Cysteine, Glutathione, and Thiol Redox Balance in Astrocytes

{kind=link}
{kind=link}
Abstract
:1. Introduction
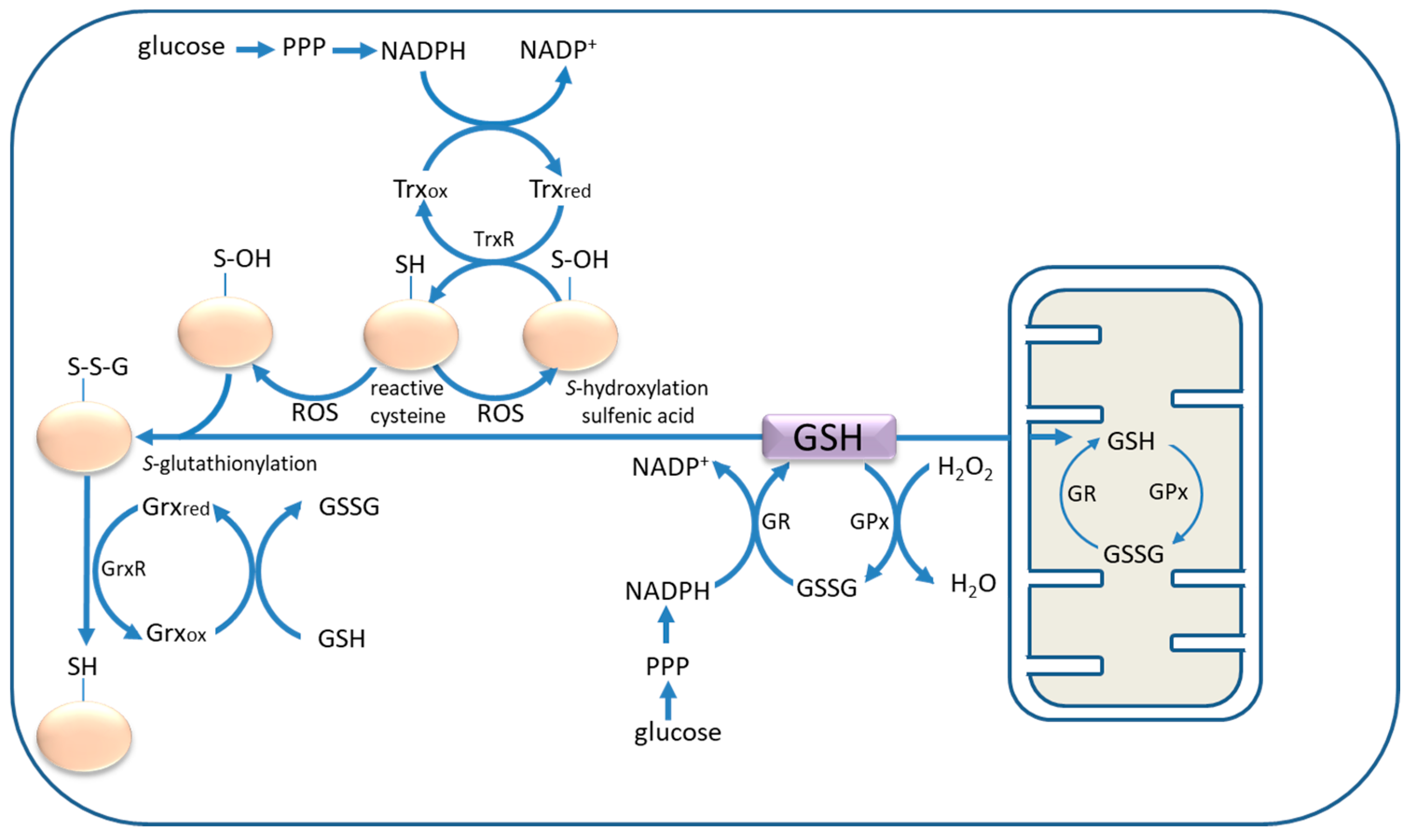
2. Cysteine and Glutathione: In Situ Antioxidants and Neuronal Protection
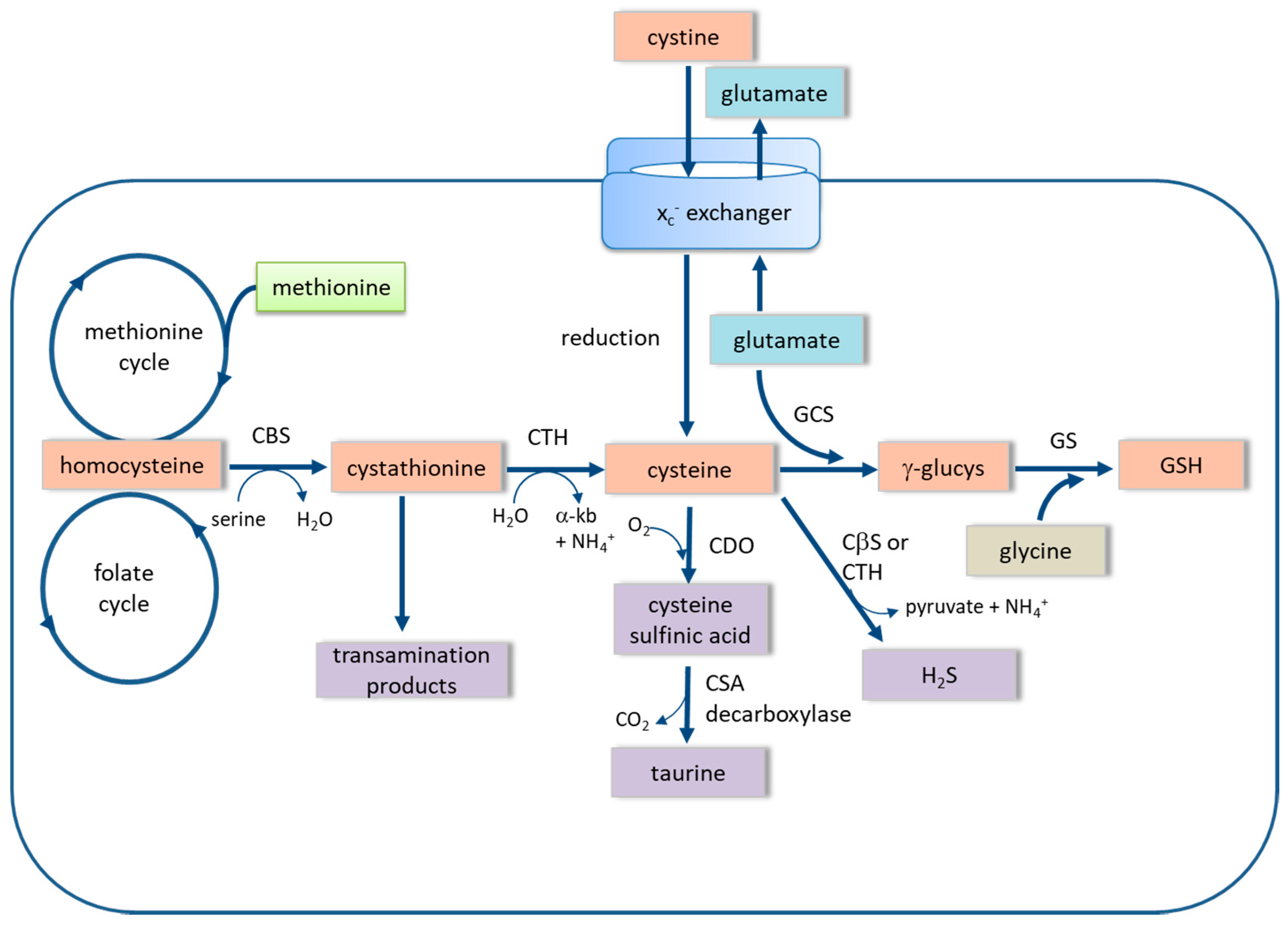
3. The xc− Cystine-Glutamate Exchanger
4. The Trans-Sulfuration Pathway
5. Astrocyte Activation and Thiol Antioxidants
6. Cysteamine: A Potential Therapeutic?
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Banerjee, R. Redox outside the box: Linking extracellular redox remodelling with intracellular redox metabolism. J. Biol. Chem. 2011, 287, 4397–4402. [Google Scholar] [CrossRef] [PubMed]
- García-Nogales, P.; Almeida, A.; Fernández, E.; Medina, J.M.; Bolaños, J.P. Induction of glucose-6-phosphate dehydrogenase by lipopolysaccharide contributes to preventing nitric oxide-mediated glutathione depletion in cultured rat astrocytes. J. Neurochem. 1999, 72, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Iizumi, T.; Takahashi, S.; Mashima, K.; Minami, K.; Izawa, Y.; Abe, T.; Hishiki, T.; Suematsu, M.; Kajimura, M.; Suzuki, N. A possible role of microglia-derived nitric oxide by lipopolysaccharide in activation of astroglial pentose-phosphate pathway via the Keap1/Nrf2 system. J. Neuroinflamm. 2016, 13, 99–119. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Carvey, P.M.; Ling, Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006, 1090, 35–44. [Google Scholar] [CrossRef] [PubMed]
- McBean, G.J.; Aslan, M.; Griffiths, H.R.; Torrão, R.C. Thiol redox homeostasis in neurodegenerative disease. Redox Biol. 2015, 5, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Kosaka, K.; Maruyama, A.; Satoh, T.; Harada, N.; Yoshida, H.; Satoh, K.; Yamamoto, M.; Itoh, K. Nrf2 regulates NGR mRNA induction by carnosic acid in T98G glioblastoma cells and normal human astrocytes. J. Biochem. 2011, 150, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Eftekharpour, E.; Holmgren, A.; Juurlink, B.H. Thioredoxin reductase and glutathione synthesis is upregulated by t-butylhydroquinone in cortical astrocytes but not in cortical neurons. Glia 2000, 31, 241–248. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, K.; Zhang, L.; Li, L.; Zhao, J. Thioredoxin 1 protects astrocytes from oxidative stress by maintaining perioxiredoxin activity. Mol. Med. Rep. 2016, 13, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Kranich, O.; Dringen, R.; Sandberg, M.; Hamprecht, B. Utilization of cysteine and cysteine precursors for the synthesis of glutathione in astroglial cultures: Preference for cystine. Glia 1998, 22, 11–18. [Google Scholar] [CrossRef]
