
Skeletal Muscle Injury in Chronic Kidney Disease—From Histologic Changes to Molecular Mechanisms and to Novel Therapies

Abstract
:1. Introduction
2. Skeletal Muscle Atrophy in CKD
3. Satellite Cells in CKD
4. Skeletal Muscle Fibrosis in CKD
5. Intramuscular Fat in CKD
6. Skeletal Muscle Inflammation in CKD
7. Mechanisms of CKD-Associated Sarcopenia
8. Phosphate Metabolism and CKD
9. Phosphate and Sarcopenia
10. FGF23 and Sarcopenia
11. Klotho and Sarcopenia
12. PTH and Sarcopenia
13. Vitamin D and Sarcopenia
14. The Effects of Dialysis and Kidney Transplantation on CKD-Associated Sarcopenia
15. Exercise as Therapy for CKD-Associated Sarcopenia
16. Nutrition as Therapy for CKD-Associated Sarcopenia
17. Pharmacological Interventions to Protect from CKD-Associated Sarcopenia
18. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murphy, D.; McCulloch, C.E.; Lin, F.; Banerjee, T.; Bragg-Gresham, J.L.; Eberhardt, M.S.; Morgenstern, H.; Pavkov, M.E.; Saran, R.; Powe, N.R.; et al. Trends in Prevalence of Chronic Kidney Disease in the United States. Ann. Intern. Med. 2016, 165, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Vart, P.; Powe, N.R.; McCulloch, C.E.; Centers for Disease Control and Prevention Chronic Kidney Disease Surveillance Team. National trends in the prevalence of chronic kidney disease among racial/ethnic and socioeconomic status groups, 1988–2016. JAMA Netw. Open 2020, 3, e207932. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.-J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef] [PubMed]
- Moorthi, R.N.; Avin, K.G. Clinical relevance of sarcopenia in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Mohanasundaram, S.; Fernando, E. Uremic Sarcopenia. Indian J. Nephrol. 2022, 32, 399–405. [Google Scholar] [CrossRef] [PubMed]
- de Souza, V.A.; Oliveira, D.; Barbosa, S.R.; Corrêa, J.O.D.A.; Colugnati, F.A.B.; Mansur, H.N.; Fernandes, N.M.d.S.; Bastos, M.G. Sarcopenia in patients with chronic kidney disease not yet on dialysis: Analysis of the prevalence and associated factors. PLoS ONE 2017, 12, e0176230. [Google Scholar] [CrossRef] [PubMed]
- Rolland, Y.; Czerwinski, S.; Abellan Van Kan, G.; Morley, J.E.; Cesari, M.; Onder, G.; Woo, J.; Baumgartner, R.; Pillard, F.; Boirie, Y.; et al. Sarcopenia: Its assessment, etiology, pathogenesis, consequences and future perspectives. J. Nutr. Health Aging 2008, 12, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, F.; Carrero, J.J.; Rodrigues, J.C.; Bigogno, F.G.; Fetter, R.L.; Avesani, C.M. Prevalence of sarcopenia in elderly maintenance hemodialysis patients: The impact of different diagnostic criteria. J. Nutr. Health Aging 2014, 18, 710–717. [Google Scholar] [CrossRef] [PubMed]
- An, J.N.; Kim, J.K.; Lee, H.S.; Kim, S.G.; Kim, H.J.; Song, Y.R. Late stage 3 chronic kidney disease is an independent risk factor for sarcopenia, but not proteinuria. Sci. Rep. 2021, 11, 18472. [Google Scholar] [CrossRef]
- Kim, J.-K.; Kim, S.G.; Oh, J.-E.; Lee, Y.-K.; Noh, J.-W.; Kim, H.J.; Song, Y.R. Impact of sarcopenia on long-term mortality and cardiovascular events in patients undergoing hemodialysis. Korean J. Intern. Med. 2019, 34, 599–607. [Google Scholar] [CrossRef]
- Moon, S.J.; Kim, T.H.; Yoon, S.Y.; Chung, J.H.; Hwang, H.J. Relationship between Stage of Chronic Kidney Disease and Sarcopenia in Korean Aged 40 Years and Older Using the Korea National Health and Nutrition Examination Surveys (KNHANES IV-2, 3, and V-1, 2), 2008–2011. PLoS ONE 2015, 10, e0130740. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.A.; Cordeiro, A.C.; Avesani, C.M.; Carrero, J.J.; Lindholm, B.; Amparo, F.C.; Amodeo, C.; Cuppari, L.; Kamimura, M.A. Sarcopenia in chronic kidney disease on conservative therapy: Prevalence and association with mortality. Nephrol. Dial. Transplant. 2015, 30, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.-D.; Zhang, H.-Z.; Zhang, Y.; Yang, S.-P.; Lin, M.; Zhang, Y.-M.; Wu, J.-B.; Hong, F.-Y.; Chen, W.-X. Relationship between chronic kidney disease and sarcopenia. Sci. Rep. 2021, 11, 20523. [Google Scholar] [CrossRef] [PubMed]
- Troutman, A.D.; Arroyo, E.; Lim, K.; Moorthi, R.N.; Avin, K.G. Skeletal Muscle Complications in Chronic Kidney Disease. Curr. Osteoporos. Rep. 2022, 20, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, L.M.; Vidal, A.; Aguilera-Tejero, E.; Rivero, J.L. Muscle plasticity is influenced by renal function and caloric intake through the FGF23-vitamin D axis. Am. J. Physiol. Cell Physiol. 2023, 324, C14–C28. [Google Scholar] [CrossRef] [PubMed]
- Reese, P.P.; Cappola, A.R.; Shults, J.; Townsend, R.R.; Gadegbeku, C.A.; Anderson, C.; Baker, J.F.; Carlow, D.; Sulik, M.J.; Lo, J.C.; et al. Physical performance and frailty in chronic kidney disease. Am. J. Nephrol. 2013, 38, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Kruse, N.T.; Buzkova, P.; Barzilay, J.I.; Valderrabano, R.J.; Robbins, J.A.; Fink, H.A.; Jalal, D.I. Association of skeletal muscle mass, kidney disease and mortality in older men and women: The cardiovascular health study. Aging 2020, 12, 21023–21036. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.; Krasnoff, J.; Da Silva, M.; Hsu, C.-Y.; Frassetto, L.; Johansen, K.L.; Painter, P. Physical functioning in patients with chronic kidney disease. J. Nephrol. 2008, 21, 550–559. [Google Scholar] [PubMed]
- Ekramzadeh, M.; Santoro, D.; Kopple, J.D. The Effect of Nutrition and Exercise on Body Composition, Exercise Capacity, and Physical Functioning in Advanced CKD Patients. Nutrients 2022, 14, 2129. [Google Scholar] [CrossRef]
- Segura-Orti, E.; Gordon, P.L.; Doyle, J.W.; Johansen, K.L. Correlates of Physical Functioning and Performance Across the Spectrum of Kidney Function. Clin. Nurs. Res. 2018, 27, 579–596. [Google Scholar] [CrossRef]
- Sterky, E.; Stegmayr, B.G. Elderly patients on haemodialysis have 50% less functional capacity than gender- and age-matched healthy subjects. Scand. J. Urol. Nephrol. 2005, 39, 423–430. [Google Scholar] [CrossRef]
- Kim, J.K.; Choi, S.R.; Choi, M.J.; Kim, S.G.; Lee, Y.K.; Noh, J.W.; Kim, H.J.; Song, Y.R. Prevalence of and factors associated with sarcopenia in elderly patients with end-stage renal disease. Clin. Nutr. 2014, 33, 64–68. [Google Scholar] [CrossRef]
- Johansen, K.L. The Frail Dialysis Population: A Growing Burden for the Dialysis Community. Blood Purif. 2015, 40, 288–292. [Google Scholar] [CrossRef]
- Markossian, T.W.; Samra, M.K.; Huisingh-Scheetz, M.; Wadhwa, A. Framework to reduce frailty in hemodialysis patients. Nephrol. Dial. Transplant. 2022, 37, 1396–1399. [Google Scholar] [CrossRef]
- Elder, M.; Moonen, A.; Crowther, S.; Aleksova, J.; Center, J.; Elder, G.J. Chronic kidney disease-related sarcopenia as a prognostic indicator in elderly haemodialysis patients. BMC Nephrol. 2023, 24, 138. [Google Scholar] [CrossRef]
- Chatzipetrou, V.; Bégin, M.-J.; Hars, M.; Trombetti, A. Sarcopenia in Chronic Kidney Disease: A Scoping Review of Prevalence, Risk Factors, Association with Outcomes, and Treatment. Calcif. Tissue Int. 2022, 110, 1–31. [Google Scholar] [CrossRef]
- Watson, E.L.; Major, R.W.; Wilkinson, T.J.; Greening, N.J.; Gould, D.W.; Barratt, J.; Smith, A.C. The association of muscle size, strength and exercise capacity with all-cause mortality in non-dialysis-dependent CKD patients. Clin. Physiol. Funct. Imaging 2020, 40, 399–406. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Carrero, J.J.; von Walden, F.; Ikizler, T.A.; Nader, G.A. Muscle wasting in end-stage renal disease promulgates premature death: Established, emerging and potential novel treatment strategies. Nephrol. Dial. Transplant. 2016, 31, 1070–1077. [Google Scholar] [CrossRef]
- Gracia-Iguacel, C.; González-Parra, E.; Perez-Gomez, M.V.; Mahillo, I.; Egido, J.; Ortiz, A.; Carrero, J.J. Prevalence of protein-energy wasting syndrome and its association with mortality in haemodialysis patients in a centre in Spain. Nefrología 2013, 33, 495–505. [Google Scholar] [CrossRef]
- Roshanravan, B.; Robinson-Cohen, C.; Patel, K.V.; Ayers, E.; Littman, A.J.; de Boer, I.H.; Ikizler, T.A.; Himmelfarb, J.; Katzel, L.I.; Kestenbaum, B.; et al. Association between physical performance and all-cause mortality in CKD. J. Am. Soc. Nephrol. 2013, 24, 822–830. [Google Scholar] [CrossRef]
- Chang, Y.-T.; Wu, H.-L.; Guo, H.-R.; Cheng, Y.-Y.; Tseng, C.-C.; Wang, M.-C.; Lin, C.-Y.; Sung, J.-M. Handgrip strength is an independent predictor of renal outcomes in patients with chronic kidney diseases. Nephrol. Dial. Transplant. 2011, 26, 3588–3595. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Chmielewski, M.; Axelsson, J.; Snaedal, S.; Heimbürger, O.; Bárány, P.; Suliman, M.E.; Lindholm, B.; Stenvinkel, P.; Qureshi, A.R. Muscle atrophy, inflammation and clinical outcome in incident and prevalent dialysis patients. Clin. Nutr. 2008, 27, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Martinson, M.; Ikizler, T.A.; Morrell, G.; Wei, G.; Almeida, N.; Marcus, R.L.; Filipowicz, R.; Greene, T.H.; Beddhu, S. Associations of body size and body composition with functional ability and quality of life in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2014, 9, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, H.S.; Neri, S.G.R.; Oliveira, J.S.; Bennett, P.N.; Viana, J.L.; Lima, R.M. Association between sarcopenia and clinical outcomes in chronic kidney disease patients: A systematic review and meta-analysis. Clin. Nutr. 2022, 41, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Kittiskulnam, P.; Chertow, G.M.; Carrero, J.J.; Delgado, C.; Kaysen, G.A.; Johansen, K.L. Sarcopenia and its individual criteria are associated, in part, with mortality among patients on hemodialysis. Kidney Int. 2017, 92, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Giglio, J.; Kamimura, M.A.; Lamarca, F.; Rodrigues, J.; Santin, F.; Avesani, C.M. Association of Sarcopenia With Nutritional Parameters, Quality of Life, Hospitalization, and Mortality Rates of Elderly Patients on Hemodialysis. J. Ren. Nutr. 2018, 28, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ma, Y.; Lin, F.; Zhao, J.; Xiong, J. Frailty and mortality among patients with chronic kidney disease and end-stage renal disease: A systematic review and meta-analysis. Int. Urol. Nephrol. 2020, 52, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Fouque, D.; Kalantar-Zadeh, K.; Kopple, J.; Cano, N.; Chauveau, P.; Cuppari, L.; Franch, H.; Guarnieri, G.; Ikizler, T.A.; Kaysen, G.; et al. A proposed nomenclature and diagnostic criteria for protein-energy wasting in acute and chronic kidney disease. Kidney Int. 2008, 73, 391–398. [Google Scholar] [CrossRef]
- Noori, N.; Kopple, J.D.; Kovesdy, C.P.; Feroze, U.; Sim, J.J.; Murali, S.B.; Luna, A.; Gomez, M.; Luna, C.; Bross, R.; et al. Mid-arm muscle circumference and quality of life and survival in maintenance hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2010, 5, 2258–2268. [Google Scholar] [CrossRef]
- KKalantar-Zadeh, K.; Streja, E.; Molnar, M.Z.; Lukowsky, L.R.; Krishnan, M.; Kovesdy, C.P.; Greenland, S. Mortality prediction by surrogates of body composition: An examination of the obesity paradox in hemodialysis patients using composite ranking score analysis. Am. J. Epidemiol. 2012, 175, 793–803. [Google Scholar] [CrossRef]
- Ziolkowski, S.L.; Long, J.; Baker, J.F.; Chertow, G.M.; Leonard, M.B. Relative sarcopenia and mortality and the modifying effects of chronic kidney disease and adiposity. J. Cachexia Sarcopenia Muscle 2019, 10, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Beddhu, S.; Pappas, L.M.; Ramkumar, N.; Samore, M. Effects of body size and body composition on survival in hemodialysis patients. J. Am. Soc. Nephrol. 2003, 14, 2366–2372. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.H.; Lee, D.H.; Min, J.; Jeon, J.Y. Handgrip Strength as a Predictor of All-Cause Mortality in Patients With Chronic Kidney Disease Undergoing Dialysis: A Meta-Analysis of Prospective Cohort Studies. J. Ren. Nutr. 2019, 29, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Fanzani, A.; Conraads, V.M.; Penna, F.; Martinet, W. Molecular and cellular mechanisms of skeletal muscle atrophy: An update. J. Cachexia Sarcopenia Muscle 2012, 3, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef] [PubMed]
- Schardong, J.; Marcolino, M.A.Z.; Plentz, R.D.M. Muscle Atrophy in Chronic Kidney Disease. Adv. Exp. Med. Biol. 2018, 1088, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Stenvinkel, P.; Cuppari, L.; Ikizler, T.A.; Kalantar-Zadeh, K.; Kaysen, G.; Mitch, W.E.; Price, S.R.; Wanner, C.; Wang, A.Y.; et al. Etiology of the protein-energy wasting syndrome in chronic kidney disease: A consensus statement from the International Society of Renal Nutrition and Metabolism (ISRNM). J. Ren. Nutr. 2013, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Qureshi, A.R.; Axelsson, J.; Avesani, C.M.; E Suliman, M.; Kato, S.; Bárány, P.; Snaedal-Jonsdottir, S.; Alvestrand, A.; Heimbürger, O.; et al. Comparison of nutritional and inflammatory markers in dialysis patients with reduced appetite. Am. J. Clin. Nutr. 2007, 85, 695–701. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Heimbürger, O.; Paultre, F.; Diczfalusy, U.; Wang, T.; Berglund, L.; Jogestrand, T. Strong association between malnutrition, inflammation, and atherosclerosis in chronic renal failure. Kidney Int. 1999, 55, 1899–1911. [Google Scholar] [CrossRef]
- Stenvinkel, P. Inflammation in end-stage renal disease: The hidden enemy. Nephrology 2006, 11, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Ketteler, M.; Johnson, R.J.; Lindholm, B.; Pecoits-Filho, R.; Riella, M.; Heimbürger, O.; Cederholm, T.; Girndt, M. IL-10, IL-6, and TNF-alpha: Central factors in the altered cytokine network of uremia—The good, the bad, and the ugly. Kidney Int. 2005, 67, 1216–1233. [Google Scholar] [CrossRef] [PubMed]
- Simões e Silva, A.C.; Oliveira, E.A.; Cheung, W.W.; Mak, R.H. Redox Signaling in Chronic Kidney Disease-Associated Cachexia. Antioxidants 2023, 12, 945. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Mitch, W.E.; Price, S.R. Pathophysiological mechanisms leading to muscle loss in chronic kidney disease. Nat. Rev. Nephrol. 2022, 18, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Price, S.R.; Gooch, J.L.; Donaldson, S.K.; Roberts-Wilson, T.K. Muscle atrophy in chronic kidney disease results from abnormalities in insulin signaling. J. Ren. Nutr. 2010, 20 (Suppl. S5), S24–S28. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.A.; Baker, L.A.; Graham-Brown, M.P.M.; Watson, E.L. Skeletal muscle wasting in chronic kidney disease: The emerging role of microRNAs. Nephrol. Dial. Transplant. 2020, 35, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.-C.; Yao, W.-C.; Liu, Y.-L.; Yang, H.-J.; Liao, M.-T.; Chong, K.; Peng, C.-H.; Lu, K.-C. The Potential Influence of Uremic Toxins on the Homeostasis of Bones and Muscles in Chronic Kidney Disease. Biomedicines 2023, 11, 2076. [Google Scholar] [CrossRef] [PubMed]
- Price, S.R.; Mitch, W.E.; Garibotto, G. Muscle Atrophy in CKD: A Historical Perspective of Advancements in Its Understanding. J. Ren. Nutr. 2022, 33, S88–S92. [Google Scholar] [CrossRef]
- Mak, R.H.; Ikizler, A.T.; Kovesdy, C.P.; Raj, D.S.; Stenvinkel, P.; Kalantar-Zadeh, K. Wasting in chronic kidney disease. J. Cachexia Sarcopenia Muscle 2011, 2, 9–25. [Google Scholar] [CrossRef]
- Avesani, C.M.; de Abreu, A.M.; Ribeiro, H.S.; Brismar, T.B.; Stenvinkel, P.; Sabatino, A.; Lindholm, B. Muscle fat infiltration in chronic kidney disease: A marker related to muscle quality, muscle strength and sarcopenia. J. Nephrol. 2023, 36, 895–910. [Google Scholar] [CrossRef]
- Richter, B.; Faul, C. FGF23 Actions on Target Tissues—With and without Klotho. Front. Endocrinol. 2018, 9, 189. [Google Scholar] [CrossRef]
- Baehr, L.M.; Hughes, D.C.; Waddell, D.S.; Bodine, S.C. SnapShot: Skeletal muscle atrophy. Cell 2022, 185, 1618.e1. [Google Scholar] [CrossRef]
- Organ, J.M.; Srisuwananukorn, A.; Price, P.; Joll, J.E.; Biro, K.C.; Rupert, J.E.; Chen, N.X.; Avin, K.G.; Moe, S.M.; Allen, M.R. Reduced skeletal muscle function is associated with decreased fiber cross-sectional area in the Cy/+ rat model of progressive kidney disease. Nephrol. Dial. Transplant. 2016, 31, 223–230. [Google Scholar] [CrossRef]
- Jubrias, S.A.; Odderson, I.R.; Esselman, P.C.; Conley, K.E. Decline in isokinetic force with age: Muscle cross-sectional area and specific force. Pflugers Arch. 1997, 434, 246–253. [Google Scholar] [CrossRef]
