

Ring-Opening Polymerization of Cyclohexene Oxide and Cycloaddition with CO2 Catalyzed by Amine Triphenolate Iron(III) Complexes
Abstract
:
1. Introduction
2. Results and Discussion
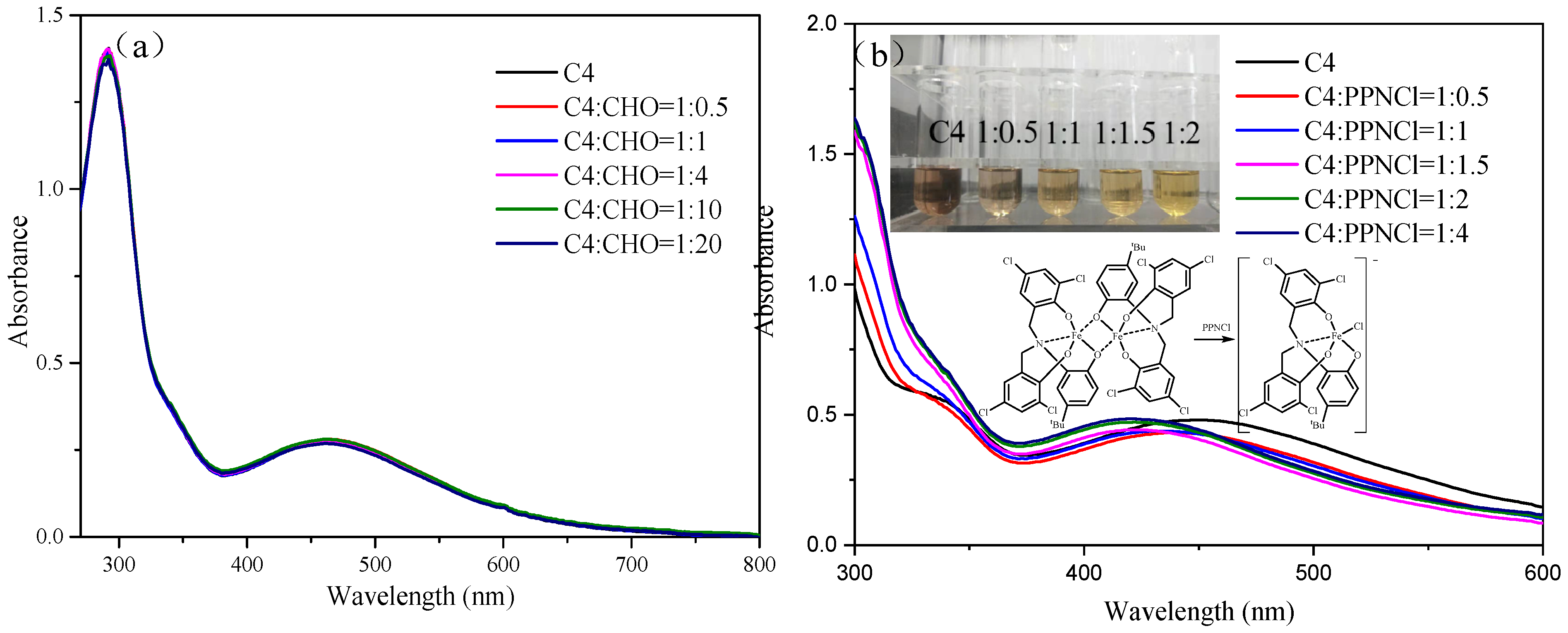
2.1. Synthesis and Characterization of Fe(III) Amine Triphenolate Complexes
2.2. Catalytic Performance of Complexes C1–C4 for the Ring-Opening Polymerization of CHO
2.3. Reaction Kinetic Studies
2.4. Catalytic Mechanism
2.5. Analysis of Polymerization Products
2.6. Catalytic Performance of Complexes C1–C4 for the Reaction of CHO with CO2
2.7. Kinetic Studies of Reactions
2.8. Mechanism of the C4/TBAB-Catalyzed CHO/CO2 Cycloaddition Reaction
3. Experimental Section
3.1. General Experimental Conditions
3.2. Synthesis of Ligands and Characterization
3.2.1. Synthesis of Ligand-1 (R=Cl)
3.2.2. Synthesis of Ligand-2 (R=H)
3.2.3. Synthesis of Ligand-3 (R=Me)
3.2.4. Synthesis of Ligand-4 (R=tBu)
3.3. Synthesis of Complexes and Characterization
3.3.1. Synthesis of Complex-1 (R=Cl)
3.3.2. Synthesis of Complex-2 (R=H)
3.3.3. Synthesis of Complex-3 (R=Me)
3.3.4. Synthesis of Complex-4 (R=tBu)
3.4. General Procedure for the Ring-Opening Polymerization of Cyclohexene Oxide
3.5. General Method for the Cycloaddition of CHO and CO2
3.6. UV Titration Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, Z.; Wu, X.; Guan, D.; Chen, X.; Dai, J.; Gu, Y.; She, S.; Zhou, W.; Shao, Z. One pot-synthesized Ag/Ag-doped CeO2 nanocomposite with rich and stable 3D interfaces and Ce3+ for efficient carbon dioxide electroreduction. ACS Appl. Mater. Interfaces 2021, 13, 59993–60001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, C.; Zhang, P.; Liang, Z. Study of preparation and properties of stereoregular poly(cyclohexenylene carbonate). Molecules 2023, 28, 5235. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xie, Y.F.; Wang, C.; Li, S.J.; Wei, D.H.; Li, M.; Dai, B. Cooperative multifunctional organocatalysts for ambient conversion of carbon dioxide into cyclic carbonates. ACS Catal. 2018, 8, 9945–9957. [Google Scholar] [CrossRef]
- Zhang, Q.; Yuan, H.; Lin, X.; Fukaya, N.; Fujitani, T.; Sato, K.; Choi, J. Calcium carbide as a dehydrating agent for the synthesis of carbamates, glycerol carbonate, and cyclic carbonates from carbon dioxide. Green Chem. 2020, 22, 4231–4239. [Google Scholar] [CrossRef]
- Mao, H.; Guo, M.; Fu, H.; Yin, K.; Jin, M.; Wang, C.; Zhao, Y.; Dong, Z.; Liu, J. A biomass-ligand-based Ru(iii) complex as a catalyst for cycloaddition of CO2 and epoxides to cyclic carbonates and a study of the mechanism. Eur. J. Inorg. Chem. 2023, 26, e202200624. [Google Scholar] [CrossRef]
