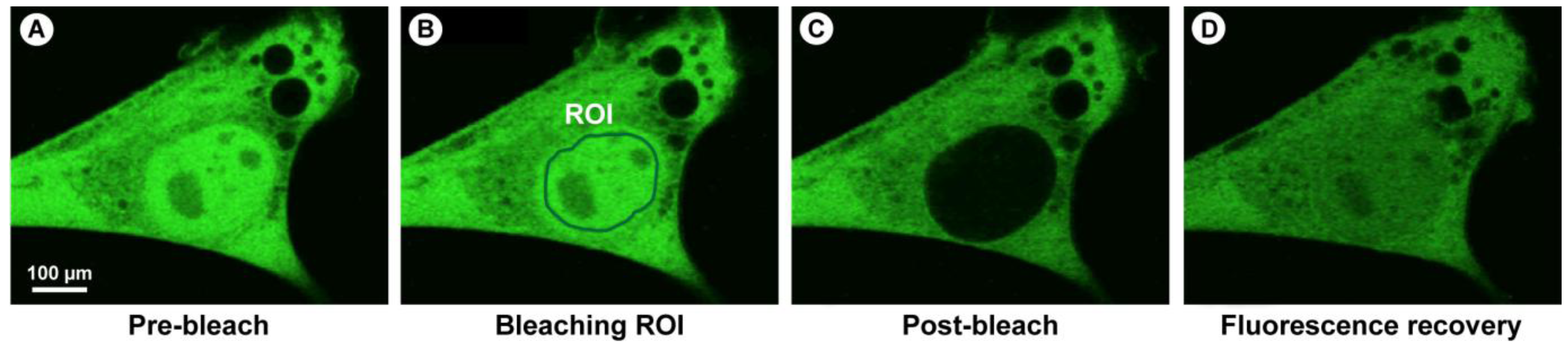
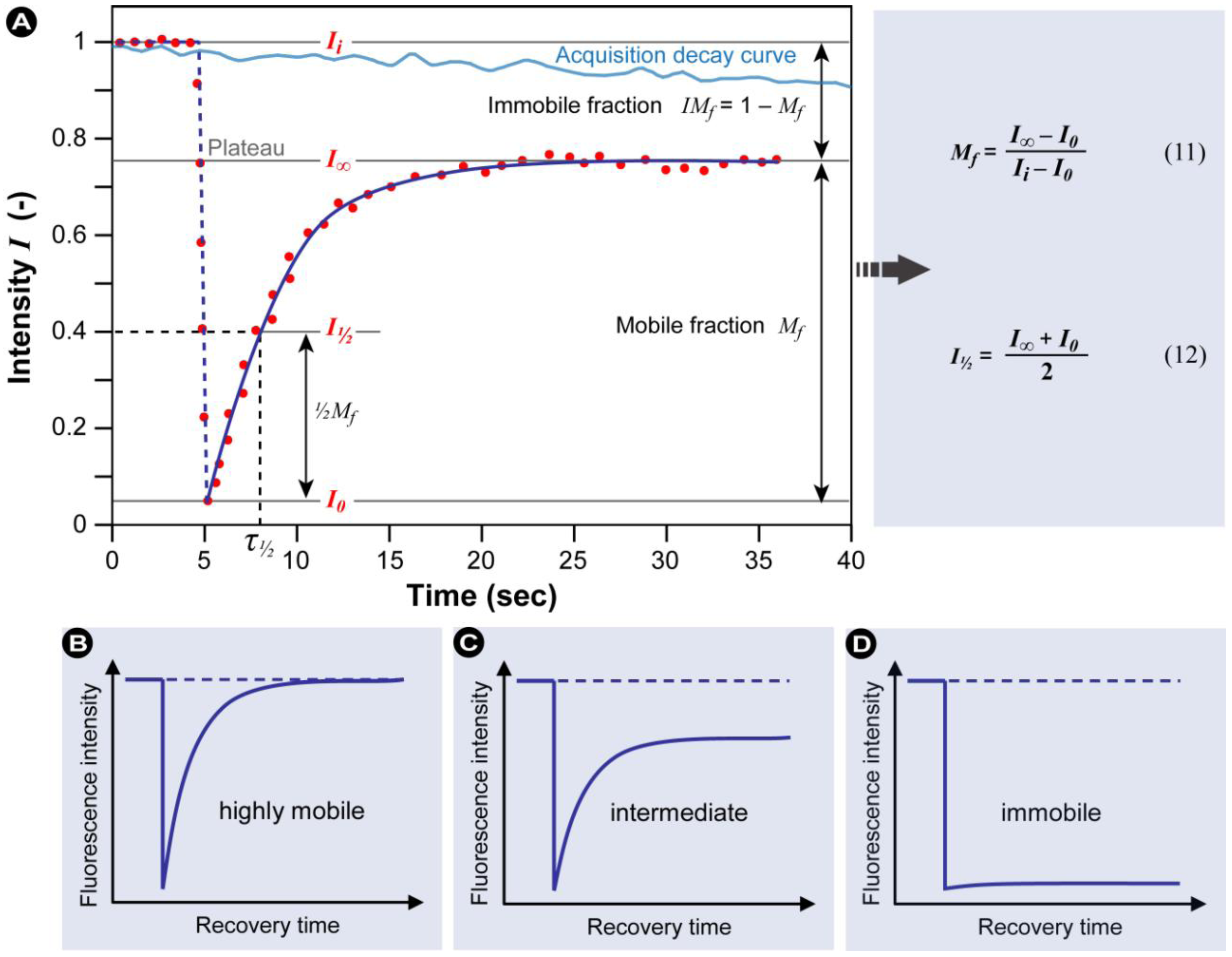
2.1.1. The Basic Principles of FRAP


 (7)
(7) (10)
(10)
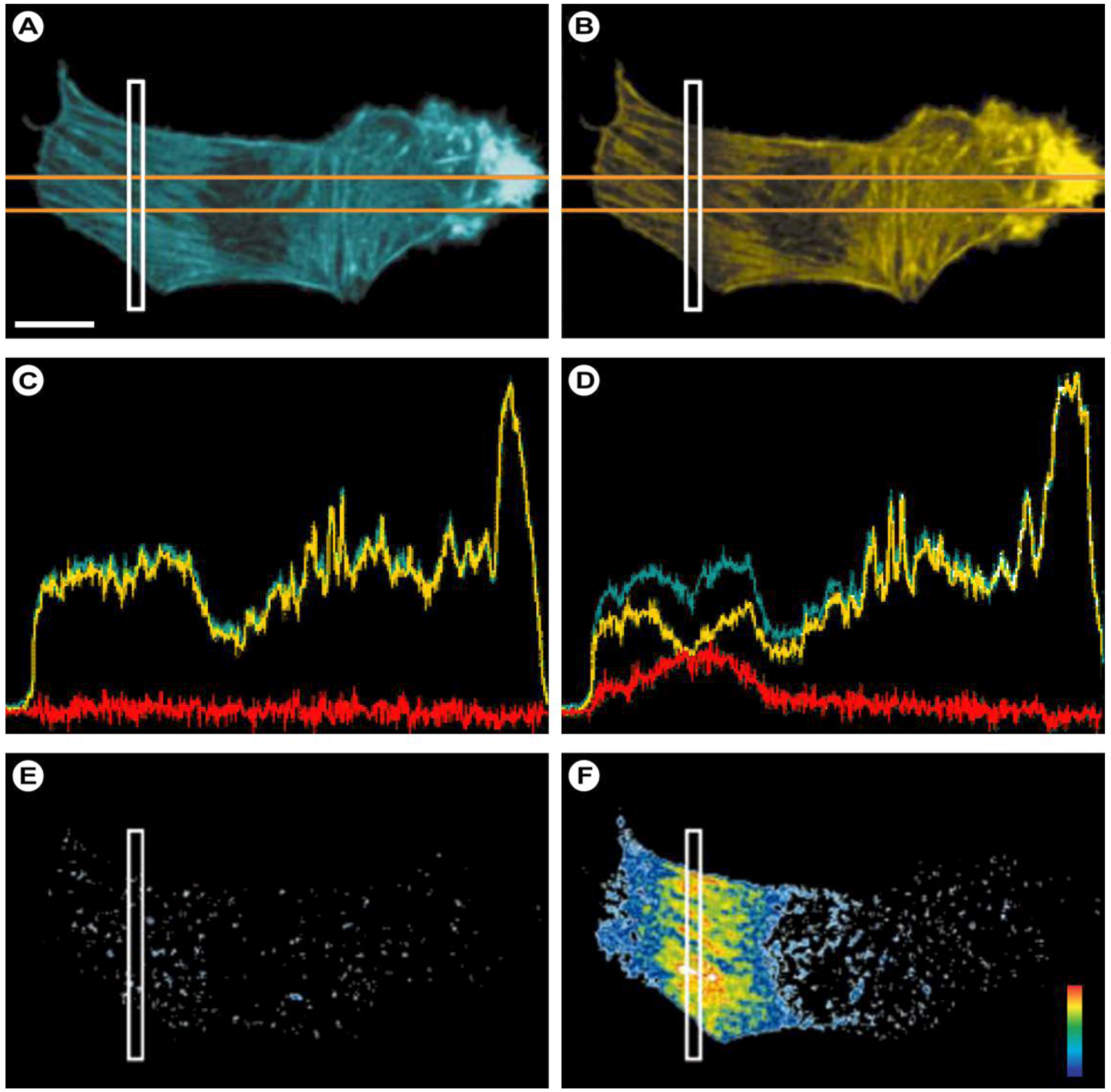
2.1.2. Practical Aspects and CLSM-Specific Considerations
 (13)
(13)2.1.3. Inverse FRAP (iFRAP) in Cell Biology

2.1.4. Summary of the Steps to Perform in FRAP Experiments
- 1) Definition of the cell region to be bleached (ROI).
- 2) Acquisition of control images to measure intensity before bleaching.
- 3) Brief illumination of the bleach region with very high laser intensity. Ideally the bleaching event should be ultra-short, followed by subsequent image acquisition without time delay.
- 4) Recording the progress of fluorescence recovery in the bleached area with high temporal resolution.
- 5) Changes in intensity in the bleached region represent the sum of all movements of the fluorescent molecules, whether passive (e.g., diffusion) or active (e.g., transport).
2.1.5. Potential Complications and Pitfalls
- 1) Living cells often move during the experiment, thus after the experiment and before the regions for analysis are defined, it is recommended to use an ‘alignment’-algorithm to compensate for these movements.
- 2) As the total amount of excitable fluorochromes present in the cell or structure under examination is reduced over time through the bleaching event, a control region must be measured and the recovery curve must be corrected for the overall loss in fluorescence.
- 3) When bleaching a region in a three-dimensional sample, fluorochromes above and below the focal plane are also bleached. The bleached volume can only be assumed to have a conical shape if microscope objectives with a low numerical aperture are used. It should be accounted for that in most cases when objectives with high numerical aperture are applied, the bleached structure is far more complex than visible in the focal plane.
- 4) In some instances the final FRAP result is determined by the size of the ROI. It is therefore important to include a control to exclude this.
- 5) If low levels of fluorochromes are present, a higher intensity is needed to obtain sufficient signal. Corrections for potentially high acquisition bleaching may result in incorrect FRAP results when an immobile fraction is present. If an immobile fraction is present, correction is difficult, because the immobile fraction contributes more to the loss in fluorescence than the mobile fraction. The immobile fraction is continuously illuminated, unlike the mobile fraction which has more freedom and diffuses freely.
- 6) When bleached and fluorescent molecules exchange with compartments distant from the bleach region, a secondary recovery will be recorded that partly overlaps the initial recovery. This leads to an apparent slowdown of the proteins’ mobility and a general underestimation of the mobility, which is especially problematic when proteins accumulate in foci, e.g., during DNA damage repair.
- 7) Fluorochrome intermittency (blinking) or reversible photobleaching may cause flawed FRAP results. This is especially a problem in FPs, since it has been shown that several of these, foremost GFP, rapidly switch between a dark non-fluorescent state and a fluorescent state [128], which causes an apparent erratic stroboscopic effect. The time that GFP spends in the dark state is independent of the laser settings, whereas the fluorescent state is distinctly dependent on the settings [129,130]. Partially the fluorescence recovery after bleaching is caused by the decreased number of fluorochromes in the dark state, since the bleach pulse is much higher in intensity than the monitoring after bleaching [122].
- 8) Because photo-induced cross-linking may occur (free radical induced cross-linking reactions), it is important to check the dependence of the recovery rate on different bleaching intensities.
- 9) Repeating FRAP on the same spot constitutes an important control to exclude differences in the FRAP result due to photo-damage. A higher recovery shows the presence of a “real” immobile fraction, whilst a similar recovery indicates photo-damage.

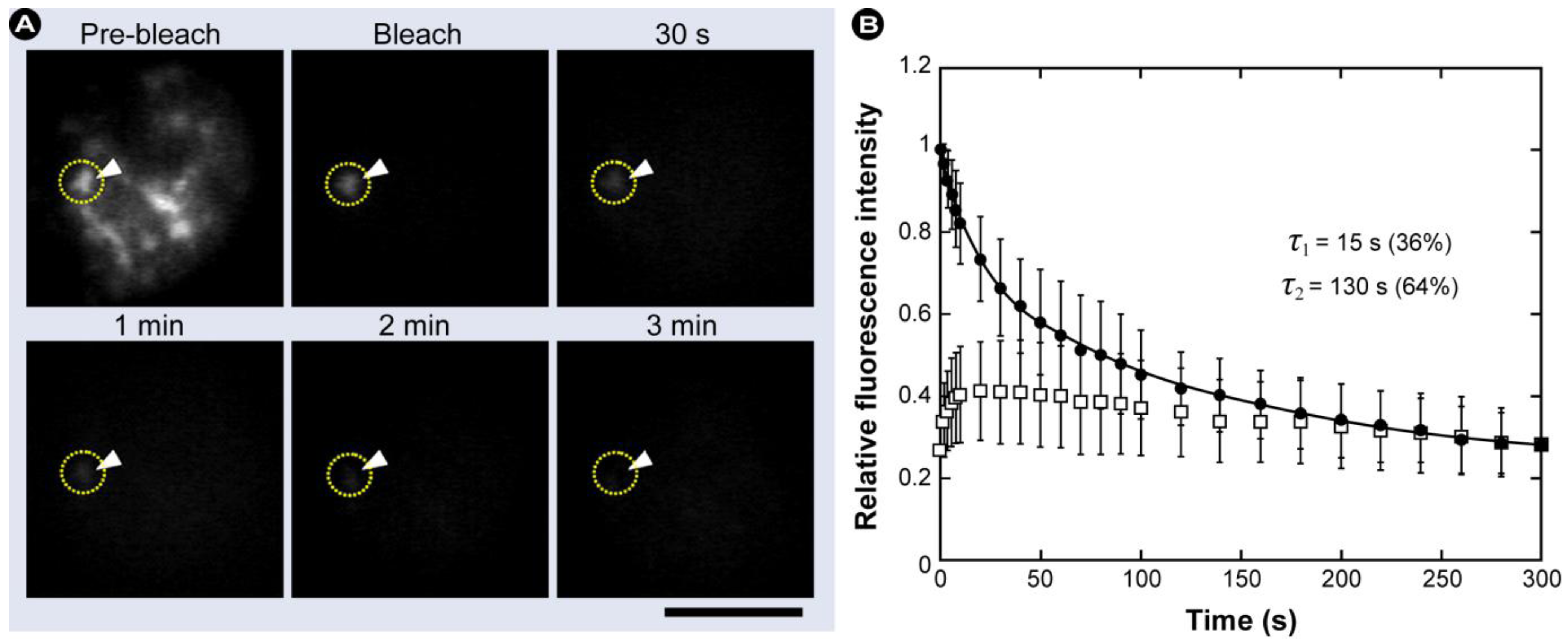
2.2. Fluorescence Loss in Photobleaching (FLIP)
2.2.1. The Basic Principles of FLIP

2.2.2. Summary of the Steps to Perform in FLIP Experiments
- 1) Definition of the cell region to be bleached (ROI).