- Chen, L.; Nelson, D.R.; Zhao, Y.; Cui, Z.; Johnston, J.A. Relationship between muscle mass and muscle strength, and the impact of comorbidities: A population-based, cross-sectional study of older adults in the United States. BMC Geriatr. 2013, 13, 74. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.-M.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Tamaki, M.; Miyashita, K.; Wakino, S.; Mitsuishi, M.; Hayashi, K.; Itoh, H. Chronic kidney disease reduces muscle mitochondria and exercise endurance and its exacerbation by dietary protein through inactivation of pyruvate dehydrogenase. Kidney Int. 2014, 85, 1330–1339. [Google Scholar] [CrossRef]
- Xu, J.; Li, R.; Workeneh, B.; Dong, Y.; Wang, X.; Hu, Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012, 82, 401–411. [Google Scholar] [CrossRef]
- Su, Z.; Klein, J.D.; Du, J.; Franch, H.A.; Zhang, L.; Hassounah, F.; Hudson, M.B.; Wang, X.H. Chronic kidney disease induces autophagy leading to dysfunction of mitochondria in skeletal muscle. Am. J. Physiol. Ren. Physiol. 2017, 312, F1128–F1140. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Gu, L.J.; Zhu, N.; Wang, L.; Cai, M.C.; Jia, J.S.; Rong, S.; Yuan, W.J. Calpain 6 inhibits autophagy in inflammatory environments: A preliminary study on myoblasts and a chronic kidney disease rat model. Int. J. Mol. Med. 2021, 48, 194. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Q.; Chen, Z.; Wang, Y.; Gamboa, J.L.; Ikizler, T.A.; Garibotto, G.; Mitch, W.E. Mechanisms Regulating Muscle Protein Synthesis in CKD. J. Am. Soc. Nephrol. 2020, 31, 2573–2587. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, C.; Zhang, A.; Cai, H.; Price, S.R.; Wang, X.H. MicroRNA-23a and MicroRNA-27a Mimic Exercise by Ameliorating CKD-Induced Muscle Atrophy. J. Am. Soc. Nephrol. 2017, 28, 2631–2640. [Google Scholar] [CrossRef]
- Kawao, N.; Kawaguchi, M.; Ohira, T.; Ehara, H.; Mizukami, Y.; Takafuji, Y.; Kaji, H. Renal failure suppresses muscle irisin expression, and irisin blunts cortical bone loss in mice. J. Cachexia Sarcopenia Muscle 2022, 13, 758–771. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, J.; Dong, Y.; Tweardy, D.J.; Dong, Y.; Garibotto, G.; Mitch, W.E. Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013, 18, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.L.; Wang, X.; England, B.K.; Price, S.R.; Ding, X.; Mitch, W.E. The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J. Clin. Investig. 1996, 97, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- May, R.C.; Kelly, R.A.; Mitch, W.E. Mechanisms for defects in muscle protein metabolism in rats with chronic uremia. Influence of metabolic acidosis. J. Clin. Investig. 1987, 79, 1099–1103. [Google Scholar] [CrossRef]
- Wang, X.H.; Du, J.; Klein, J.D.; Bailey, J.L.; Mitch, W.E. Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int. 2009, 76, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.T.; Yang, Y.J.; Huang, R.H.; Zhang, Z.H.; Lin, X. Myostatin Activates the Ubiquitin-Proteasome and Autophagy-Lysosome Systems Contributing to Muscle Wasting in Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2015, 2015, 684965. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Wang, M.; Liu, L.; You, H.; Wu, X.; Liu, Y.; Wang, Y.; Lu, L.; Xiao, W.; Wei, L. Calycosin inhibited autophagy and oxidative stress in chronic kidney disease skeletal muscle atrophy by regulating AMPK/SKP2/CARM1 signalling pathway. J. Cell. Mol. Med. 2020, 24, 11084–11099. [Google Scholar] [CrossRef]
- Peng, H.; Cao, J.; Yu, R.; Danesh, F.; Wang, Y.; Mitch, W.E.; Xu, J.; Hu, Z. CKD Stimulates Muscle Protein Loss Via Rho-associated Protein Kinase 1 Activation. J. Am. Soc. Nephrol. 2016, 27, 509–519. [Google Scholar] [CrossRef]
- Qian, F.-Y.; Li, Z.-L.; Guo, Y.-D.; Gao, H.-C.; Gu, L.-H.; Le, K.; Xie, C.-M.; Wang, B.; Zhang, Z.-J. Hypoxia-inducible factor-prolyl hydroxylase inhibitor ameliorates myopathy in a mouse model of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2019, 317, F1265–F1273. [Google Scholar] [CrossRef]
- Kim, K.; Anderson, E.M.; Thome, T.; Lu, G.; Salyers, Z.R.; Cort, T.A.; O’Malley, K.A.; Scali, S.T.; Ryan, T.E. Skeletal myopathy in CKD: A comparison of adenine-induced nephropathy and 5/6 nephrectomy models in mice. Am. J. Physiol. Ren. Physiol. 2021, 321, F106–F119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, X.H.; Wang, H.; Du, J.; Mitch, W.E. Satellite cell dysfunction and impaired IGF-1 signaling cause CKD-induced muscle atrophy. J. Am. Soc. Nephrol. 2010, 21, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rajan, V.; Lin, E.; Hu, Z.; Han, H.Q.; Zhou, X.; Song, Y.; Min, H.; Wang, X.; Du, J.; et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011, 25, 1653–1663. [Google Scholar] [CrossRef] [PubMed]
- Enoki, Y.; Watanabe, H.; Arake, R.; Sugimoto, R.; Imafuku, T.; Tominaga, Y.; Ishima, Y.; Kotani, S.; Nakajima, M.; Tanaka, M.; et al. Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress-mediated expression of myostatin and atrogin-1. Sci. Rep. 2016, 6, 32084. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Komaba, H.; Garcia, A.P.; Economopoulos, K.P.; Liu, W.; Lanske, B.; Hodin, R.A.; Spiegelman, B.M. PTH/PTHrP Receptor Mediates Cachexia in Models of Kidney Failure and Cancer. Cell Metab. 2016, 23, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.R.; Zhan, C.D.; Haddad, F.; Vaziri, N.D. Voluntary exercise during chronic renal failure in rats. Med. Sci. Sports Exerc. 2005, 37, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Bundschu, H.D.; Pfeilsticker, D.; Matthews, C.; Ritz, E. Myopathy in experimental uremia. Res. Exp. Med. 1975, 165, 205–212. [Google Scholar] [CrossRef]
- Flisinski, M.; Brymora, A.; Elminowskawenda, G.; Bogucka, J.; Walasik, K.; Stefanska, A.; Odrowąz, G.; Sypniewska; Manitius, J. Influence of different stages of experimental chronic kidney disease on rats locomotor and postural skeletal muscles microcirculation. Ren. Fail. 2008, 30, 443–451. [Google Scholar] [CrossRef]
- Flisinski, M.; Brymora, A.; Elminowska-Wenda, G.; Bogucka, J.; Walasik, K.; Stefanska, A.; Strozecki, P.; Manitius, J. Morphometric analysis of muscle fibre types in rat locomotor and postural skeletal muscles in different stages of chronic kidney disease. J. Physiol. Pharmacol. 2014, 65, 567–576. [Google Scholar]
- Taes, Y.E.; Speeckaert, M.; Bauwens, E.; De Buyzere, M.R.; Libbrecht, J.; Lameire, N.H.; Delanghe, J.R. Effect of dietary creatine on skeletal muscle myosin heavy chain isoform expression in an animal model of uremia. Nephron Exp. Nephrol. 2004, 96, e103–e110. [Google Scholar] [CrossRef] [PubMed]
- Andres-Hernando, A.; Cicerchi, C.; Garcia, G.E.; Orlicky, D.J.; Stenvinkel, P.; Johnson, R.J.; Lanaspa, M.A. Phosphate depletion in insulin-insensitive skeletal muscle drives AMPD activation and sarcopenia in chronic kidney disease. iScience 2023, 26, 106355. [Google Scholar] [CrossRef]
- Benoit, B.; Beau, A.; Bres, É.; Chanon, S.; Pinteur, C.; Vieille-Marchiset, A.; Jalabert, A.; Zhang, H.; Garg, P.; Strigini, M.; et al. Treatment with fibroblast growth factor 19 increases skeletal muscle fiber size, ameliorates metabolic perturbations and hepatic inflammation in 5/6 nephrectomized mice. Sci. Rep. 2023, 13, 5520. [Google Scholar] [CrossRef]
- Liu, X.; Yu, R.; Sun, L.; Garibotto, G.; Lin, X.; Wang, Y.; Thomas, S.S.; Li, R.; Hu, Z. The nuclear phosphatase SCP4 regulates FoxO transcription factors during muscle wasting in chronic kidney disease. Kidney Int. 2017, 92, 336–348. [Google Scholar] [CrossRef]
- Enoki, Y.; Nagai, T.; Hamamura, Y.; Osa, S.; Nakamura, H.; Taguchi, K.; Watanabe, H.; Maruyama, T.; Matsumoto, K. The G protein-coupled receptor ligand apelin-13 ameliorates skeletal muscle atrophy induced by chronic kidney disease. J. Cachexia Sarcopenia Muscle 2023, 14, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Olauson, H.; Lindberg, K.; Amin, R.; Edvardsson, K.; Lindholm, B.; Andersson, G.; Wernerson, A.; Sabbagh, Y.; Schiavi, S.; et al. A novel model of adenine-induced tubulointerstitial nephropathy in mice. BMC Nephrol. 2013, 14, 116. [Google Scholar] [CrossRef]
- Berru, F.N.; Gray, S.E.; Thome, T.; Kumar, R.A.; Salyers, Z.R.; Coleman, M.; Le, D.; O’malley, K.; Ferreira, L.F.; Berceli, S.A.; et al. Chronic kidney disease exacerbates ischemic limb myopathy in mice via altered mitochondrial energetics. Sci. Rep. 2019, 9, 15547. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, K.; Wakino, S.; Irie, J.; Miyamoto, J.; Matsui, A.; Tajima, T.; Itoh, T.; Oshima, Y.; Yoshifuji, A.; Kimura, I.; et al. Contribution of uremic dysbiosis to insulin resistance and sarcopenia. Nephrol. Dial. Transplant. 2020, 35, 1501–1517. [Google Scholar] [CrossRef] [PubMed]
- Momb, B.A.; Patino, E.; Akchurin, O.M.; Miller, M.S. Iron Supplementation Improves Skeletal Muscle Contractile Properties in Mice with CKD. Kidney360 2022, 3, 843–858. [Google Scholar] [CrossRef]
- Thome, T.; Kumar, R.A.; Burke, S.K.; Khattri, R.B.; Salyers, Z.R.; Kelley, R.C.; Coleman, M.D.; Christou, D.D.; Hepple, R.T.; Scali, S.T.; et al. Impaired muscle mitochondrial energetics is associated with uremic metabolite accumulation in chronic kidney disease. JCI Insight 2020, 6, e139826. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.; Stevens, J.; Binder–Macleod, S.A. Human Skeletal Muscle Fiber Type Classifications. Phys. Ther. 2001, 81, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Brocca, L.; Toniolo, L.; Reggiani, C.; Bottinelli, R.; Sandri, M.; Pellegrino, M.A. FoxO-dependent atrogenes vary among catabolic conditions and play a key role in muscle atrophy induced by hindlimb suspension. J. Physiol. 2017, 595, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, L.M.; Peralta-Ramírez, A.; López, I.; Chamizo, V.E.; Pineda, C.; Rodríguez-Ortiz, M.E.; Rodríguez, M.; Aguilera-Tejero, E.; Rivero, J.-L.L.; Qian, F.-Y.; et al. Slow- and fast-twitch hindlimb skeletal muscle phenotypes 12 wk after (5/6) nephrectomy in Wistar rats of both sexes. Am. J. Physiol. Ren. Physiol. 2015, 309, F638–F647. [Google Scholar] [CrossRef] [PubMed]
- Avin, K.G.; Allen, M.R.; Chen, N.X.; Srinivasan, S.; O’neill, K.D.; Troutman, A.D.; Mast, G.; Swallow, E.A.; Brown, M.B.; Wallace, J.M.; et al. Voluntary Wheel Running Has Beneficial Effects in a Rat Model of CKD-Mineral Bone Disorder (CKD-MBD). J. Am. Soc. Nephrol. 2019, 30, 1898–1909. [Google Scholar] [CrossRef] [PubMed]
- Deger, S.M.; Hung, A.M.; Gamboa, J.L.; Siew, E.D.; Ellis, C.D.; Booker, C.; Sha, F.; Li, H.; Bian, A.; Stewart, T.G.; et al. Systemic inflammation is associated with exaggerated skeletal muscle protein catabolism in maintenance hemodialysis patients. JCI Insight 2017, 2, e95185. [Google Scholar] [CrossRef] [PubMed]
- Solagna, F.; Tezze, C.; Lindenmeyer, M.T.; Lu, S.; Wu, G.; Liu, S.; Zhao, Y.; Mitchell, R.; Meyer, C.; Omairi, S.; et al. Pro-cachectic factors link experimental and human chronic kidney disease to skeletal muscle wasting programs. J. Clin. Investig. 2021, 131, e135821. [Google Scholar] [CrossRef] [PubMed]
- Avin, K.G.; Chen, N.X.; Organ, J.M.; Zarse, C.; O’neill, K.; Conway, R.G.; Konrad, R.J.; Bacallao, R.L.; Allen, M.R.; Moe, S.M. Skeletal Muscle Regeneration and Oxidative Stress Are Altered in Chronic Kidney Disease. PLoS ONE 2016, 11, e0159411. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.; Meehan, D.T.; Grunkemeyer, J.A.; Kornak, J.M.; Sayers, R.; Hunter, W.J.; Samuelson, G.C. Collagen COL4A3 knockout: A mouse model for autosomal Alport syndrome. Genes Dev. 1996, 10, 2981–2992. [Google Scholar] [CrossRef]
- Czaya, B.; Heitman, K.; Campos, I.; Yanucil, C.; Kentrup, D.; Westbrook, D.; Gutierrez, O.; Babitt, J.L.; Jung, G.; Salusky, I.B.; et al. Hyperphosphatemia increases inflammation to exacerbate anemia and skeletal muscle wasting independently of FGF23-FGFR4 signaling. eLife 2022, 11, e74782. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Gu, L.J.; Huang, J.; Cai, M.C.; Yu, H.L.; Zhang, W.; Bao, J.F.; Yuan, W.J. CKD autophagy activation and skeletal muscle atrophy-a preliminary study of mitophagy and inflammation. Eur. J. Clin. Nutr. 2019, 73, 950–960. [Google Scholar] [CrossRef]
- Souweine, J.-S.; Gouzi, F.; Badia, E.; Pomies, P.; Garrigue, V.; Morena, M.; Hayot, M.; Mercier, J.; Ayoub, B.; Le Quintrec, M.; et al. Skeletal Muscle Phenotype in Patients Undergoing Long-Term Hemodialysis Awaiting Kidney Transplantation. Clin. J. Am. Soc. Nephrol. 2021, 16, 1676–1685. [Google Scholar] [CrossRef]
- Baker, L.A.; O’Sullivan, T.F.; Robinson, K.A.; Graham-Brown, M.P.; Major, R.W.; Ashford, R.U.; Smith, A.C.; Philp, A.; Watson, E.L. Primary skeletal muscle cells from chronic kidney disease patients retain hallmarks of cachexia in vitro. J. Cachexia Sarcopenia Muscle 2022, 13, 1238–1249. [Google Scholar] [CrossRef]
- Verzola, D.; Bonanni, A.; Sofia, A.; Montecucco, F.; D’Amato, E.; Cademartori, V.; Parodi, E.L.; Viazzi, F.; Venturelli, C.; Brunori, G.; et al. Toll-like receptor 4 signalling mediates inflammation in skeletal muscle of patients with chronic kidney disease. J. Cachexia Sarcopenia Muscle 2017, 8, 131–144. [Google Scholar] [CrossRef]
- John, S.G.; Sigrist, M.K.; Taal, M.W.; McIntyre, C.W. Natural history of skeletal muscle mass changes in chronic kidney disease stage 4 and 5 patients: An observational study. PLoS ONE 2013, 8, e65372. [Google Scholar] [CrossRef]
- McIntyre, C.W.; Selby, N.M.; Sigrist, M.; Pearce, L.E.; Mercer, T.H.; Naish, P.F. Patients receiving maintenance dialysis have more severe functionally significant skeletal muscle wasting than patients with dialysis-independent chronic kidney disease. Nephrol. Dial. Transplant. 2006, 21, 2210–2216. [Google Scholar] [CrossRef]
- Clyne, N.; Esbjornsson, M.; Jansson, E.; Jogestrand, T.; Lins, L.E.; Pehrsson, S.K. Effects of renal failure on skeletal muscle. Nephron 1993, 63, 395–399. [Google Scholar] [CrossRef]
- Lewis, M.I.; Fournier, M.; Wang, H.; Storer, T.W.; Casaburi, R.; Cohen, A.H.; Kopple, J.D.; Abramowitz, M.K.; Paredes, W.; Zhang, K.; et al. Metabolic and morphometric profile of muscle fibers in chronic hemodialysis patients. J. Appl. Physiol. 2012, 112, 72–78. [Google Scholar] [CrossRef]
- Wang, H.; Casaburi, R.; Taylor, W.E.; Aboellail, H.; Storer, T.W.; Kopple, J.D. Skeletal muscle mRNA for IGF-IEa, IGF-II, and IGF-I receptor is decreased in sedentary chronic hemodialysis patients. Kidney Int. 2005, 68, 352–361. [Google Scholar] [CrossRef]
- Verzola, D.; Procopio, V.; Sofia, A.; Villaggio, B.; Tarroni, A.; Bonanni, A.; Mannucci, I.; De Cian, F.; Gianetta, E.; Saffioti, S.; et al. Apoptosis and myostatin mRNA are upregulated in the skeletal muscle of patients with chronic kidney disease. Kidney Int. 2011, 79, 773–782. [Google Scholar] [CrossRef]
- Castaneda, C.; Gordon, P.L.; Parker, R.C.; Uhlin, K.L.; Roubenoff, R.; Levey, A.S. Resistance training to reduce the malnutrition-inflammation complex syndrome of chronic kidney disease. Am. J. Kidney Dis. 2004, 43, 607–616. [Google Scholar] [CrossRef]
- Sabatino, A.; Regolisti, G.; Delsante, M.; Di Motta, T.; Cantarelli, C.; Pioli, S.; Grassi, G.; Batini, V.; Gregorini, M.; Fiaccadori, E. Noninvasive evaluation of muscle mass by ultrasonography of quadriceps femoris muscle in End-Stage Renal Disease patients on hemodialysis. Clin. Nutr. 2019, 38, 1232–1239. [Google Scholar] [CrossRef]
- Gould, D.W.; Watson, E.L.; Wilkinson, T.J.; Wormleighton, J.; Xenophontos, S.; Viana, J.L.; Smith, A.C. Ultrasound assessment of muscle mass in response to exercise training in chronic kidney disease: A comparison with MRI. J. Cachexia Sarcopenia Muscle 2019, 10, 748–755. [Google Scholar] [CrossRef]
- Wilkinson, T.J.; Gore, E.F.; Vadaszy, N.; Nixon, D.G.D.; Watson, E.L.; Smith, A.C. Utility of Ultrasound as a Valid and Accurate Diagnostic Tool for Sarcopenia: Sex-Specific Cutoff Values in Chronic Kidney Disease. J. Ultrasound Med. 2021, 40, 457–467. [Google Scholar] [CrossRef]
- de Souza, V.A.; Oliveira, D.; Cupolilo, E.N.; Miranda, C.S.; Colugnati, F.A.B.; Mansur, H.N.; Fernandes, N.M.d.S.; Bastos, M.G. Rectus femoris muscle mass evaluation by ultrasound: Facilitating sarcopenia diagnosis in pre-dialysis chronic kidney disease stages. Clinics 2018, 73, e392. [Google Scholar] [CrossRef]
- Holloszy, J.O. The biology of aging. Mayo Clin. Proc. 2000, 75, S3–S8, discussion S8–S9. [Google Scholar] [CrossRef]
- Wang, X.H.; Mitch, W.E. Muscle wasting from kidney failure—A model for catabolic conditions. Int. J. Biochem. Cell Biol. 2013, 45, 2230–2238. [Google Scholar] [CrossRef]
- Gamboa, J.L.; Roshanravan, B.; Towse, T.; Keller, C.A.; Falck, A.M.; Yu, C.; Frontera, W.R.; Brown, N.J.; Ikizler, T.A. Skeletal Muscle Mitochondrial Dysfunction Is Present in Patients with CKD before Initiation of Maintenance Hemodialysis. Clin. J. Am. Soc. Nephrol. 2020, 15, 926–936. [Google Scholar] [CrossRef]
- Kestenbaum, B.; Gamboa, J.; Liu, S.; Ali, A.S.; Shankland, E.; Jue, T.; Giulivi, C.; Smith, L.R.; Himmelfarb, J.; de Boer, I.H.; et al. Impaired skeletal muscle mitochondrial bioenergetics and physical performance in chronic kidney disease. JCI Insight 2020, 5, e133289. [Google Scholar] [CrossRef]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef]