- Qu, K.; Lee, S.W.; Bian, J.S.; Low, C.M.; Wong, P.T. Hydrogen sulfide: Neurochemistry and neurobiology. Neurochem. Int. 2008, 52, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Garg, S.K.; Banerjee, R. Taurine biosynthesis by neurons and astrocytes. J. Biol. Chem. 2011, 286, 32002–32010. [Google Scholar] [CrossRef] [PubMed]
- Roede, J.R.; Uppal, K.; Laing, Y.; Promislow, D.E.; Watchman, L.M.; Jones, D.P. Characterization of plasma thiol redox potential in a common marmoset model of aging. Redox Biol. 2013, 1, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yudkoff, M.; Pleasure, D.; Cregar, L.; Lin, Z.-P.; Nissim, I.; Stern, J.; Nissim, I. Glutathione turnover in cultured astrocytes: Studies with [15N]glutamate. J. Neurochem. 1990, 55, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Makar, T.K.; Nedergaard, M.; Preuss, A.; Gelbard, A.S.; Perumal, A.S.; Cooper, A.J.L. Vitamin E, ascorbate, glutathione, glutathione disulfide and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: Evidence that astrocytes play an important role in antioxidative processes in the brain. J. Neurochem. 1994, 62, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Raps, S.P.; Lai, J.C.; Hertz, L.; Cooper, A.J. Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res. 1989, 493, 389–401. [Google Scholar] [CrossRef]
- Dringen, R.; Kranich, O.; Hamprecht, B. The gamma-glutamyl transpeptidase inhibitor, acivicin preserves glutathione released by astroglial cells in culture. Neurochem. Res. 1997, 22, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [PubMed]
- Orlowski, M.; Meister, A. The gamma-glutamyl cycle: A possible transport system for amino acids. Proc. Natl. Acad. Sci. USA 1970, 67, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.; McKenna, M.C. Glutamate metabolism in the brain focussing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Tsai, M.J.; Chang, Y.F.; Schwarcz, R.; Brookes, N. Characterization of l-alpha-aminoadipic acid transport in cultured astrocytes. Brain Res. 1996, 741, 166–173. [Google Scholar] [CrossRef]
- O’Connor, E.; Devesa, A.; García, C.; Puertes, I.R.; Pellín, A.; Viña, J.R. Biosynthesis and maintenance of GSH in primary astrocyte cultures: Role of l-cystine and ascorbate. Brain Res. 1995, 680, 157–163. [Google Scholar] [CrossRef]
- Sato, H.; Shiiya, A.; Kimata, M.; Maebara, K.; Tamba, M.; Sakakura, Y.; Makino, N.; Sugiyama, F.; Yagami, K.; Moriguchi, T.; et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 2005, 280, 37429. [Google Scholar] [CrossRef] [PubMed]
- De Bundel, D.; Schalier, A.; Loyens, E.; Fernando, R.; Miyashita, H.; van Liefferinge, J.; Vermoesen, K.; Bannai, S.; Sato, H.; Michotte, Y.; et al. Loss of system xc− does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J. Neurosci. 2011, 31, 5792–5803. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchanger modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system xc−: Cystine supplier and beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Banjac, A.; Perisic, T.; Sato, H.; Seiler, A.; Bannai, S.; Weiss, N.; Kölle, P.; Tsoep, K.; Issels, R.D.; Daniel, P.T.; et al. The cystine/cysteine cycle: A redox cycle regulating susceptibility versus resistance to cell death. Oncogene 2008, 27, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.W.; Yuan, J.F.; Yang, H.M.; Wang, S.T.; Zhang, C.G.; Sun, L.L.; Yang, H.; Zhang, H. Extracellular cysteine (Cys)/cystine (CySS) redox regulates metabotropic glutamate receptor 5 activity. Biochemie 2012, 94, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Soria, F.N.; Pérez-Samartín, A.; Martin, A.; Gona, K.B.; Llop, J.; Szczupak, B.; Chara, J.C.; Matute, C.; Domercq, M. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J. Clin. Investig. 2014, 124, 3645–3655. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Ankeny, D.P.; Garg, S.K.; Wei, P.; Guan, Z.; Lai, W.; McTigue, D.M.; Banerjee, R.; Popovich, P.G. System xc− regulates microglia and macrophage glutamate excitotoxicity in vivo. Exp. Neurol. 2012, 233, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Piani, D.; Fontana, A. Involvement of the cystine transport system xc− in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J. Immunol. 1994, 152, 3578–3585. [Google Scholar] [PubMed]
- Mysona, B.; Dun, Y.; Duplantier, J.; Ganapathy, V.; Smith, S.B. Effects of hyperglycaemia and oxidative stress on the glutamate transporters GLAST and system xc− in mouse retinal Müller glial cells. Cell Tissue Res. 2009, 335, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Jiminez-Blasco, D.; Santofimia-Castaño, P.; Gonzalez, S.; Almeida, A.; Bolaños, J.P. Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5-Nrf2 pathway. Cell Death Differ. 2015, 22, 1877–1889. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [PubMed]
- Lewerenz, J.; Albrecht, P.; Tien, M.L.; Henke, N.; Karumbayaram, S.; Kornblum, H.I.; Wiedau-Pazos, M.; Schubert, D.; Maher, P.; Methner, A. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J. Neurochem. 2009, 111, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Dowell, J.A.; Johnson, J.A. Mechanisms of Nrf2 protection in astrocytes as identified by quantitative proteomics and siRNA screening. PLoS ONE 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocystes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef] [PubMed]
- Sieb, T.M.; Patel, S.A.; Bridges, R.J. Regulation of the system xc−-cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia 2011, 59, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Rothstein, J.D.; Sontheimer, H. Compromised glutamate transport in human glioma cells: Reduction and mis-localisation of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J. Neurosci. 1999, 19, 10767–10777. [Google Scholar] [PubMed]