- 2) Acquisition of control images to measure intensity before bleaching.
- 3) Brief repeated illumination of the bleach region with very high laser intensity.
- 4) Recording the progress of fluorescence decay in the adjacent non-bleached area with high temporal resolution, ideally simultaneously with bleaching.
- 5) Changes in intensity in the non-bleached region represent the sum of all movements of the fluorescent molecules, whether passive (e.g., diffusion) or active (e.g., transport).
- 6) The decay time (half-decay period) is a measure of the speed of protein movement.
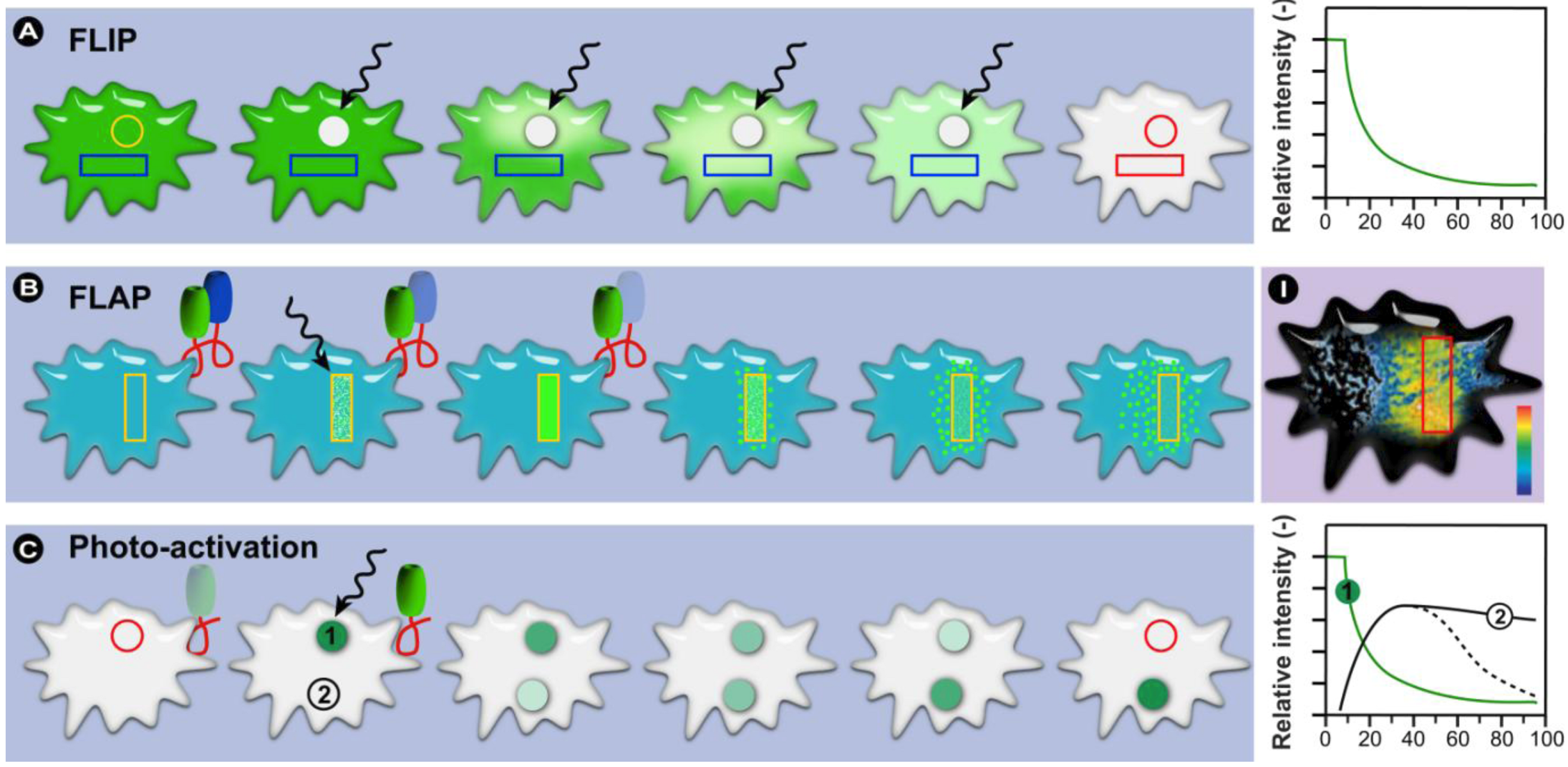
2.3. Fluorescence Localization after Photobleaching (FLAP) and Photo-Activation Methods

3. Energy Transfer Methods for Inter- and Intra-Molecular Interaction Measurements
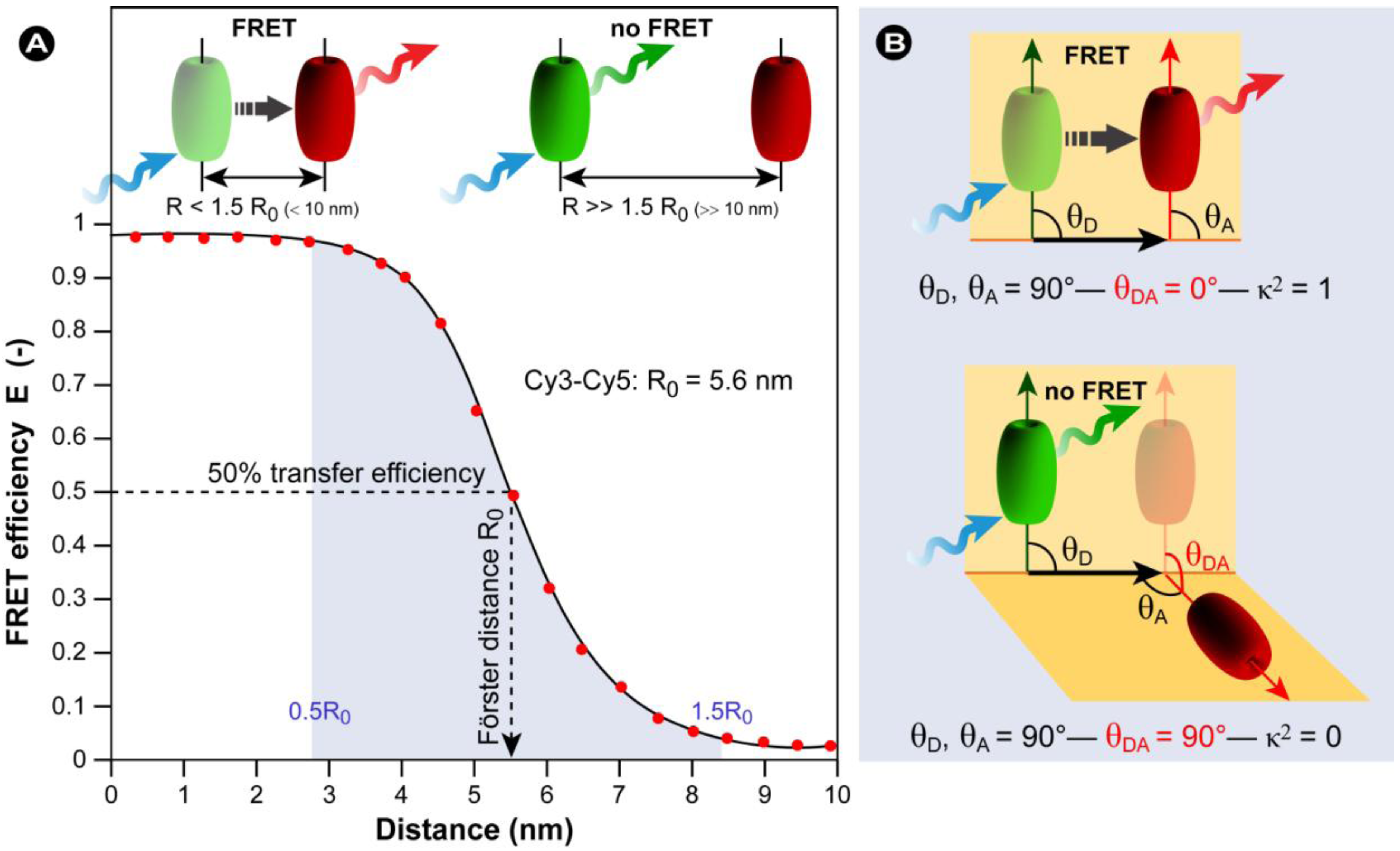
3.1. Förster Resonance Energy Transfer (FRET)

 (14)
(14) (15a)
(15a) (15b)
(15b) (nm) (16)
(nm) (16) (17)
(17) (18)
(18)
3.2. FRET Couples
 |
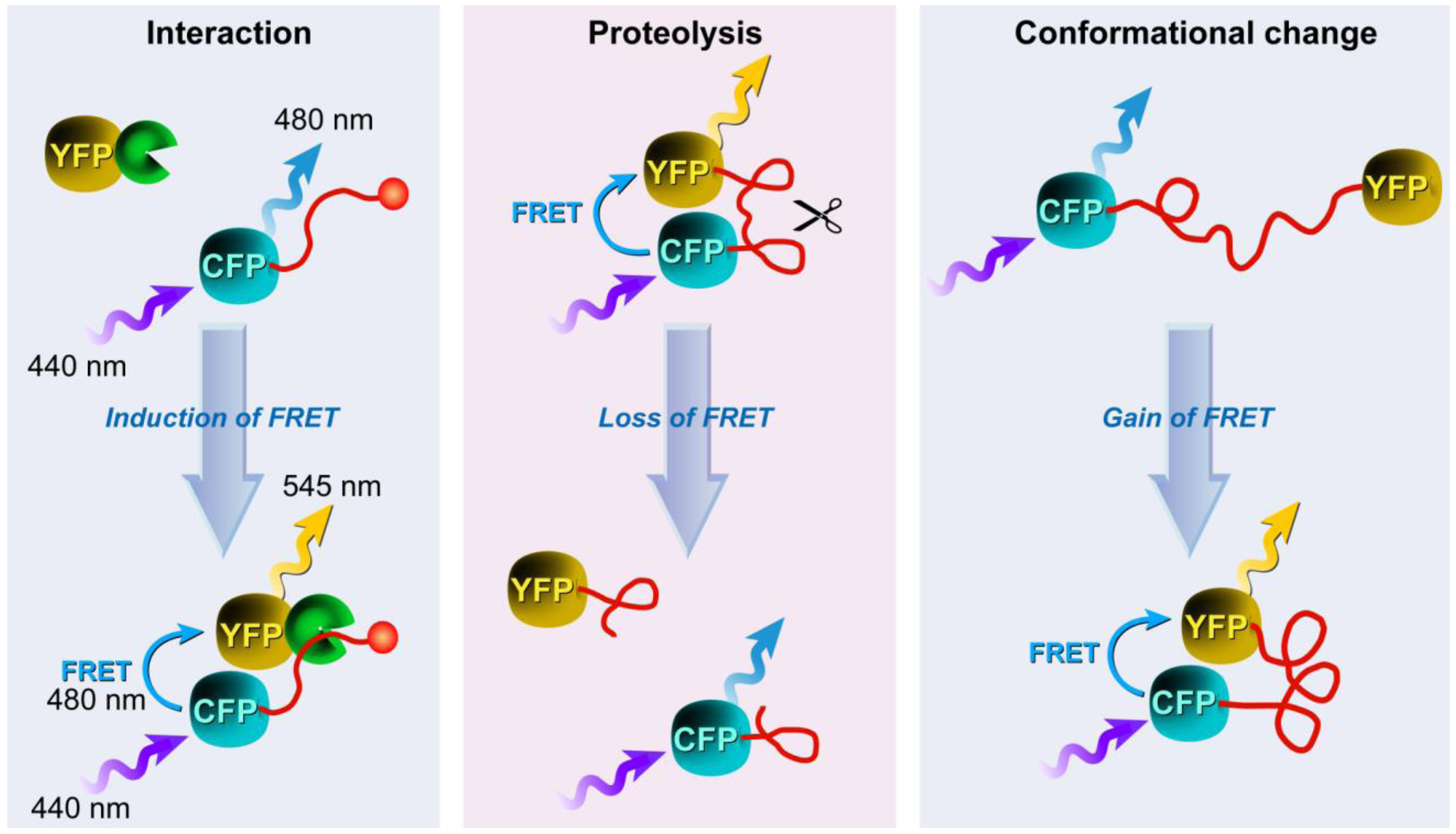
3.3. Applications of FRET in Cell Biology

3.4. Approaches to FRET Imaging
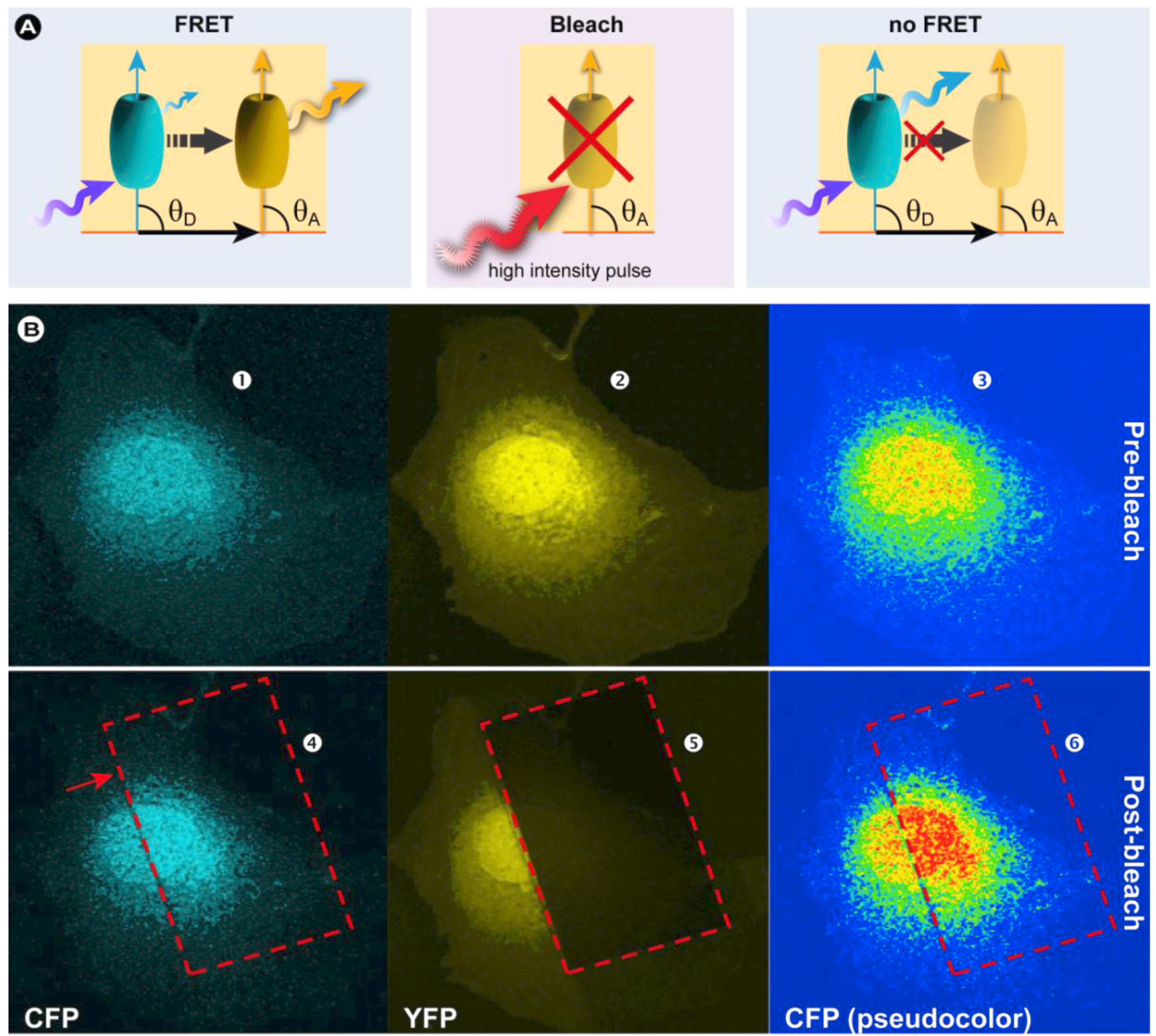
3.4.1. Donor and Acceptor Photobleaching
3.4.1.1. Basic Principles
 (19)
(19)

3.4.1.2. Summary of the Steps to Perform in Acceptor-photobleaching Experiments
- 1) Choose an appropriate FRET couple to perform the experiments.