- Bailey, J.L.; Zheng, B.; Hu, Z.; Price, S.R.; Mitch, W.E. Chronic Kidney Disease Causes Defects in Signaling through the Insulin Receptor Substrate/Phosphatidylinositol 3-Kinase/Akt Pathway: Implications for muscle atrophy. J. Am. Soc. Nephrol. 2006, 17, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Karalaki, M.; Fili, S.; Philippou, A.; Koutsilieris, M. Muscle regeneration: Cellular and molecular events. In Vivo 2009, 23, 779–796. [Google Scholar] [PubMed]
- Grefte, S.; Kuijpers-Jagtman, A.M.; Torensma, R.; Von den Hoff, J.W. Skeletal Muscle Development and Regeneration. Stem Cells Dev. 2007, 16, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Molina, T.; Fabre, P.; Dumont, N.A. Fibro-adipogenic progenitors in skeletal muscle homeostasis, regeneration and diseases. Open Biol. 2021, 11, 210110. [Google Scholar] [CrossRef]
- Brightwell, C.R.; Kulkarni, A.S.; Paredes, W.; Zhang, K.; Perkins, J.B.; Gatlin, K.J.; Custodio, M.; Farooq, H.; Zaidi, B.; Pai, R.; et al. Muscle fibrosis and maladaptation occur progressively in CKD and are rescued by dialysis. JCI Insight 2021, 6, e150112. [Google Scholar] [CrossRef]
- Mahdy, M.A.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef]
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef]
- Fry, C.S.; Lee, J.D.; Jackson, J.R.; Kirby, T.J.; Stasko, S.A.; Liu, H.; McCarthy, J.J.; Peterson, C.A.; Dupont-Versteegden, E.E. Regulation of the muscle fiber microenvironment by activated satellite cells during hypertrophy. FASEB J. 2014, 28, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Dong, Y.; Chen, Z.; Mitch, W.E.; Zhang, L. The pathway to muscle fibrosis depends on myostatin stimulating the differentiation of fibro/adipogenic progenitor cells in chronic kidney disease. Kidney Int. 2017, 91, 119–128. [Google Scholar] [CrossRef]
- Alcalde-Estévez, E.; Sosa, P.; Asenjo-Bueno, A.; Plaza, P.; Olmos, G.; Naves-Díaz, M.; Rodríguez-Puyol, D.; López-Ongil, S.; Ruiz-Torres, M.P. Uraemic toxins impair skeletal muscle regeneration by inhibiting myoblast proliferation, reducing myogenic differentiation, and promoting muscular fibrosis. Sci. Rep. 2021, 11, 512. [Google Scholar] [CrossRef]
- Abramowitz, M.K.; Paredes, W.; Zhang, K.; Brightwell, C.R.; Newsom, J.N.; Kwon, H.-J.; Custodio, M.; Buttar, R.S.; Farooq, H.; Zaidi, B.; et al. Skeletal muscle fibrosis is associated with decreased muscle inflammation and weakness in patients with chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2018, 315, F1658–F1669. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-K.; Kim, C.-H. Quality Matters as Much as Quantity of Skeletal Muscle: Clinical Implications of Myosteatosis in Cardiometabolic Health. Endocrinol. Metab. 2021, 36, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Addison, O.; Marcus, R.L.; LaStayo, P.C.; Ryan, A.S. Intermuscular Fat: A Review of the Consequences and Causes. Int. J. Endocrinol. 2014, 2014, 309570. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.L.; Addison, O.; Kidde, J.P.; Dibble, L.E.; Lastayo, P.C. Skeletal muscle fat infiltration: Impact of age, inactivity, and exercise. J. Nutr. Health Aging 2010, 14, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Gueugneau, M.; Coudy-Gandilhon, C.; Theron, L.; Meunier, B.; Barboiron, C.; Combaret, L.; Taillandier, D.; Polge, C.; Attaix, D.; Picard, B.; et al. Skeletal Muscle Lipid Content and Oxidative Activity in Relation to Muscle Fiber Type in Aging and Metabolic Syndrome. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2015, 70, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.; Goodpaster, B.H.; Kritchevsky, S.; Newman, A.B.; Nevitt, M.; Rubin, S.M.; Simonsick, E.M.; Harris, T.B.; for the Health ABC Study. Muscle Mass, Muscle Strength, and Muscle Fat Infiltration as Predictors of Incident Mobility Limitations in Well-Functioning Older Persons. J. Gerontol. Ser. A 2005, 60, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Hilton, T.N.; Tuttle, L.J.; Bohnert, K.L.; Mueller, M.J.; Sinacore, D.R. Excessive Adipose Tissue Infiltration in Skeletal Muscle in Individuals With Obesity, Diabetes Mellitus, and Peripheral Neuropathy: Association With Performance and Function. Phys. Ther. 2008, 88, 1336–1344. [Google Scholar] [CrossRef]
- Engelke, K.; Ghasemikaram, M.; Chaudry, O.; Uder, M.; Nagel, A.M.; Jakob, F.; Kemmler, W. The effect of ageing on fat infiltration of thigh and paraspinal muscles in men. Aging Clin. Exp. Res. 2022, 34, 2089–2098. [Google Scholar] [CrossRef]
- Al Saedi, A.; Debruin, D.A.; Hayes, A.; Hamrick, M. Lipid metabolism in sarcopenia. Bone 2022, 164, 116539. [Google Scholar] [CrossRef]
- Johansen, K.L.; Shubert, T.; Doyle, J.; Soher, B.; Sakkas, G.K.; Kent-Braun, J.A. Muscle atrophy in patients receiving hemodialysis: Effects on muscle strength, muscle quality, and physical function. Kidney Int. 2003, 63, 291–297. [Google Scholar] [CrossRef]
- Lindholm, B.; Alvestrand, A.; Hultman, E.; Ström, J.B. Muscle Water and Electrolytes in Patients Undergoing Continuous Ambulatory Peritoneal Dialysis. Acta Medica Scand. 1986, 219, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-L.; Ding, T.-T.; Lu, S.; Xu, Y.; Tian, J.; Hu, W.-F.; Zhang, J.-Y. Muscle mass loss and intermuscular lipid accumulation were associated with insulin resistance in patients receiving hemodialysis. Chin. Med. J. 2013, 126, 4612–4617. [Google Scholar] [CrossRef] [PubMed]
- Cheema, B.; Abas, H.; Smith, B.; O’Sullivan, A.J.; Chan, M.; Patwardhan, A.; Kelly, J.; Gillin, A.; Pang, G.; Lloyd, B.; et al. Investigation of skeletal muscle quantity and quality in end-stage renal disease. Nephrology 2010, 15, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, T.J.; Gould, D.W.; Nixon, D.G.D.; Watson, E.L.; Smith, A.C. Quality over quantity? Association of skeletal muscle myosteatosis and myofibrosis on physical function in chronic kidney disease. Nephrol. Dial. Transplant. 2019, 34, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Delmonico, M.J.; Harris, T.B.; Visser, M.; Park, S.W.; Conroy, M.B.; Velasquez-Mieyer, P.; Boudreau, R.; Manini, T.M.; Nevitt, M.; Newman, A.B.; et al. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 2009, 90, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Yajima, T. Skeletal muscle density measured by computed tomography as a predictor of mortality in patients receiving hemodialysis. J. Nephrol. 2022, 35, 1535–1537. [Google Scholar] [CrossRef] [PubMed]
- Keddar, M.; Muylle, T.; Carrie, E.; Trefois, P.; Nachit, M.; Crott, R.; Christiaens, C.; Bammens, B.; Jadoul, M.; Goffin, E.; et al. Non-invasive Quantification of Fat Deposits in Skeletal Muscle Predicts Cardiovascular Outcome in Kidney Failure. Front. Physiol. 2020, 11, 130. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, L. The Many Roles of Macrophages in Skeletal Muscle Injury and Repair. Front. Cell Dev. Biol. 2022, 10, 952249. [Google Scholar] [CrossRef]
- Belizário, J.E.; Fontes-Oliveira, C.C.; Borges, J.P.; Kashiabara, J.A.; Vannier, E. Skeletal muscle wasting and renewal: A pivotal role of myokine IL-6. SpringerPlus 2016, 5, 619. [Google Scholar] [CrossRef]
- Haddad, F.; Zaldivar, F.; Cooper, D.M.; Adams, G.R. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005, 98, 911–917. [Google Scholar] [CrossRef] [PubMed]
- McIntire, K.L.; Chen, Y.; Sood, S.; Rabkin, R. Acute uremia suppresses leucine-induced signal transduction in skeletal muscle. Kidney Int. 2014, 85, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.W.; Zheng, R.; Hao, S.; Wang, Z.; Gonzalez, A.; Zhou, P.; Hoffman, H.M.; Mak, R.H. The role of IL-1 in adipose browning and muscle wasting in CKD-associated cachexia. Sci. Rep. 2021, 11, 15141. [Google Scholar] [CrossRef] [PubMed]
- Garibotto, G.; Sofia, A.; Procopio, V.; Villaggio, B.; Tarroni, A.; Di Martino, M.; Cappelli, V.; Gandolfo, M.; Aloisi, F.; De Cian, F.; et al. Peripheral tissue release of interleukin-6 in patients with chronic kidney diseases: Effects of end-stage renal disease and microinflammatory state. Kidney Int. 2006, 70, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.S.; Dominic, E.A.; Pai, A.; Osman, F.; Morgan, M.; Pickett, G.; Shah, V.O.; Ferrando, A.; Moseley, P. Skeletal muscle, cytokines, and oxidative stress in end-stage renal disease. Kidney Int. 2005, 68, 2338–2344. [Google Scholar] [CrossRef] [PubMed]
- Cobo, G.; Lindholm, B.; Stenvinkel, P. Chronic inflammation in end-stage renal disease and dialysis. Nephrol. Dial. Transplant. 2018, 33 (Suppl. S3), iii35–iii40. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, C.L.; Carrero, J.J.; Stenvinkel, P. Recent insights in inflammation-associated wasting in patients with chronic kidney disease. Contrib. Nephrol. 2011, 171, 120–126. [Google Scholar] [CrossRef]
- Ikizler, T.A. Nutrition, inflammation and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2008, 17, 162–167. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Alvestrand, A. Inflammation in End-stage Renal Disease: Sources, Consequences, and Therapy. Semin. Dial. 2002, 15, 329–337. [Google Scholar] [CrossRef]
- Kaizu, Y.; Ohkawa, S.; Odamaki, M.; Ikegaya, N.; Hibi, I.; Miyaji, K.; Kumagai, H. Association between inflammatory mediators and muscle mass in long-term hemodialysis patients. Am. J. Kidney Dis. 2003, 42, 295–302. [Google Scholar] [CrossRef]
- Johansen, K.L.; Kaysen, G.A.; Young, B.S.; Hung, A.M.; da Silva, M.; Chertow, G.M. Longitudinal study of nutritional status, body composition, and physical function in hemodialysis patients. Am. J. Clin. Nutr. 2003, 77, 842–846. [Google Scholar] [CrossRef]
- Delano, M.J.; Moldawer, L.L. The Origins of Cachexia in Acute and Chronic Inflammatory Diseases. Nutr. Clin. Pract. 2006, 21, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Xu, T.; Wang, Y.; Wu, C.; Wang, L.; Yang, X.; Sun, H. The role of inflammatory factors in skeletal muscle injury. Biotarget 2018, 2, 7. [Google Scholar] [CrossRef]
- Ziemkiewicz, N.; Hilliard, G.; Pullen, N.A.; Garg, K. The Role of Innate and Adaptive Immune Cells in Skeletal Muscle Regeneration. Int. J. Mol. Sci. 2021, 22, 3265. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Lindholm, B.; Heimbürger, O. Novel Approaches in an Integrated Therapy of Inflammatory-Associated Wasting in End-Stage Renal Disease. Semin. Dial. 2004, 17, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Degaspari, S.; Tzanno-Martins, C.B.; Fujihara, C.K.; Zatz, R.; Branco-Martins, J.P.; Viel, T.A.; Buck, H.d.S.; Orellana, A.M.M.; Böhmer, A.E.; Lima, L.d.S.; et al. Altered KLOTHO and NF-κB-TNF-α Signaling Are Correlated with Nephrectomy-Induced Cognitive Impairment in Rats. PLoS ONE 2015, 10, e0125271. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Tokumoto, M.; Tatsumoto, N.; Taniguchi, M.; Noguchi, H.; Nakano, T.; Masutani, K.; Ooboshi, H.; Tsuruya, K.; Kitazono, T. Phosphate overload directly induces systemic inflammation and malnutrition as well as vascular calcification in uremia. Am. J. Physiol. Ren. Physiol. 2014, 306, F1418–F1428. [Google Scholar] [CrossRef]
- Mendieta-Condado, E.; Villaseñor-Tapia, E.C.; Gálvez-Gastelum, F.J.; Yáñez-Sánchez, I.; Pizano-Martínez, O.; Canales-Aguirre, A.; Márquez-Aguirre, A.L. Effects of Etanercept on TNF-α Inhibition in Rats with Adenine-Induced Chronic Kidney Disease. BioMed Res. Int. 2022, 2022, 1–8. [Google Scholar] [CrossRef]
- Iida, M.; Ohtomo, S.; Wada, N.A.; Ueda, O.; Tsuboi, Y.; Kurata, A.; Jishage, K.-I.; Horiba, N. TNF-α induces Claudin-1 expression in renal tubules in Alport mice. PLoS ONE 2022, 17, e0265081. [Google Scholar] [CrossRef]
- Lee, B.T.; Ahmed, F.A.; Hamm, L.L.; Teran, F.J.; Chen, C.-S.; Liu, Y.; Shah, K.; Rifai, N.; Batuman, V.; Simon, E.E.; et al. Association of C-reactive protein, tumor necrosis factor-alpha, and interleukin-6 with chronic kidney disease. BMC Nephrol. 2015, 16, 77. [Google Scholar] [CrossRef]
- Mitch, W.E.; Du, J.; Bailey, J.L.; Price, S.R. Mechanisms Causing Muscle Proteolysis in Uremia: The Influence of Insulin and Cytokines. Miner. Electrolyte Metab. 1999, 25, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Costelli, P.; Baccino, F.M. Mechanisms of skeletal muscle depletion in wasting syndromes: Role of ATP-ubiquitin-dependent proteolysis. Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Layne, M.D.; Farmer, S.R. Tumor Necrosis Factor-α and Basic Fibroblast Growth Factor Differentially Inhibit the Insulin-like Growth Factor-I Induced Expression of Myogenin in C2C12 Myoblasts. Exp. Cell Res. 1999, 249, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Langen, R.C.; Schols, A.M.; Kelders, M.C.; Wouters, E.F.; Janssen-Heininger, Y.M. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor-κΒ. FASEB J. 2001, 15, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-P.; Schwartz, R.J. TNF-α regulates early differentiation of C2C12 myoblasts in an autocrine fashion. FASEB J. 2001, 15, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Llovera, M.; García-Martínez, C.; López-Soriano, J.; Carbó, N.; Agell, N.; López-Soriano, F.J.; Argiles, J.M. Role of TNF receptor 1 in protein turnover during cancer cachexia using gene knockout mice. Mol. Cell. Endocrinol. 1998, 142, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-P.; Reid, M.B. NF-κB mediates the protein loss induced by TNF-α in differentiated skeletal muscle myotubes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1165–R1170. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B.; Li, Y.-P. Tumor necrosis factor-α and muscle wasting: A cellular perspective. Respir. Res. 2001, 2, 269–272. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Panguluri, S.K.; Gupta, S.K.; Dahiya, S.; Lundy, R.F.; Kumar, A. Tumor Necrosis Factor-α Regulates Distinct Molecular Pathways and Gene Networks in Cultured Skeletal Muscle Cells. PLoS ONE 2010, 5, e13262. [Google Scholar] [CrossRef]
- Pecoits-Filho, R.; Heimbürger, O.; Bárány, P.; Suliman, M.; Fehrman-Ekholm, I.; Lindholm, B.; Stenvinkel, P. Associations between circulating inflammatory markers and residual renal function in CRF patients. Am. J. Kidney Dis. 2003, 41, 1212–1218. [Google Scholar] [CrossRef]
- Pelosi, M.; De Rossi, M.; Barberi, L.; Musarò, A. IL-6 Impairs Myogenic Differentiation by Downmodulation of p90RSK/eEF2 and mTOR/p70S6K Axes, without Affecting AKT Activity. BioMed Res. Int. 2014, 2014, 206026. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Berardinelli, M.G.; Forcina, L.; Ascenzi, F.; Rizzuto, E.; Sandri, M.; De Benedetti, F.; Scicchitano, B.M.; Musarò, A. Sustained Systemic Levels of IL-6 Impinge Early Muscle Growth and Induce Muscle Atrophy and Wasting in Adulthood. Cells 2021, 10, 1816. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.A.; Baltgalvis, K.A. Interleukin 6 as a Key Regulator of Muscle Mass during Cachexia. Exerc. Sport Sci. Rev. 2010, 38, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Cantini, M.; Massimino, M.; Rapizzi, E.; Rossini, K.; Catani, C.; Dallalibera, L.; Carraro, U. Human Satellite Cell-Proliferation in Vitro Is Regulated by Autocrine Secretion of IL-6 Stimulated by a Soluble Factor(s) Released by Activated Monocytes. Biochem. Biophys. Res. Commun. 1995, 216, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, S. Amphioxus IGF-like peptide induces mouse muscle cell development via binding to IGF receptors and activating MAPK and PI3K/Akt signaling pathways. Mol. Cell. Endocrinol. 2011, 343, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, H.; Xu, Y.; Yu, D.; Li, D.; Liu, X.; Du, W. Insulin-like growth factor-1 (IGF-1) promotes myoblast proliferation and skeletal muscle growth of embryonic chickens via the PI3K/Akt signalling pathway. Cell Biol. Int. 2015, 39, 910–922. [Google Scholar] [CrossRef] [PubMed]
- Bando, H.; Zhang, C.; Takada, Y.; Yamasaki, R.; Saito, S. Impaired secretion of growth hormone-releasing hormone, growth hormone and IGF-I in elderly men. Acta Endocrinol. 1991, 124, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Nilsson, E.; Lindholm, B.; Heimbürger, O.; Barany, P.; Stenvinkel, P.; Qureshi, A.R.; Chen, J. Low-Plasma Insulin-like Growth Factor-1 Associates with Increased Mortality in Chronic Kidney Disease Patients with Reduced Muscle Strength. J. Ren. Nutr. 2023, 33, 298–306. [Google Scholar] [CrossRef]
- Ding, H.; Gao, X.L.; Hirschberg, R.; Vadgama, J.V.; Kopple, J.D. Impaired actions of insulin-like growth factor 1 on protein Synthesis and degradation in skeletal muscle of rats with chronic renal failure. Evidence for a postreceptor defect. J. Clin. Investig. 1996, 97, 1064–1075. [Google Scholar] [CrossRef]
