
The Histone Deacetylase Family: Structural Features and Application of Combined Computational Methods


1
Dipartimento di Scienze della Salute, Campus “S. Venuta”, Università degli Studi “Magna Græcia” di Catanzaro, Viale Europa, 88100 Catanzaro, Italy
2
Net4Science S.r.l., Università degli Studi “Magna Græcia” di Catanzaro, Viale Europa, 88100 Catanzaro, Italy
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2024, 17(5), 620; https://doi.org/10.3390/ph17050620
Submission received: 18 April 2024
/
Revised: 3 May 2024
/
Accepted: 8 May 2024
/
Published: 10 May 2024
(This article belongs to the Special Issue Selective Histone Deacetylase Isoforms as Potential Therapeutic Targets)
Abstract
:Histone deacetylases (HDACs) are crucial in gene transcription, removing acetyl groups from histones. They also influence the deacetylation of non-histone proteins, contributing to the regulation of various biological processes. Thus, HDACs play pivotal roles in various diseases, including cancer, neurodegenerative disorders, and inflammatory conditions, highlighting their potential as therapeutic targets. This paper reviews the structure and function of the four classes of human HDACs. While four HDAC inhibitors are currently available for treating hematological malignancies, numerous others are undergoing clinical trials. However, their non-selective toxicity necessitates ongoing research into safer and more efficient class-selective or isoform-selective inhibitors. Computational methods have aided the discovery of HDAC inhibitors with the desired potency and/or selectivity. These methods include ligand-based approaches, such as scaffold hopping, pharmacophore modeling, three-dimensional quantitative structure–activity relationships, and structure-based virtual screening (molecular docking). Moreover, recent developments in the field of molecular dynamics simulations, combined with Poisson–Boltzmann/molecular mechanics generalized Born surface area techniques, have improved the prediction of ligand binding affinity. In this review, we delve into the ways in which these methods have contributed to designing and identifying HDAC inhibitors.
1. Introduction
Histone deacetylases (HDACs), also known as lysine deacetylases (KDACs), are zinc (Zn2+)-dependent or nicotinamide adenine dinucleotide (NAD+)-dependent proteolytic enzymes. They are involved in transcriptional repression and chromatin condensation mechanisms through controlling the acetylation state of lysine side chains in histone tails [1]. Specifically, the HDAC protein family normally removes the acetate moiety from acetylated ε-amino groups of histone lysine and other non-histone proteins, thus regulating and modulating several pivotal biological signaling pathways [2]. After deacetylation, the positively charged N-terminal residues of amino acids interact with DNA phosphate groups, leading to the repression of gene transcription. By contrast, acetyltransferases (HATs) carry out the acetylation of histone lysines, thus neutralizing the positive charge of the lysine residue and relaxing the chromatin structure [3].
Moreover, HDACs can indirectly regulate other post-translational modifications (PTMs) by releasing the acetyl group from lysine so that other PTMs, for instance ubiquitination, can mark on the loci [4].
The enzymatic activities of HDAC isoforms are responsible for the maintenance of several normal physiological processes, such as cell proliferation, apoptosis, neurogenesis, and epigenetic regulations. An imbalance between histone acetylation and deacetylation can lead to a variety of diseases, including neurodegenerative and cardiovascular disorders, autoimmune diseases, metabolic disorders, diabetes, and cancer [5,6].
Due to their contributions in these pathophysiological conditions, HDACs have become attractive and significant targets, especially in cancer research [7,8]. Specifically, irregular HDAC expression and its abnormal deacetylation activity have become a very popular and interesting topic for the prevention of cancer generation and progression [9,10].
Contemporary advancements in computational methods for designing HDAC inhibitors highlight innovative and multifaceted screening and design strategies. These approaches markedly improve the quality of identified compounds as potential HDAC inhibitors. Specifically, the use of multilayered computational methods reduces the risk of false positive hits, enhancing the ability to select specific inhibitors through diverse filtering and scoring functions. Thus, this review aims to investigate how the integration of diverse approaches and methodologies has substantially enhanced the reliability of compounds identified in the domain of HDAC inhibitor discovery.
1.1. Classification of HDAC Family
So far, according to their 3D structure, function, and sequence homology, 18 isoforms of human HDACs have been identified. Based on their intracellular localization and tissue distribution specificity, they can be divided into four subfamilies (Class I, Class II, Class III, and Class IV) characterized by different biological functions (Figure 1) [11].