- 2) Acquire images of the donor in the presence of the acceptor (IDA) and of the acceptor at low laser intensity (pre-bleach).
- 3) Draw a ROI within the image, corresponding to the bleaching area and the part in which the FRET efficiency will be calculated
- 4) Zoom in on the ROI and photobleach the acceptor with high laser intensity.
- 5) Zoom out to the original magnification and re-record the donor (ID) and acceptor images (post-bleach).
- 6) By utilizing an algorithm that corrects for SBT and other unwanted artifacts, the FRET signal can be consolidated. Furthermore, background subtraction, filtering, and noise reduction will improve image quality.
- 7) Use cross-correlation to align the images.
- 8) Calculate the FRET efficiency according to Equation 19
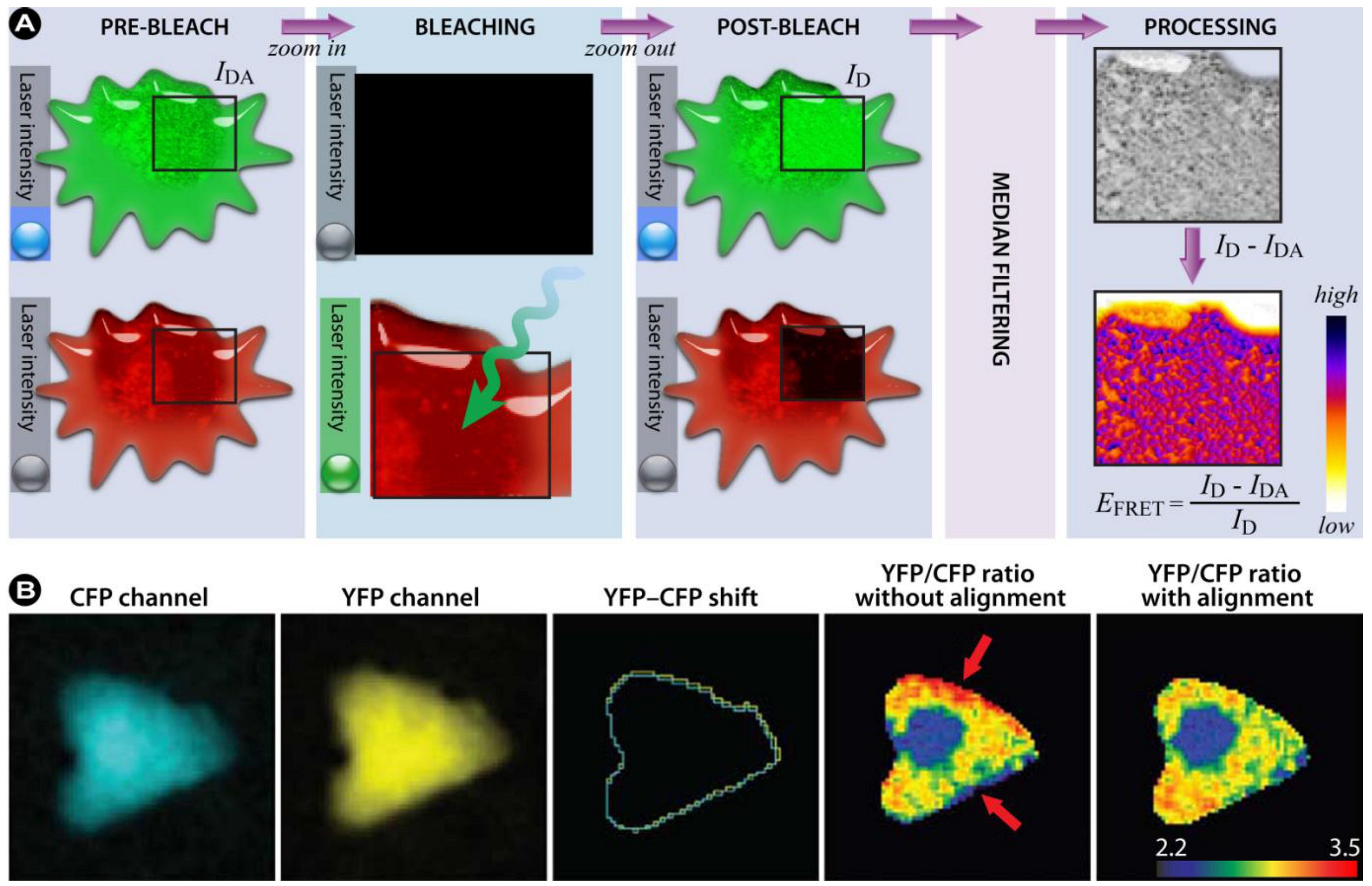
3.4.2. Sensitized Emission
 (20)
(20) (21)
(21) represents a background-subtracted donor image taken in the FRET channel;
represents a background-subtracted donor image taken in the FRET channel;  is a background-subtracted donor image taken using the donor channel. The same notation is used for the acceptor fluorochrome and the ratios in Equation 21 are used to correct the FRET channel intensities pixel-by-pixel. In this method the F c is corrected for the donor and acceptor contribution to the final signal measurement with the FRET settings, but is not normalized for donor and acceptor concentrations. The method proposed by Gordon et al. [203] extends the Youvan method and corrects for the concentration of the donor and acceptor. The Gordon FRET correction can therefore be expressed as:
is a background-subtracted donor image taken using the donor channel. The same notation is used for the acceptor fluorochrome and the ratios in Equation 21 are used to correct the FRET channel intensities pixel-by-pixel. In this method the F c is corrected for the donor and acceptor contribution to the final signal measurement with the FRET settings, but is not normalized for donor and acceptor concentrations. The method proposed by Gordon et al. [203] extends the Youvan method and corrects for the concentration of the donor and acceptor. The Gordon FRET correction can therefore be expressed as: (22)
(22) (23)
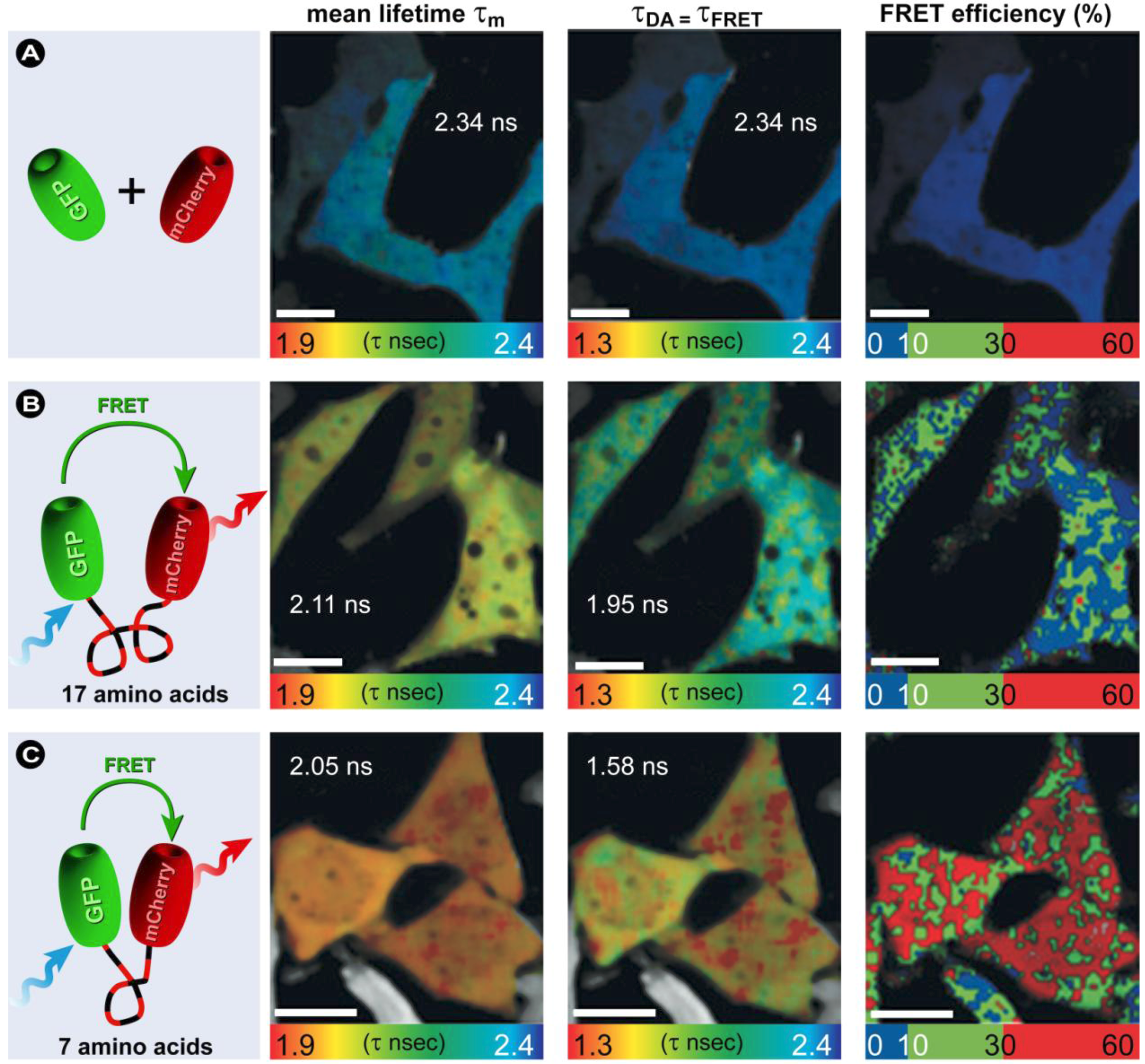
(23)3.4.3. Fluorescence Lifetime Imaging Microscopy (FLIM)
 (24)
(24)
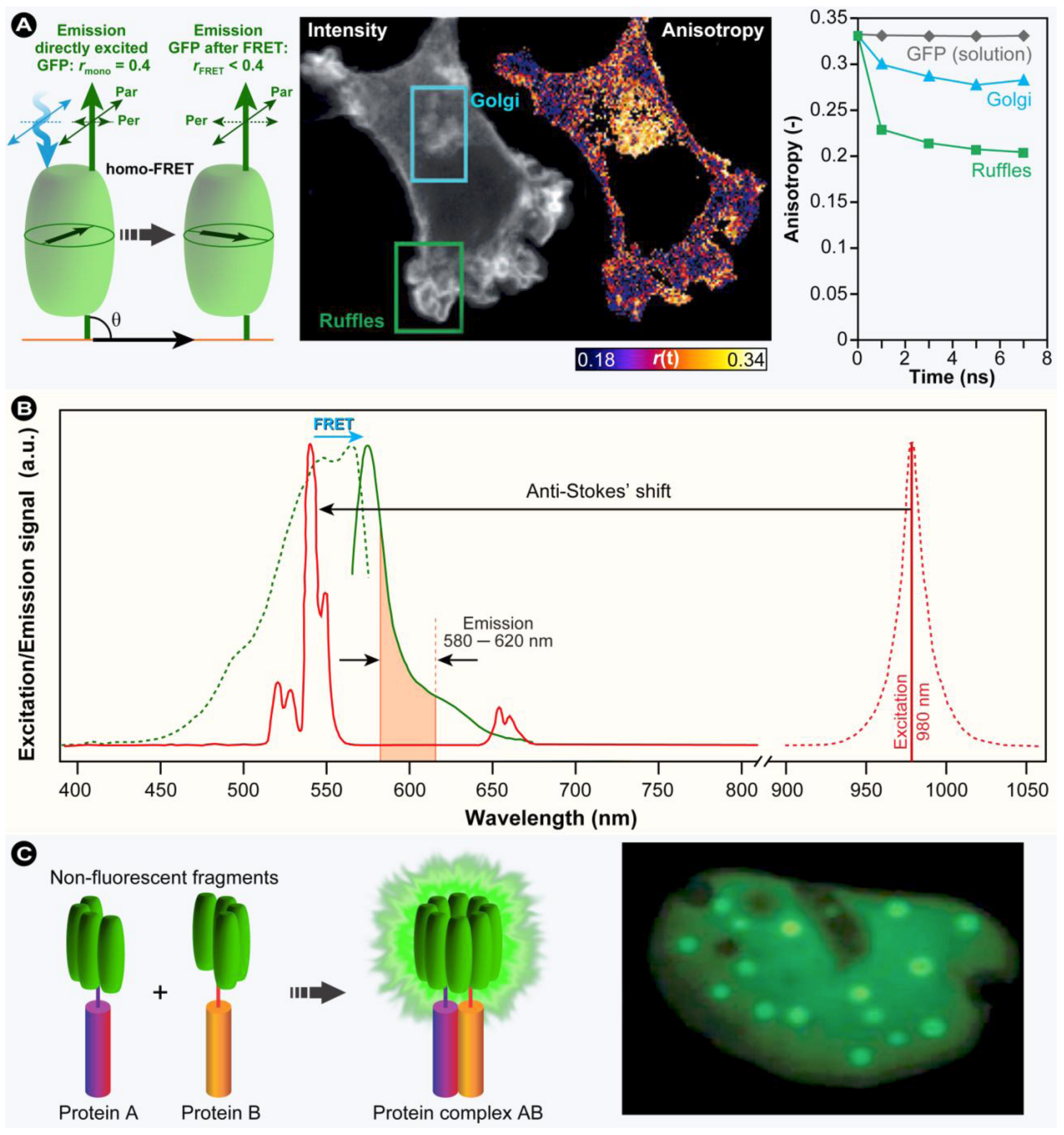
3.4.4. Polarization Anisotropy Imaging
 (25)
(25)3.5. Homo-FRET versus Hetero-FRET

3.6. Advances in Protein-Interaction Methods
3.6.1. Upconversion FRET
3.6.2. FRET Frustration
3.6.3. Photochromic FRET
3.6.4. Single-Molecule-FRET and Switchable-FRET
3.6.5. Bimolecular Fluorescence Complementation
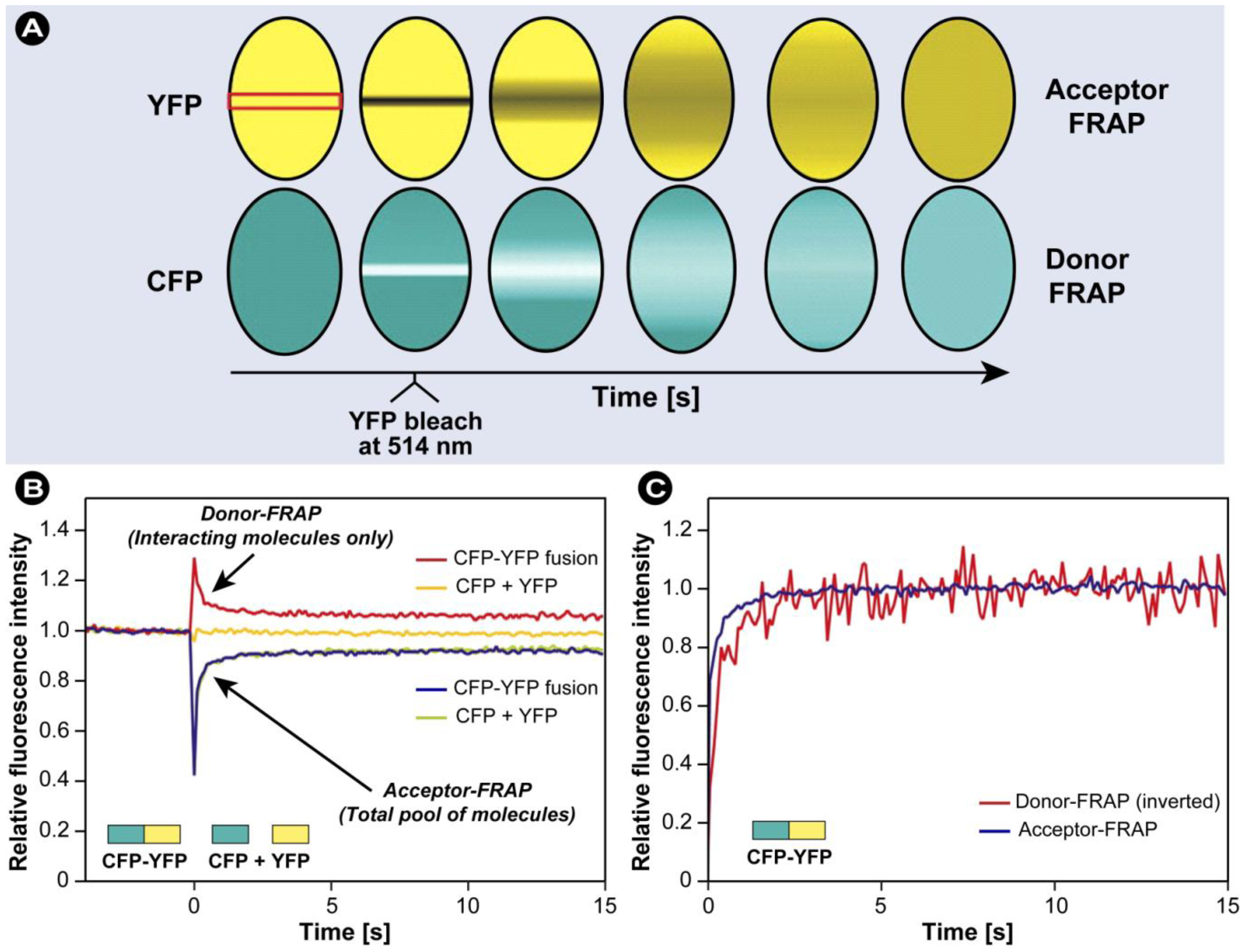
3.7. Combination of FRAP and FRET
- 1) A narrow strip across the nucleus was scanned at 458 nm excitation at 100 ms intervals and low laser power, and donor (CFP) and acceptor (YFP) signals were both acquired.