- Thomas, S.S.; Wu, J.; Davogustto, G.; Holliday, M.W.; Eckel-Mahan, K.; Verzola, D.; Garibotto, G.; Hu, Z.; Mitch, W.E.; Taegtmeyer, H. SIRPα Mediates IGF1 Receptor in Cardiomyopathy Induced by Chronic Kidney Disease. Circ. Res. 2022, 131, 207–221. [Google Scholar] [CrossRef]
- Spoto, B.; Pisano, A.; Zoccali, C. Insulin resistance in chronic kidney disease: A systematic review. Am. J. Physiol. Ren. Physiol. 2016, 311, F1087–F1108. [Google Scholar] [CrossRef]
- Nishikawa, H.; Asai, A.; Fukunishi, S.; Nishiguchi, S.; Higuchi, K. Metabolic Syndrome and Sarcopenia. Nutrients 2021, 13, 3519. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.L. Insulin Resistance and Muscle Metabolism in Chronic Kidney Disease. ISRN Endocrinol. 2013, 2013, 329606. [Google Scholar] [CrossRef] [PubMed]
- Siew, E.D.; Ikizler, T.A. Determinants of insulin resistance and its effects on protein metabolism in patients with advanced chronic kidney disease. Contrib. Nephrol. 2008, 161, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Deger, S.M.; Hewlett, J.R.; Gamboa, J.; Ellis, C.D.; Hung, A.M.; Siew, E.D.; Mamnungu, C.; Sha, F.; Bian, A.; Stewart, T.G.; et al. Insulin resistance is a significant determinant of sarcopenia in advanced kidney disease. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E1108–E1120. [Google Scholar] [CrossRef] [PubMed]
- Garibotto, G.; Sofia, A.; Russo, R.; Paoletti, E.; Bonanni, A.; Parodi, E.L.; Viazzi, F.; Verzola, D. Insulin sensitivity of muscle protein metabolism is altered in patients with chronic kidney disease and metabolic acidosis. Kidney Int. 2015, 88, 1419–1426. [Google Scholar] [CrossRef]
- Siew, E.; Pupim, L.; Majchrzak, K.; Shintani, A.; Flakoll, P.; Ikizler, T. Insulin resistance is associated with skeletal muscle protein breakdown in non-diabetic chronic hemodialysis patients. Kidney Int. 2007, 71, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Langley, B.; Berry, C.; Sharma, M.; Kirk, S.; Bass, J.; Kambadur, R. Myostatin, a Negative Regulator of Muscle Growth, Functions by Inhibiting Myoblast Proliferation. J. Biol. Chem. 2000, 275, 40235–40243. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Han, H.Q.; Zhou, X.; Mitch, W.E.; Goldberg, A.L. Myostatin/activin pathway antagonism: Molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol. 2013, 45, 2333–2347. [Google Scholar] [CrossRef]
- Amthor, H.; Macharia, R.; Navarrete, R.; Schuelke, M.; Brown, S.C.; Otto, A.; Voit, T.; Muntoni, F.; Vrbóva, G.; Partridge, T.; et al. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc. Natl. Acad. Sci. USA 2007, 104, 1835–1840. [Google Scholar] [CrossRef]
- Amirouche, A.; Durieux, A.-C.; Banzet, S.; Koulmann, N.; Bonnefoy, R.; Mouret, C.; Bigard, X.; Peinnequin, A.; Freyssenet, D. Down-Regulation of Akt/Mammalian Target of Rapamycin Signaling Pathway in Response to Myostatin Overexpression in Skeletal Muscle. Endocrinology 2009, 150, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Barisione, C.; Picciotto, D.; Garibotto, G.; Koppe, L. Emerging role of myostatin and its inhibition in the setting of chronic kidney disease. Kidney Int. 2019, 95, 506–517. [Google Scholar] [CrossRef]
- Yano, S.; Nagai, A.; Isomura, M.; Yamasaki, M.; Kijima, T.; Takeda, M.; Hamano, T.; Nabika, T. Relationship between Blood Myostatin Levels and Kidney Function:Shimane CoHRE Study. PLoS ONE 2015, 10, e0141035. [Google Scholar] [CrossRef]
- Bataille, S.; Chauveau, P.; Fouque, D.; Aparicio, M.; Koppe, L. Myostatin and muscle atrophy during chronic kidney disease. Nephrol. Dial. Transplant. 2020, 36, 1986–1993. [Google Scholar] [CrossRef] [PubMed]
- Bataille, S.; Dou, L.; Bartoli, M.; Sallée, M.; Aniort, J.; Ferkak, B.; Chermiti, R.; McKay, N.; Da Silva, N.; Burtey, S.; et al. Mechanisms of myostatin and activin A accumulation in chronic kidney disease. Nephrol. Dial. Transplant. 2022, 37, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-C.; Liu, Y.-M.; Liao, M.-T.; Zheng, C.-M.; Lu, C.-L.; Liu, W.-C.; Hung, K.-C.; Lin, S.-M.; Lu, K.-C. Indoxyl sulfate mediates low handgrip strength and is predictive of high hospitalization rates in patients with end-stage renal disease. Front. Med. 2023, 10, 1023383. [Google Scholar] [CrossRef]
- Sato, E.; Mori, T.; Mishima, E.; Suzuki, A.; Sugawara, S.; Kurasawa, N.; Saigusa, D.; Miura, D.; Morikawa-Ichinose, T.; Saito, R.; et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci. Rep. 2016, 6, 36618. [Google Scholar] [CrossRef] [PubMed]
- Thome, T.; Salyers, Z.R.; Kumar, R.A.; Hahn, D.; Berru, F.N.; Ferreira, L.F.; Scali, S.T.; Ryan, T.E. Uremic metabolites impair skeletal muscle mitochondrial energetics through disruption of the electron transport system and matrix dehydrogenase activity. Am. J. Physiol. Cell Physiol. 2019, 317, C701–C713. [Google Scholar] [CrossRef]
- Enoki, Y.; Watanabe, H.; Arake, R.; Fujimura, R.; Ishiodori, K.; Imafuku, T.; Nishida, K.; Sugimoto, R.; Nagao, S.; Miyamura, S.; et al. Potential therapeutic interventions for chronic kidney disease-associated sarcopenia via indoxyl sulfate-induced mitochondrial dysfunction. J. Cachexia Sarcopenia Muscle 2017, 8, 735–747. [Google Scholar] [CrossRef]
- Koppe, L.; Pillon, N.J.; Vella, R.E.; Croze, M.L.; Pelletier, C.C.; Chambert, S.; Massy, Z.; Glorieux, G.; Vanholder, R.; Dugenet, Y.; et al. p-Cresyl Sulfate Promotes Insulin Resistance Associated with CKD. J. Am. Soc. Nephrol. 2013, 24, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Beavers, K.M.; Beavers, D.P.; Serra, M.C.; Bowden, R.G.; Wilson, R.L. Low relative skeletal muscle mass indicative of sarcopenia is associated with elevations in serum uric acid levels: Findings from NHANES III. J. Nutr. Heal. Aging 2009, 13, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Toyoguchi, T.; Inage, K.; Fujimoto, K.; Orita, S.; Suzuki, M.; Kanamoto, H.; Abe, K.; Norimoto, M.; Umimura, T.; et al. Advanced glycation end products are associated with sarcopenia in older women: Aging marker dynamics. J. Women Aging 2019, 33, 328–340. [Google Scholar] [CrossRef]
- Mori, H.; Kuroda, A.; Ishizu, M.; Ohishi, M.; Takashi, Y.; Otsuka, Y.; Taniguchi, S.; Tamaki, M.; Kurahashi, K.; Yoshida, S.; et al. Association of accumulated advanced glycation end-products with a high prevalence of sarcopenia and dynapenia in patients with type 2 diabetes. J. Diabetes Investig. 2021, 10, 1332–1340. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef]
- Griggs, R.C.; Kingston, W.; Jozefowicz, R.F.; Herr, B.E.; Forbes, G.; Halliday, D. Effect of testosterone on muscle mass and muscle protein synthesis. J. Appl. Physiol. 1989, 66, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Qureshi, A.R.; Nakashima, A.; Arver, S.; Parini, P.; Lindholm, B.; Bárány, P.; Heimbürger, O.; Stenvinkel, P. Prevalence and clinical implications of testosterone deficiency in men with end-stage renal disease. Nephrol. Dial. Transplant. 2011, 26, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Cigarrán, S.; Pousa, M.; Castro, M.J.; González, B.; Martínez, A.; Barril, G.; Aguilera, A.; Coronel, F.; Stenvinkel, P.; Carrero, J.J. Endogenous Testosterone, Muscle Strength, and Fat-Free Mass in Men With Chronic Kidney Disease. J. Ren. Nutr. 2013, 23, e89–e95. [Google Scholar] [CrossRef]
- Garibotto, G.; Russo, R.; Sofia, A.; Sala, M.R.; Robaudo, C.; Moscatelli, P.; Deferrari, G.; Tizianello, A. Skeletal muscle protein synthesis and degradation in patients with chronic renal failure. Kidney Int. 1994, 45, 1432–1439. [Google Scholar] [CrossRef]
- Garibotto, G.; Russo, R.; Sofia, A.; Sala, M.R.; Sabatino, C.; Moscatelli, P.; Deferrari, G.; Tizianello, A. Muscle protein turnover in chronic renal failure patients with metabolic acidosis or normal acid-base balance. Miner. Electrolyte Metab. 1996, 22, 58–61. [Google Scholar]
- van Vliet, S.; Skinner, S.K.; Beals, J.W.; Pagni, B.A.; Fang, H.-Y.; Ulanov, A.V.; Li, Z.; Paluska, S.A.; Mazzulla, M.; West, D.W.; et al. Dysregulated Handling of Dietary Protein and Muscle Protein Synthesis After Mixed-Meal Ingestion in Maintenance Hemodialysis Patients. Kidney Int. Rep. 2018, 3, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Adey, D.; Kumar, R.; McCarthy, J.T.; Nair, K.S. Reduced synthesis of muscle proteins in chronic renal failure. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E219–E225. [Google Scholar] [CrossRef] [PubMed]
- Alvestrand, A.; Bergström, J.; Fürst, P.; Germanis, G.; Widstam, U. Effect of essential amino acid supplementation on muscle and plasma free amino acids in chronic uremia. Kidney Int. 1978, 14, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, J.; Furst, P.; Noree, L.O. Treatment of chronic uremic patients with protein-poor diet and oral supply of essential amino acids. I. Nitrogen balance studies. Clin. Nephrol. 1975, 3, 187–194. [Google Scholar] [PubMed]
- Marceca, G.P.; Nigita, G.; Calore, F.; Croce, C.M. MicroRNAs in Skeletal Muscle and Hints on Their Potential Role in Muscle Wasting During Cancer Cachexia. Front. Oncol. 2020, 10, 607196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Li, M.; Wang, B.; Klein, J.D.; Price, S.R.; Wang, X.H. miRNA-23a/27a attenuates muscle atrophy and renal fibrosis through muscle-kidney crosstalk. J. Cachexia Sarcopenia Muscle 2018, 9, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Virkki, L.V.; Biber, J.; Murer, H.; Forster, I.C. Phosphate transporters: A tale of two solute carrier families. Am. J. Physiol. Ren. Physiol. 2007, 293, F643–F654. [Google Scholar] [CrossRef] [PubMed]
- Chande, S.; Bergwitz, C. Role of phosphate sensing in bone and mineral metabolism. Nat. Rev. Endocrinol. 2018, 14, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Komaba, H.; Fukagawa, M. Phosphate—A poison for humans? Kidney Int. 2016, 90, 753–763. [Google Scholar] [CrossRef]
- Quarles, L.D. Endocrine functions of bone in mineral metabolism regulation. J. Clin. Investig. 2008, 118, 3820–3828. [Google Scholar] [CrossRef]
- Quarles, L.D. Role of FGF23 in vitamin D and phosphate metabolism: Implications in chronic kidney disease. Exp. Cell Res. 2012, 318, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.G.; Sezer, S.; Massy, Z.A.; Johansson, L.; Cozzolino, M.; Fouque, D. The role of phosphate in kidney disease. Nat. Rev. Nephrol. 2017, 13, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.S.; Slatopolsky, E. Phosphate Toxicity in CKD: The Killer among Us. Clin. J. Am. Soc. Nephrol. 2016, 11, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, M.; Spriet, L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 817–828. [Google Scholar] [CrossRef]
- Wallimann, T.; Wyss, M.; Brdiczka, D.; Nicolay, K.; Eppenberger, H.M. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: The ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 1992, 281 Pt 1, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Westerblad, H. Role of phosphate and calcium stores in muscle fatigue. J. Physiol. 2001, 536 Pt 3, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Abraham, K.A.; Brault, J.J.; Terjung, R.L. Phosphate uptake and PiT-1 protein expression in rat skeletal muscle. Am. J. Physiol. Cell Physiol. 2004, 287, C73–C78. [Google Scholar] [CrossRef] [PubMed]
- Chande, S.; Caballero, D.; Ho, B.B.; Fetene, J.; Serna, J.; Pesta, D.; Nasiri, A.; Jurczak, M.; Chavkin, N.W.; Hernando, N.; et al. Slc20a1/Pit1 and Slc20a2/Pit2 are essential for normal skeletal myofiber function and survival. Sci. Rep. 2020, 10, 3069. [Google Scholar] [CrossRef]
- Pesta, D.H.; Tsirigotis, D.N.; Befroy, D.E.; Caballero, D.; Jurczak, M.J.; Rahimi, Y.; Cline, G.W.; Dufour, S.; Birkenfeld, A.L.; Rothman, D.L.; et al. Hypophosphatemia promotes lower rates of muscle ATP synthesis. FASEB J. 2016, 30, 3378–3387. [Google Scholar] [CrossRef]
- Hettleman, B.D.; Sabina, R.L.; Drezner, M.K.; Holmes, E.W.; Swain, J.L. Defective adenosine triphosphate synthesis. An explanation for skeletal muscle dysfunction in phosphate-deficient mice. J. Clin. Investig. 1983, 72, 582–589. [Google Scholar] [CrossRef]
- Knochel, J.P.; Barcenas, C.; Cotton, J.R.; Fuller, T.J.; Haller, R.; Carter, N.W. Hypophosphatemia and Rhabdomyolysis. J. Clin. Investig. 1978, 62, 1240–1246. [Google Scholar] [CrossRef]
- Veilleux, L.-N.; Cheung, M.; Ben Amor, M.; Rauch, F. Abnormalities in Muscle Density and Muscle Function in Hypophosphatemic Rickets. J. Clin. Endocrinol. Metab. 2012, 97, E1492–E1498. [Google Scholar] [CrossRef]
- Romagnoli, C.; Iantomasi, T.; Brandi, M.L. Impact of X-Linked Hypophosphatemia on Muscle Symptoms. Genes 2022, 13, 2415. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, L.M.; López, I.; Peralta-Ramírez, A.; Pineda, C.; Chamizo, V.E.; Rodríguez, M.; Aguilera-Tejero, E.; Rivero, J.-L.L. High-phosphorus diet maximizes and low-dose calcitriol attenuates skeletal muscle changes in long-term uremic rats. J. Appl. Physiol. 2016, 120, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.-H.; Liu, S.-T.; Huang, S.-M.; Salter, D.M.; Lee, H.-S.; Hsu, Y.-J. High phosphate induces skeletal muscle atrophy and suppresses myogenic differentiation by increasing oxidative stress and activating Nrf2 signaling. Aging 2020, 12, 21446–21468. [Google Scholar] [CrossRef]
- Peri-Okonny, P.A.; Baskin, K.K.; Iwamoto, G.; Mitchell, J.H.; Smith, S.A.; Kim, H.K.; Szweda, L.I.; Bassel-Duby, R.; Fujikawa, T.; Castorena, C.M.; et al. High-Phosphate Diet Induces Exercise Intolerance and Impairs Fatty Acid Metabolism in Mice. Circulation 2019, 139, 1422–1434. [Google Scholar] [CrossRef]
- Amitani, H.; Chiba, S.; Amitani, M.; Michihara, S.; Takemoto, R.; Han, L.; Fujita, N.; Takahashi, R.; Inui, A. Impact of Ninjin’yoeito on frailty and short life in klotho-hypomorphic (kl/kl) mice. Front. Pharmacol. 2022, 13, 973897. [Google Scholar] [CrossRef] [PubMed]
- Fujitsuka, N.; Asakawa, A.; Morinaga, A.; Amitani, M.S.; Amitani, H.; Katsuura, G.; Sawada, Y.; Sudo, Y.; Uezono, Y.; Mochiki, E.; et al. Increased ghrelin signaling prolongs survival in mouse models of human aging through activation of sirtuin1. Mol. Psychiatry 2016, 21, 1613–1623. [Google Scholar] [CrossRef]
- Nakatani, T.; Sarraj, B.; Ohnishi, M.; Densmore, M.J.; Taguchi, T.; Goetz, R.; Mohammadi, M.; Lanske, B.; Razzaque, M.S. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. FASEB J. 2007, 23, 433–441. [Google Scholar] [CrossRef]
- Erem, S.; Razzaque, M.S. Dietary phosphate toxicity: An emerging global health concern. Histochem. Cell Biol. 2018, 150, 711–719. [Google Scholar] [CrossRef]
- Wada, E.; Yoshida, M.; Kojima, Y.; Nonaka, I.; Ohashi, K.; Nagata, Y.; Shiozuka, M.; Date, M.; Higashi, T.; Nishino, I.; et al. Dietary Phosphorus Overload Aggravates the Phenotype of the Dystrophin-Deficient mdx Mouse. Am. J. Pathol. 2014, 184, 3094–3104. [Google Scholar] [CrossRef] [PubMed]
- Sosa, P.; Alcalde-Estevez, E.; Plaza, P.; Troyano, N.; Alonso, C.; Martínez-Arias, L.; Aroeira, A.E.d.M.; Rodriguez-Puyol, D.; Olmos, G.; López-Ongil, S.; et al. Hyperphosphatemia Promotes Senescence of Myoblasts by Impairing Autophagy Through Ilk Overexpression, A Possible Mechanism Involved in Sarcopenia. Aging Dis. 2018, 9, 769–784. [Google Scholar] [CrossRef]
- Sosa, P.; Alcalde-Estévez, E.; Asenjo-Bueno, A.; Plaza, P.; Carrillo-López, N.; Olmos, G.; López-Ongil, S.; Ruiz-Torres, M.P. Aging-related hyperphosphatemia impairs myogenic differentiation and enhances fibrosis in skeletal muscle. J. Cachexia Sarcopenia Muscle 2021, 12, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- Alcalde-Estévez, E.; Sosa, P.; Asenjo-Bueno, A.; Plaza, P.; Valenzuela, P.L.; Naves-Díaz, M.; Olmos, G.; López-Ongil, S.; Ruiz-Torres, M.P. Dietary phosphate restriction prevents the appearance of sarcopenia signs in old mice. J. Cachexia Sarcopenia Muscle 2023, 14, 1060–1074. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Kao, T.W.; Wang, C.C.; Chen, Y.J.; Wu, C.J.; Chen, W.L. Exploring the link between metabolic syndrome and risk of dysmobility syndrome in elderly population. PLoS ONE 2018, 13, e0207608. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Kao, T.W.; Chou, C.W.; Wu, C.J.; Yang, H.F.; Lai, C.H.; Wu, L.W.; Chen, W.L. Exploring the Link between Serum Phosphate Levels and Low Muscle Strength, Dynapenia, and Sarcopenia. Sci. Rep. 2018, 8, 3573. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, M.; Kosaki, K.; Matsui, M.; Okabe, N.; Saito, C.; Yamagata, K.; Kuro-O, M.; Maeda, S. Association of circulating calciprotein particle levels with skeletal muscle mass and strength in middle-aged and older adults. Hypertens. Res. 2022, 45, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Giacona, J.M.; Afridi, A.; Petric, U.B.; Johnson, T.; Pastor, J.; Ren, J.; Sandon, L.; Malloy, C.; Pandey, A.; Shah, A.; et al. Association between dietary phosphate intake and skeletal muscle energetics in adults without cardiovascular disease. J. Appl. Physiol. 2024, 136, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Veloso, M.P.; Coelho, V.A.; Sekercioglu, N.; Moyses, R.M.A.; Elias, R.M. Phosphate is associated with frailty in older patients with chronic kidney disease not on dialysis. Int. Urol. Nephrol. 2024. online ahead of print. [Google Scholar] [CrossRef]