- Hussong, C.; Langanke, J.; Leitner, W. A green route to polyurethanes: Oxidative carbonylation of industrially relevant aromatic diamines by CO2-based methyl formate. Green Chem. 2020, 22, 8260–8270. [Google Scholar] [CrossRef]
- Guo, L.; Lamb, K.J.; North, M. Recent developments in organocatalysed transformations of epoxides and carbon dioxide into cyclic carbonates. Green Chem. 2021, 23, 77–118. [Google Scholar] [CrossRef]
- Guo, W.; Gómez, J.E.; Cristòfol, À.; Xie, J.; Kleij, A.W. Katalytische umwandlung von funktionalisierten cyclischen organischen carbonaten. Angew. Chem. 2018, 130, 13928–13941. [Google Scholar] [CrossRef]
- Muthuramalingam, S.; Sankaralingam, M.; Velusamy, M.; Mayilmurugan, R. Catalytic conversion of atmospheric CO2 into organic carbonates by nickel(ii) complexes of diazepane-based n4 ligands. Inorg. Chem. 2019, 58, 12975–12985. [Google Scholar] [CrossRef]
- Song, H.; Wang, Y.; Xiao, M.; Liu, L.; Liu, Y.; Liu, X.; Gai, H. Design of novel poly(ionic liquids) for the conversion of CO2 to cyclic carbonates under mild conditions without solvent. ACS Sustain. Chem. Eng. 2019, 7, 9489–9497. [Google Scholar] [CrossRef]
- Gao, P.; Zhao, Z.; Chen, L.; Yuan, D.; Yao, Y. Dinuclear aluminum poly(phenolate) complexes as efficient catalysts for cyclic carbonate synthesis. Organometallics 2016, 35, 1707–1712. [Google Scholar] [CrossRef]
- Taherimehr, M.; Al-Amsyar, S.M.; Whiteoak, C.J.; Kleij, A.W.; Pescarmona, P.P. High activity and switchable selectivity in the synthesis of cyclic and polymeric cyclohexene carbonates with iron amino triphenolate catalysts. Green Chem. 2013, 15, 3083–3090. [Google Scholar] [CrossRef]
- Pescarmona, P.P.; Taherimehr, M. Challenges in the catalytic synthesis of cyclic and polymeric carbonates from epoxides and CO2. Catal. Sci. Technol. 2012, 2, 2169–2187. [Google Scholar] [CrossRef]
- Suresh, L.; Lalrempuia, R.; Ekeli, J.B.; Gillis-D’Hamers, F.; Törnroos, K.W.; Jensen, V.R.; Le Roux, E. Unsaturated and benzannulated n-heterocyclic carbene complexes of titanium and hafnium: Impact on catalysts structure and performance in copolymerization of cyclohexene oxide with CO2. Molecules 2020, 25, 4364. [Google Scholar] [CrossRef] [PubMed]
- Ertürk, E.; Tezeren, M.A.; Tilki, T.; Erdogan, T.; Gören, A.C. Polymerization of epoxides catalyzed by a mixed-valent iron trifluoroacetate [Fe3O(O2CCF3)6(H2O)3]. Polym. Int. 2012, 61, 795–799. [Google Scholar] [CrossRef]
- Sinhababu, S.; Radzhabov, M.R.; Telser, J.; Mankad, N.P. Cooperative activation of CO2 and epoxide by a heterobinuclear Al–Fe complex via radical pair mechanisms. J. Am. Chem. Soc. 2022, 144, 3210–3221. [Google Scholar] [CrossRef] [PubMed]
- Sârbu, T.; Beckman, E.J. Homopolymerization and copolymerization of cyclohexene oxide with carbon dioxide using zinc and aluminum catalysts. Macromolecules 1999, 32, 6904–6912. [Google Scholar] [CrossRef]