- 2) After 40 scans, specifically YFP was photobleached with a high-intensity 100 ms pulse at 514 nm.
- 3) Acquisition of the acceptor and donor signals in the bleached strip was resumed at 458 nm, but at considerably lower laser intensity.

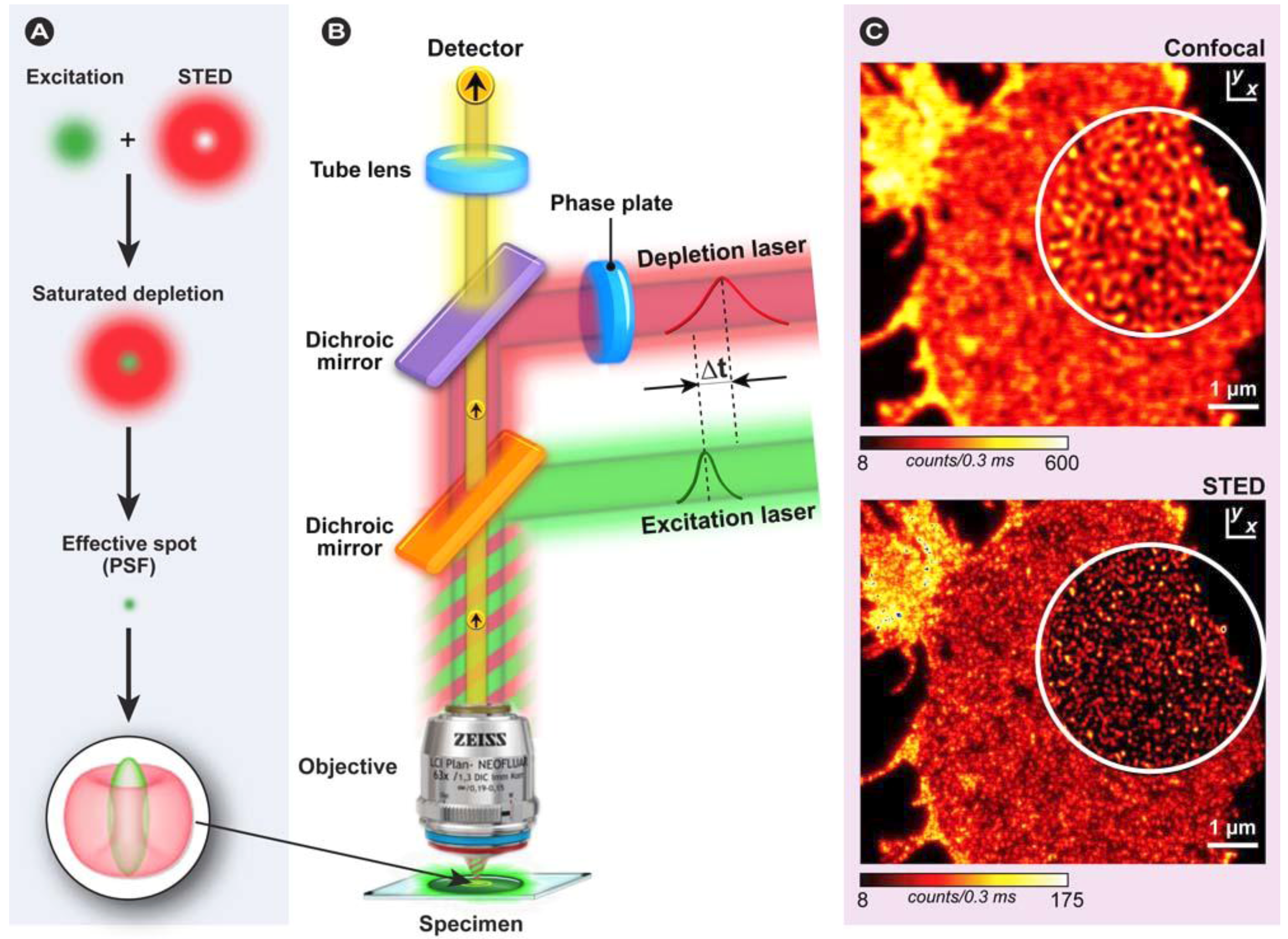
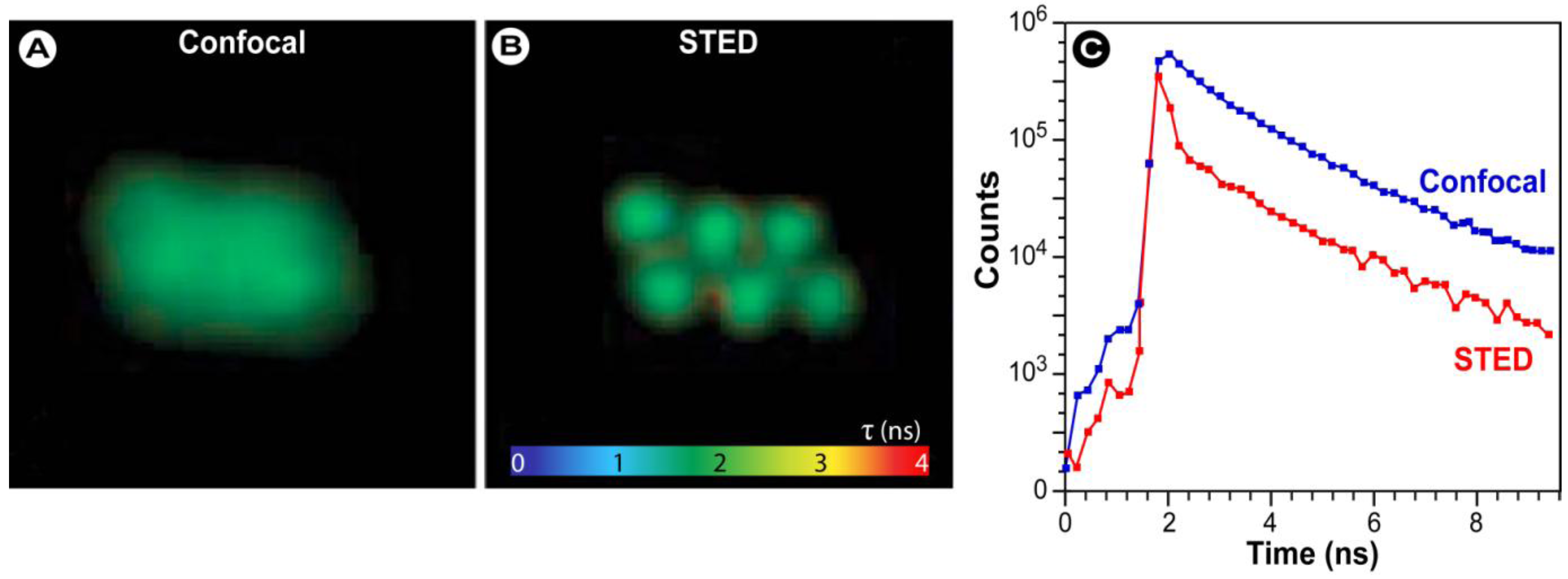
3.8. Super-Resolution and FRET Microscopy


4. Concluding Remarks
Acknowledgements
References
- Phillips, M.A.; Gran, M.L.; Peppas, N.A. Targeted nanodelivery of drugs and diagnostics. Nano Today 2010, 5, 143–159. [Google Scholar]
- Planck, M. Ueber das Gesetz der Energieverteilung im Normalspectrum (On the Law of energy distribution in the normal spectrum). Annalen der Physik 1901, 309, 553–563. [Google Scholar]
- Kasha, M. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 1950, 14–19. [Google Scholar]
- Binnemans, K. Lanthanide-based luminescent hybrid materials. Chem. Rev. 2009, 109, 4283–4374. [Google Scholar]
- Gunnlaugsson, T.; Leonard, J.P. Responsive lanthanide luminescent cyclen complexes: from switching/sensing to supramolecular architectures. Chem. Commun. 2005, 3114–3131. [Google Scholar]
- Berezin, M.Y.; Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar]
- Hemmila, I.; Laitala, V. Progress in lanthanides as luminescent probes. J. Fluoresc. 2005, 15, 529–542. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed; Springer Science: New York, NY, USA, 2006. [Google Scholar]
- Stokes, G.G. On the change of refrangibility of light. Philos. Trans. R. Soc. Lond. 1852, 142, 463–562. [Google Scholar]
- Wang, Y.L. Fluorescence Microscopy of Living Cells in Culture. Part A. Fluorescent Analogs, Labelling Cells, and Basic Microscopy. Methods in Cell Biology; Academic Press: San Diego, State abbr., USA, 1989; Volume 29. [Google Scholar]
- Lakowicz, J.R.; Szmacinski, H.; Nowaczyk, K.; Berndt, K.W.; Johnson, M. Fluorescence lifetime imaging. Anal. Biochem. 1992, 202, 316–230. [Google Scholar]
- Williams, A.T.R.; Winfield, S.A.; Miller, J.N. Relative fluorescence quantum yields using a computer controlled luminescence spectrometer. Analyst 1983, 108, 1067. [Google Scholar]
- Chatterjee, D.K.; Gnanasammandhan, M.K.; Zhang, Y. Small upconverting fluorescent nanoparticles for biomedical applications. Small 2010, 6, 2781–2795. [Google Scholar]
- Mansfield, J.R.; Gossage, K.W.; Hoyt, C.C.; Levenson, R.M. Autofluorescence removal, multiplexing, and automated analysis methods for in-vivo fluorescence imaging. J. Biomed. Opt. 2005, 10, 041207. [Google Scholar]
- Förster, T. Energiewanderung und Fluoreszenz (Energy transfer and fluorescence). Naturwissenschaften 1946, 6, 166–175. [Google Scholar]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz (Inter-molecular energy transfer and fluorescence). Annalen der Physik 1948, 2, 55–75. [Google Scholar]
- Heimstädt, O. The fluorescence microscope (Das Fluoreszenzmikroskop). Z Wiss Mikrosk 1911, 28, 330–337. [Google Scholar]
- Coons, A.H.; Creech, H.J.; Jones, R.N. Immunological properties of an antibody containing a fluorescent group. Proc. Soc. Expt. Biol. Med. 1941, 47, 200–202. [Google Scholar]
- Einstein, A. On the quantum theory of radiation. Physikalische Zeitschrift 1917, 18, 121–128. [Google Scholar]
- Schawlow, A.L.; Townes, C.H. Infrared and optical masers. Phys. Rev. 1958, 112, 1940–1949. [Google Scholar]
- Solon, L.R.; Aronson, R.; Gould, G. Physiological implications of laser beams. Science 1961, 134, 1506–1508. [Google Scholar]
- Chalfie, M. GFP: Lighting up life (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2009, 48, 5603–5611. [Google Scholar]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar]
- Shimomura, O. Discovery of green fluorescent protein (GFP) (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2009, 48, 5590–5602. [Google Scholar]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell Comp. Physiol. 1962, 59, 223–239. [Google Scholar]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar]
- Tsien, R.Y. Constructing and exploiting the fluorescent protein paintbox (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2009, 48, 5612–5626. [Google Scholar]
- Ormö, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal structure of the Aequorea victoria green fluorescent protein. Science 1996, 273, 1392–1395. [Google Scholar]
- Yang, F.; Moss, L.G.; Phillips, G.N., Jr. The molecular structure of green fluorescent protein. Nat. Biotechnol. 1996, 14, 1246–1251. [Google Scholar]
- Alivisatos, A.P. Semiconductor clusters, nanocrystals, and quantum dots. Science 1996, 271, 933–937. [Google Scholar]
- Alivisatos, A.P.; Gu, W.W.; Larabell, C. Quantum dots as cellular probes. Annu. Rev. Biomed. Eng. 2005, 7, 55–76. [Google Scholar]
- Drummen, G.P. Quantum dots-from synthesis to applications in biomedicine and life sciences. Int. J. Mol. Sci. 2010, 11, 154–163. [Google Scholar]
- Dahan, M.; Laurence, T.; Pinaud, F.; Chemla, D.S.; Alivisatos, A.P.; Sauer, M.; Weiss, S. Time-gated biological imaging by use of colloidal quantum dots. Opt. Lett. 2001, 26, 825–827. [Google Scholar]
- Mitchell, A.C.; Dad, S.; Morgan, C.G. Selective detection of luminescence from semiconductor quantum dots by nanosecond time-gated imaging with a colour-masked CCD detector. J. Microsc. 2008, 230, 172–176. [Google Scholar]
- van Driel, A.F.; Allan, G.; Delerue, C.; Lodahl, P.; Vos, W.L.; Vanmaekelbergh, D. Frequency-dependent spontaneous emission rate from CdSe and CdTe nanocrystals: influence of dark states. Phys. Rev. Lett. 2005, 95, 236804. [Google Scholar]
- Medintz, I.L.; Mattoussi, H. Quantum dot-based resonance energy transfer and its growing application in biology. Phys. Chem. Chem. Phys. 2009, 11, 17–45. [Google Scholar]
- Biju, V.; Itoh, T.; Anas, A.; Sujith, A.; Ishikawa, M. Semiconductor quantum dots and metal nanoparticles: syntheses, optical properties, and biological applications. Anal. Bioanal. Chem. 2008, 391, 2469–2495. [Google Scholar]
- Roduner, E. Size matters: Why nanomaterials are different. Chem. Soc. Rev. 2006, 35, 583–592. [Google Scholar]
- Pawley, J.B. Handbook of Biological Confocal Microscopy, 3rd ed; Springer US: New York, NY, USA, 2006. [Google Scholar]
- Swift, S.R.; Trinkle-Mulcahy, L. Basic principles of FRAP, FLIM and FRET. Proc. Royal Mic. Soc. 2004, 39, 3–10. [Google Scholar]
- Taylor, D.L.; Salmon, E.D. Basic fluorescence microscopy. Methods Cell Biol. 1989, 29, 207–237. [Google Scholar]
- Lichtman, J.W.; Conchello, J.A. Fluorescence microscopy. Nat. Methods 2005, 2, 910–919. [Google Scholar]
- Abbe, E., VII. On the Estimation of Aperture in the Microscope. J R Microsc. Soc. 1881, 1, 388–423. [Google Scholar]
- Airy, G.B. On the diffraction of an object-glass with circular aperture. Trans. Cambridge Phil. Soc. 1835, 5, 283–291. [Google Scholar]
- Diaspro, A.; Bianchini, P.; Vicidomini, G.; Faretta, M.; Ramoino, P.; Usai, C. Multi-photon excitation microscopy. BioMed. Eng. OnLine 2006, 5, 36. [Google Scholar]
- Stelzer, E.H.K. Contrast, resolution, pixelation, dynamic range and signal-to-noise ratio: Fundamental limits to resolution in fluorescence light microscopy. J. Microsc. 1998, 189, 15–24. [Google Scholar]
- Jonkman, J.E.N.; Stelzer, E.H.K. Resolution and contrast in confocal and two-photon microscopy. In Handbook of Biological Confocal Microscopy; Diaspro, A., Ed.; Wiley-Liss: New York, NY, USA, 2002; pp. 101–125. [Google Scholar]
- Abbe, E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung (Contributions to the theory of the microscope and microscopical obsercation). Archiv für Mikroskopische Anatomie 1873, 9, 413–418. [Google Scholar]
- Minsky, M. Microscopy apparatus. US Patent 3013467, 1961. [Google Scholar]
- Paddock, S.W. Principles and practices of laser scanning confocal microscopy. Mol. Biotechnol. 2000, 16, 127–149. [Google Scholar]
- Wright, S.J.; Wright, D.J. Introduction to confocal microscopy. Methods Cell Biol. 2002, 70, 1–85. [Google Scholar]
- Göppert-Mayer, M. Über Elementarakte mit zwei Quantensprüngen (on elementary acts with two quantum jumps). Annalen der Physik 1931, 401, 273–294. [Google Scholar]
- Kaiser, W.; Garret, C.G. Two-photon excitation in CaF2 : Eu2+. Phys. Rev. Lett. 1961, 7, 229–231. [Google Scholar]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73–76. [Google Scholar]
- Konig, K. Multiphoton microscopy in life sciences. J. Microsc. 2000, 200, 83–104. [Google Scholar]
- Durr, N.J.; Weisspfennig, C.T.; Holfeld, B.A.; Ben-Yakar, A. Maximum imaging depth of two-photon autofluorescence microscopy in epithelial tissues. J. Biomed. Opt. 2011, 16, 026008. [Google Scholar]
- Beaurepaire, E.; Oheim, M.; Mertz, J. Ultra-deep two-photon fluorescence excitation in turbid media. Opt. Commun. 2001, 188, 25–29. [Google Scholar]
- Theer, P.; Hasan, M.T. Denk, WTwo-photon imaging to a depth of 1000 µm in living brains by use of a Ti:Al2O3 regenerative amplifier. Opt. Lett. 2003, 28, 1022–1024. [Google Scholar] [CrossRef]
- Zipfel, W.R.; Williams, R.M.; Webb, W.W. Nonlinear magic: Multiphoton microscopy in the biosciences. Nat. Biotechnol. 2003, 21, 1369–1377. [Google Scholar]
- Campagnola, P. Second harmonic generation imaging microscopy: Applications to diseases diagnostics. Anal. Chem. 2011, 83, 3224–3231. [Google Scholar]
- Campagnola, P.J.; Loew, L.M. Second-harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues and organisms. Nat. Biotechnol. 2003, 21, 1356–1360. [Google Scholar]
- Auzel, F. Upconversion and anti-Stokes processes with f and d ions in solids. Chem. Rev. 2004, 104, 139–173. [Google Scholar]
- Gamelin, D.; Gudel, H. Upconversion processes in transition metal and rare earth metal systems. In Transition Metal and Rare Earth Compounds: Topics in Current Chemistry; Yersin, H., Ed.; Springer: Heidelberg, Germany, 2001; Volume 214, pp. 1–56. [Google Scholar]
- Suyver, J.F.; Aebischer, A.; Biner, D.; Gerner, P.; Grimm, J.; Heer, S.; Krämer, K.W.; Reinhard, C.; Güdel, H.U. Novel materials doped with trivalent lanthanides and transition metal ions showing near-infrared to visible photon upconversion. Opt. Mater. 2005, 27, 1111–1130. [Google Scholar]
- Auzel, F.E. Up-Conversion in Rare-Earth-Doped Systems: Past, Present and Future; SPIE: Kazan, Russia, 2002; p. 179. [Google Scholar]
- Soukka, T.; Rantanen, T.; Kuningas, K. Photon upconversion in homogeneous fluorescence-based bioanalytical assays. Ann. NY Acad. Sci. 2008, 1130, 188–200. [Google Scholar]
- Drobizhev, M.; Makarov, N.S.; Tillo, S.E.; Hughes, T.E.; Rebane, A. Two-photon absorption properties of fluorescent proteins. Nat. Methods 2011, 8, 393–399. [Google Scholar]
- Benninger, R.K.; Hao, M.; Piston, D.W. Multi-photon excitation imaging of dynamic processes in living cells and tissues. Rev. Physiol. Biochem. Pharmacol. 2008, 160, 71–92. [Google Scholar]
- Diaspro, A.; Chirico, G.; Collini, M. Two-photon fluorescence excitation and related techniques in biological microscopy. Q Rev. Biophys. 2005, 38, 97–166. [Google Scholar]
- Dunn, K.W.; Young, P.A. Principles of multiphoton microscopy. Nephron. Exp. Nephrol. 2006, 103, e33–e40. [Google Scholar]
- Piston, D.W. Imaging living cells and tissues by two-photon excitation microscopy. Trends Cell Biol. 1999, 9, 66–69. [Google Scholar]