- Smith, I.C.; Collao, N.; Herzog, W. The effects of inorganic phosphate on contractile function of slow skeletal muscle fibres are length-dependent. Biochem. Biophys. Res. Commun. 2020, 533, 818–823. [Google Scholar] [CrossRef]
- Allen, D.G.; Trajanovska, S. The multiple roles of phosphate in muscle fatigue. Front. Physiol. 2012, 3, 463. [Google Scholar] [CrossRef]
- Raimann, A.; Dangl, A.; Javanmardi, A.; Greber-Platzer, S.; Egerbacher, M.; Pietschmann, P.; Haeusler, G. Elevation of phosphate levels impairs skeletal myoblast differentiation. Cell Tissue Res. 2020, 382, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Sonou, T.; Ohya, M.; Kawakami, K.; Yashiro, M.; Mima, T.; Negi, S.; Shigematsu, T. High phosphate levels promote muscle atrophy via myostatin expression in differentiated L6 myotubes. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Yang, M.; Bao, J.-F.; Gu, L.-J.; Yu, H.-L.; Yuan, W.-J. Phosphate stimulates myotube atrophy through autophagy activation: Evidence of hyperphosphatemia contributing to skeletal muscle wasting in chronic kidney disease. BMC Nephrol. 2018, 19, 45. [Google Scholar] [CrossRef]
- Westerblad, H.; Allen, D.G.; Lännergren, J. Muscle Fatigue: Lactic Acid or Inorganic Phosphate the Major Cause? News Physiol. Sci. 2002, 17, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.B. Phosphate toxicity and SERCA2a dysfunction in sudden cardiac arrest. FASEB J. 2023, 37, e23030. [Google Scholar] [CrossRef]
- Kunkler, K.J.; Everett, L.M.; Breedlove, D.K.; Kempson, S.A. Insulin stimulates sodium-dependent phosphate transport by osteoblast-like cells. Am. J. Physiol. 1991, 260 Pt 1, E751–E755. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.I.; McAteer, J.A.; Kempson, S.A. Insulin stimulates phosphate transport in opossum kidney epithelial cells. Am. J. Physiol. 1990, 258 Pt 2, F1592–F1598. [Google Scholar] [CrossRef]
- Whaley-Connell, A.; Sowers, J.R. Insulin Resistance in Kidney Disease: Is There a Distinct Role Separate from That of Diabetes or Obesity. Cardiorenal Med. 2017, 8, 41–49. [Google Scholar] [CrossRef]
- Umakanthan, M.; Li, J.W.; Sud, K.; Duque, G.; Guilfoyle, D.; Cho, K.; Brown, C.; Boersma, D.; Komala, M.G. Prevalence and Factors Associated with Sarcopenia in Patients on Maintenance Dialysis in Australia—A Single Centre, Cross-Sectional Study. Nutrients 2021, 13, 3284. [Google Scholar] [CrossRef]
- Chazot, G.; Lemoine, S.; Kocevar, G.; Kalbacher, E.; Sappey-Marinier, D.; Rouvière, O.; Juillard, L. Intracellular Phosphate and ATP Depletion Measured by Magnetic Resonance Spectroscopy in Patients Receiving Maintenance Hemodialysis. J. Am. Soc. Nephrol. 2021, 32, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Agoro, R.; White, K.E. Regulation of FGF23 production and phosphate metabolism by bone–kidney interactions. Nat. Rev. Nephrol. 2023, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Agoro, R.; Ni, P.; Noonan, M.L.; White, K.E. Osteocytic FGF23 and Its Kidney Function. Front. Endocrinol. 2020, 11, 592. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.-C.; Moe, O.W.; et al. Regulation of Fibroblast Growth Factor-23 Signaling by Klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Wacker, M.J.; Touchberry, C.D.; Silswal, N.; Brotto, L.; Elmore, C.J.; Bonewald, L.F.; Andresen, J.; Brotto, M. Skeletal Muscle, but not Cardiovascular Function, Is Altered in a Mouse Model of Autosomal Recessive Hypophosphatemic Rickets. Front. Physiol. 2016, 7, 173. [Google Scholar] [CrossRef] [PubMed]
- Aono, Y.; Hasegawa, H.; Yamazaki, Y.; Shimada, T.; Fujita, T.; Yamashita, T.; Fukumoto, S. Anti-FGF-23 neutralizing antibodies ameliorate muscle weakness and decreased spontaneous movement of Hyp mice. J. Bone Miner. Res. 2011, 26, 803–810. [Google Scholar] [CrossRef]
- Castillo, R.F.; Pérez, R.G.; González, A.L. Beneficial effects of physical exercise on the osteo-renal Klotho-FGF-23 axis in Chronic Kidney Disease: A systematic review with meta-analysis. Int. J. Med Sci. 2024, 21, 332–340. [Google Scholar] [CrossRef]
- Neves, R.V.P.; Corrêa, H.L.; Deus, L.A.; Reis, A.L.; Souza, M.K.; Simões, H.G.; Navalta, J.W.; Moraes, M.R.; Prestes, J.; Rosa, T.S. Dynamic not isometric training blunts osteo-renal disease and improves the sclerostin/FGF23/Klotho axis in maintenance hemodialysis patients: A randomized clinical trial. J. Appl. Physiol. 2021, 130, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, H.; Ishigaki, S.; Kinoshita-Katahashi, N.; Niwa, H.; Yasuda, H.; Kumagai, H.; Furuya, R. Plasma levels of fibroblast growth factor-23 are associated with muscle mass in haemodialysis patients. Nephrology 2014, 19, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Egund, L.; Paulin, T.; Ekstubbe, H.; Bartosch, P.; Malmgren, L. Longitudinal Measurements of FGF23, Sarcopenia, Frailty and Fracture in Older Community Dwelling Women. J. Frailty Aging 2023, 12, 166–174. [Google Scholar] [CrossRef]
- Li, D.-J.; Fu, H.; Zhao, T.; Ni, M.; Shen, F.-M. Exercise-stimulated FGF23 promotes exercise performance via controlling the excess reactive oxygen species production and enhancing mitochondrial function in skeletal muscle. Metabolism 2016, 65, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Buskermolen, J.; van der Meijden, K.; Furrer, R.; Mons, D.-J.; van Essen, H.W.; Heijboer, A.C.; Lips, P.; Jaspers, R.T.; Bravenboer, N. Effects of different training modalities on phosphate homeostasis and local vitamin D metabolism in rat bone. PeerJ 2019, 7, e6184. [Google Scholar] [CrossRef] [PubMed]
- Kerschan-Schindl, K.; Skenderi, K.; Wahl-Figlash, K.; Gelles, K.; Föger-Samwald, U.; Thalmann, M.; Tsironi, M.; Szekeres, T.; Pietschmann, P. Increased serum levels of fibroblast growth factor 23 after an ultradistance run. J. Sci. Med. Sport 2021, 24, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; Corsetti, R.; Lanteri, P.; Grasso, D.; Vianello, E.; Marazzi, M.G.; Graziani, R.; Colombini, A.; Galliera, E.; Romanelli, M.M.C.; et al. Reciprocal regulation of calcium-/phosphate-regulating hormones in cyclists during the Giro d’Italia 3-week stage race. Scand. J. Med. Sci. Sports 2014, 24, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Emrich, I.E.; Baier, M.; Zawada, A.M.; Meyer, T.; Fliser, D.; Scharhag, J.; Heine, G.H. Plasma FGF23 does not rise during physical exercise as a physiological model of sympathetic activation. Clin. Res. Cardiol. 2019, 108, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Gardinier, J.D.; Al-Omaishi, S.; Morris, M.D.; Kohn, D.H. PTH signaling mediates perilacunar remodeling during exercise. Matrix Biol. 2016, 52–54, 162–175. [Google Scholar] [CrossRef]
- Jia, W.-H.; Wang, N.-Q.; Yin, L.; Chen, X.; Hou, B.-Y.; Wang, J.-H.; Qiang, G.-F.; Chan, C.B.; Yang, X.-Y.; Du, G.-H. Effects of fasting on the expression pattern of FGFs in different skeletal muscle fibre types and sexes in mice. Biol. Sex Differ. 2020, 11, 9. [Google Scholar] [CrossRef]
- Ochi, E.; Barrington, A.; Wehling-Henricks, M.; Avila, M.; Kuro-O, M.; Tidball, J.G. Klotho regulates the myogenic response of muscle to mechanical loading and exercise. Exp. Physiol. 2023, 108, 1531–1547. [Google Scholar] [CrossRef] [PubMed]
- Kahn, D.; Macias, E.; Zarini, S.; Garfield, A.; Berry, K.Z.; Gerszten, R.; Schoen, J.; Cree-Green, M.; Bergman, B.C. Quantifying the inflammatory secretome of human intermuscular adipose tissue. Physiol. Rep. 2022, 10, e15424. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Kazamel, M.; Benatar, M.; Wuu, J.; Kwon, Y.; Kwan, T.; Jiang, N.; Kentrup, D.; Faul, C.; Alesce, L.; et al. FGF23, a novel muscle biomarker detected in the early stages of ALS. Sci. Rep. 2021, 11, 12062. [Google Scholar] [CrossRef] [PubMed]
- Welc, S.S.; Wehling-Henricks, M.; Kuro-O, M.; Thomas, K.A.; Tidball, J.G. Modulation of Klotho expression in injured muscle perturbs Wnt signalling and influences the rate of muscle growth. Exp. Physiol. 2020, 105, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Avin, K.G.; Vallejo, J.A.; Chen, N.X.; Wang, K.; Touchberry, C.D.; Brotto, M.; Dallas, S.L.; Moe, S.M.; Wacker, M.J. Fibroblast growth factor 23 does not directly influence skeletal muscle cell proliferation and differentiation or ex vivo muscle contractility. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E594–E604. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; Kenny, R.; McMahon, L.P. FGF23 promotes myoblast differentation but attenuates myogenic differentiation. Kidney Int. Rep. 2023, 8, S277. [Google Scholar] [CrossRef]
- Zhao, P.; Hoffman, E.P. Embryonic myogenesis pathways in muscle regeneration. Dev. Dyn. 2004, 229, 380–392. [Google Scholar] [CrossRef]
- Zhao, P.; Caretti, G.; Mitchell, S.; McKeehan, W.L.; Boskey, A.L.; Pachman, L.M.; Sartorelli, V.; Hoffman, E.P. Fgfr4 Is Required for Effective Muscle Regeneration in Vivo. Delineation of a MyoD-Tead2-Fgfr4 transcriptional pathway. J. Biol. Chem. 2006, 281, 429–438. [Google Scholar] [CrossRef]
- Taylor Vi, J.G.T.; Cheuk, A.T.; Tsang, P.S.; Chung, J.-Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.-R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 119, 3395–3407. [Google Scholar] [CrossRef]
- Sato, C.; Iso, Y.; Mizukami, T.; Otabe, K.; Sasai, M.; Kurata, M.; Sanbe, T.; Sekiya, I.; Miyazaki, A.; Suzuki, H. Fibroblast growth factor-23 induces cellular senescence in human mesenchymal stem cells from skeletal muscle. Biochem. Biophys. Res. Commun. 2016, 470, 657–662. [Google Scholar] [CrossRef]
- Li, S.; Chen, J.; Wei, P.; Zou, T.; You, J. Fibroblast Growth Factor 21: A Fascinating Perspective on the Regulation of Muscle Metabolism. Int. J. Mol. Sci. 2023, 24, 16951. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Sherrier, M.; Li, H. Skeletal Muscle and Bone—Emerging Targets of Fibroblast Growth Factor-21. Front. Physiol. 2021, 12, 625287. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. What’s So Special about FGF19—Unique Effects Reported on Skeletal Muscle Mass and Function. Cell Metab. 2017, 26, 287–288. [Google Scholar] [CrossRef]
- Miller, P.D. Chronic kidney disease and the skeleton. Bone Res. 2014, 2, 14044. [Google Scholar] [CrossRef]
- Leal, D.V.; Ferreira, A.; Watson, E.L.; Wilund, K.R.; Viana, J.L. Muscle-Bone Crosstalk in Chronic Kidney Disease: The Potential Modulatory Effects of Exercise. Calcif. Tissue Int. 2021, 108, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Avin, K.G.; Hughes, M.C.; Chen, N.X.; Srinivasan, S.; O’neill, K.D.; Evan, A.P.; Bacallao, R.L.; Schulte, M.L.; Moorthi, R.N.; Gisch, D.L.; et al. Skeletal muscle metabolic responses to physical activity are muscle type specific in a rat model of chronic kidney disease. Sci. Rep. 2021, 11, 9788. [Google Scholar] [CrossRef] [PubMed]
- Lara-Castillo, N.; Johnson, M.L. Bone-Muscle Mutual Interactions. Curr. Osteoporos. Rep. 2020, 18, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, L.; Wang, D.; Aiqudsy, L.; Jiang, J.X.; Xu, H.; Shang, P. Muscle-bone crosstalk and potential therapies for sarco-osteoporosis. J. Cell. Biochem. 2019, 120, 14262–14273. [Google Scholar] [CrossRef]
- Ewendt, F.; Feger, M.; Föller, M. Myostatin regulates the production of fibroblast growth factor 23 (FGF23) in UMR106 osteoblast–like cells. Pflugers Arch. 2021, 473, 969–976. [Google Scholar] [CrossRef]
- Elsurer Afsar, R.; Afsar, B.; Ikizler, T.A. Fibroblast Growth Factor 23 and Muscle Wasting: A Metabolic Point of View. Kidney Int. Rep. 2023, 8, 1301–1314. [Google Scholar] [CrossRef]
- Jurina, A.; Kasumović, D.; Delimar, V.; Kanižaj, T.F.; Japjec, M.; Dujmović, T.; Lovrenčić, M.V.; Starešinić, M. Fibroblast growth factor 23 and its role in bone diseases. Growth Factors 2023, 42, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mencke, R.; Olauson, H.; Hillebrands, J.L. Effects of Klotho on fibrosis and cancer: A renal focus on mechanisms and therapeutic strategies. Adv. Drug Deliv. Rev. 2017, 121, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of Aging in Mice by the Hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Li, Q.; Faul, C. Fibroblast growth factor 23, klotho and heparin. Curr. Opin. Nephrol. Hypertens. 2023, 32, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Zhang, J.; Quiñones, H.; Griffith, C.; Kuro-O, M.; Moe, O.W. Klotho Deficiency Causes Vascular Calcification in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 124–136. [Google Scholar] [CrossRef]
- Neyra, J.A.; Hu, M.C.; Moe, O.W. Klotho in Clinical Nephrology: Diagnostic and Therapeutic Implications. Clin. J. Am. Soc. Nephrol. 2020, 16, 162–176. [Google Scholar] [CrossRef]
- Yamauchi, M.; Hirohashi, Y.; Torigoe, T.; Matsumoto, Y.; Yamashita, K.; Kayama, M.; Sato, N.; Yotsuyanagi, T. Wound healing delays in α-Klotho-deficient mice that have skin appearance similar to that in aged humans—Study of delayed wound healing mechanism. Biochem. Biophys. Res. Commun. 2016, 473, 845–852. [Google Scholar] [CrossRef]
- Sahu, A.; Mamiya, H.; Shinde, S.N.; Cheikhi, A.; Winter, L.L.; Vo, N.V.; Stolz, D.; Roginskaya, V.; Tang, W.-Y.; St. Croix, C.; et al. Age-related declines in α-Klotho drive progenitor cell mitochondrial dysfunction and impaired muscle regeneration. Nat. Commun. 2018, 9, 4859. [Google Scholar] [CrossRef]
- Ahrens, H.E.; Huettemeister, J.; Schmidt, M.; Kaether, C.; von Maltzahn, J. Klotho expression is a prerequisite for proper muscle stem cell function and regeneration of skeletal muscle. Skelet. Muscle 2018, 8, 20. [Google Scholar] [CrossRef]
- Patel, M.; Donaldson, A.; Lewis, A.; Natanek, S.; Lee, J.; Andersson, Y.; Haji, G.; Jackson, S.; Bolognese, B.; Foley, J.; et al. Klotho and smoking—An interplay influencing the skeletal muscle function deficits that occur in COPD. Respir. Med. 2016, 113, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Li, Z.; Lindsey, C.; Wang, Y.; Welc, S.S.; Ramos, J.N.; Khanlou, N.; Kuro-O, M.; Tidball, J.G. Klotho gene silencing promotes pathology in the mdx mouse model of Duchenne muscular dystrophy. Hum. Mol. Genet. 2016, 25, 2465–2482. [Google Scholar] [CrossRef] [PubMed]
- McKee, C.M.; Chapski, D.J.; Vondriska, T.M.; Tidball, J.G.; Wehling-Henricks, M.; Rosa-Garrido, M.; Kuro-O, M. The anti-aging protein Klotho affects early postnatal myogenesis by downregulating Jmjd3 and the canonical Wnt pathway. FASEB J. 2022, 36, e22192. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Welc, S.S.; Samengo, G.; Rinaldi, C.; Lindsey, C.; Wang, Y.; Lee, J.; Kuro-O, M.; Tidball, J.G. Macrophages escape Klotho gene silencing in the mdx mouse model of Duchenne muscular dystrophy and promote muscle growth and increase satellite cell numbers through a Klotho-mediated pathway. Hum. Mol. Genet. 2017, 27, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, Y.; Ohtsubo, H.; Munekane, A.; Ohkubo, K.; Murakami, T.; Fujino, M.; Nishimatsu, S.-I.; Hagiwara, H.; Nishimura, H.; Kaneko, R.; et al. Circulating α-Klotho Counteracts Transforming Growth Factor-β–Induced Sarcopenia. Am. J. Pathol. 2023, 193, 591–607. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, M.; Nakatani, T.; Lanske, B.; Razzaque, M.S. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1α-hydroxylase. Kidney Int. 2009, 75, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, M.; Razzaque, M.S. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010, 24, 3562–3571. [Google Scholar] [CrossRef] [PubMed]
- Avin, K.G.; Coen, P.M.; Huang, W.; Stolz, D.B.; Sowa, G.A.; Dubé, J.J.; Goodpaster, B.H.; O’Doherty, R.M.; Ambrosio, F. Skeletal muscle as a regulator of the longevity protein, Klotho. Front. Physiol. 2014, 5, 189. [Google Scholar] [CrossRef]
- Clemens, Z.; Sivakumar, S.; Pius, A.; Sahu, A.; Shinde, S.; Mamiya, H.; Luketich, N.; Cui, J.; Dixit, P.; Hoeck, J.D.; et al. The biphasic and age-dependent impact of klotho on hallmarks of aging and skeletal muscle function. eLife 2021, 10, e61138. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Turner, M.C. Klotho, the Greek goddess controlling the fate of skeletal muscle satellite cells. Exp. Physiol. 2023, 108, 1451–1452. [Google Scholar] [CrossRef]
- Sahu, A.; Clemens, Z.J.; Shinde, S.N.; Sivakumar, S.; Pius, A.; Bhatia, A.; Picciolini, S.; Carlomagno, C.; Gualerzi, A.; Bedoni, M.; et al. Regulation of aged skeletal muscle regeneration by circulating extracellular vesicles. Nat. Aging 2021, 1, 1148–1161. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.-J.; Chu, M.M.; Toussaint, N.D.; Cai, M.M.; Hewitson, T.D.; Holt, S.G. High-intensity physical exercise increases serum α-klotho levels in healthy volunteers. J. Circ. Biomarkers 2018, 7, 1849454418794582. [Google Scholar] [CrossRef] [PubMed]