- Plommer, H.; Reim, I.; Kerton, F.M. Ring-opening polymerization of cyclohexene oxide using aluminum amine–phenolate complexes. Dalton Trans. 2015, 44, 12098–12102. [Google Scholar] [CrossRef] [PubMed]
- Braune, W.; Okuda, J. An efficient method for controlled propylene oxide polymerization: The significance of bimetallic activation in aluminum lewis acids. Angew. Chem. Int. Ed. 2003, 42, 64–68. [Google Scholar] [CrossRef]
- Rodriguez, C.G.; Ferrier, R.C., Jr.; Helenic, A.; Lynd, N.A. Ring-opening polymerization of epoxides: Facile pathway to functional polyethers via a versatile organoaluminum initiator. Macromolecules 2017, 50, 3121–3130. [Google Scholar] [CrossRef]
- Roy, S.S.; Sarkar, S.; Antharjanam, P.K.S.; Chakraborty, D. Ring-opening copolymerization of CO2 with epoxides catalyzed by binary catalysts containing half salen aluminum compounds and quaternary phosphonium salt. Mol. Catal. 2023, 540, 113053. [Google Scholar] [CrossRef]
- Takeda, N.; Inoue, S. Polymerization of 1,2-epoxypropane and copolymerization with carbon dioxide catalyzed by metalloporphyrins. Die Makromol. Chem. 1978, 179, 1377–1381. [Google Scholar] [CrossRef]
- Shaik, M.; Chidara, V.K.; Abbina, S.; Du, G. Zinc amido-oxazolinate catalyzed ring opening copolymerization and terpolymerization of maleic anhydride and epoxides. Molecules 2020, 25, 4044. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ohkawara, T.; Nozaki, K. Manganese-corrole complexes as versatile catalysts for the ring-opening homo- and co-polymerization of epoxide. Chem. A Eur. J. 2014, 20, 4789–4795. [Google Scholar] [CrossRef] [PubMed]
- Peretti, K.L.; Ajiro, H.; Cohen, C.T.; Lobkovsky, E.B.; Coates, G.W. A highly active, isospecific cobalt catalyst for propylene oxide polymerization. J. Am. Chem. Soc. 2005, 127, 11566–11567. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, H.; Peretti, K.L.; Lobkovsky, E.B.; Coates, G.W. On the mechanism of isospecific epoxide polymerization by salen cobalt(iii) complexes: Evidence for solid-state catalysis. Dalton Trans. 2009, 41, 8828–8830. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, K.; Murphy, J.N.; Kozak, C.M. Chromium amino-bis(phenolate) complexes as catalysts for ring-opening polymerization of cyclohexene oxide. Macromolecules 2019, 52, 7403–7412. [Google Scholar] [CrossRef]
- Cui, D.; Nishiura, M.; Hou, Z. Alternating copolymerization of cyclohexene oxide and carbon dioxide catalyzed by organo rare earth metal complexes. Macromolecules 2005, 38, 4089–4095. [Google Scholar] [CrossRef]
- Thiam, M.; Spassky, N. Polymerization of cyclohexene oxide using yttrium isopropoxide and a bimetallic yttrium-aluminium isopropoxide as initiators. Macromol. Chem. Phys. 1999, 200, 2107–2110. [Google Scholar] [CrossRef]