Class I subfamily includes HDAC1, HDAC2, HDAC3, and HDAC8. These isoforms are widely expressed in various tissues [12] and behave as repressors of gene transcription. HDAC1 and HDAC2 are highly homologous and are involved in cell proliferation, cell cycle regulation, and apoptosis [13]. HDAC3 plays a crucial role in cell cycle and DNA damage response [14], while HDAC8 is responsible for smooth muscle cell differentiation [15]. The Class II subfamily, which includes HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10, can be further divided into two groups: Class IIa (HDAC4, HDAC5, HDAC7, and HDAC9) and Class IIb (HDAC6 and HDAC10) [16]. Class IIa has only one catalytic domain, while Class IIb members show two catalytic domains [17]. HDAC4 and HDAC5, which are members of Class IIa, are expressed in the brain, heart, and skeletal muscle (Kee et al., 2022), while HDAC7 is predominantly present in the heart, lung, placenta, pancreas, skeletal muscle, and thymus [18]. HDAC9 is mainly expressed in the brain and skeletal muscle [19]. On the contrary, HDAC6 and HDAC10, which are representative of Class IIb, are expressed in the heart, skeletal muscle and brain [20], and in the liver, spleen, and kidney, respectively [21]. HDAC11, the only Class IV enzyme, shows a high catalytic efficiency as a fatty acid acylase and is present in the brain, heart, kidney, testis, and skeletal muscle [22].
The Class III subfamily is known as Sirtuins, due to its strong resemblance to the yeast Sir2 protein. It contains seven members (SIRT-7) and uses NAD+ for its ADP-ribosyltransferase and histone deacetylase enzymatic activities [23]. The SIR2 regulator family is divided into four subclasses: I, II, III, and IV. Subclass I consists of SIRT1, SIRT2, and SIRT3 proteins; subclass II contains SIRT4 protein; subclass III includes SIRT5 protein; and subclass IV comprise SIRT6 and SIRT7 proteins [24]. SIRT1, SIRT6, and SIRT7 are mainly present in the nucleus, SIRT2 in the cytoplasm, while SIRT3, SIRT4, and SIRT5 are located in the mitochondria [25]. Classes I, II, and IV HDACs exert their catalytic activity by means of Zn2+ ions and show a high homology of their catalytic core structural domain [26,27], with more remarkable variations in the sequences and structures outside the catalytic domain. By contrast, Class III HDACs are totally different from other HDACs [28,29], since they are NAD+-dependent Sir2 super proteins and their deacetylase reaction does not need Zn2+ direct involvement [30].
1.2. HDAC Structure and Function
HDACs typically include a core structural domain known as the HDAC structural domain, which consists of two highly conserved isoforms: the HDAC N-terminal structural domain and the HDAC central structural domain [6]. Within the HDAC structural domain, some catalytic sites, such as zinc ions and arginine residues, are crucial for their catalytic activity [31]. On the other hand, the HDAC C-terminal structural domain represents a more miscellaneous region, characterized by variations in length and amino acid sequences among different HDAC types [32]. Moreover, HDACs can form complexes with various proteins, including cell cycle regulatory proteins and transcription factors [33].
The earliest identified members of the Class I HDAC family include Rpd3 in budding yeast, along with HDACs1–3 and 8 all tumbling into this class. Their N-terminus contains the catalytic structural domains and exhibits a sequence conservation of 40–70% with the catalytic domain of yeast Rpd3 [34]. Also, HDACs1-3 possess C-terminal extensions of varying lengths, which can be subject to phosphorylation, enhancing their deacetylase activity and influencing the formation of co-inhibitory complexes [35]. These three isozymes are found within large multiprotein complexes [36]. HDAC1 and HDAC2 are exclusively localized in the nucleus, while HDAC3 contains a nuclear localization signal (NLS) region and a nuclear export signal (NES) region. Its localization may vary depending on cell type and environmental conditions [37]. However, all three isozymes collectively play a significant role as nuclear deacetylases.