- Axelrod, D.; Koppel, D.E.; Schlessinger, J.; Elson, E.; Webb, W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976, 16, 1055–1069. [Google Scholar]
- Koppel, D.E.; Axelrod, D.; Schlessinger, J.; Elson, E.L.; Webb, W.W. Dynamics of fluorescence marker concentration as a probe of mobility. Biophys. J. 1976, 16, 1315–1329. [Google Scholar]
- Liebman, P.A.; Entine, G. Lateral diffusion of visual pigment in photorecptor disk membranes. Science 1974, 185, 457–459. [Google Scholar]
- Poo, M.; Cone, R.A. Lateral diffusion of rhodopsin in the photoreceptor membrane. Nature 1974, 247, 438–441. [Google Scholar]
- Jacobson, K.; Ishihara, A.; Inman, R. Lateral diffusion of proteins in membranes. Annu. Rev. Physiol. 1987, 49, 163–175. [Google Scholar]
- Axelrod, D. Lateral motion of membrane proteins and biological function. J. Membr. Biol. 1983, 75, 1–10. [Google Scholar]
- Peters, R.; Beck, K. Translational diffusion in phospholipid monolayers measured by fluorescence microphotolysis. Proc. Natl. Acad. Sci. USA 1983, 80, 7183–7187. [Google Scholar]
- Jovin, T.M.; Vaz, W.L. Rotational and translational diffusion in membranes measured by fluorescence and phosphorescence methods. Methods Enzymol 1989, 172, 471–513. [Google Scholar]
- Tocanne, J.F.; Dupou-Cezanne, L.; Lopez, A.; Tournier, J.F. Lipid lateral diffusion and membrane organization. FEBS Lett. 1989, 257, 10–16. [Google Scholar]
- Reits, E.A.; Neefjes, J.J. From fixed to FRAP: Measuring protein mobility and activity in living cells. Nat. Cell Biol. 2001, 3, E145–E147. [Google Scholar]
- Carrero, G.; McDonald, D.; Crawford, E.; de Vries, G.; Hendzel, M.J. Using FRAP and mathematical modeling to determine the in vivo kinetics of nuclear proteins. Methods 2003, 29, 14–28. [Google Scholar]
- Kimura, H.; Hieda, M.; Cook, P.R. Measuring histone and polymerase dynamics in living cells. Methods Enzymol 2004, 375, 381–393. [Google Scholar]
- Houtsmuller, A.B.; Vermeulen, W. Macromolecular dynamics in living cell nuclei revealed by fluorescence redistribution after photobleaching. Histochem. Cell Biol. 2001, 115, 13–21. [Google Scholar]
- White, J.; Stelzer, E. Photobleaching GFP reveals protein dynamics inside live cells. Trends Cell Biol. 1999, 9, 61–65. [Google Scholar]
- Shaw, S.L.; Kamyar, R.; Ehrhardt, D.W. Sustained microtubule treadmilling in Arabidopsis cortical arrays. Science 2003, 300, 1715–1718. [Google Scholar]
- Kapustina, M.; Vitriol, E.; Elston, T.C.; Loew, L.M.; Jacobson, K. Modeling capping protein FRAP and CALI experiments reveals in vivo regulation of actin dynamics. Cytoskeleton (Hoboken) 2010, 67, 519–534. [Google Scholar]
- Goodson, H.V.; Dzurisin, J.S.; Wadsworth, P. Methods for expressing and analyzing GFP-tubulin and GFP-microtubule-associated proteins. Cold Spring Harb. Protoc. 2010, 2010, pdb top85. [Google Scholar]
- Smith, A.J.; Pfeiffer, J.R.; Zhang, J.; Martinez, A.M.; Griffiths, G.M.; Wilson, B.S. Microtubule-dependent transport of secretory vesicles in RBL-2H3 cells. Traffic 2003, 4, 302–312. [Google Scholar]
- Tagawa, A.; Mezzacasa, A.; Hayer, A.; Longatti, A.; Pelkmans, L.; Helenius, A. Assembly and trafficking of caveolar domains in the cell: Caveolae as stable, cargo-triggered, vesicular transporters. J. Cell Biol. 2005, 170, 769–779. [Google Scholar]
- Seabrooke, S.; Qiu, X.; Stewart, B.A. Nonmuscle myosin II helps regulate synaptic vesicle mobility at the Drosophila neuromuscular junction. BMC Neurosci. 2010, 11, 37. [Google Scholar]
- Monetta, P.; Slavin, I.; Romero, N.; Alvarez, C. Rab1b interacts with GBF1 and modulates both ARF1 dynamics and COPI association. Mol Biol Cell 2007, 18, 2400–2410. [Google Scholar]
- von Wichert, G.; Haimovich, B.; Feng, G.S.; Sheetz, M.P. Force-dependent integrin-cytoskeleton linkage formation requires downregulation of focal complex dynamics by Shp2. EMBO J. 2003, 22, 5023–5035. [Google Scholar]
- Zhang, X.; Tee, Y.H.; Heng, J.K.; Zhu, Y.; Hu, X.; Margadant, F.; Ballestrem, C.; Bershadsky, A.; Griffiths, G.; Yu, H. Kinectin-mediated endoplasmic reticulum dynamics supports focal adhesion growth in the cellular lamella. J. Cell Sci. 2010, 123, 3901–3912. [Google Scholar]
- Howell, B.J.; Moree, B.; Farrar, E.M.; Stewart, S.; Fang, G.; Salmon, E.D. Spindle checkpoint protein dynamics at kinetochores in living cells. Curr. Biol. 2004, 14, 953–964. [Google Scholar]
- Pockwinse, S.M.; Kota, K.P.; Quaresma, A.J.; Imbalzano, A.N.; Lian, J.B.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Nickerson, J.A. Live cell imaging of the cancer-related transcription factor RUNX2 during mitotic progression. J. Cell Physiol. 2010, 226, 1383–1389. [Google Scholar]
- Tripathi, K.; Parnaik, V.K. Differential dynamics of splicing factor SC35 during the cell cycle. J. Biosci. 2008, 33, 345–354. [Google Scholar]
- Dundr, M.; Hoffmann-Rohrer, U.; Hu, Q.; Grummt, I.; Rothblum, L.I.; Phair, R.D.; Misteli, T. A kinetic framework for a mammalian RNA polymerase in vivo. Science 2002, 298, 1623–1626. [Google Scholar]
- Stasevich, T.J.; Mueller, F.; Brown, D.T.; McNally, J.G. Dissecting the binding mechanism of the linker histone in live cells: an integrated FRAP analysis. EMBO J. 2010, 29, 1225–1234. [Google Scholar]
- Festenstein, R.; Pagakis, S.N.; Hiragami, K.; Lyon, D.; Verreault, A.; Sekkali, B.; Kioussis, D. Modulation of heterochromatin protein 1 dynamics in primary Mammalian cells. Science 2003, 299, 719–721. [Google Scholar]
- Hitakomate, E.; Hood, F.E.; Sanderson, H.S.; Clarke, P.R. The methylated N-terminal tail of RCC1 is required for stabilisation of its interaction with chromatin by Ran in live cells. BMC Cell Biol. 2010, 11, 43. [Google Scholar]
- Mueller, F.; Mazza, D.; Stasevich, T.J.; McNally, J.G. FRAP and kinetic modeling in the analysis of nuclear protein dynamics: What do we really know? Curr. Opin. Cell Biol. 2010, 22, 403–411. [Google Scholar] [CrossRef]
- Shav-Tal, Y.; Darzacq, X.; Shenoy, S.M.; Fusco, D.; Janicki, S.M.; Spector, D.L.; Singer, R.H. Dynamics of single mRNPs in nuclei of living cells. Science 2004, 304, 1797–1800. [Google Scholar]
- Kota, K.P.; Wagner, S.R.; Huerta, E.; Underwood, J.M.; Nickerson, J.A. Binding of ATP to UAP56 is necessary for mRNA export. J. Cell Sci. 2008, 121, 1526–1537. [Google Scholar]
- van Royen, M.E.; Zotter, A.; Ibrahim, S.M.; Geverts, B.; Houtsmuller, A.B. Nuclear proteins: finding and binding target sites in chromatin. Chromosome Res 2010, 19, 83–98. [Google Scholar]
- Elsner, M.; Hashimoto, H.; Simpson, J.C.; Cassel, D.; Nilsson, T.; Weiss, M. Spatiotemporal dynamics of the COPI vesicle machinery. EMBO Rep. 2003, 4, 1000–1004. [Google Scholar]
- Balse, E.; El-Haou, S.; Dillanian, G.; Dauphin, A.; Eldstrom, J.; Fedida, D.; Coulombe, A.; Hatem, S.N. Cholesterol modulates the recruitment of Kv1.5 channels from Rab11-associated recycling endosome in native atrial myocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 14681–14686. [Google Scholar]
- Kenworthy, A.K. Fluorescence-based methods to image palmitoylated proteins. Methods 2006, 40, 198–205. [Google Scholar]
- Giese, B.; Au-Yeung, C.K.; Herrmann, A.; Diefenbach, S.; Haan, C.; Kuster, A.; Wortmann, S.B.; Roderburg, C.; Heinrich, P.C.; Behrmann, I.; Muller-Newen, G. Long term association of the cytokine receptor gp130 and the Janus kinase Jak1 revealed by FRAP analysis. J. Biol. Chem. 2003, 278, 39205–39213. [Google Scholar]
- Rotblat, B.; Belanis, L.; Liang, H.; Haklai, R.; Elad-Zefadia, G.; Hancock, J.F.; Kloog, Y.; Plowman, S.J. H-Ras nanocluster stability regulates the magnitude of MAPK signal output. PLoS One 2010, 5, e11991. [Google Scholar]
- Kambara, T.; Komaba, S.; Ikebe, M. Human myosin III is a motor having an extremely high affinity for actin. J. Biol. Chem. 2006, 281, 37291–37301. [Google Scholar]
- Beaudouin, J.; Mora-Bermudez, F.; Klee, T.; Daigle, N.; Ellenberg, J. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys. J. 2006, 90, 1878–1894. [Google Scholar]
- Ellenberg, J.; Siggia, E.D.; Moreira, J.E.; Smith, C.L.; Presley, J.F.; Worman, H.J.; Lippincott-Schwartz, J. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 1997, 138, 1193–1206. [Google Scholar]
- Mueller, F.; Wach, P.; McNally, J.G. Evidence for a common mode of transcription factor interaction with chromatin as revealed by improved quantitative fluorescence recovery after photobleaching. Biophys. J. 2008, 94, 3323–3339. [Google Scholar]
- Phair, R.D.; Gorski, S.A.; Misteli, T. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol. 2004, 375, 393–414. [Google Scholar]
- Sbalzarini, I.F.; Mezzacasa, A.; Helenius, A.; Koumoutsakos, P. Effects of organelle shape on fluorescence recovery after photobleaching. Biophys. J. 2005, 89, 1482–1492. [Google Scholar]
- Soumpasis, D.M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys. J. 1983, 41, 95–97. [Google Scholar]
- Sprague, B.L.; McNally, J.G. FRAP analysis of binding: proper and fitting. Trends Cell Biol. 2005, 15, 84–91. [Google Scholar]
- Sprague, B.L.; Pego, R.L.; Stavreva, D.A.; McNally, J.G. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 2004, 86, 3473–3495. [Google Scholar]
- Dundr, M.; Misteli, T.; Olson, M.O. The dynamics of postmitotic reassembly of the nucleolus. J. Cell Biol. 2000, 150, 433–446. [Google Scholar]
- Zimmer, M. Green fluorescent protein (GFP): Applications, structure, and related photophysical behavior. Chem. Rev. 2002, 102, 759–781. [Google Scholar]
- Houtsmuller, A.B. Fluorescence recovery after photobleaching: Application to nuclear proteins. Adv. Biochem. Eng. Biotechnol. 2005, 95, 177–199. [Google Scholar]
- Swaminathan, R.; Hoang, C.P.; Verkman, A.S. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: Cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J. 1997, 72, 1900–1907. [Google Scholar]
- Patterson, G.H.; Knobel, S.M.; Sharif, W.D.; Kain, S.R.; Piston, D.W. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 1997, 73, 2782–2790. [Google Scholar]
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 2002, 297, 1873–1877. [Google Scholar]
- Klonis, N.; Rug, M.; Harper, I.; Wickham, M.; Cowman, A.; Tilley, L. Fluorescence photobleaching analysis for the study of cellular dynamics. Eur. Biophys. J. 2002, 31, 36–51. [Google Scholar]
- Ishihama, Y.; Tadakuma, H.; Tani, T.; Funatsu, T. The dynamics of pre-mRNAs and poly(A)+ RNA at speckles in living cells revealed by iFRAP studies. Exp. Cell Res. 2008, 314, 748–762. [Google Scholar]
- Dickson, R.M.; Cubitt, A.B.; Tsien, R.Y.; Moerner, W.E. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 1997, 388, 355–358. [Google Scholar]
- Garcia-Parajo, M.F.; Segers-Nolten, G.M.; Veerman, J.A.; Greve, J.; van Hulst, N.F. Real-time light-driven dynamics of the fluorescence emission in single green fluorescent protein molecules. Proc. Natl. Acad. Sci. USA 2000, 97, 7237–7242. [Google Scholar]
- Moerner, W.E.; Peterman, E.J.; Brasselet, S.; Kummer, S.; Dickson, R.M. Optical methods for exploring dynamics of single copies of green fluorescent protein. Cytometry 1999, 36, 232–238. [Google Scholar]
- Cole, N.B.; Smith, C.L.; Sciaky, N.; Terasaki, M.; Edidin, M.; Lippincott-Schwartz, J. Diffusional mobility of Golgi proteins in membranes of living cells. Science 1996, 273, 797–801. [Google Scholar]