- Hirahata, W.; Thomas, R.M.; Lobkovsky, E.B.; Coates, G.W. Enantioselective polymerization of epoxides: A highly active and selective catalyst for the preparation of stereoregular polyethers and enantiopure epoxides. J. Am. Chem. Soc. 2008, 130, 17658–17659. [Google Scholar] [CrossRef]
- Thomas, R.M.; Widger, P.C.B.; Ahmed, S.M.; Jeske, R.C.; Hirahata, W.; Lobkovsky, E.B.; Coates, G.W. Enantioselective epoxide polymerization using a bimetallic cobalt catalyst. J. Am. Chem. Soc. 2010, 132, 16520–16525. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Poater, A.; Childers, M.I.; Widger, P.C.B.; Lapointe, A.M.; Lobkovsky, E.B.; Coates, G.W.; Cavallo, L. Enantioselective polymerization of epoxides using biaryl-linked bimetallic cobalt catalysts: A mechanistic study. J. Am. Chem. Soc. 2013, 135, 18901–18911. [Google Scholar] [CrossRef] [PubMed]
- Egorova, K.S.; Ananikov, V.P. Which metals are green for catalysis? Comparison of the toxicities of Ni, Cu, Fe, Pd, Pt, Rh, and Au salts. Angew. Chem. Int. Ed. 2016, 55, 12150–12162. [Google Scholar] [CrossRef] [PubMed]
- Bolm, C.; Legros, J.; Le Paih, J.; Zani, L. Iron-catalyzed reactions in organic synthesis. Chem. Rev. 2004, 104, 6217–6254. [Google Scholar] [CrossRef]
- Fürstner, A. Iron catalysis in organic synthesis: A critical assessment of what it takes to make this base metal a multitasking champion. ACS Cent. Sci. 2016, 2, 778–789. [Google Scholar] [CrossRef]
- Della Monica, F.; Leone, M.; Buonerba, A.; Grassi, A.; Milione, S.; Capacchione, C. CO2 cycloaddition to epoxides promoted by bis-thioether-phenolate Fe(ii) and Fe(iii) complexes. Mol. Catal. 2018, 460, 46–52. [Google Scholar] [CrossRef]
- Tong, L.H.; Wong, Y.; Pascu, S.I.; Dilworth, J.R. Synthesis, structures and catalytic properties of iron(iii) complexes with asymmetric n-capped tripodal NO3 ligands and a pentadentate N2O3 ligand. Dalton Trans. 2008, 35, 4784–4791. [Google Scholar] [CrossRef]
- Whiteoak, C.J.; Martin, E.; Belmonte, M.M.; Benet-Buchholz, J.; Kleij, A.W. An efficient iron catalyst for the synthesis of five- and six-membered organic carbonates under mild conditions. Adv. Synth. Catal. 2012, 354, 469–476. [Google Scholar] [CrossRef]
- Sanford, M.J.; Peña Carrodeguas, L.; Van Zee, N.J.; Kleij, A.W.; Coates, G.W. Alternating copolymerization of propylene oxide and cyclohexene oxide with tricyclic anhydrides: Access to partially renewable aliphatic polyesters with high glass transition temperatures. Macromolecules 2016, 49, 6394–6400. [Google Scholar] [CrossRef]
