
Palladium-Catalyzed Arylations towards 3,6-Diaryl-1,3a,6a-triazapentalenes and Evaluation of Their Fluorescence Properties
 ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results and Discussion
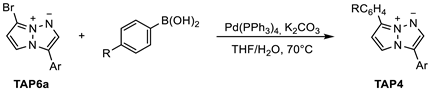
2.1. Synthesis
2.2. Photophysical Measurements
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Instruments
3.3. Synthesis
3.3.1. Palladium-Catalyzed CH Arylations
3.3.2. Brominations
3.3.3. Palladium-Catalyzed Suzuki Coupling Reactions
3.3.4. Saponification
3.3.5. Experimental Details and Characterization Data
3.4. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Opsomer, T.; Dehaen, W. Developments in the chemistry of 1,3a,6a-triazapentalenes and their fused analogs. In Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 2022; pp. 25–70. ISBN 0065-2725. [Google Scholar]
- Wang, Y.; Opsomer, T.; Dehaen, W. Bicyclic 1,3a,6a-Triazapentalene Chromophores: Synthesis, Spectroscopy and Their Use as Fluorescent Sensors and Probes. Chemosensors 2021, 9, 16. [Google Scholar] [CrossRef]
- Verbelen, B.; Dehaen, W. Two-Step Synthesis of Fluorescent 3-Arylated 1,3a,6a-Triazapentalenes via a Three-Component Triazolization Reaction. Org. Lett. 2016, 18, 6412–6415. [Google Scholar] [CrossRef] [PubMed]
- Namba, K.; Osawa, A.; Ishizaka, S.; Kitamura, N.; Tanino, K. Direct Synthesis of Fluorescent 1,3a,6a-Triazapentalene Derivatives via Click–Cyclization–Aromatization Cascade Reaction. J. Am. Chem. Soc. 2011, 133, 11466–11469. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Wang, D.; Chen, Y.; Yan, W.; Geise, N.R.; Sharma, S.; Li, H.; Petersen, J.L.; Li, M.; Shi, X. Facile synthesis of fluorescent active triazapentalenes through gold-catalyzed triazole–alkyne cyclization. Chem. Commun. 2014, 50, 7303–7305. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.; Hiebel, M.-A.; Guillaumet, G.; Pasquinet, E.; Suzenet, F. Intramolecular Metal-Free N−N Bond Formation with Heteroaromatic Amines: Mild Access to Fused-Triazapentalene Derivatives. Chem. Eur. J. 2020, 26, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Kamada, R.; Tano, F.; Kudoh, F.; Kimura, N.; Chuman, Y.; Osawa, A.; Namba, K.; Tanino, K.; Sakaguchi, K. Effective Cellular Morphology Analysis for Differentiation Processes by a Fluorescent 1,3a,6a-Triazapentalene Derivative Probe in Live Cells. PLoS ONE 2016, 11, e0160625. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Otani, A.; Inokuma, T.; Tsuji, D.; Mukaiyama, H.; Nakayama, A.; Itoh, K.; Otaka, A.; Tanino, K.; Namba, K. Development of a 1,3a,6a-triazapentalene derivative as a compact and thiol-specific fluorescent labeling reagent. Commun. Chem. 2020, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Osawa, A.; Watanabe, T.; Murata, Y.; Nakayama, A.; Namba, K. Development of 1,3a,6a-triazapentalene-labeled enterobactin as a fluorescence quenching sensor of iron ion. Tetrahedron Lett. 2017, 58, 1961–1964. [Google Scholar] [CrossRef]
- Sawada, J.; Osawa, A.; Takeuchi, T.; Kaneda, M.; Oishi, S.; Fujii, N.; Asai, A.; Tanino, K.; Namba, K. Functional 1,3a,6a-triazapentalene scaffold: Design of fluorescent probes for kinesin spindle protein (KSP). Bioorg. Med. Chem. Lett. 2016, 26, 5765–5769. [Google Scholar] [CrossRef]
- Namba, K.; Mera, A.; Osawa, A.; Sakuda, E.; Kitamura, N.; Tanino, K. One-Pot Synthesis of Highly Fluorescent 2,5-Disubstituted-1,3a,6a-triazapentalene. Org. Lett. 2012, 14, 5554–5557. [Google Scholar] [CrossRef]