- King, K.E.; McCormick, J.J.; Notley, S.R.; Fujii, N.; Kenny, G.P. Serum klotho concentrations in young and older men during prolonged exercise in temperate and hot conditions. Curr. Aging Sci. 2022, 15, 180–185. [Google Scholar] [CrossRef]
- Santos-Dias, A.; MacKenzie, B.; Oliveira-Junior, M.C.; Moyses, R.M.; Consolim-Colombo, F.M.; Vieira, R.P. Longevity protein klotho is induced by a single bout of exercise. Br. J. Sports Med. 2017, 51, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Miyaki, A.; Akazawa, N.; Choi, Y.; Ra, S.-G.; Tanahashi, K.; Kumagai, H.; Oikawa, S.; Maeda, S. Aerobic exercise training increases plasma Klotho levels and reduces arterial stiffness in postmenopausal women. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H348–H355. [Google Scholar] [CrossRef] [PubMed]
- Middelbeek, R.J.W.; Motiani, P.; Brandt, N.; Nigro, P.; Zheng, J.; Virtanen, K.A.; Kalliokoski, K.K.; Hannukainen, J.C.; Goodyear, L.J. Exercise intensity regulates cytokine and klotho responses in men. Nutr. Diabetes 2021, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Lomas, G.; Plaza-Florido, A.; De-La-O, A.; Castillo, M.J.; Amaro-Gahete, F.J. Exercise-induced changes in plasma S-Klotho levels are associated with the obtained enhancements of heart rate variability in sedentary middle-aged adults: The FIT-AGEING study. J. Physiol. Biochem. 2024, 80, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.; Luan, J.; Hu, F.; Zhao, Y.; Lv, B.; Wang, W.; Xia, M.; Zhao, X.; Lao, K. Aerobic exercise-stimulated Klotho upregulation extends life span by attenuating the excess production of reactive oxygen species in the brain and kidney. Exp. Ther. Med. 2018, 16, 3511–3517. [Google Scholar] [CrossRef]
- Baghaiee, B.; Karimi, P.; Siahkouhian, M.; Pescatello, L.S. Moderate aerobic exercise training decreases middle-aged induced pathologic cardiac hypertrophy by improving Klotho expression, MAPK signaling pathway, and oxidative stress status in Wistar rats. Iran. J. Basic Med. Sci. 2018, 21, 911–919. [Google Scholar]
- Rao, Z.; Zheng, L.; Huang, H.; Feng, Y.; Shi, R. α-Klotho Expression in Mouse Tissues Following Acute Exhaustive Exercise. Front. Physiol. 2019, 10, 1498. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Cappola, A.R.; Sun, K.; Bandinelli, S.; Dalal, M.; Crasto, C.; Guralnik, J.M.; Ferrucci, L. Relationship of low plasma klotho with poor grip strength in older community-dwelling adults: The InCHIANTI study. Eur. J. Appl. Physiol. 2012, 112, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Crasto, C.L.; Semba, R.D.; Sun, K.; Cappola, A.R.; Bandinelli, S.; Ferrucci, L. Relationship of Low-Circulating “Anti-Aging” Klotho Hormone with Disability in Activities of Daily Living among Older Community-Dwelling Adults. Rejuvenation Res. 2012, 15, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Ferrucci, L.; Sun, K.; Simonsick, E.; Turner, R.; Miljkovic, I.; Harris, T.; Schwartz, A.V.; Asao, K.; Kritchevsky, S.; et al. Low Plasma Klotho Concentrations and Decline of Knee Strength in Older Adults. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Shardell, M.; Semba, R.D.; Kalyani, R.R.; Hicks, G.E.; Bandinelli, S.; Ferrucci, L. Serum 25-Hydroxyvitamin D, Plasma Klotho, and Lower-Extremity Physical Performance Among Older Adults: Findings From the InCHIANTI Study. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, P.L.; Cobo, F.; Diez-Vega, I.; Sánchez-Hernández, R.; Pedrero-Chamizo, R.; Verde-Rello, Z.; González-Gross, M.; Santiago, C.; Ruiz, M.P. Physical performance, plasma S-klotho, and all-cause mortality in elderly dialysis patients: A prospective cohort study. Exp. Gerontol. 2019, 122, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Pavik, I.; Jaeger, P.; Ebner, L.; Wagner, C.A.; Petzold, K.; Spichtig, D.; Poster, D.; Wüthrich, R.P.; Russmann, S.; Serra, A.L. Secreted Klotho and FGF23 in chronic kidney disease Stage 1 to 5: A sequence suggested from a cross-sectional study. Nephrol. Dial. Transplant. 2013, 28, 352–359. [Google Scholar] [CrossRef]
- Shimamura, Y.; Hamada, K.; Inoue, K.; Ogata, K.; Ishihara, M.; Kagawa, T.; Inoue, M.; Fujimoto, S.; Ikebe, M.; Yuasa, K.; et al. Serum levels of soluble secreted α-Klotho are decreased in the early stages of chronic kidney disease, making it a probable novel biomarker for early diagnosis. Clin. Exp. Nephrol. 2012, 16, 722–729. [Google Scholar] [CrossRef]
- Fakhrpour, R.; Khosroshahi, H.H.T.; Ebrahim, K.; Ahmadizad, S.; Abbasnejad, M.; Abbasi, M.M.; Ghanbari, A.; Yaghoobi, S.F. Effect of Sixteen Weeks Combined Training on FGF-23, Klotho, and Fetuin-A Levels in Patients on Maintenance Hemodialysis. Iran J. Kidney Dis. 2020, 14, 212–218. [Google Scholar]
- Kemper, B.; Habener, J.F.; Rich, A.; Potts, J.T., Jr. Parathyroid secretion: Discovery of a major calcium-dependent protein. Science 1974, 184, 167–169. [Google Scholar] [CrossRef]
- Kemper, B.; Habener, J.F.; Potts, J.T., Jr.; Rich, A. Proparathyroid hormone: Identification of a biosynthetic precursor to parathyroid hormone. Proc. Natl. Acad. Sci. USA 1972, 69, 643–647. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, R.; Hamilton, J.; Kent, G.; Shofstall, R.; Cohn, D. The degradation of proparathormone and parathormone by parathyroid and liver cathepsin B. J. Biol. Chem. 1979, 254, 4428–4433. [Google Scholar] [CrossRef] [PubMed]
- Jüppner, H.; Abou-Samra, A.-B.; Freeman, M.; Kong, X.F.; Schipani, E.; Richards, J.; Kolakowski, L.F.; Hock, J.; Potts, J.T.; Kronenberg, H.M.; et al. A G Protein-Linked Receptor for Parathyroid Hormone and Parathyroid Hormone-Related Peptide. Science 1991, 254, 1024–1026. [Google Scholar] [CrossRef] [PubMed]
- Behar, V.; Pines, M.; Nakamoto, C.; Greenberg, Z.; Bisello, A.; Stueckle, S.M.; Bessalle, R.; Usdin, T.B.; Chorev, M.; Rosenblatt, M.; et al. The human PTH2 receptor: Binding and signal transduction properties of the stably expressed recombinant receptor. Endocrinology 1996, 137, 2748–2757. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.P.; Romero, G.; Friedman, P.A.; Gardella, T.J. Molecular basis of parathyroid hormone receptor signaling and trafficking: A family B GPCR paradigm. Cell. Mol. Life Sci. 2010, 68, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, K.D.; Weber, M.; Piper, H.M. Parathyroid hormone induces protein kinase C but not adenylate cyclase in adult cardiomyocytes and regulates cyclic AMP levels via protein kinase C-dependent phosphodiesterase activity. Biochem. J. 1995, 310 Pt 2, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.C.; Bilezikian, J.P. Parathyroid hormone: Anabolic and catabolic actions on the skeleton. Curr. Opin. Pharmacol. 2015, 22, 41–50. [Google Scholar] [CrossRef]
- Isakova, T.; Anderson, C.A.M.; Leonard, M.B.; Xie, D.; Gutiérrez, O.M.; Rosen, L.K.; Theurer, J.; Bellovich, K.; Steigerwalt, S.P.; Tang, I.; et al. Diuretics, calciuria and secondary hyperparathyroidism in the Chronic Renal Insufficiency Cohort. Nephrol. Dial. Transplant. 2011, 26, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Bakris, G.L.; Molitch, M.; Smulders, M.; Tian, J.; Williams, L.A.; Andress, D.L. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: Results of the study to evaluate early kidney disease. Kidney Int. 2007, 71, 31–38. [Google Scholar] [CrossRef]
- Lishmanov, A.; Dorairajan, S.; Pak, Y.; Chaudhary, K.; Chockalingam, A. Elevated serum parathyroid hormone is a cardiovascular risk factor in moderate chronic kidney disease. Int. Urol. Nephrol. 2012, 44, 541–547. [Google Scholar] [CrossRef]
- Natoli, J.L.; Boer, R.; Nathanson, B.H.; Miller, R.M.; Chiroli, S.; Goodman, W.G.; Belozeroff, V. Is there an association between elevated or low serum levels of phosphorus, parathyroid hormone, and calcium and mortality in patients with end stage renal disease? A meta-analysis. BMC Nephrol. 2013, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- De Boer, I.H.; Gorodetskaya, I.; Young, B.; Hsu, C.Y.; Chertow, G.M. The severity of secondary hyperparathyroidism in chronic renal insufficiency is GFR-dependent, race-dependent, and associated with cardiovascular disease. J. Am. Soc. Nephrol. 2002, 13, 2762–2769. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.; Deeg, D.J.H.; Lips, P. Low Vitamin D and High Parathyroid Hormone Levels as Determinants of Loss of Muscle Strength and Muscle Mass (Sarcopenia): The Longitudinal Aging Study Amsterdam. J. Clin. Endocrinol. Metab. 2003, 88, 5766–5772. [Google Scholar] [CrossRef] [PubMed]
- Bislev, L.S.; Rødbro, L.L.; Sikjær, T.; Rejnmark, L. Effects of Elevated Parathyroid Hormone Levels on Muscle Health, Postural Stability and Quality of Life in Vitamin D-Insufficient Healthy Women: A Cross-Sectional Study. Calcif. Tissue Int. 2019, 105, 642–650. [Google Scholar] [CrossRef] [PubMed]
- de Souza Genaro, P.; de Medeiros Pinheiro, M.; Szejnfeld, V.L.; Martini, L.A. Secondary hyperparathyroidism and its relationship with sarcopenia in elderly women. Arch. Gerontol. Geriatr. 2015, 60, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Patten, B.M.; Bilezikian, J.P.; Mallette, L.E.; Prince, A.; Engel, W.K.; Aurbach, G.D. Neuromuscular Disease in Primary Hyperparathyroidism. Ann. Intern. Med. 1974, 80, 182–193. [Google Scholar] [CrossRef]
- Minisola, S.; Arnold, A.; Belaya, Z.; Brandi, M.L.; Clarke, B.L.; Hannan, F.M.; Hofbauer, L.C.; Insogna, K.L.; Lacroix, A.; Liberman, U.; et al. Epidemiology, Pathophysiology, and Genetics of Primary Hyperparathyroidism. J. Bone Miner. Res. 2022, 37, 2315–2329. [Google Scholar] [CrossRef] [PubMed]
- Passeri, E.; Bugiardini, E.; Sansone, V.; Valaperta, R.; Costa, E.; Ambrosi, B.; Meola, G.; Corbetta, S. Vitamin D, parathyroid hormone and muscle impairment in myotonic dystrophies. J. Neurol. Sci. 2013, 331, 132–135. [Google Scholar] [CrossRef]
- Garber, A.J. Effects of Parathyroid Hormone on Skeletal Muscle Protein and Amino Acid Metabolism in the Rat. J. Clin. Investig. 1983, 71, 1806–1821. [Google Scholar] [CrossRef]
- Baczynski, R.; Massry, S.G.; Magott, M.; El-Belbessi, S.; Kohan, R.; Brautbar, N. Effect of parathyroid hormone on energy metabolism of skeletal muscle. Kidney Int. 1985, 28, 722–727. [Google Scholar] [CrossRef]
- Thomas, S.S.; Mitch, W.E. Parathyroid hormone stimulates adipose tissue browning: A pathway to muscle wasting. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, C.; Brandi, M.L. Muscle Physiopathology in Parathyroid Hormone Disorders. Front. Med. 2021, 8, 764346. [Google Scholar] [CrossRef] [PubMed]
- Rendina-Ruedy, E.; Rosen, C.J. Parathyroid hormone (PTH) regulation of metabolic homeostasis: An old dog teaches us new tricks. Mol. Metab. 2022, 60, 101480. [Google Scholar] [CrossRef] [PubMed]
- Jimeno-Fraile, J.; Cao, H.; Sancho-Insenser, J.; Lorente-Poch, L.; Sitges-Serra, A. Muscle strength, physical performance, and metabolic changes after subtotal parathyroidectomy for secondary hyperparathyroidism. Surgery 2021, 169, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; White, J.P.; Kleiner, S.; Kazak, L.; Cohen, P.; Baracos, V.E.; Spiegelman, B.M. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 2014, 513, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Sato, C.; Miyakoshi, N.; Kasukawa, Y.; Nozaka, K.; Tsuchie, H.; Nagahata, I.; Yuasa, Y.; Abe, K.; Saito, H.; Shoji, R.; et al. Teriparatide and exercise improve bone, skeletal muscle, and fat parameters in ovariectomized and tail-suspended rats. J. Bone Miner. Metab. 2021, 39, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Brent, M.B.; Brüel, A.; Thomsen, J.S. PTH (1–34) and growth hormone in prevention of disuse osteopenia and sarcopenia in rats. Bone 2018, 110, 244–253. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Grynpas, M.; Mitchell, J. Intermittent PTH treatment improves bone and muscle in glucocorticoid treated Mdx mice: A model of Duchenne Muscular Dystrophy. Bone 2019, 121, 232–242. [Google Scholar] [CrossRef]
- Kimura, S.; Yoshioka, K. Parathyroid hormone and parathyroid hormone type-1 receptor accelerate myocyte differentiation. Sci. Rep. 2014, 4, 5066. [Google Scholar] [CrossRef]
- Romagnoli, C.; Zonefrati, R.; Lucattelli, E.; Innocenti, M.; Civinini, R.; Iantomasi, T.; Brandi, M.L. In Vitro Effects of PTH (1-84) on Human Skeletal Muscle-Derived Satellite Cells. Biomedicines 2023, 11, 1017. [Google Scholar] [CrossRef]
- Lavie, C.J.; Lee, J.H.; Milani, R.V. Vitamin D and Cardiovascular Disease: Will It Live Up to its Hype? J. Am. Coll. Cardiol. 2011, 58, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Haussler, C.A.; Hsieh, D.; Hsieh, J.-C.; Jurutka, P.W. Molecular Mechanisms of Vitamin D Action. Calcif. Tissue Int. 2013, 92, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Halfon, M.; Phan, O.; Teta, D. Vitamin D: A Review on Its Effects on Muscle Strength, the Risk of Fall, and Frailty. BioMed Res. Int. 2015, 2015, 953241. [Google Scholar] [CrossRef] [PubMed]
- Pojednic, R.M.; Ceglia, L. The Emerging Biomolecular Role of Vitamin D in Skeletal Muscle. Exerc. Sport Sci. Rev. 2014, 42, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Beaudart, C.; Buckinx, F.; Rabenda, V.; Gillain, S.; Cavalier, E.; Slomian, J.; Petermans, J.; Reginster, J.-Y.; Bruyère, O. The Effects of Vitamin D on Skeletal Muscle Strength, Muscle Mass, and Muscle Power: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Clin. Endocrinol. Metab. 2014, 99, 4336–4345. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, P.B.; Joseph, C.; Angioi, M. Effects of vitamin D supplementation on upper and lower body muscle strength levels in healthy individuals. A systematic review with meta-analysis. J. Sci. Med. Sport 2015, 18, 575–580. [Google Scholar] [CrossRef]
- Redzic, M.; Lewis, R.M.; Thomas, D.T. Relationship between 25-hydoxyvitamin D, muscle strength, and incidence of injury in healthy adults: A systematic review. Nutr. Res. 2013, 33, 251–258. [Google Scholar] [CrossRef]
- Stockton, K.A.; Mengersen, K.; Paratz, J.D.; Kandiah, D.; Bennell, K.L. Effect of vitamin D supplementation on muscle strength: A systematic review and meta-analysis. Osteoporos. Int. 2011, 22, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Rodman, J.S.; Baker, T. Changes in the kinetics of muscle contraction in vitamin D-depleted rats. Kidney Int. 1978, 13, 189–193. [Google Scholar] [CrossRef]
- Bhat, M.; Kalam, R.; Qadri, S.S.; Madabushi, S.; Ismail, A. Vitamin D Deficiency-Induced Muscle Wasting Occurs through the Ubiquitin Proteasome Pathway and Is Partially Corrected by Calcium in Male Rats. Endocrinology 2013, 154, 4018–4029. [Google Scholar] [CrossRef]
- Cheung, W.W.; Ding, W.; Hoffman, H.M.; Wang, Z.; Hao, S.; Zheng, R.; Gonzalez, A.; Zhan, J.-Y.; Zhou, P.; Li, S.; et al. Vitamin D ameliorates adipose browning in chronic kidney disease cachexia. Sci. Rep. 2020, 10, 14175. [Google Scholar] [CrossRef] [PubMed]
- Vogt, B.P.; Caramori, J.C.T. Vitamin D and skeletal muscle: A narrative review focusing on chronic kidney disease and dialysis. Hemodial. Int. 2021, 25, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.L.; Doyle, J.W.; Johansen, K.L. Association of 1,25-Dihydroxyvitamin D Levels With Physical Performance and Thigh Muscle Cross-sectional Area in Chronic Kidney Disease Stage 3 and 4. J. Ren. Nutr. 2012, 22, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Rolland, Y.; Dray, C.; Vellas, B.; Barreto, P.D.S. Current and investigational medications for the treatment of sarcopenia. Metabolism 2023, 149, 155597. [Google Scholar] [CrossRef] [PubMed]
- A Bischoff, H.; Stähelin, H.B.; Dick, W.; Akos, R.; Knecht, M.; Salis, C.; Nebiker, M.; Theiler, R.; Pfeifer, M.; Begerow, B.; et al. Effects of Vitamin D and Calcium Supplementation on Falls: A Randomized Controlled Trial. J. Bone Miner. Res. 2003, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Ceglia, L.; Niramitmahapanya, S.; da Silva Morais, M.; Rivas, D.A.; Harris, S.S.; Bischoff-Ferrari, H.; Fielding, R.A.; Dawson-Hughes, B. A Randomized Study on the Effect of Vitamin D3 Supplementation on Skeletal Muscle Morphology and Vitamin D Receptor Concentration in Older Women. J. Clin. Endocrinol. Metab. 2013, 98, E1927–E1935. [Google Scholar] [CrossRef]
- Tabrizi, R.; Hallajzadeh, J.; Mirhosseini, N.; Lankarani, K.B.; Maharlouei, N.; Akbari, M.; Asemi, Z. The effects of vitamin D supplementation on muscle function among postmenopausal women: A systematic review and meta-analysis of randomized controlled trials. EXCLI J. 2019, 18, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Nagano, N.; Urakawa, I.; Yamazaki, Y.; Iijima, K.; Fujita, T.; Yamashita, T.; Fukumoto, S.; Shimada, T. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010, 78, 975–980. [Google Scholar] [CrossRef]
- Gunta, S.S.; Thadhani, R.I.; Mak, R.H. The effect of vitamin D status on risk factors for cardiovascular disease. Nat. Rev. Nephrol. 2013, 9, 337–347. [Google Scholar] [CrossRef]