- Shi, Z.; Jiang, Q.; Song, Z.; Wang, Z.; Gao, C. Dinuclear iron(iii) complexes bearing phenylene-bridged bis(amino triphenolate) ligands as catalysts for the copolymerization of cyclohexene oxide with carbon dioxide or phthalic anhydride. Polym. Chem. 2018, 9, 4733–4743. [Google Scholar] [CrossRef]
- Whiteoak, C.J.; Gjoka, B.; Martin, E.; Belmonte, M.M.; Escudero-Adán, E.C.; Zonta, C.; Licini, G.; Kleij, A.W. Reactivity control in iron(iii) amino triphenolate complexes: Comparison of monomeric and dimeric complexes. Inorg. Chem. 2012, 51, 10639–10649. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, C.; Chisholm, M.H. Influence of the metal (Al, Cr, and Co) and the substituents of the porphyrin in controlling the reactions involved in the copolymerization of propylene oxide and carbon dioxide by porphyrin metal(iii) complexes. 2. Chromium chemistry. Inorg. Chem. 2012, 51, 12041–12052. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Zhang, L.; Kyran, S.J.; Liu, B.; Duan, Z.; Darensbourg, D.J. Copolymerization of carbon dioxide and cyclohexene oxide catalyzed by chromium complexes bearing semirigid [ONSO]-type ligands. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 1938–1944. [Google Scholar] [CrossRef]
- Zevaco, T.A.; Sypien, J.K.; Janssen, A.; Walter, O.; Dinjus, E. Synthesis, structural characterisation of new oligomeric alkyl aluminium (2,2′-methylene-p-chloro-bisphenoxides) and application as catalysts in polymerisation reactions involving cyclohexene oxide. J. Organomet. Chem. 2007, 692, 1963–1973. [Google Scholar] [CrossRef]
- Sokolovicz, Y.C.A.; Buonerba, A.; Capacchione, C.; Dagorne, S.; Grassi, A. Perfluoroaryl zinc catalysts active in cyclohexene oxide homopolymerization and alternating copolymerization with carbon dioxide. Catalysts 2022, 12, 970. [Google Scholar] [CrossRef]
- Liu, B.; Li, H.; Ha, C.; Kim, I.; Yan, W. Ring-opening polymerization of ε-caprolactone and cyclohexene oxide initiated by aluminum β-ketoamino complexes: Steric and electronic effect of 3-position substituents of the ligands. Macromol. Res. 2008, 16, 441–445. [Google Scholar] [CrossRef]
- Chang, H.; Li, D.; Cao, T.; Li, Q.; Bu, Z.; Zhao, W.; Lin, T. Solvent-free ring-opening polymerization of cyclohexene oxide catalyzed by palladium chloride. Polym. Adv. Technol. 2018, 29, 1870–1874. [Google Scholar] [CrossRef]
- Lu, X.; Shi, L.; Wang, Y.; Zhang, R.; Zhang, Y.; Peng, X.; Zhang, Z.; Li, B. Design of highly active binary catalyst systems for CO2/epoxide copolymerization: polymer selectivity, enantioselectivity, and stereochemistry control. J. Am. Chem. Soc. 2006, 128, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Elmas, S.; Subhani, M.A.; Harrer, M.; Leitner, W.; Sundermeyer, J.; Müller, T.E. Highly active Cr(iii) catalysts for the reaction of CO2 with epoxides. Catal. Sci. Technol. 2014, 4, 1652–1657. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, Z.; Ding, K. Intramolecularly dinuclear magnesium complex catalyzed copolymerization of cyclohexene oxide with CO2 under ambient CO2 pressure: kinetics and mechanism. Macromolecules 2006, 39, 128–137. [Google Scholar] [CrossRef]