- Nakayama, A.; Nishio, S.; Otani, A.; Mera, A.; Osawa, A.; Tanino, K.; Namba, K. Substituent Effect at the C4-Position of 1,3a,6a-Triazapentalene. Chem. Pharm. Bull. 2016, 64, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Mera, A.; Ito, M.; Nakayama, A.; Namba, K. Synthesis of 2,6-Disubstituted-1,3a,6a-Triazapentalenes and Their Fluorescence Properties. Chem. Lett. 2017, 46, 539–542. [Google Scholar] [CrossRef]
- Namba, K.; Osawa, A.; Nakayama, A.; Mera, A.; Tano, F.; Chuman, Y.; Sakuda, E.; Taketsugu, T.; Sakaguchi, K.; Kitamura, N.; et al. Synthesis of yellow and red fluorescent 1,3a,6a-triazapentalenes and the theoretical investigation of their optical properties. Chem. Sci. 2015, 6, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Hirobe, M.; Okamoto, T. Mesoionic 1,3a,6a-triazapentalenes. Tet. Lett. 1978, 19, 1291–1294. [Google Scholar] [CrossRef]
- Prakash, R.; Opsomer, T.; Dehaen, W. Triazolization of Enolizable Ketones with Primary Amines: A General Strategy toward Multifunctional 1,2,3-Triazoles. Chem. Rec. 2021, 21, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Opsomer, T.; Van Meervelt, L.; Dehaen, W. Ring-Degenerate Rearrangement Resulting from the Azo Coupling Reaction of a 3-Aryl-1,3a,6a-triazapentalene. J. Org. Chem. 2020, 85, 9434–9439. [Google Scholar] [CrossRef]
- Sirbu, D.; Chopin, N.; Martinić, I.; Ndiaye, M.; Eliseeva, S.V.; Hiebel, M.-A.; Petoud, S.; Suzenet, F. Pyridazino-1,3a,6a-Triazapentalenes as Versatile Fluorescent Probes: Impact of Their Post-Functionalization and Application for Cellular Imaging. Int. J. Mol. Sci. 2021, 22, 6645. [Google Scholar] [CrossRef] [PubMed]
- Verbelen, B.; Leen, V.; Wang, L.; Boens, N.; Dehaen, W. Direct palladium-catalysed C-H arylation of BODIPY dyes at the 3- and 3,5-positions. Chem. Commun. 2012, 48, 9129–9131. [Google Scholar] [CrossRef]
- Gorelsky, S.I.; Lapointe, D.; Fagnou, K. Analysis of the Palladium-Catalyzed (Aromatic)C–H Bond Metalation–Deprotonation Mechanism Spanning the Entire Spectrum of Arenes. J. Org. Chem. 2012, 77, 658–668. [Google Scholar] [CrossRef]
- Mendiola, J.; Castellote, I.; Alvarez-Builla, J.; Fernández-Gadea, J.; Gómez, A.; Vaquero, J.J. Palladium-catalyzed arylation and heteroarylation of azolopyrimidines. J. Org. Chem. 2006, 71, 1254–1257. [Google Scholar] [CrossRef]
- Pivsa-Art, S.; Satoh, T.; Kawamura, Y.; Miura, M.; Nomura, M. Palladium-Catalyzed Arylation of Azole Compounds with Aryl Halides in the Presence of Alkali Metal Carbonates and the Use of Copper Iodide in the Reaction. Bull. Chem. Soc. Jpn. 1998, 71, 467–473. [Google Scholar] [CrossRef]
- Marquet, A.; Jacques, J. Halogenations selectives au moyen des perhalogenures de phenyltrimethylammonium. Tetrahedron Lett. 1959, 1, 24–26. [Google Scholar] [CrossRef]
- Jacques, J.; Marquet, A. Selective α-Bromination of an Aralkyl Ketone with Phenyltrimethylammonium Tribromide: 2-Bromoacetyl-6-Methoxynaphthalene and 2,2-Dibromoacetyl-6-Methoxynaphthalene. Org. Synth. 1973, 53, 111. [Google Scholar] [CrossRef]
- Ni, J.-S.; Lu, G.-H. Natural protoberberine alkaloid–montmorillonite nanocomposite powders with AIE features for visualizing high-resolution latent fingerprints. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2023, 300, 122908. [Google Scholar] [CrossRef]
- Zha, M.; Lin, X.; Ni, J.-S.; Li, Y.; Zhang, Y.; Zhang, X.; Wang, L.; Li, K. An Ester-Substituted Semiconducting Polymer with Efficient Nonradiative Decay Enhances NIR-II Photoacoustic Performance for Monitoring of Tumor Growth. Angew. Chem. Int. Ed. 2020, 59, 23268–23276. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. On the Theory of Shifts and Broadening of Electronic Spectra of Polar Solutes in Polar Media. J. Chem. Phys. 1965, 43, 1261–1274. [Google Scholar] [CrossRef]