- Chen, D.; Huang, S. Nucleolar components involved in ribosome biogenesis cycle between the nucleolus and nucleoplasm in interphase cells. J. Cell Biol. 2001, 153, 169–176. [Google Scholar]
- Nehls, S.; Snapp, E.L.; Cole, N.B.; Zaal, K.J.; Kenworthy, A.K.; Roberts, T.H.; Ellenberg, J.; Presley, J.F.; Siggia, E.; Lippincott-Schwartz, J. Dynamics and retention of misfolded proteins in native ER membranes. Nat. Cell Biol. 2000, 2, 288–295. [Google Scholar]
- Phair, R.D.; Misteli, T. High mobility of proteins in the mammalian cell nucleus. Nature 2000, 404, 604–609. [Google Scholar]
- Koster, M.; Frahm, T.; Hauser, H. Nucleocytoplasmic shuttling revealed by FRAP and FLIP technologies. Curr. Opin. Biotechnol. 2005, 16, 28–34. [Google Scholar]
- Politz, J.C. Use of caged fluorochromes to track macromolecular movement in living cells. Trends Cell Biol. 1999, 9, 284–287. [Google Scholar]
- Puliti, D.; Warther, D.; Orange, C.; Specht, A.; Goeldner, M. Small photoactivatable molecules for controlled fluorescence activation in living cells. Bioorg. Med. Chem. 2011, 19, 1023–1029. [Google Scholar]
- Lukyanov, K.A.; Chudakov, D.M.; Lukyanov, S.; Verkhusha, V.V. Innovation: Photoactivatable fluorescent proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 885–891. [Google Scholar]
- Ehrlicher, A.J.; Nakamura, F.; Hartwig, J.H.; Weitz, D.A.; Stossel, T.P. Mechanical strain in actin networks regulates FilGAP and integrin binding to filamin A. Nature 2011, 478, 260–263. [Google Scholar]
- Dunn, G.A.; Dobbie, I.M.; Monypenny, J.; Holt, M.R.; Zicha, D. Fluorescence localization after photobleaching (FLAP): a new method for studying protein dynamics in living cells. J. Microsc. 2002, 205, 109–112. [Google Scholar]
- Dunn, G.A.; Holt, M.R.; Soong, D.Y.; Gray, C.; Zicha, D. Fluorescence localization after photobleaching (FLAP). Curr. Protoc. Cell Biol. 2004, Chapter 21, Unit 21 2. [Google Scholar]
- Gerlich, D.; Beaudouin, J.; Kalbfuss, B.; Daigle, N.; Eils, R.; Ellenberg, J. Global chromosome positions are transmitted through mitosis in mammalian cells. Cell 2003, 112, 751–764. [Google Scholar]
- Ando, R.; Hama, H.; Yamamoto-Hino, M.; Mizuno, H.; Miyawaki, A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 12651–12656. [Google Scholar]
- Gurskaya, N.G.; Verkhusha, V.V.; Shcheglov, A.S.; Staroverov, D.B.; Chepurnykh, T.V.; Fradkov, A.F.; Lukyanov, S.; Lukyanov, K.A. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol. 2006, 24, 461–465. [Google Scholar]
- Henderson, J.N.; Remington, S.J. The kindling fluorescent protein: A transient photoswitchable marker. Physiology (Bethesda) 2006, 21, 162–170. [Google Scholar] [CrossRef]
- Tian, Z.; Wu, W.; Li, A.D. Photoswitchable fluorescent nanoparticles: Preparation, properties and applications. Chemphyschem 2009, 10, 2577–2591. [Google Scholar]
- Dedecker, P.; Hotta, J.-I.; Flors, C.; Sliwa, M.; Uji-I, H.; Roeffaers, M.B.J.; Ando, R.; Mizuno, H.; Miyawaki, A.; Hofkens, J. Subdiffraction imaging through the selective donut-mode depletion of thermally stable photoswitchable fluorophores: Numerical analysis and application to the fluorescent protein Dronpa. J. Am. Chem. Soc. 2007, 129, 16132–16141. [Google Scholar]
- Hofmann, M.; Eggeling, C.; Jakobs, S.; Hell, S.W. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 17565–17569. [Google Scholar]
- Grotjohann, T.; Testa, I.; Leutenegger, M.; Bock, H.; Urban, N.T.; Lavoie-Cardinal, F.; Willig, K.I.; Eggeling, C.; Jakobs, S.; Hell, S.W. Diffraction-unlimited all-optical imaging and writing with a photochromic GFP. Nature 2011, 478, 204–208. [Google Scholar]
- Brakemann, T.; Stiel, A.C.; Weber, G.; Andresen, M.; Testa, I.; Grotjohann, T.; Leutenegger, M.; Plessmann, U.; Urlaub, H.; Eggeling, C.; Wahl, M.C.; Hell, S.W.; Jakobs, S. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nat. Biotech. 2011, 29, 942–947. [Google Scholar]
- Stryer, L. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 1978, 47, 819–846. [Google Scholar]
- Stryer, L.; Haugland, R.P. Energy transfer: a spectroscopic ruler. Proc. Natl. Acad. Sci. USA 1967, 58, 719–726. [Google Scholar]
- Haugland, R.P.; Yguerabide, J.; Stryer, L. Dependence of the kinetics of singlet-singlet energy transfer on spectral overlap. Proc. Natl. Acad. Sci. USA 1969, 63, 23–30. [Google Scholar]
- Elangovan, M.; Wallrabe, H.; Chen, Y.; Day, R.N.; Barroso, M.; Periasamy, A. Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy. Methods 2003, 29, 58–73. [Google Scholar]
- Gu, Y.; Di, W.L.; Kelsell, D.P.; Zicha, D. Quantitative fluorescence resonance energy transfer (FRET) measurement with acceptor photobleaching and spectral unmixing. J. Microsc. 2004, 215, 162–173. [Google Scholar]
- Mills, J.D.; Stone, J.R.; Rubin, D.G.; Melon, D.E.; Okonkwo, D.O.; Periasamy, A.; Helm, G.A. Illuminating protein interactions in tissue using confocal and two-photon excitation fluorescent resonance energy transfer microscopy. J. Biomed. Opt. 2003, 8, 347–356. [Google Scholar]
- Thaler, C.; Vogel, S.S. Quantitative linear unmixing of CFP and YFP from spectral images acquired with two-photon excitation. Cytometry A 2006, 69, 904–911. [Google Scholar]
- Wallrabe, H.; Chen, Y.; Periasamy, A.; Barroso, M. Issues in confocal microscopy for quantitative FRET analysis. Microsc. Res. Tech. 2006, 69, 196–206. [Google Scholar]
- Feige, J.N.; Sage, D.; Wahli, W.; Desvergne, B.; Gelman, L. PixFRET, an ImageJ plug-in for FRET calculation that can accommodate variations in spectral bleed-throughs. Microsc. Res. Tech. 2005, 68, 51–58. [Google Scholar]
- Bates, M.; Huang, B.; Dempsey, G.T.; Zhuang, X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science 2007, 317, 1749–1753. [Google Scholar]
- Shroff, H.; Galbraith, C.G.; Galbraith, J.A.; White, H.; Gillette, J.; Olenych, S.; Davidson, M.W.; Betzig, E. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc. Natl. Acad. Sci. USA 2007, 104, 20308–20313. [Google Scholar]
- Lundin, K.; Blomberg, K.; Nordstrom, T.; Lindqvist, C. Development of a time-resolved fluorescence resonance energy transfer assay (cell TR-FRET) for protein detection on intact cells. Anal. Biochem. 2001, 299, 92–97. [Google Scholar]
- Selvin, P.R.; Hearst, J.E. Luminescence energy-transfer using a terbium chelate—Improvements on fluorescence energy transfer. Proc. Natl. Acad. Sci. USA 1994, 91, 10024–10028. [Google Scholar]
- Callis, P.R.; Vivian, J.T. Understanding the variable fluorescence quantum yield of tryptophan in proteins using QM-MM simulations. Quenching by charge transfer to the peptide backbone. Chem. Phys. Lett. 2003, 369, 409–414. [Google Scholar] [CrossRef]
- Garcia-Mira, M.M.; Sadqi, M.; Fischer, N.; Sanchez-Ruiz, J.M.; Munoz, V. Experimental identification of downhill protein folding. Science 2002, 298, 2191–2195. [Google Scholar]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar]
- Karasawa, S.; Araki, T.; Nagai, T.; Mizuno, H.; Miyawaki, A. Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochem. J. 2004, 381, 307–312. [Google Scholar]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef]
- Rizzo, M.A.; Springer, G.H.; Granada, B.; Piston, D.W. An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 2004, 22, 445–449. [Google Scholar]
- Kremers, G.J.; Goedhart, J.; van Munster, E.B.; Gadella, T.W., Jr. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Forster radius. Biochemistry 2006, 45, 6570–6580. [Google Scholar]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar]
- Cantor, C.R.; Schimmel, P.R. Biophysical Chemistry Part II: Techniques for the Study of Biological Structure and Function; W.H. Freeman & Co.: San Francisco, CA, USA, 1980; p. 846. [Google Scholar]
- Koushik, S.V.; Blank, P.S.; Vogel, S.S. Anomalous surplus energy transfer observed with multiple FRET acceptors. PLoS One 2009, 4, e8031. [Google Scholar]
- Sun, Y.; Wallrabe, H.; Booker, C.F.; Day, R.N.; Periasamy, A. Three-color spectral FRET microscopy localizes three interacting proteins in living cells. Biophys. J. 2010, 99, 1274–1283. [Google Scholar]
- Johnson, I.; Spence, M.T.Z. Molecular Probes Handbook,A Guide to Fluorescent Probes and Labeling Technologies, 11th ed; Molecular Probes: Eugene, OR, USA, 2010. [Google Scholar]
- Clapp, A.R.; Medintz, I.L.; Fisher, B.R.; Anderson, G.P.; Mattoussi, H. Can luminescent quantum dots be efficient energy acceptors with organic dye donors? J. Am. Chem. Soc. 2005, 127, 1242–1250. [Google Scholar]
- So, M.K.; Xu, C.; Loening, A.M.; Gambhir, S.S.; Rao, J. Self-illuminating quantum dot conjugates for in vivo imaging. Nat. Biotechnol. 2006, 24, 339–343. [Google Scholar]
- Clapp, A.R.; Medintz, I.L.; Mauro, J.M.; Fisher, B.R.; Bawendi, M.G.; Mattoussi, H. Fluorescence resonance energy transfer between quantum dot donors and dye-labeled protein acceptors. J. Am. Chem. Soc. 2004, 126, 301–310. [Google Scholar]
- Clapp, A.R.; Medintz, I.L.; Mattoussi, H. Förster resonance energy transfer investigations using quantum-dot fluorophores. Chemphyschem 2006, 7, 47–57. [Google Scholar]
- Barroso, M.M. Quantum dots in cell biology. J. Histochem. Cytochem. 2011, 59, 237–251. [Google Scholar]
- Jares-Erijman, E.A.; Jovin, T.M. FRET imaging. Nat. Biotechnol. 2003, 21, 1387–1395. [Google Scholar]
- Borst, J.W.; Visser, A.J.W.G. Fluorescence lifetime imaging microscopy in life sciences. Meas. Sci. Technol. 2010, 21, 102002. [Google Scholar]
- Liu, R.; Ren, D.; Liu, Y.; Deng, Y.; Sun, B.; Zhang, Q.; Guo, X. Biosensors of DsRed as FRET partner with CFP or GFP for quantitatively imaging induced activation of Rac, Cdc42 in living cells. Mol. Imaging. Biol. 2011, 13, 424–431. [Google Scholar]
- Del Pozo, M.A.; Kiosses, W.B.; Alderson, N.B.; Meller, N.; Hahn, K.M.; Schwartz, M.A. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat. Cell Biol. 2002, 4, 232–239. [Google Scholar]
- Wang, H.; Nakata, E.; Hamachi, I. Recent progress in strategies for the creation of protein-based fluorescent biosensors. Chembiochem 2009, 10, 2560–2577. [Google Scholar]
- Frommer, W.B.; Davidson, M.W.; Campbell, R.E. Genetically encoded biosensors based on engineered fluorescent proteins. Chem. Soc. Rev. 2009, 38, 2833–2841. [Google Scholar]
- Aoki, K.; Komatsu, N.; Hirata, E.; Kamioka, Y.; Matsuda, M. Stable expression of FRET biosensors: a new light in cancer research. Cancer Sci. 2011, 103, 614–619. [Google Scholar]
- Varghese, S.S.; Zhu, Y.; Davis, T.J.; Trowell, S.C. FRET for lab-on-a-chip devices—Current trends and future prospects. Lab Chip 2010, 10, 1355–1364. [Google Scholar]
- Welch, C.M.; Elliott, H.; Danuser, G.; Hahn, K.M. Imaging the coordination of multiple signalling activities in living cells. Nat. Rev. Mol. Cell Biol. 2011, 12, 749–756. [Google Scholar]
- Larson, D.R.; Ma, Y.M.; Vogt, V.M.; Webb, W.W. Direct measurement of Gag-Gag interaction during retrovirus assembly with FRET and fluorescence correlation spectroscopy. J. Cell Biol. 2003, 162, 1233–1244. [Google Scholar]
- Hiller, D.A.; Fogg, J.M.; Martin, A.M.; Beechem, J.M.; Reich, N.O.; Perona, J.J. Simultaneous DNA binding and bending by EcoRV endonuclease observed by real-time fluorescence. Biochemistry 2003, 42, 14375–14385. [Google Scholar]
- Shchyolkina, A.K.; Kaluzhny, D.N.; Borisova, O.F.; Hawkins, M.E.; Jernigan, R.L.; Jovin, T.M.; Arndt-Jovin, D.J.; Zhurkin, V.B. Formation of an intramolecular triple-stranded DNA structure monitored by fluorescence of 2-aminopurine or 6-methylisoxanthopterin. Nucleic Acids Res. 2004, 32, 432–440. [Google Scholar]