- Paddock, R.L.; Nguyen, S.T. Chemical CO2 fixation: Cr(iii) salen complexes as highly efficient catalysts for the coupling of CO2 and epoxides. J. Am. Chem. Soc. 2001, 123, 11498–11499. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Yang, H.; Geng, Y.; Wei, Z.; Wang, L.; Guo, C. Novel, recyclable supramolecular metal complexes for the synthesis of cyclic carbonates from epoxides and CO2 under solvent-free conditions. J. CO2 Util. 2017, 17, 243–255. [Google Scholar] [CrossRef]
- Kollenz, G.; Holzer, S.; Kappe, C.O.; Dalvi, T.S.; Fabian, W.M.F.; Sterk, H.; Wong, M.W.; Wentrup, C. Preparation and chemistry of an unexpectedly stable α-oxoketene-pyridine zwitterion, 2,2-bis(tert-butylcarbonyl)-1-[4-(dimethylamino)pyridinio]ethen-1-olate. Eur. J. Org. Chem. 2001, 2001, 1315–1322. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Mackiewicz, R.M.; Billodeaux, D.R. Pressure dependence of the carbon dioxide/cyclohexene oxide coupling reaction catalyzed by chromium salen complexes. Optimization of the comonomer-alternating enchainment pathway. Organometallics 2005, 24, 144–148. [Google Scholar] [CrossRef]
- Basu, D.; Allard, M.M.; Xavier, F.R.; Heeg, M.J.; Schlegel, H.B.; Verani, C.N. Modulation of electronic and redox properties in phenolate-rich cobalt(iii) complexes and their implications for catalytic proton reduction. Dalton Trans. 2015, 44, 3454–3466. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | CHO/Cat | Temp. (°C) | Time (min) | Yield (Conv. a) (%) | TOF (h−1) | Mn (kg/mol) | Đ |
|---|---|---|---|---|---|---|---|---|
| 1 | C1 | 10,000/1 | 25 | 11 | 45.7 (46.9) | 12,474 | 25.3 | 1.78 |
| 2 | C2 | 10,000/1 | 25 | 12 | 48.5 (52.9) | 12,134 | 26.4 | 1.74 |
| 3 | C3 | 10,000/1 | 25 | 13 | 47.8 (51.8) | 11,050 | 25.2 | 1.80 |
| 4 | C4 | 10,000/1 | 25 | 15 | 56.3 (56.9) | 11,262 | 23.4 | 1.88 |
| 5 | C1 | 10,000/1 | 35 | 8 | 53.2 | 19,950 | 21.9 | 1.80 |
| 6 | C2 | 10,000/1 | 35 | 11 | 48.0 | 13,101 | 20.8 | 1.95 |
| 7 | C3 | 10,000/1 | 35 | 12 | 48.4 | 12,110 | 21.2 | 1.93 |
| 8 | C4 | 10,000/1 | 35 | 12 | 50.9 | 12,701 | 22.5 | 1.89 |
| 9 b | C4 | 10,000/1 | 25 | 60 | 0 | 0 | - | - |
| 10 c | C4 | 10,000/1 | 25 | 60 | 0 | 0 | - | - |
| 11 | C1 | 5000/1 | 25 | 3.5 | 53.1 | 22,773 | 23.4 | 1.83 |
| 12 | C1 | 20,000/1 | 25 | 16 | 16.2 | 6062 | 22.9 | 2.02 |
| 13 | C1 | 10,000/1 | 35 | 8 | 56.2 | 21,058 | 19.1 | 2.08 |
| 14 | C1 | 20,000/1 | 35 | 17 | 48.6 | 17,165 | 18.7 | 2.10 |