- Verhoeven, J.W.; Wegewijs, B.; Kroon, J.; Rettschnick, R.P.H.; Paddon-Row, M.N.; Oliver, A.M. Rigid donor—Bridge—Acceptor systems: Photoinduced charge transfer in solution and in a supersonic jet. J. Photochem. Photobiol. A Chem. 1994, 82, 161–170. [Google Scholar] [CrossRef]
- Tominaga, K.; Walker, G.C.; Kang, T.J.; Barbara, P.F.; Fonseca, T. Reaction rates in the phenomenological adiabatic excited-state electron-transfer theory. J. Phys. Chem. 1991, 95, 10485–10492. [Google Scholar] [CrossRef]
- Opsomer, T.; Van Hoof, M.; D’Angelo, A.; Dehaen, W. 1,2,3-Triazole-Mediated Synthesis of 1-Methyleneisoquinolines: A Three-Step Synthesis of Papaverine and Analogues. Org. Lett. 2020, 22, 3596–3600. [Google Scholar] [CrossRef]
- CrysAlis PRO; Agilent Technologies UK Ltd.: Oxfordshire, UK, 2012.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Compound | R | Reaction Time | Yield (%) b | ||
| TAP4 | TAP5 | TAP1 | ||||
| 1 | a | H | 72 h | 57 e (21) c | 14 e | - d |
| 2 | b | OCH3 | 48 h | 40 e (29) c | - f | - d |
| 3 | c | COOEt | 72 h | - g (21) c | - g | - d |
| 4 | d | N(CH3)2 | 72 h | - d | - d | 44 |
| 5 | e | OC6H13 | 48 h | - e (29) c | - f | - d |
 | |||
|---|---|---|---|
| TAP1 (Equiv) | PTAB (Equiv) | Yield (%) | |
| TAP6a | TAP6b | ||
| 6 | 1 | 77 | - a |
| 1 | 2.3 | - a | 85 |
 | |||
|---|---|---|---|
| Entry | Compound | R | Yield (%) b |
| 1 | a | H | 94 |
| 2 | b | OMe | 89 |
| 3 | c | COOEt | 46 |
| 4 | d | N(CH3)2 | 87 |
| TAP | λabs, max (nm) | λem, max (nm) | Stokes Shift (cm−1) | FWHMem (cm−1) | φ (%) | |
|---|---|---|---|---|---|---|
| 1 [3] | 399 | 476 | 4050 ± 50 | 79 | ||
| DCM (RT) | 4a | 424 | 503 | 3700 ± 50 | 3420 ± 50 | 12 |
| 4b | 428 | 523 | 4240 ± 50 | 3610 ± 50 | 28 | |
| 4c | 441 | 476 | 1670 ± 50 | 2570 ± 50 | 1 | |
| 4e | 430 | 525 | 4210 ± 50 | 3660 ± 50 | 27 | |
| 5a | 427 | 529 | 4520 ± 50 | 3730 ± 50 | 14 | |
| ACN (RT) | 7 | 394 | 512 | 5850 ± 50 | 4950 ± 50 | 2 |
| 8 | 418 | 522 | 4770 ± 50 | 4020 ± 50 | 4 | |
| MeTHF (77 K) | 4a | 439 | 457 | 900 ± 50 | 2290 ± 50 | |
| 4b | 452 | 466 | 660 ± 50 | 2190 ± 50 | ||
| 4c | 453 | 462 | 430 ± 50 | 1770 ± 50 | ||
| 4e | 452 | 466 | 660 ± 50 | 2090 ± 50 | ||
| 5a | 445 | 467 | 1060 ± 50 | 2330 ± 50 | ||
| 7 | 409 | 423 | 810 ± 50 | 2200 ± 50 | ||
| 8 | 433 | 445 | 620 ± 50 | 2050 ± 50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Opsomer, T.; de Jong, F.; Verhaeghe, D.; Mulier, M.; Van Meervelt, L.; Van der Auweraer, M.; Dehaen, W. Palladium-Catalyzed Arylations towards 3,6-Diaryl-1,3a,6a-triazapentalenes and Evaluation of Their Fluorescence Properties. Molecules 2024, 29, 2229. https://doi.org/10.3390/molecules29102229
Wang Y, Opsomer T, de Jong F, Verhaeghe D, Mulier M, Van Meervelt L, Van der Auweraer M, Dehaen W. Palladium-Catalyzed Arylations towards 3,6-Diaryl-1,3a,6a-triazapentalenes and Evaluation of Their Fluorescence Properties. Molecules. 2024; 29(10):2229. https://doi.org/10.3390/molecules29102229
Chicago/Turabian StyleWang, Yingchun, Tomas Opsomer, Flip de Jong, Davy Verhaeghe, Maarten Mulier, Luc Van Meervelt, Mark Van der Auweraer, and Wim Dehaen. 2024. "Palladium-Catalyzed Arylations towards 3,6-Diaryl-1,3a,6a-triazapentalenes and Evaluation of Their Fluorescence Properties" Molecules 29, no. 10: 2229. https://doi.org/10.3390/molecules29102229