- Gordon, P.L.; Sakkas, G.K.; Doyle, J.W.; Shubert, T.; Johansen, K.L. Relationship Between Vitamin D and Muscle Size and Strength in Patients on Hemodialysis. J. Ren. Nutr. 2007, 17, 397–407. [Google Scholar] [CrossRef]
- Shoben, A.B.; Rudser, K.D.; de Boer, I.H.; Young, B.; Kestenbaum, B. Association of Oral Calcitriol with Improved Survival in Nondialyzed CKD. J. Am. Soc. Nephrol. 2008, 19, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Wolf, M.; Ofsthun, M.N.; Lazarus, J.M.; Hernán, M.A.; Camargo, C.A.; Thadhani, R. Activated Injectable Vitamin D and Hemodialysis Survivall: A Historical Cohort Study. J. Am. Soc. Nephrol. 2005, 16, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Akagawa, M.; Miyakoshi, N.; Kasukawa, Y.; Ono, Y.; Yuasa, Y.; Nagahata, I.; Sato, C.; Tsuchie, H.; Nagasawa, H.; Hongo, M.; et al. Effects of activated vitamin D, alfacalcidol, and low-intensity aerobic exercise on osteopenia and muscle atrophy in type 2 diabetes mellitus model rats. PLoS ONE 2018, 13, e0204857. [Google Scholar] [CrossRef] [PubMed]
- Bass, J.J.; Nakhuda, A.; Deane, C.S.; Brook, M.S.; Wilkinson, D.J.; Phillips, B.E.; Philp, A.; Tarum, J.; Kadi, F.; Andersen, D.; et al. Overexpression of the vitamin D receptor (VDR) induces skeletal muscle hypertrophy. Mol. Metab. 2020, 42, 101059. [Google Scholar] [CrossRef]
- Bass, J.J.; Kazi, A.A.; Deane, C.S.; Nakhuda, A.; Ashcroft, S.P.; Brook, M.S.; Wilkinson, D.J.; Phillips, B.E.; Philp, A.; Tarum, J.; et al. The mechanisms of skeletal muscle atrophy in response to transient knockdown of the vitamin D receptor in vivo. J. Physiol. 2021, 599, 963–979. [Google Scholar] [CrossRef] [PubMed]
- Girgis, C.M.; Cha, K.M.; Houweling, P.J.; Rao, R.; Mokbel, N.; Lin, M.; Clifton-Bligh, R.J.; Gunton, J.E. Vitamin D Receptor Ablation and Vitamin D Deficiency Result in Reduced Grip Strength, Altered Muscle Fibers, and Increased Myostatin in Mice. Calcif. Tissue Int. 2015, 97, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Makanae, Y.; Ogasawara, R.; Sato, K.; Takamura, Y.; Matsutani, K.; Kido, K.; Shiozawa, N.; Nakazato, K.; Fujita, S. Acute bout of resistance exercise increases vitamin D receptor protein expression in rat skeletal muscle. Exp. Physiol. 2015, 100, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, K.R.; Cruzat, V.; Carlessi, R.; Newsholme, P. Mechanisms of vitamin D action in skeletal muscle. Nutr. Res. Rev. 2019, 32, 192–204. [Google Scholar] [CrossRef]
- Girgis, C.M.; Clifton-Bligh, R.J.; Mokbel, N.; Cheng, K.; Gunton, J.E. Vitamin D Signaling Regulates Proliferation, Differentiation, and Myotube Size in C2C12 Skeletal Muscle Cells. Endocrinology 2014, 155, 347–357. [Google Scholar] [CrossRef]
- Okuno, H.; Kishimoto, K.N.; Hatori, M.; Itoi, E. 1α,25-dihydroxyvitamin D3 enhances fast-myosin heavy chain expression in differentiated C2C12 myoblasts. Cell Biol. Int. 2012, 36, 441–447. [Google Scholar] [CrossRef]
- Garcia, L.A.; King, K.K.; Ferrini, M.G.; Norris, K.C.; Artaza, J.N. 1,25(OH)2Vitamin D3 Stimulates Myogenic Differentiation by Inhibiting Cell Proliferation and Modulating the Expression of Promyogenic Growth Factors and Myostatin in C2C12 Skeletal Muscle Cells. Endocrinology 2011, 152, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kishimoto, K.N.; Okuno, H.; Saito, H.; Itoi, E. Vitamin D receptor gene silencing effects on differentiation of myogenic cell lines. Muscle Nerve 2014, 49, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Romeu Montenegro, K.; Carlessi, R.M.; Cruzat, V.F.; Newsholme, P. Effects of vitamin D on primary human skeletal muscle cell proliferation, differentiation, protein synthesis and bioenergetics. J. Steroid Biochem. Mol. Biol. 2019, 193, 105423. [Google Scholar] [CrossRef] [PubMed]
- Capiati, D.; Benassati, S.; Boland, R.L. 1,25(OH)2-vitamin D3 induces translocation of the vitamin D receptor (VDR) to the plasma membrane in skeletal muscle cells. J. Cell. Biochem. 2002, 86, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.J.; Sharples, A.P.; Polydorou, I.; Alwan, N.; Donovan, T.; Tang, J.; Fraser, W.D.; Cooper, R.G.; Morton, J.P.; Stewart, C.; et al. A systems-based investigation into vitamin D and skeletal muscle repair, regeneration, and hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E1019–E1031. [Google Scholar] [CrossRef] [PubMed]
- Alliband, K.H.; Kozhevnikova, S.V.; Parr, T.; Jethwa, P.H.; Brameld, J.M. In vitro Effects of Biologically Active Vitamin D on Myogenesis: A Systematic Review. Front. Physiol. 2021, 12, 736708. [Google Scholar] [CrossRef] [PubMed]
- Wimalawansa, S.J. Associations of vitamin D with insulin resistance, obesity, type 2 diabetes, and metabolic syndrome. J. Steroid Biochem. Mol. Biol. 2018, 175, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, R.A.; Ashby, V.B.; Milford, E.L.; Ojo, A.O.; Ettenger, R.E.; Agodoa, L.Y.C.; Held, P.J.; Port, F.K. Comparison of Mortality in All Patients on Dialysis, Patients on Dialysis Awaiting Transplantation, and Recipients of a First Cadaveric Transplant. N. Engl. J. Med. 1999, 341, 1725–1730. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M. Forging Forward with 10 Burning Questions on FGF23 in Kidney Disease. J. Am. Soc. Nephrol. 2010, 21, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Pupim, L.B.; Brouillette, J.R.; Levenhagen, D.K.; Farmer, K.; Hakim, R.M.; Flakoll, P.J.; Raj, D.S.C.; Adeniyi, O.; Dominic, E.A.; et al. Hemodialysis stimulates muscle and whole body protein loss and alters substrate oxidation. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E107–E116. [Google Scholar] [CrossRef]
- Bohé, J.; Low, A.; Wolfe, R.R.; Rennie, M.J. Human Muscle Protein Synthesis is Modulated by Extracellular, Not Intramuscular Amino Acid Availability: A Dose-Response Study. J. Physiol. 2003, 552 Pt 1, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Alvestrand, A.; Wahren, J.; Bergström, J. Effect of in vivo contact between blood and dialysis membranes on protein catabolism in humans. Kidney Int. 1990, 38, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Roshanravan, B.; Zelnick, L.R.; Djucovic, D.; Gu, H.; Alvarez, J.A.; Ziegler, T.R.; Gamboa, J.L.; Utzschneider, K.; Kestenbaum, B.; Himmelfarb, J.; et al. Chronic kidney disease attenuates the plasma metabolome response to insulin. JCI Insight 2018, 3, e122219. [Google Scholar] [CrossRef] [PubMed]
- Dienemann, T.; Ziolkowski, S.L.; Bender, S.; Goral, S.; Long, J.; Baker, J.F.; Shults, J.; Zemel, B.S.; Reese, P.P.; Wilson, F.P.; et al. Changes in Body Composition, Muscle Strength, and Fat Distribution Following Kidney Transplantation. Am. J. Kidney Dis. 2021, 78, 816–825. [Google Scholar] [CrossRef]
- van Vliet, I.M.; Post, A.; Kremer, D.; Boslooper-Meulenbelt, K.; van der Veen, Y.; de Jong, M.F.; Pol, R.A.; Jager-Wittenaar, H.; Navis, G.J.; Bakker, S.J.; et al. Muscle mass, muscle strength and mortality in kidney transplant recipients: Results of the TransplantLines Biobank and Cohort Study. J. Cachexia Sarcopenia Muscle 2022, 13, 2932–2943. [Google Scholar] [CrossRef] [PubMed]
- Kosoku, A.; Ishihara, T.; Iwai, T.; Nishide, S.; Kabei, K.; Maeda, K.; Kumada, N.; Uchida, J. The Change in Muscle Mass Among Kidney Transplant Recipients: A Prospective Cohort Study. Transplant. Proc. 2022, 54, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Steffl, M.; Bohannon, R.W.; Sontakova, L.; Tufano, J.J.; Shiells, K.; Holmerova, I. Relationship between sarcopenia and physical activity in older people: A systematic review and meta-analysis. Clin. Interv. Aging 2017, 12, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Bishop, N.C.; Burton, J.O.; Graham-Brown, M.P.M.; Stensel, D.J.; Viana, J.L.; Watson, E.L. Exercise and chronic kidney disease: Potential mechanisms underlying the physiological benefits. Nat. Rev. Nephrol. 2023, 19, 244–256. [Google Scholar] [CrossRef]
- KDIGO Guidelines. Chapter 3: Management of progression and complications of CKD. Kidney Int. Suppl. 2013, 3, 73–90. [Google Scholar] [CrossRef]
- Roshanravan, B.; Gamboa, J.; Wilund, K. Exercise and CKD: Skeletal Muscle Dysfunction and Practical Application of Exercise to Prevent and Treat Physical Impairments in CKD. Am. J. Kidney Dis. 2017, 69, 837–852. [Google Scholar] [CrossRef]
- Baker, L.A.; March, D.S.; Wilkinson, T.J.; Billany, R.E.; Bishop, N.C.; Castle, E.M.; Chilcot, J.; Davies, M.D.; Graham-Brown, M.P.M.; Greenwood, S.A.; et al. Clinical practice guideline exercise and lifestyle in chronic kidney disease. BMC Nephrol. 2022, 23, 75. [Google Scholar] [CrossRef] [PubMed]
- Koufaki, P.; Greenwood, S.; Painter, P.; Mercer, T. The BASES expert statement on exercise therapy for people with chronic kidney disease. J. Sports Sci. 2015, 33, 1902–1907. [Google Scholar] [CrossRef]
- Smart, N.A.; Williams, A.D.; Levinger, I.; Selig, S.; Howden, E.; Coombes, J.S.; Fassett, R.G. Exercise & Sports Science Australia (ESSA) position statement on exercise and chronic kidney disease. J. Sci. Med. Sport 2013, 16, 406–411. [Google Scholar] [CrossRef]
- Nishi, H.; Takemura, K.; Higashihara, T.; Inagi, R. Uremic Sarcopenia: Clinical Evidence and Basic Experimental Approach. Nutrients 2020, 12, 1814. [Google Scholar] [CrossRef]
- Watson, E.L.; Baker, L.A.; Wilkinson, T.J.; Gould, D.W.; Major, R.W.; Ashford, R.U.; Philp, A.; Smith, A.C.; Graham-Brown, M.P. Reductions in skeletal muscle mitochondrial mass are not restored following exercise training in patients with chronic kidney disease. FASEB J. 2020, 34, 1755–1767. [Google Scholar] [CrossRef]
- Barcellos, F.C.; Santos, I.S.; Umpierre, D.; Bohlke, M.; Hallal, P.C. Effects of exercise in the whole spectrum of chronic kidney disease: A systematic review. Clin. Kidney J. 2015, 8, 753–765. [Google Scholar] [CrossRef]
- Izumi, A.; Kitamura, M.; Izawa, K.P. Effects of Exercise Training on Delaying Disease Progression in Patients with Chronic Kidney Disease: A Review of the Literature. Rev. Recent Clin. Trials 2016, 11, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Cheema, B.S.; Chan, D.; Fahey, P.; Atlantis, E. Effect of Progressive Resistance Training on Measures of Skeletal Muscle Hypertrophy, Muscular Strength and Health-Related Quality of Life in Patients with Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Sports Med. 2014, 44, 1125–1138. [Google Scholar] [CrossRef]
- da Silva, S.F.; Pereira, A.A.; da Silva, W.A.H.; Simôes, R.; Neto, J.d.R.B. Physical therapy during hemodialyse in patients with chronic kidney disease. J. Bras. Nefrol. 2013, 35, 170–176. [Google Scholar] [CrossRef]
- Oliveros, R.M.; Avendano, M.; Bunout, D.; Hirsch, S.; De La Maza, M.P.; Pedreros, C.; Mueller, H. A pilot study on physical training of patients in hemodialysis. Rev. Med. Chil. 2011, 139, 1046–1053. [Google Scholar]
- Watson, E.L.; Baker, L.A.; Wilkinson, T.J.; Gould, D.W.; Xenophontos, S.; Graham-Brown, M.; Major, R.W.; Ashford, R.U.; Viana, J.L.; Smith, A.C. Inflammation and physical dysfunction: Responses to moderate intensity exercise in chronic kidney disease. Nephrol. Dial. Transplant. 2022, 37, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Robinson-Cohen, C.; Ellis, C.; Headley, S.A.; Tuttle, K.; Wood, R.J.; Evans, E.E.; Milch, C.M.; Moody, K.A.; Germain, M.; et al. Metabolic Effects of Diet and Exercise in Patients with Moderate to Severe CKD: A Randomized Clinical Trial. J. Am. Soc. Nephrol. 2018, 29, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.; Tang, Y.; Tan, L.; Tan, J.; Ge, L.; Qin, W. Aerobic exercise in adults with chronic kidney disease (CKD): A meta-analysis. Int. Urol. Nephrol. 2019, 51, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, H.L.; Neves, R.V.P.; Deus, L.A.; Maia, B.C.H.; Maya, A.T.; Tzanno-Martins, C.; Souza, M.K.; Silva, J.A.B.; Haro, A.S.; Costa, F.; et al. Low-load resistance training with blood flow restriction prevent renal function decline: The role of the redox balance, angiotensin 1–7 and vasopressin(✰,✰✰). Physiol. Behav. 2021, 230, 113295. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, H.L.; Moura, S.R.G.; Neves, R.V.P.; Tzanno-Martins, C.; Souza, M.K.; Haro, A.S.; Costa, F.; Silva, J.A.B.; Stone, W.; Honorato, F.S.; et al. Resistance training improves sleep quality, redox balance and inflammatory profile in maintenance hemodialysis patients: A randomized controlled trial. Sci. Rep. 2020, 10, 11708. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.L.; Viana, J.L.; Wimbury, D.; Martin, N.; Greening, N.J.; Barratt, J.; Smith, A.C. The Effect of Resistance Exercise on Inflammatory and Myogenic Markers in Patients with Chronic Kidney Disease. Front. Physiol. 2017, 8, 541. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, V.S.; Rao, M.; Menon, V.; Gordon, P.L.; Pilichowska, M.; Castaneda, F.; Castaneda-Sceppa, C. Resistance Training Increases Muscle Mitochondrial Biogenesis in Patients with Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 996–1002. [Google Scholar] [CrossRef]
- Dong, Z.-J.; Zhang, H.-L.; Yin, L.-X. Effects of intradialytic resistance exercise on systemic inflammation in maintenance hemodialysis patients with sarcopenia: A randomized controlled trial. Int. Urol. Nephrol. 2019, 51, 1415–1424. [Google Scholar] [CrossRef]
- Vezzoli, A.; Mrakic-Sposta, S.; Montorsi, M.; Porcelli, S.; Vago, P.; Cereda, F.; Longo, S.; Maggio, M.; Narici, M. Moderate Intensity Resistive Training Reduces Oxidative Stress and Improves Muscle Mass and Function in Older Individuals. Antioxidants 2019, 8, 431. [Google Scholar] [CrossRef]
- Theilen, N.T.; Jeremic, N.; Weber, G.J.; Tyagi, S.C. Exercise preconditioning diminishes skeletal muscle atrophy after hindlimb suspension in mice. J. Appl. Physiol. 2018, 125, 999–1010. [Google Scholar] [CrossRef]
- Sovatzidis, A.; Chatzinikolaou, A.; Fatouros, I.G.; Panagoutsos, S.; Draganidis, D.; Nikolaidou, E.; Avloniti, A.; Michailidis, Y.; Mantzouridis, I.; Batrakoulis, A.; et al. Intradialytic Cardiovascular Exercise Training Alters Redox Status, Reduces Inflammation and Improves Physical Performance in Patients with Chronic Kidney Disease. Antioxidants 2020, 9, 868. [Google Scholar] [CrossRef] [PubMed]
- Gadelha, A.B.; Cesari, M.; Corrêa, H.L.; Neves, R.V.P.; Sousa, C.V.; Deus, L.A.; Souza, M.K.; Reis, A.L.; Moraes, M.R.; Prestes, J.; et al. Effects of pre-dialysis resistance training on sarcopenia, inflammatory profile, and anemia biomarkers in older community-dwelling patients with chronic kidney disease: A randomized controlled trial. Int. Urol. Nephrol. 2021, 53, 2137–2147. [Google Scholar] [CrossRef]
- Kouidi, E.; Grekas, D.; Deligiannis, A.; Tourkantonis, A. Outcomes of long-term exercise training in dialysis patients: Comparison of two training programs. Clin. Nephrol. 2004, 61 (Suppl. S3), S31–S38. [Google Scholar] [PubMed]
- Uchiyama, K.; Adachi, K.; Muraoka, K.; Nakayama, T.; Oshida, T.; Yasuda, M.; Hishikawa, A.; Minakuchi, H.; Miyashita, K.; Tokuyama, H.; et al. Home-based aerobic exercise and resistance training for severe chronic kidney disease: A randomized controlled trial. J. Cachexia Sarcopenia Muscle 2021, 12, 1789–1802. [Google Scholar] [CrossRef]
- Majchrzak, K.M.; Pupim, L.B.; Flakoll, P.J.; Ikizler, T.A. Resistance exercise augments the acute anabolic effects of intradialytic oral nutritional supplementation. Nephrol. Dial. Transplant. 2008, 23, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D.; Wang, H.; Casaburi, R.; Fournier, M.; Lewis, M.I.; Taylor, W.; Storer, T.W. Exercise in Maintenance Hemodialysis Patients Induces Transcriptional Changes in Genes Favoring Anabolic Muscle. J. Am. Soc. Nephrol. 2007, 18, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sood, S.; Biada, J.; Roth, R.; Rabkin, R. Increased workload fully activates the blunted IRS-1/PI3-kinase/Akt signaling pathway in atrophied uremic muscle. Kidney Int. 2008, 73, 848–855. [Google Scholar] [CrossRef]
- Sun, D.; Chen, Y.; Rabkin, R. Work-induced changes in skeletal muscle IGF-1 and myostatin gene expression in uremia. Kidney Int. 2006, 70, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Cheema, B.; Abas, H.; Smith, B.; O’Sullivan, A.; Chan, M.; Patwardhan, A.; Kelly, J.; Gillin, A.; Pang, G.; Lloyd, B.; et al. Progressive Exercise for Anabolism in Kidney Disease (PEAK): A Randomized, Controlled Trial of Resistance Training during Hemodialysis. J. Am. Soc. Nephrol. 2007, 18, 1594–1601. [Google Scholar] [CrossRef]
- Martin-Alemañy, G.; Perez-Navarro, M.; Wilund, K.R.; García-Villalobos, G.; Gómez-Guerrero, I.; Cantú-Quintanilla, G.; Reyes-Caldelas, M.A.; Espinosa-Cuevas, A.; Escobedo, G.; Medeiros, M.; et al. Effect of Intradialytic Oral Nutritional Supplementation with or without Exercise Improves Muscle Mass Quality and Physical Function in Hemodialysis Patients: A Pilot Study. Nutrients 2022, 14, 2946. [Google Scholar] [CrossRef]