| 15 | C1 | 40,000/1 | 35 | 60 | 28.9 | 5780 | 17.6 | 2.18 |
| 16 | C1 | 10,000/1 | 45 | 3 | 38.0 | 38,000 | 19.1 | 2.08 |
| 17 | C1 | 10,000/1 | 55 | 2.5 | 36.0 | 42,911 | 17.8 | 2.26 |
| Entry | Solvent | Volume (mL) | Time (min) | Yield (%) | TOF (h−1) | Mn (kg/mol) | Đ |
|---|---|---|---|---|---|---|---|
| 1 | THF | 0.5 | 45 | - | - | - | - |
| 2 | Hexane | 0.5 | 45 | 48.8 | 3251 | 20.2 | 1.98 |
| 3 | Toluene | 0.5 | 30 | 49.4 | 4937 | 21.7 | 1.93 |
| 4 | Toluene | 1 | 45 | 33.9 | 2257 | 20.8 | 1.96 |
| 5 | Dichloromethane | 0.5 | 30 | 55.5 | 5545 | 25.5 | 1.82 |
| 6 | Dichloromethane | 1 | 45 | 61.1 | 4072 | 24.3 | 1.85 |
| 7 | Dichloromethane | 1 | 90 | 69.0 | 2300 | 21.5 | 1.96 |
| 8 | Dichloromethane | 1 | 135 | 70.8 | 1573 | 20.9 | 2.10 |
| 9 | 1,2-Dichloroethane | 0.5 | 30 | 58.9 | 5892 | 24.8 | 1.85 |
| Entry | Cat. | Cocat | CHO/Cat/Cocat | Conv. (%) b | PCHC/cis-CHC c | PCHO (%) |
|---|---|---|---|---|---|---|
| 1 | C4 | - | 2000/1/0 | 20 | - | 99 |
| 2 | - | TBAB | 2000/0/8 | 17 | 1/99 | - |
| 3 | C4 | PPNCl | 2000/1/8 | 67 | 1/99 | <1 |
| 5 | C4 | TBAB | 2000/1/2 | 46 | 2/98 | 2 |
| 6 | C4 | TBAB | 2000/1/4 | 74 | 1/99 | <1 |
| 7 | C4 | TBAB | 2000/1/8 | 80 | 1/99 | <1 |
| 8 | C1 | TBAB | 2000/1/8 | 71 | 1/99 | <1 |
| 9 | C2 | TBAB | 2000/1/8 | 73 | 1/99 | <1 |
| 10 | C3 | TBAB | 2000/1/8 | 74 | 1/99 | <1 |
| Entry | T (°C) | Time (h) | P (MPa) | Conv. (%) | PCHC/cis-CHC (%) | PCHO (%) |
|---|---|---|---|---|---|---|
| 1 | 100 | 4 | 3 | 37 | 1/99 | <1 |
| 2 | 100 | 8 | 3 | 50 | 1/99 | <1 |
| 3 | 100 | 12 | 3 | 65 | 1/99 | <1 |
| 4 | 100 | 16 | 3 | 80 | 1/99 | <1 |
| 5 a | 100 | 12 | 3 | 48 | 1/99 | <1 |
| 6 b | 100 | 12 | 3 | 77 | 1/99 | <1 |
| 7 | 80 | 12 | 3 | 47 | 2/98 | <1 |
| 8 | 120 | 12 | 3 | 85 | 1/99 | <1 |
| 9 | 100 | 12 | 0.1 | 30 | 1/99 | 3 |
| 10 | 100 | 12 | 1.5 | 40 | 1/99 | <1 |
| 11 | 100 | 12 | 4.5 | 68 | 1/99 | <1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Li, S.; Dai, X.; Gao, S.; Song, Z.; Jiang, Q. Ring-Opening Polymerization of Cyclohexene Oxide and Cycloaddition with CO2 Catalyzed by Amine Triphenolate Iron(III) Complexes. Molecules 2024, 29, 2139. https://doi.org/10.3390/molecules29092139
Li P, Li S, Dai X, Gao S, Song Z, Jiang Q. Ring-Opening Polymerization of Cyclohexene Oxide and Cycloaddition with CO2 Catalyzed by Amine Triphenolate Iron(III) Complexes. Molecules. 2024; 29(9):2139. https://doi.org/10.3390/molecules29092139
Chicago/Turabian StyleLi, Peng, Sixuan Li, Xin Dai, Shifeng Gao, Zhaozheng Song, and Qingzhe Jiang. 2024. "Ring-Opening Polymerization of Cyclohexene Oxide and Cycloaddition with CO2 Catalyzed by Amine Triphenolate Iron(III) Complexes" Molecules 29, no. 9: 2139. https://doi.org/10.3390/molecules29092139