- Riven, I.; Kalmanzon, E.; Segev, L.; Reuveny, E. Conformational rearrangements associated with the gating of the G protein-coupled potassium channel revealed by FRET microscopy. Neuron 2003, 38, 225–235. [Google Scholar]
- Wouters, F.S.; Bastiaens, P.I.; Wirtz, K.W.; Jovin, T.M. FRET microscopy demonstrates molecular association of non-specific lipid transfer protein (nsL-TP) with fatty acid oxidation enzymes in peroxisomes. EMBO J. 1998, 17, 7179–7189. [Google Scholar]
- Bacskai, B.J.; Skoch, J.; Hickey, G.A.; Allen, R.; Hyman, B.T. Fluorescence resonance energy transfer determinations using multiphoton fluorescence lifetime imaging microscopy to characterize amyloid-beta plaques. J. Biomed. Opt. 2003, 8, 368–375. [Google Scholar]
- Valentin, G.; Verheggen, C.; Piolot, T.; Neel, H.; Coppey-Moisan, M.; Bertrand, E. Photoconversion of YFP into a CFP-like species during acceptor photobleaching FRET experiments. Nat. Methods 2005, 2, 801–801. [Google Scholar]
- Kremers, G.-J.; Hazelwood, K.L.; Murphy, C.S.; Davidson, M.W.; Piston, D.W. Photoconversion in orange and red fluorescent proteins. Nat. Methods 2009, 6, 355–358. [Google Scholar]
- Van Munster, E.B.; Kremers, G.J.; Adjobo-Hermans, M.J.; Gadella, T.W., Jr. Fluorescence resonance energy transfer (FRET) measurement by gradual acceptor photobleaching. J. Microsc. 2005, 218, 253–262. [Google Scholar]
- Kardash, E.; Bandemer, J.; Raz, E. Imaging protein activity in live embryos using fluorescence resonance energy transfer biosensors. Nat. Protoc. 2011, 6, 1835–1846. [Google Scholar]
- van Rheenen, J.; Langeslag, M.; Jalink, K. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys. J. 2004, 86, 2517–2529. [Google Scholar]
- Youvan, D.C.; Silva, C.M.; Bylina, E.J.; Coleman, W.J.; Dilworth, M.R.; Yang, M.M. Calibration of fuorescence resonance energy transfer in microscopy using genetically engineered GFP derivatives on nickel chelating beads. Biotechnology et alia 1997, 3, 1–18. [Google Scholar]
- Gordon, G.W.; Berry, G.; Liang, X.H.; Levine, B.; Herman, B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998, 74, 2702–2713. [Google Scholar]
- Xia, Z.; Liu, Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 2001, 81, 2395–2402. [Google Scholar]
- Persechini, A.; Lynch, J.A.; Romoser, V.A. Novel fluorescent indicator proteins for monitoring free intracellular CaM2+. Cell Calcium 1997, 22, 209, Cell Calcium 216.. [Google Scholar]
- Berney, C.; Danuser, G. FRET or no FRET: A quantitative comparison. Biophys. J. 2003, 84, 3992–4010. [Google Scholar]
- Chen, H.; Puhl, H.L., 3rd; Koushik, S.V.; Vogel, S.S.; Ikeda, S.R. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys. J. 2006, 91, L39–L41. [Google Scholar] [CrossRef]
- Rizzo, M.A.; Springer, G.; Segawa, K.; Zipfel, W.R.; Piston, D.W. Optimization of pairings and detection conditions for measurement of FRET between cyan and yellow fluorescent proteins. Microsc. Microanal. 2006, 12, 238–254. [Google Scholar]
- Venetta, B.D. Microscope phase fluorometer for determining the fluorescence lifetimes of fluorochromes. Rev. Sci. Instrum. 1959, 30, 450–457. [Google Scholar]
- Wang, X.F.; Uchida, T.; Minami, S. A fluorescence lifetime distribution measurement system based on phase-resolved detection using an image sissector tube. Appl. Spectrosc. 1989, 43, 840–845. [Google Scholar]
- Oida, T.; Sako, Y.; Kusumi, A. Fluorescence lifetime imaging microscopy (flimscopy). Methodology development and application to studies of endosome fusion in single cells. Biophys. J. 1993, 64, 676–685. [Google Scholar] [CrossRef]
- Van Munster, E.B.; Gadella, T.W., Jr. phiFLIM: A new method to avoid aliasing in frequency-domain fluorescence lifetime imaging microscopy. J. Microsc. 2004, 213, 29–38. [Google Scholar]
- Bastiaens, P.I.; Squire, A. Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends Cell Biol. 1999, 9, 48–52. [Google Scholar]
- van Munster, E.B.; Gadella, T.W., Jr. Suppression of photobleaching-induced artifacts in frequency-domain FLIM by permutation of the recording order. Cytometry A 2004, 58, 185–194. [Google Scholar]
- Pawley, J.B. Handbook of Biological Confocal Microscopy, 2nd ed; Springer US: New York, NY, USA, 1995; pp. 267–280. [Google Scholar]
- Becker, W.; Bergmann, A.; Biskup, C.; Kelbauskas, L.; Zimmer, T.; Klocker; Benndorf, K. High resolution TCSPC lifetime imaging, Multiphoton Microscopy in the Biomedical Sciences III, 2003; Periasamy, A., So, P., Eds.; BIOS, Proceedings of SPIE, 2003; pp. 175–184. [Google Scholar]
- Llères, D.; Swift, S.; Lamond, A.I. Detecting protein-protein interactions in vivo with FRET using multiphoton fluorescence lifetime imaging microscopy (FLIM). Curr. Protoc. Cytom. 2007, Chapter 12, Unit12 10. [Google Scholar]
- Parsons, M.; Messent, A.J.; Humphries, J.D.; Deakin, N.O.; Humphries, M.J. Quantification of integrin receptor agonism by fluorescence lifetime imaging. J. Cell Sci. 2008, 121, 265–271. [Google Scholar]
- Kinoshita, K.; Goryo, K.; Takada, M.; Tomokuni, Y.; Aso, T.; Okuda, H.; Shuin, T.; Fukumura, H.; Sogawa, K. Ternary complex formation of pVHL, elongin B and elongin C visualized in living cells by a fluorescence resonance energy transfer-fluorescence lifetime imaging microscopy technique. FEBS J. 2007, 274, 5567–5575. [Google Scholar]
- Jose, M.; Nair, D.K.; Reissner, C.; Hartig, R.; Zuschratter, W. Photophysics of Clomeleon by FLIM: discriminating excited state reactions along neuronal development. Biophys. J. 2007, 92, 2237–2254. [Google Scholar]
- Auksorius, E.; Boruah, B.R.; Dunsby, C.; Lanigan, P.M.; Kennedy, G.; Neil, M.A.; French, P.M. Stimulated emission depletion microscopy with a supercontinuum source and fluorescence lifetime imaging. Opt. Lett. 2008, 33, 113–115. [Google Scholar]
- Seah, L.K.; Wang, P.; Murukeshan, V.M.; Chao, Z.X. Application of fluorescence lifetime imaging (FLIM) in latent finger mark detection. Forensic. Sci. Int. 2006, 160, 109–114. [Google Scholar]
- Elder, A.D.; Matthews, S.M.; Swartling, J.; Yunus, K.; Frank, J.H.; Brennan, C.M.; Fisher, A.C.; Kaminski, C.F. Application of frequency-domain Fluorescence Lifetime Imaging Microscopy as a quantitative analytical tool for microfluidic devices. Opt. Express 2006, 14, 5456–5467. [Google Scholar]
- Robinson, T.; Valluri, P.; Manning, H.B.; Owen, D.M.; Munro, I.; Talbot, C.B.; Dunsby, C.; Eccleston, J.F.; Baldwin, G.S.; Neil, M.A.; de Mello, A.J.; French, P.M. Three-dimensional molecular mapping in a microfluidic mixing device using fluorescence lifetime imaging. Opt. Lett. 2008, 33, 1887–1889. [Google Scholar]
- Bunt, G.; Wouters, F.S. Visualization of molecular activities inside living cells with fluorescent labels. Int. Rev. Cytol. 2004, 237, 205–277. [Google Scholar]
- Piston, D.W.; Kremers, G.J. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar]
- Wouters, F.S. The physics and biology of fluorescence microscopy in the life sciences. Contemp. Phys. 2006, 47, 239–255. [Google Scholar]
- Digman, M.A.; Caiolfa, V.R.; Zamai, M.; Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophys. J. 2008, 94, L14–L16. [Google Scholar]
- Wouters, F.S.; Esposito, A. Quantitative analysis of fluorescence lifetime imaging made easy. HFSP J. 2008, 2, 7–11. [Google Scholar]
- Fereidouni, F.; Esposito, A.; Blab, G.A.; Gerritsen, H.C. A modified phasor approach for analyzing time-gated fluorescence lifetime images. J. Microsc. 2011, 244, 248–258. [Google Scholar]
- Mattheyses, A.L.; Hoppe, A.D.; Axelrod, D. Polarized fluorescence resonance energy transfer microscopy. Biophys. J. 2004, 87, 2787–2797. [Google Scholar]
- Bader, A.N.; Hofman, E.G.; van Bergen en Henegouwen, P.M.P.; Gerritsen, H.C. Imaging of protein cluster sizes by means of confocal time-gated fluorescence anisotropy microscopy. Opt. Express 2007, 15, 6934–6945. [Google Scholar]
- Rajaram, N.; Kerppola, T.K. Synergistic transcription activation by Maf and Sox and their subnuclear localization are disrupted by a mutation in Maf that causes cataract. Mol. Cell. Biol. 2004, 24, 5694–5709. [Google Scholar]
- Bader, A.N.; Hofman, E.G.; Voortman, J.; en Henegouwen, P.M.; Gerritsen, H.C. Homo-FRET imaging enables quantification of protein cluster sizes with subcellular resolution. Biophys. J. 2009, 97, 2613–2622. [Google Scholar]
- Loura, L.M.; Prieto, M. FRET in membrane biophysics: An overview. Front. Physiol. 2011, 2, 82. [Google Scholar]
- Zhang, P.; Rogelj, S.; Nguyen, K.; Wheeler, D. Design of a highly sensitive and specific nucleotide sensor based on photon upconverting particles. J. Am. Chem. Soc. 2006, 128, 12410–12411. [Google Scholar]
- Kuningas, K.; Rantanen, T.; Ukonaho, T.; Lovgren, T.; Soukka, T. Homogeneous assay technology based on upconverting phosphors. Anal. Chem. 2005, 77, 7348–7355. [Google Scholar]
- Kuningas, K.; Ukonaho, T.; Pakkila, H.; Rantanen, T.; Rosenberg, J.; Lovgren, T.; Soukka, T. Upconversion fluorescence resonance energy transfer in a homogeneous immunoassay for estradiol. Anal. Chem. 2006, 78, 4690–4696. [Google Scholar]
- Wang, L.; Yan, R.; Huo, Z.; Zeng, J.; Bao, J.; Wang, X.; Peng, Q.; Li, Y. Fluorescence resonant energy transfer biosensor based on upconversion-luminescent nanoparticles. Angew. Chem. Int. Ed. Engl. 2005, 44, 6054–6057. [Google Scholar]
- Rantanen, T.; Järvenpää, M.-L.; Vuojola, J.; Kuningas, K.; Soukka, T. Fluorescence-quenching-based enzyme-activity assay by using photon upconversion. Angew. Chem. Int. Ed. Engl. 2008, 120, 3871–3873. [Google Scholar]
- Kuningas, K.; Pakkila, H.; Ukonaho, T.; Rantanen, T.; Lovgren, T.; Soukka, T. Upconversion fluorescence enables homogeneous immunoassay in whole blood. Clin. Chem. 2007, 53, 145–146. [Google Scholar]
- Jiang, S.; Zhang, Y. Upconversion nanoparticle-based FRET system for study of siRNA in live cells. Langmuir 2010, 26, 6689–6694. [Google Scholar]
- Schönle, A.; Hänninen, P.E.; Hell, S.W. Nonlinear fluorescence through intermolecular energy transfer and resolution increase in fluorescence microscopy. Annalen der Physik 1999, 8, 115–133. [Google Scholar]
- Gopich, I.; Szabo, A. Theory of photon statistics in single-molecule Förster resonance energy transfer. J. Chem. Phys. 2005, 122, 14707–14718. [Google Scholar]
- Hofkens, J.; Cotlet, M.; Vosch, T.; Tinnefeld, P.; Weston, K.D.; Ego, C.; Grimsdale, A.; Mullen, K.; Beljonne, D.; Bredas, J.L.; Jordens, S.; Schweitzer, G.; Sauer, M.; De Schryver, F. Revealing competitive Förster-type resonance energy-transfer pathways in single bichromophoric molecules. Proc. Natl. Acad. Sci. USA 2003, 100, 13146–13151. [Google Scholar]
- Vosch, T.; Cotlet, M.; Hofkens, J.; Van Der Biest, K.; Lor, M.; Weston, K.; Tinnefeld, P.; Sauer, M.; Latterini, L.; Müllen, K.; De Schryver, F.C. Probing Förster type energy pathways in a first generation rigid dendrimer bearing two perylene imide chromophores. J. Phys. Chem. A 2003, 107, 6920–6931. [Google Scholar]
- Tinnefeld, P.; Buschmann, V.; Weston, K.D.; Sauer, M. Direct observation of collective blinking and energy transfer in a bichromophoric system. J. Phys. Chem. A 2002, 107, 323–327. [Google Scholar]
- Vogelsang, J.; Cordes, T.; Forthmann, C.; Steinhauer, C.; Tinnefeld, P. Intrinsically resolution enhancing probes for confocal microscopy. Nano Lett. 2010, 10, 672–679. [Google Scholar]