- Qi, Z.; Liu, W.; Lu, J. The mechanisms underlying the beneficial effects of exercise on bone remodeling: Roles of bone-derived cytokines and microRNAs. Prog. Biophys. Mol. Biol. 2016, 122, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Klein, J.D.; Hassounah, F.; Cai, H.; Zhang, C.; Xu, P.; Wang, X.H. Low-Frequency Electrical Stimulation Attenuates Muscle Atrophy in CKD—A Potential Treatment Strategy. J. Am. Soc. Nephrol. 2015, 26, 626–635. [Google Scholar] [CrossRef]
- Pupim, L.B.; Flakoll, P.J.; Brouillette, J.R.; Levenhagen, D.K.; Hakim, R.M.; Ikizler, T.A. Intradialytic parenteral nutrition improves protein and energy homeostasis in chronic hemodialysis patients. J. Clin. Investig. 2002, 110, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Pupim, L.B.; Majchrzak, K.M.; Flakoll, P.J.; Ikizler, T.A. Intradialytic Oral Nutrition Improves Protein Homeostasis in Chronic Hemodialysis Patients with Deranged Nutritional Status. J. Am. Soc. Nephrol. 2006, 17, 3149–3157. [Google Scholar] [CrossRef] [PubMed]
- Cano, N.D.J.; Fouque, D.; Roth, H.; Aparicio, M.; Azar, R.; Canaud, B.; Chauveau, P.; Combe, C.; Laville, M.; Leverve, X.M. Intradialytic parenteral nutrition does not improve survival in malnourished hemodialysis patients: A 2-year multicenter, prospective, randomized study. J. Am. Soc. Nephrol. 2007, 18, 2583–2591. [Google Scholar] [CrossRef]
- Castaneda, C.; Gordon, P.L.; Uhlin, K.L.; Levey, A.S.; Kehayias, J.J.; Dwyer, J.T.; Fielding, R.A.; Roubenoff, R.; Singh, M.F. Resistance Training to Counteract the Catabolism of a Low-Protein Diet in Patients with Chronic Renal Insufficiency. A Randomized, Controlled Trial. Ann. Intern. Med. 2001, 135, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Song, S.H. Sarcopenia in chronic kidney disease: From bench to bedside. Korean J. Intern. Med. 2023, 38, 303–321. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Dong, Y.; Chen, Z.; Eckols, T.K.; Kasembeli, M.M.; Tweardy, D.J.; Mitch, W.E. Pharmacokinetics and pharmacodynamics of TTI-101, a STAT3 inhibitor that blocks muscle proteolysis in rats with chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2020, 319, F84–F92. [Google Scholar] [CrossRef]
- Guebre-Egziabher, F.; Juillard, L.; Boirie, Y.; Laville, M.; Beaufrère, B.; Fouque, D. Short-Term Administration of a Combination of Recombinant Growth Hormone and Insulin-Like Growth Factor-I Induces Anabolism in Maintenance Hemodialysis. J. Clin. Endocrinol. Metab. 2009, 94, 2299–2305. [Google Scholar] [CrossRef]
- Hansen, T.B.; Gram, J.; Jensen, P.B.; Kristiansen, J.H.; Ekelund, B.; Christiansen, J.S.; Pedersen, F.B. Influence of growth hormone on whole body and regional soft tissue composition in adult patients on hemodialysis. A double-blind, randomized, placebo-controlled study. Clin. Nephrol. 2000, 53, 99–107. [Google Scholar]
- Anker, M.S.; von Haehling, S.; Springer, J. Blocking myostatin: Muscle mass equals muscle strength? J. Cachexia Sarcopenia Muscle 2020, 11, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, L.; Gandhi, R.; Warden, S.J.; Poiraudeau, S.; Myers, S.L.; Benson, C.T.; Hu, L.; Ahmad, Q.I.; Linnemeier, P.; Gomez, E.V.; et al. A Phase 2 Randomized Study Investigating the Efficacy and Safety of Myostatin Antibody LY2495655 versus Placebo in Patients Undergoing Elective Total Hip Arthroplasty. J. Frailty Aging 2016, 5, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Lee, Y.-S. Myostatin Inhibitors: Panacea or Predicament for Musculoskeletal Disorders? J. Bone Metab. 2020, 27, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Attie, K.M.; Borgstein, N.G.; Yang, Y.; Condon, C.H.; Wilson, D.M.; Pearsall, A.E.; Kumar, R.; Willins, D.A.; Seehra, J.S.; Sherman, M.L. A single ascending-dose study of muscle regulator ACE-031 in healthy volunteers. Muscle Nerve 2012, 47, 416–423. [Google Scholar] [CrossRef]
- Campbell, C.; McMillan, H.J.; Mah, J.K.; Tarnopolsky, M.; Selby, K.; McClure, T.; Wilson, D.M.; Sherman, M.L.; Escolar, D.; Attie, K.M. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve 2017, 55, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Morvan, F.; Rondeau, J.-M.; Zou, C.; Minetti, G.; Scheufler, C.; Scharenberg, M.; Jacobi, C.; Brebbia, P.; Ritter, V.; Toussaint, G.; et al. Blockade of activin type II receptors with a dual anti-ActRIIA/IIB antibody is critical to promote maximal skeletal muscle hypertrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 12448–12453. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, R.; You, H.; Li, J.; Liu, Y.; Li, Q.; Wu, X.; Huang, J.; Cai, X.; Wang, M.; et al. Formononetin ameliorates muscle atrophy by regulating myostatin-mediated PI3K/Akt/FoxO3a pathway and satellite cell function in chronic kidney disease. J. Cell. Mol. Med. 2021, 25, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Falcao-Pires, I.; Ladeiras-Lopes, R.; Leite-Moreira, A.F. The apelinergic system: A promising therapeutic target. Expert Opin. Ther. Targets 2010, 14, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Vinel, C.; Lukjanenko, L.; Batut, A.; Deleruyelle, S.; Pradère, J.-P.; Le Gonidec, S.; Dortignac, A.; Geoffre, N.; Pereira, O.; Karaz, S.; et al. The exerkine apelin reverses age-associated sarcopenia. Nat. Med. 2018, 24, 1360–1371. [Google Scholar] [CrossRef]
- Chapman, F.A.; Nyimanu, D.; Maguire, J.J.; Davenport, A.P.; Newby, D.E.; Dhaun, N. The therapeutic potential of apelin in kidney disease. Nat. Rev. Nephrol. 2021, 17, 840–853. [Google Scholar] [CrossRef]
- Ason, B.; Chen, Y.; Guo, Q.; Hoagland, K.M.; Chui, R.W.; Fielden, M.; Sutherland, W.; Chen, R.; Zhang, Y.; Mihardja, S.; et al. Cardiovascular response to small-molecule APJ activation. JCI Insight. 2020, 5, e132898. [Google Scholar] [CrossRef] [PubMed]
- Asai, M.; Kumakura, S.; Kikuchi, M. Review of the efficacy of AST-120 (KREMEZIN®) on renal function in chronic kidney disease patients. Ren. Fail. 2019, 41, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Su, P.-Y.; Lee, Y.-H.; Kuo, L.-N.; Chen, Y.-C.; Chen, C.; Kang, Y.-N.; Chang, E.H. Efficacy of AST-120 for Patients With Chronic Kidney Disease: A Network Meta-Analysis of Randomized Controlled Trials. Front. Pharmacol. 2021, 12, 676345. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Ishimori, N.; Takada, S.; Saito, A.; Kadoguchi, T.; Furihata, T.; Fukushima, A.; Matsushima, S.; Yokota, T.; Kinugawa, S.; et al. AST-120 ameliorates lowered exercise capacity and mitochondrial biogenesis in the skeletal muscle from mice with chronic kidney disease via reducing oxidative stress. Nephrol. Dial. Transplant. 2015, 30, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Cha, R.; Kang, S.H.; Han, M.Y.; An, W.S.; Kim, S.; Kim, J.C. Effects of AST-120 on muscle health and quality of life in chronic kidney disease patients: Results of RECOVERY study. J. Cachexia Sarcopenia Muscle 2022, 13, 397–408. [Google Scholar] [CrossRef]
- Abramowitz, M.K.; Melamed, M.L.; Bauer, C.; Raff, A.C.; Hostetter, T.H. Effects of Oral Sodium Bicarbonate in Patients with CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Kurajoh, M.; Mori, K.; Miyabe, M.; Matsufuji, S.; Kizu, A.; Tsujimoto, Y.; Emoto, M. Xanthine Oxidoreductase Inhibitor Use Associated With Reduced Risk of Sarcopenia and Severe Sarcopenia in Patients Undergoing Hemodialysis. Front. Med. 2022, 9, 817578. [Google Scholar] [CrossRef]
- Johansen, K.L.; Painter, P.L.; Sakkas, G.K.; Gordon, P.; Doyle, J.; Shubert, T. Effects of Resistance Exercise Training and Nandrolone Decanoate on Body Composition and Muscle Function among Patients Who Receive Hemodialysis. J. Am. Soc. Nephrol. 2006, 17, 2307–2314. [Google Scholar] [CrossRef] [PubMed]
- Johansen, K.L.; Mulligan, K.; Schambelan, M. Anabolic Effects of Nandrolone Decanoate in Patients Receiving Dialysis: A Randomized Controlled Tria. JAMA 1999, 281, 1275–1281. [Google Scholar] [CrossRef]
- Supasyndh, O.; Satirapoj, B.; Aramwit, P.; Viroonudomphol, D.; Chaiprasert, A.; Thanachatwej, V.; Vanichakarn, S.; Kopple, J.D. Effect of Oral Anabolic Steroid on Muscle Strength and Muscle Growth in Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2013, 8, 271–279. [Google Scholar] [CrossRef]
- Dalton, J.T.; Barnette, K.G.; Bohl, C.E.; Hancock, M.L.; Rodriguez, D.; Dodson, S.T.; Morton, R.A.; Steiner, M.S. The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: Results of a double-blind, placebo-controlled phase II trial. J. Cachexia Sarcopenia Muscle 2011, 2, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.; Prado, C.M.M.; Johnston, M.A.; Gralla, R.J.; Taylor, R.P.; Hancock, M.L.; Dalton, J.T. Study Design and Rationale for the Phase 3 Clinical Development Program of Enobosarm, a Selective Androgen Receptor Modulator, for the Prevention and Treatment of Muscle Wasting in Cancer Patients (POWER Trials). Curr. Oncol. Rep. 2016, 18, 37. [Google Scholar] [CrossRef]
- Chan, S.; Au, K.; Francis, R.S.; Mudge, D.W.; Johnson, D.W.; Pillans, P.I. Phosphate binders in patients with chronic kidney disease. Aust. Prescr. 2017, 40, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Al-Makki, A.; Shepler, B. Tenapanor: A new treatment option for hyperphosphatemia in end stage kidney disease. J. Pharm. Pharm. Sci. 2022, 25, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Rosenbaum, D.P.; Leonsson-Zachrisson, M.; Åstrand, M.; Johansson, S.; Knutsson, M.; Langkilde, A.M.; Chertow, G.M. Effect of Tenapanor on Serum Phosphate in Patients Receiving Hemodialysis. J. Am. Soc. Nephrol. 2017, 28, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Rosenbaum, D.P.; Yan, A.; Chertow, G.M. Efficacy and Safety of Tenapanor in Patients with Hyperphosphatemia Receiving Maintenance Hemodialysis: A Randomized Phase 3 Trial. J. Am. Soc. Nephrol. 2019, 30, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Taskapan, H.; Baysal, O.; Karahan, D.; Durmus, B.; Altay, Z.; Ulutas, O. Vitamin D and muscle strength, functional ability and balance in peritoneal dialysis patients with vitamin D deficiency. Clin. Nephrol. 2011, 76, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Uto, N.S.; Amitani, H.; Atobe, Y.; Sameshima, Y.; Sakaki, M.; Rokot, N.; Ataka, K.; Amitani, M.; Inui, A. Herbal Medicine Ninjin’yoeito in the Treatment of Sarcopenia and Frailty. Front. Nutr. 2018, 5, 126. [Google Scholar] [CrossRef] [PubMed]
- Ranjit, R.; Van Remmen, H.; Ahn, B. Acylated Ghrelin Receptor Agonist HM01 Decreases Lean Body and Muscle Mass, but Unacylated Ghrelin Protects against Redox-Dependent Sarcopenia. Antioxidants 2022, 11, 2358. [Google Scholar] [CrossRef]
- Busquets, S.; Serpe, R.; Sirisi, S.; Toledo, M.; Coutinho, J.; Martínez, R.; Orpí, M.; López-Soriano, F.J.; Argilés, J.M. Megestrol acetate: Its impact on muscle protein metabolism supports its use in cancer cachexia. Clin. Nutr. 2010, 29, 733–737. [Google Scholar] [CrossRef]
- Mariam, Z.; Niazi, S.K. Glucagon-like peptide agonists: A prospective review. Endocrinol. Diabetes Metab. 2024, 7, e462. [Google Scholar] [CrossRef]
- Michos, E.D.; Bakris, G.L.; Rodbard, H.W.; Tuttle, K.R. Glucagon-like peptide-1 receptor agonists in diabetic kidney disease: A review of their kidney and heart protection. Am. J. Prev. Cardiol. 2023, 14, 100502. [Google Scholar] [CrossRef]
- Sattar, N.; Lee, M.M.Y.; Kristensen, S.L.; Branch, K.R.H.; Del Prato, S.; Khurmi, N.S.; Lam, C.S.P.; Lopes, R.D.; McMurray, J.J.V.; Pratley, R.E.; et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021, 9, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-J.; Wu, C.-Y.; Jenq, C.-C.; Lee, T.-H.; Tsai, C.-Y.; Tu, H.-T.; Huang, Y.-T.; Yen, C.-L.; Yen, T.-H.; Chen, Y.-C.; et al. Association of Glucagon-Like Peptide-1 Receptor Agonist vs Dipeptidyl Peptidase-4 Inhibitor Use With Mortality Among Patients With Type 2 Diabetes and Advanced Chronic Kidney Disease. JAMA Netw. Open 2022, 5, e221169. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, T.-H.; Tsai, M.-L.; Wu, V.C.-C.; Tseng, C.-J.; Lin, M.-S.; Li, Y.-R.; Chang, C.-H.; Chou, T.-S.; Tsai, T.-H.; et al. The cardiovascular and renal effects of glucagon-like peptide 1 receptor agonists in patients with advanced diabetic kidney disease. Cardiovasc. Diabetol. 2023, 22, 60. [Google Scholar] [CrossRef]
- Jastreboff, A.M.; Aronne, L.J.; Stefanski, A. Tirzepatide Once Weekly for the Treatment of Obesity. Reply. N. Engl. J. Med. 2022, 387, 1433–1435. [Google Scholar] [CrossRef]
- Wilding, J.P.H.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.; Wadden, T.A.; et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N. Engl. J. Med. 2021, 384, 989–1002. [Google Scholar] [CrossRef]
- Arnold, C. After obesity drugs’ success, companies rush to preserve skeletal muscle. Nat. Biotechnol. 2024, 42, 351–353. [Google Scholar] [CrossRef]
- Abdulla, H.; Phillips, B.; Wilkinson, D.; Gates, A.; Limb, M.; Jandova, T.; Bass, J.; Lewis, J.; Williams, J.; Smith, K.; et al. Effects of GLP-1 Infusion Upon Whole-body Glucose Uptake and Skeletal Muscle Perfusion During Fed-state in Older Men. J. Clin. Endocrinol. Metab. 2022, 108, 971–978. [Google Scholar] [CrossRef]
- Subaran, S.C.; Sauder, M.A.; Chai, W.; Jahn, L.A.; Fowler, D.E.; Aylor, K.W.; Basu, A.; Liu, Z. GLP-1 at physiological concentrations recruits skeletal and cardiac muscle microvasculature in healthy humans. Clin. Sci. 2014, 127, 163–170. [Google Scholar] [CrossRef]
- Hong, Y.; Lee, J.H.; Jeong, K.W.; Choi, C.S.; Jun, H. Amelioration of muscle wasting by glucagon-like peptide-1 receptor agonist in muscle atrophy. J. Cachexia Sarcopenia Muscle 2019, 10, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Kaji, K.; Nishimura, N.; Kubo, T.; Tomooka, F.; Shibamoto, A.; Suzuki, J.; Tsuji, Y.; Fujinaga, Y.; Kitagawa, K.; et al. Glucagon-like peptide-1 receptor agonist, semaglutide attenuates chronic liver disease-induced skeletal muscle atrophy in diabetic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166770. [Google Scholar] [CrossRef] [PubMed]
- Gurjar, A.A.; Kushwaha, S.; Chattopadhyay, S.; Das, N.; Pal, S.; China, S.P.; Kumar, H.; Trivedi, A.K.; Guha, R.; Chattopadhyay, N.; et al. Long acting GLP-1 analog liraglutide ameliorates skeletal muscle atrophy in rodents. Metabolism 2020, 103, 154044. [Google Scholar] [CrossRef]


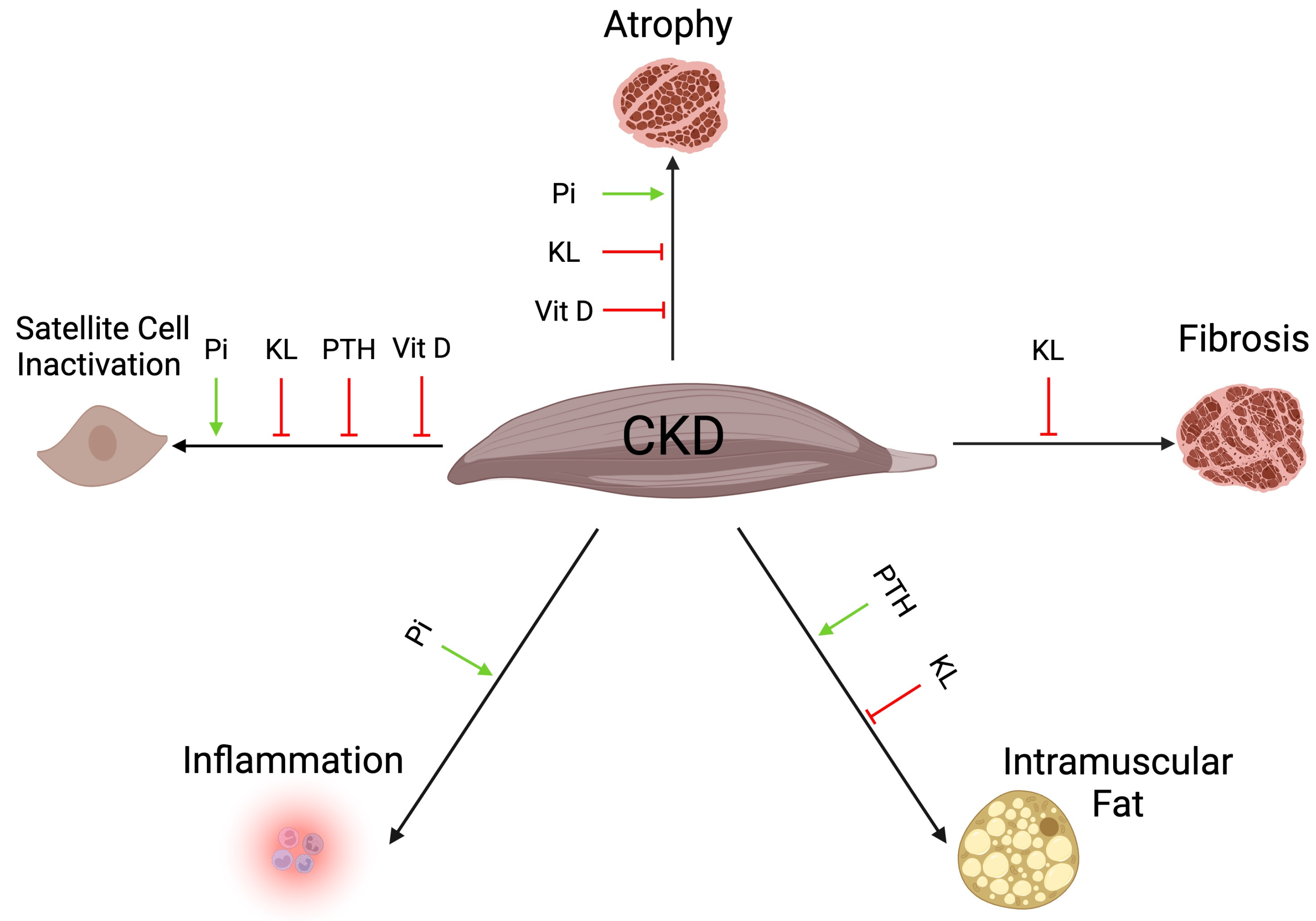
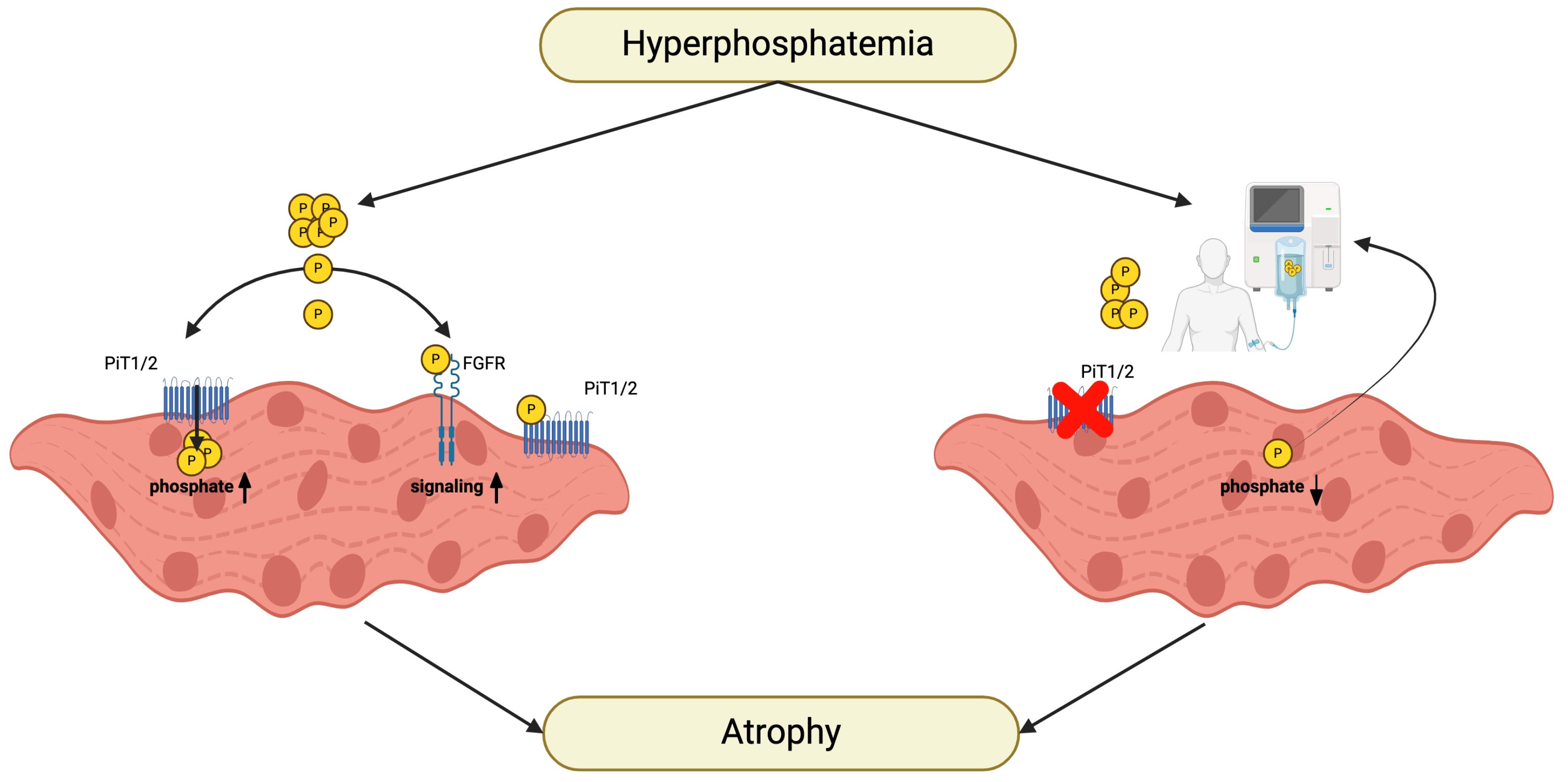

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traditional Factors | Non-Traditional Factors |
|---|---|
| Diabetes | Metabolic acidosis |
| Obesity | Anemia |
| Age | Accumulation of uremic toxins |
| Male | Low branched chain amino acid levels |
| Anorexia | High phosphate levels |
| Dietary restrictions | High FGF23 levels |
| Low physical activity | High PTH levels |
| Systemic inflammation | Low 1,25D levels |
| Insulin resistance | Low klotho/sKL levels |
| Low androgen levels | |
| High AGE levels |
| Renal replacement therapy | Kidney transplantation |
| Exercise | Resistance training |
| Endurance training | |
| Nutritional management | Low or high protein diet? |
| Supplementation of essential amino acids | |
| Low phosphate diet | |
| Interference with pro-inflammatory cytokines and their receptors and signaling | IL1R antagonist |
| IL6R antagonist | |
| IL6 antagonist | |
| STAT3 antagonist | |
| Shifting from atrophy to hypertrophy, from catabolism to anabolism | IGF1 administration |
| Blocking antibody against myostatin | |
| Soluble ActRIIB | |
| Blocking antibody against ActRIIA/B | |
| Interference with phosphate metabolism | PTH administration |
| 1,25D supplementation | |
| Phosphate binders | |
| Blockade of intestinal phosphate uptake | |
| Other pharmacologic interventions | AST-120 (absorbance of uremic toxins) |
| Sodium bicarbonate (neutralization of acidosis) | |
| Apelin, APJ receptor agonists (improving glucose and lipid metabolism) | |
| Iron supplementation (tackle anemia) | |
| Anabolic steroids | |
| GLP-1 receptor agonists |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heitman, K.; Alexander, M.S.; Faul, C. Skeletal Muscle Injury in Chronic Kidney Disease—From Histologic Changes to Molecular Mechanisms and to Novel Therapies. Int. J. Mol. Sci. 2024, 25, 5117. https://doi.org/10.3390/ijms25105117
Heitman K, Alexander MS, Faul C. Skeletal Muscle Injury in Chronic Kidney Disease—From Histologic Changes to Molecular Mechanisms and to Novel Therapies. International Journal of Molecular Sciences. 2024; 25(10):5117. https://doi.org/10.3390/ijms25105117
Chicago/Turabian StyleHeitman, Kylie, Matthew S. Alexander, and Christian Faul. 2024. "Skeletal Muscle Injury in Chronic Kidney Disease—From Histologic Changes to Molecular Mechanisms and to Novel Therapies" International Journal of Molecular Sciences 25, no. 10: 5117. https://doi.org/10.3390/ijms25105117