- Chung, W.J.; Lyons, S.A.; Nelson, G.M.; Hamza, H.; Gladson, C.L.; Gillespie, G.Y.; Sontheimer, H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 2005, 25, 7101–7110. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Cao, Y.; Chen, Y.; Parsons, C.; Qin, Z. Targeting xCT, a cystine-glutamate transporter induces apoptosis and tumor regression for KSHV/HIV-associated lymphoma. J. Hematol. Oncol. 2014, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.Z.; Chen, G.; Wang, P.; Lu, W.H.; Zhu, C.F.; Song, M.; Yang, J.; Wen, S.; Xu, R.H.; Hu, Y.; et al. xc− inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett. 2015, 368, 88–96. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.; Sontheimer, H. Glutamate and the biology of the gliomas. Glia 2011, 59, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Savaskan, N.E.; Heckl, A.; Hahnen, E.; Engelhorn, T.; Doerfler, A.; Gansladt, O.; Nimsky, C.; Buchfelder, M.; Eyuepoglu, I.Y. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 2008, 15, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomised study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Yu, G.; Xue, Y.; Liu, Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol. Rep. 2015, 33, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Thomas, M.; Ghorpade, A.; Gendelman, H.E.; Banerjee, R. A functional transsulfuration pathway in the brain links to glutathione homeostasis. J. Biol. Chem. 2006, 281, 35785–35793. [Google Scholar] [CrossRef] [PubMed]
- Kandil, S.; Brennan, L.; McBean, G.J. Glutathione depletion causes a JNK and p38MAPK-mediated increase in expression of cystathionine-γ-lyase and upregulation of the transsulfuration pathway in C6 glioma cells. Neurochem. Int. 2010, 56, 611–619. [Google Scholar] [CrossRef] [PubMed]
- McBean, G.J. The transsulfuration pathway: A source of cysteine for glutathione in astrocytes. Amino Acids 2012, 42, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.S.; Lee, J.; Homma, T.; Kurahashi, T.; Kobayashi, S.; Nabeshima, A.; Yamada, S.; Seo, H.G.; Miyata, S.; Sato, H.; et al. xCt deficiency aggravates acetaminophen-induced hepatotoxicity under inhibition of the transsulfuration pathway. Free Radic Res. 2017, 51, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Schwab, C.; Yu, S.; McGeer, E.; McGeer, P.L. Astrocytes produce the anti-inflammatory and neuroprotective agent hydrogen sulfide. Neurobiol. Aging 2009, 10, 1523–1524. [Google Scholar] [CrossRef] [PubMed]