- Giordano, L.; Jovin, T.M.; Irie, M.; Jares-Erijman, E.A. Diheteroarylethenes as thermally stable photoswitchable acceptors in photochromic Fluorescence Resonance Energy Transfer (pcFRET). J. Am. Chem. Soc. 2002, 124, 7481–7489. [Google Scholar]
- Mao, S.; Benninger, R.K.; Yan, Y.; Petchprayoon, C.; Jackson, D.; Easley, C.J.; Piston, D.W.; Marriott, G. Optical lock-in detection of FRET using synthetic and genetically encoded optical switches. Biophys. J. 2008, 94, 4515–4524. [Google Scholar] [CrossRef]
- Subach, F.V.; Zhang, L.; Gadella, T.W.; Gurskaya, N.G.; Lukyanov, K.A.; Verkhusha, V.V. Red fluorescent protein with reversibly photoswitchable absorbance for photochromic FRET. Chem. Biol. 2010, 17, 745–755. [Google Scholar]
- Ha, T.; Enderle, T.; Ogletree, D.F.; Chemla, D.S.; Selvin, P.R.; Weiss, S. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. USA 1996, 93, 6264–6248. [Google Scholar]
- Kawashima, N.; Nakayama, K.; Itoh, K.; Itoh, T.; Ishikawa, M.; Biju, V. Reversible dimerization of EGFR revealed by single-molecule fluorescence imaging using quantum dots. Chem. Eur. J. 2010, 16, 1186–1192. [Google Scholar]
- Gadella, T.W., Jr.; Jovin, T.M. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 1995, 129, 1543–1558. [Google Scholar] [CrossRef]
- Tisler, J.; Reuter, R.; Lämmle, A.; Jelezko, F.; Balasubramanian, G.; Hemmer, P.R.; Reinhard, F.; Wrachtrup, J. Highly efficient FRET from a single nitrogen-vacancy center in nanodiamonds to a single organic molecule. ACS Nano 2011, 5, 7893–7898. [Google Scholar]
- Lee, J.; Lee, S.; Ragunathan, K.; Joo, C.; Ha, T.; Hohng, S. Single-molecule four-color FRET. Angew. Chem. Int. Ed. Engl. 2010, 49, 9922–9925. [Google Scholar]
- Uphoff, S.; Holden, S.J.; Le Reste, L.; Periz, J.; van de Linde, S.; Heilemann, M.; Kapanidis, A.N. Monitoring multiple distances within a single molecule using switchable FRET. Nat. Methods 2010, 7, 831–836. [Google Scholar]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar]
- Johannes, H.; Kristofer, G.; Mike, H.; Achillefs, N.K. Surfing on a new wave of single-molecule fluorescence methods. Physical. Biol. 2010, 7, 031001. [Google Scholar]
- Jovin, T.M.; Arndt-Jovin, D.J. FRET microscopy: digital imaging of fluorescence resonance energy transfer. Application in cell biology. In Cell Structure and Function by Microspectrofluorimetry; Kohen, E., Ploem, J.S., Hirschberg, J.G., Eds.; Academic Press: New York, NY, USA, 1989; pp. 99–117. [Google Scholar]
- Wouters, F.S.; Bastiaens, P.I. Imaging protein-protein interactions by Fluorescence Resonance Energy Transfer (FRET) microscopy. Curr. Protoc. Neurosci. 2006, Chapter 5, 5–22. [Google Scholar]
- Ai, H.W.; Hazelwood, K.L.; Davidson, M.W.; Campbell, R.E. Fluorescent protein FRET pairs for ratiometric imaging of dual biosensors. Nat. Methods 2008, 5, 401–403. [Google Scholar]
- Lippincott-Schwartz, J.; Snapp, E.; Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2001, 2, 444–456. [Google Scholar]
- Miyawaki, A. Development of probes for cellular functions using fluorescent proteins and fluorescence resonance energy transfer. Annu. Rev. Biochem. 2011, 80, 357–373. [Google Scholar]
- Richards, F.M. On the enzymic activity of subtilisin-modified ribonuclease. Proc. Natl. Acad. Sci. USA 1958, 44, 162–166. [Google Scholar]
- Hu, C.-D.; Chinenov, Y.; Kerppola, T.K. Visualization of interactions among bZIP and Rel family proteins in living cells using Bimolecular Fluorescence Complementation. Mol. cell 2002, 9, 789–798. [Google Scholar]
- Shyu, Y.J.; Hiatt, S.M.; Duren, H.M.; Ellis, R.E.; Kerppola, T.K.; Hu, C.D. Visualization of protein interactions in living Caenorhabditis elegans using bimolecular fluorescence complementation analysis. Nat. Protoc. 2008, 3, 588–596. [Google Scholar]
- Hu, C.D.; Grinberg, A.V.; Kerppola, T.K. Visualization of protein interactions in living cells using bimolecular fluorescence complementation (BiFC) analysis. Curr. Protoc. Cell Biol. 2006, Chapter 21, Unit 21 3. [Google Scholar]
- Kerppola, T.K. Bimolecular fluorescence complementation: Visualization of molecular interactions in living cells. Methods Cell Biol. 2008, 85, 431–470. [Google Scholar]
- Kerppola, T.K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar]
- Kerppola, T.K. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: characteristics of protein fragment complementation. Chem. Soc. Rev. 2009, 38, 2876–2886. [Google Scholar]
- Kerppola, T.K. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 2008, 37, 465–487. [Google Scholar]
- van Royen, M.E.; Cunha, S.M.; Brink, M.C.; Mattern, K.A.; Nigg, A.L.; Dubbink, H.J.; Verschure, P.J.; Trapman, J.; Houtsmuller, A.B. Compartmentalization of androgen receptor protein-protein interactions in living cells. J. Cell Biol. 2007, 177, 63–72. [Google Scholar]
- van Royen, M.E.; Dinant, C.; Farla, P.; Trapman, J.; Houtsmuller, A.B. FRAP and FRET methods to study nuclear receptors in living cells. Methods Mol. Biol. 2009, 505, 69–96. [Google Scholar]
- Dyba, M.; Jakobs, S.; Hell, S.W. Immunofluorescence stimulated emission depletion microscopy. Nat. Biotechnol. 2003, 21, 1303–1304. [Google Scholar]
- Bretschneider, S.; Eggeling, C.; Hell, S.W. Breaking the diffraction barrier in fluorescence microscopy by optical shelving. Phys. Rev. Lett. 2007, 98, 218103. [Google Scholar]
- Gustafsson, M.G. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA 2005, 102, 13081–13086. [Google Scholar]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008, 319, 810–813. [Google Scholar]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar]
- Stepanenko, O.V.; Shcherbakova, D.M.; Kuznetsova, I.M.; Turoverov, K.K.; Verkhusha, V.V. Modern fluorescent proteins: from chromophore formation to novel intracellular applications. Biotechniques 2011, 51, 313-314, 316, 318, passim.. [Google Scholar]
- Kner, P.; Chhun, B.B.; Griffis, E.R.; Winoto, L.; Gustafsson, M.G. Super-resolution video microscopy of live cells by structured illumination. Nat. Methods 2009, 6, 339–342. [Google Scholar]
- Moneron, G.; Hell, S.W. Two-photon excitation STED microscopy. Opt. Express 2009, 17, 14567–14573. [Google Scholar]
- Bianchini, P.; Diaspro, A. Fast scanning STED and two-photon fluorescence excitation microscopy with continuous wave beam. J. Microsc. 2012, 245, 225–228. [Google Scholar]
- Patterson, G.; Davidson, M.; Manley, S.; Lippincott-Schwartz, J. Superresolution imaging using single-molecule localization. Annu. Rev. Phys. Chem. 2010, 61, 345–367. [Google Scholar]
- Wang, Z.; Guo, W.; Li, L.; Luk'yanchuk, B.; Khan, A.; Liu, Z.; Chen, Z.; Hong, M. Optical virtual imaging at 50 nm lateral resolution with a white-light nanoscope. Nat. Commun. 2011, 2, 218. [Google Scholar]
- Grecco, H.E.; Verveer, P.J. FRET in cell biology: Still shining in the age of super-resolution? Chemphyschem 2011, 12, 484–490. [Google Scholar] [CrossRef]
- Willig, K.I.; Keller, J.; Bossi, M.; Hell, S.W. STED microscopy resolves nanoparticle assemblies. New J. Phys. 2006, 8, 106. [Google Scholar]
- Reymann, J.; Baddeley, D.; Gunkel, M.; Lemmer, P.; Stadter, W.; Jegou, T.; Rippe, K.; Cremer, C.; Birk, U. High-precision structural analysis of subnuclear complexes in fixed and live cells via spatially modulated illumination (SMI) microscopy. Chromosome Res. 2008, 16, 367–382. [Google Scholar]
- Mueller, V.; Ringemann, C.; Honigmann, A.; Schwarzmann, G.; Medda, R.; Leutenegger, M.; Polyakova, S.; Belov, V.N.; Hell, S.W.; Eggeling, C. STED nanoscopy reveals molecular details of cholesterol- and cytoskeleton-modulated lipid interactions in living cells. Biophys. J. 2011, 101, 1651–1660. [Google Scholar]
- Deng, S.; Chen, J.; Huang, Q.; Fan, C.; Cheng, Y. Saturated Förster resonance energy transfer microscopy with a stimulated emission depletion beam: A pathway toward single-molecule resolution in far-field bioimaging. Opt. Lett. 2010, 35, 3862–3864. [Google Scholar]
- Wang, L.; Jackson, W.C.; Steinbach, P.A.; Tsien, R.Y. Evolution of new nonantibody proteins via iterative somatic hypermutation. Proc. Natl. Acad. Sci. USA 2004, 101, 16745–16749. [Google Scholar]
- Shu, X.; Shaner, N.C.; Yarbrough, C.A.; Tsien, R.Y.; Remington, S.J. Novel chromophores and buried charges control color in mFruits. Biochemistry 2006, 45, 9639–9647. [Google Scholar]
- Campbell, R.E.; Tour, O.; Palmer, A.E.; Steinbach, P.A.; Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 7877–7882. [Google Scholar]
- Matz, M.V.; Fradkov, A.F.; Labas, Y.A.; Savitsky, A.P.; Zaraisky, A.G.; Markelov, M.L.; Lukyanov, S.A. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotechnol. 1999, 17, 969–973. [Google Scholar]
- Tian, Z.; Wu, W.; Li, A.D. Photoswitchable fluorescent nanoparticles: preparation, properties and applications. Chem. Phys. Chem. 2009, 10, 2577–2591. [Google Scholar]
- Tomosugi, W.; Matsuda, T.; Tani, T.; Nemoto, T.; Kotera, I.; Saito, K.; Horikawa, K.; Nagai, T. An ultramarine fluorescent protein with increased photostability and pH insensitivity. Nat. Methods 2009, 6, 351–353. [Google Scholar]
- Sakon, J.J.; Weninger, K.R. Detecting the conformation of individual proteins in live cells. Nat. Methods 2010, 7, 203–205. [Google Scholar]
- Knoblauch, M.; Hibberd, J.M.; Gray, J.C.; van Bel, A.J.E. A galinstan expansion femtosyringe for microinjection of eukaryotic organelles and prokaryotes. Nat. Biotechnol. 1999, 17, 906–909. [Google Scholar]
- Laforge, F.O.; Carpino, J.; Rotenberg, S.A.; Mirkin, M.V. Electrochemical attosyringe. Proc. Natl. Acad. Sci. USA 2007, 104, 11895–11900. [Google Scholar]
- Chen, X.; Kis, A.; Zettl, A.; Bertozzi, C.R. A cell nanoinjector based on carbon nanotubes. Proc. Natl. Acad. Sci. USA 2007, 104, 8218–8222. [Google Scholar]
- Sönnichsen, C.; Reinhard, B.M.; Liphardt, J.; Alivisatos, A.P. A molecular ruler based on plasmon coupling of single gold and silver nanoparticles. Nat. Biotechnol. 2005, 23, 741–745. [Google Scholar]
- Reinhard, B.M.; Siu, M.; Agarwal, H.; Alivisatos, A.P.; Liphardt, J. Calibration of dynamic molecular rulers based on plasmon coupling between gold nanoparticles. Nano Lett. 2005, 5, 2246–2252. [Google Scholar]
- Mathew-Fenn, R.S.; Das, R.; Silverman, J.A.; Walker, P.A.; Harbury, P.A.B. A molecular ruler for measuring quantitative distance distributions. PLoS One 2008, 3, e3229. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P.C. Advanced Fluorescence Microscopy Techniques—FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012, 17, 4047-4132. https://doi.org/10.3390/molecules17044047
Ishikawa-Ankerhold HC, Ankerhold R, Drummen GPC. Advanced Fluorescence Microscopy Techniques—FRAP, FLIP, FLAP, FRET and FLIM. Molecules. 2012; 17(4):4047-4132. https://doi.org/10.3390/molecules17044047
Chicago/Turabian StyleIshikawa-Ankerhold, Hellen C., Richard Ankerhold, and Gregor P. C. Drummen. 2012. "Advanced Fluorescence Microscopy Techniques—FRAP, FLIP, FLAP, FRET and FLIM" Molecules 17, no. 4: 4047-4132. https://doi.org/10.3390/molecules17044047
APA StyleIshikawa-Ankerhold, H. C., Ankerhold, R., & Drummen, G. P. C. (2012). Advanced Fluorescence Microscopy Techniques—FRAP, FLIP, FLAP, FRET and FLIM. Molecules, 17(4), 4047-4132. https://doi.org/10.3390/molecules17044047