- Kabil, P.; Vitvitsky, V.; Xie, P.; Banerjee, R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antiox. Redox Signal 2011, 15, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronowicka-Adamska, P.; Bentke, A.; Wróbel, M. Hydrogen sulphide generation from l-cysteine in the human glioblastoma-astrocytoma U-87 MG and neuroblastoma SHSY5Y cell lines. Acta Biochim. Pol. 2017, 64, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Enokido, Y.; Suzuki, E.; Iwasawa, K.; Namekata, K.; Okazawa, H.; Kimura, H. Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 2005, 19, 1854–1856. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, R.; Otsuguro, K.; Yamaguchi, S.; Ito, S. Neuronal regulation of expression of hydrogen sulphide-producing enzyme cystathionine-beta-synthase in rat spinal cord astrocytes. Neurosci. Res. 2015, 97, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Longoni, A.; Bellaver, B.; Bobermin, L.D.; Santos, C.L.; Nonose, Y.; Kolling, J.; Dos Santos, T.M.; de Assis, A.M.; Quincozes-Santos, A.; Wyse, A.T. Homocysteine induces glial reactivity in adult rat astrocyte cultures. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Isobe, C.; Murata, T.; Sato, C.; Terayama, Y. Increase of total homocysteine concentration in cerebrospinal fluid in patients with Alzheimer’s disease and Parkinson’s disease. Life Sci. 2005, 77, 1836–1843. [Google Scholar] [CrossRef] [PubMed]
- Hogg, N. The effect of cyst(e)ine on the auto-oxidation of homocysteine. Free Radic. Biol. Med. 1999, 27, 28–33. [Google Scholar] [CrossRef]
- Kobayashi, S.; Sato, M.; Kasakoshi, T.; Tsuitsui, T.; Sugimoto, M.; Osaki, M.; Okada, F.; Igarashi, K.; Hiratake, J.; Homma, T.; et al. Cystathionine is a novel substrate of cystine/glutamate antiporter: Implications for immune function. J. Biol. Chem. 2015, 290, 8778–8788. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem. Int. 2013, 63, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Li, Y.; Song, J.N.; Pang, H.G. Role of hydrogen sulfide in secondary neuronal injury. Neurochem. Int. 2014, 64, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Tyagi, N. Role of hydrogen sulfide in brain synaptic remodeling. Meth. Enzymol. 2015, 555, 207–229. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.L.; Fuller, S.; Maczurek, A.E.; Kersaitis, C.; Ooi, L.; Műnch, G. Chronic inflammation alters production and release of glutathione and related brain thiols in human U373 astroglial cells. Cell Mol. Neurobiol. 2013, 33, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Jackman, N.; Uliasz, T.F.; Hewett, J.A.; Hewett, S.J. Regulation of system xc− activity and expression in astrocytes by interleukin-1β: Implications for hypoxic neuronal injury. Glia 2010, 58, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; He, Y.; Hewett, S.J.; Hewett, J.A. Interleukin1β regulation of the system xc− substrate-specific subunit, xCT, in primary mouse astrocytes involves the RNA-binding protein, HuR. J. Biol. Chem. 2016, 291, 1634–1651. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Jackman, N.A.; Thorn, T.L.; Vought, V.E.; Hewett, S.J. Interleukin-1β protects astrocytes against oxidant-induced injury via an NF-κB-dependent upregulation of glutathione synthesis. Glia 2015, 63, 1568–1580. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhang, X.; Chen, H.P.; Li, L.; Xie, W.; Lan, G.; Zhao, Z.W.; Zheng, X.L.; Wang, Z.B.; Tang, C.K. Micro-RNA-186 promotes macrophage lipid accumulation and secretion of pro-inflammatory cytokines by targeting cystathionine-γ-lyase in THP-1 macrophages. Atherosclerosis 2016, 250, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Gibrat, C.; Cicchetti, F. Potential of cystamine and cysteamine in the treatment of neurodegenerative disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Justino, L.; Welner, S.A.; Tannenbaum, G.S.; Schipper, H.M. Long-term effects of cysteamine on cognitive and locomotor behaviour in rats: Relationship to hippocampal glial pathology and somatostatin levels. Brain Res. 1997, 761, 127–134. [Google Scholar] [CrossRef]
- Frankel, D.; Schipper, H.M. Cysteamine pretreatment of the astroglial substratum (mitochondrial iron sequestration) enhances PC12 cell vulnerability to oxidative injury. Exp. Neurol. 1990, 160, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Frankel, D.; Mehindate, K.; Schipper, H.M. Role of heme oxygenase-1 in the regulation of manganese superoxide dismutase in oxidatively-challenged astroglia. J. Cell. Physiol. 2000, 185, 80–86. [Google Scholar] [CrossRef]
- Manganaro, F.; Chopra, V.S.; Mydlarski, M.B.; Bernatchez, G.; Schipper, H.M. Redox perturbations in cysteamine-stressed astroglia: Implication for inclusion formation and gliosis in the ageing brain. Free Radic. Biol. Med. 1999, 19, 823–835. [Google Scholar] [CrossRef]
- Cisbani, G.; Drouin-Ouellet, J.; Gibrat, C.; Saint-Pierre, M.; Lagacé, S.; Lavallée-Bourget, M.H.; Charest, J.; Chabrat, A.; Boivin, L.; et al. Cystamine/cysteamine rescues the dopaminergic systems and shows neurorestorative properties in an animal model of Parkinson’s disease. Neurobiol. Dis. 2015, 82, 430–444. [Google Scholar] [CrossRef] [PubMed]
- Elmonem, M.A.; Veys, K.R.; Soliman, N.A.; van Dyck, M.; van den Heuvel, L.P.; Levtchenko, E. Cystinosis: A review. Orphanet J. Rare Dis. 2016, 11, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McBean, G.J. Cysteine, Glutathione, and Thiol Redox Balance in Astrocytes. Antioxidants 2017, 6, 62. https://doi.org/10.3390/antiox6030062
McBean GJ. Cysteine, Glutathione, and Thiol Redox Balance in Astrocytes. Antioxidants. 2017; 6(3):62. https://doi.org/10.3390/antiox6030062
Chicago/Turabian StyleMcBean, Gethin J. 2017. "Cysteine, Glutathione, and Thiol Redox Balance in Astrocytes" Antioxidants 6, no. 3: 62. https://doi.org/10.3390/antiox6030062
APA StyleMcBean, G. J. (2017). Cysteine, Glutathione, and Thiol Redox Balance in Astrocytes. Antioxidants, 6(3), 62. https://doi.org/10.3390/antiox6030062