HDAC8 differentiates itself from other Class I isozymes through the absence of a C-terminal extension region. Notably, HDAC8 demonstrates significant histone deacetylase activity and substrate selectivity even in its isolated form, suggesting a relatively independent functional capacity [38]. Structural analysis through crystallography reveals that the catalytic domain of Class I HDACs comprises approximately 400 amino acid residues, sharing a common structural framework. This framework consists of a core featuring eight parallel β-fold bundles forming a β-fold sheet, surrounded by more than thirteen α-helices and elongated loops stemming from the C-terminus of the β-fold, thus creating a narrow hydrophobic channel [4]. Within HDAC8, the hydrophobic channel is delineated by specific residues, including Phe152, Phe208, His180, Gly151, Met274, and Tyr306. Contrastingly, in other Class I subfamily members (HDAC1-3), most residues within this channel are conserved, except for Met274, which is substituted by leucine residues [39]. These conserved hydrophobic residues characterize the binding sites for the substrate. In the catalytic mechanism, the acetylated lysine of the substrate docks into the catalytic core pocket at the bottom of the hydrophobic channel, interacting with the zinc ion bound therein. Under normal physiological conditions, the hydrophobic channel accommodates the acetylated lysine side chain, containing four methylene groups of the substrate. At the bottom of the channel, the Zn2+ ion forms a chelate involving His180, Asp178, and Asp267 of HDAC, the acetyl group oxygen of the substrate, and the water molecule oxygen participating in the hydrolysis reaction. Aside from the substrate binding site and the zinc ion binding site, two metal ion binding sites are found in the catalytic domain of HDAC (Site 1 and Site 2) [40]. Site 1 is located close to the zinc ion binding site, while Site 2 is on the periphery of the catalytic domain, near the N-terminal end of the β-fold bundle. The presence of two metal ions within the enzyme structure is crucial in promoting its stability. Additionally, the metal ion located at Site 1 may potentially serve a functional purpose in the deacetylase reaction. This suggests that the metal ions not only provide structural support, but also contribute to the enzyme’s catalytic activity, making them essential components of the enzyme’s overall functionality. The main function of Class I HDACs is to remove acetyl groups from histones within complexes, thereby catalyzing enzymatic reactions. These complexes play a crucial role in regulating HDAC activity and specificity, and are also subject to regulation by other transcription factors. By means of this regulation, the complexes are able to bind to chromosomes and precisely target specific temporal and spatial locations for deacetylation. Besides, phosphatidylinositol, a conserved regulator within the Class I HDAC co-inhibitor complex, has been found to enhance HDAC enzymatic activity. However, it has been highlighted that effective activation of HDAC enzymatic activity requires the presence of both co-inhibitors and phosphatidylinositol. The interactions of phosphatidylinositol within the substrate binding channel alter the conformation of the channel, facilitating substrate access to the catalytic active site [41]. The involvement of polyinositol in this complex also hints at a possible connection between epigenetics and cellular metabolism. It is worth noting that HDAC8, unlike other Class I isoenzymes, has a higher catalytic efficiency for acyl-lysine substrates than for acetyl-lysine. Furthermore, HDAC8 operates independently of multiprotein complexes. What is particularly interesting is that the three-dimensional structural model of human HDAC8 in the PDB database reveals that the secondary structure consists of eleven or thirteen α-helices and eight β-folds [42]. The analysis of the PDB database reveals several distinctive features of HDAC8 compared to HDACs1-3 (Figure 2). Specifically, α-helix H1 in HDAC8 adopts a conventional α-helix structure, while loop L1 is two amino acid residues shorter than that of HDAC3. Additionally, loop L6 in HDAC8 contains a proline residue, causing the loop to be slightly distant from the catalytic site. As a result, HDAC8 maintains a relatively open catalytic pocket, which could potentially facilitate the access of substrate to its catalytic core. These structural differences make HDAC8 more compatible with its catalytic core and promote easier substrate access [43]. The elucidation of HDAC8’s crystal structure is crucial for understanding the structural dynamics and catalytic mechanisms of zinc ion-dependent HDACs, particularly within the Class I subgroup (Figure 3) [43]. Given that HDAC8 shares substantial sequence identity with HDAC1 (40%), HDAC2 (41%), and HDAC3 (41%), and that the catalytic activity centers of zinc ion-dependent HDACs are conserved, the information obtained from HDAC8’s crystal structure can provide valuable insights into this particular group of enzymes.
Enzymes belonging to Class II are further divided into two categories: Type a and Type b. The first Type encompasses HDAC4, 5, 7, and 9, while Type b comprises HDAC6 and HDAC10, which were recently discovered [46] (Figure 4). HDACs of Class IIa share a common catalytic core structural domain located at their C-terminus. They also have a unique and conserved N-terminal extension region, which contains multiple binding sites. Specifically, this region includes the MEF2 (myogenic transcription factor 2) binding site, which is crucial for inhibiting muscle cell differentiation. Additionally, the region contains several phosphorylated serine sites that regulate enzyme localization and interactions with transcription factors and co-blockers [47]. Class IIa HDACs can move easily between the nucleus and cytoplasm. The catalytic domains of Class IIa HDACs and Class I HDACs share similarities, featuring an α/β structure with several loops that form the substrate binding channel and catalytic active site. The active site contains a single zinc ion, whereas two potassium/sodium ion binding sites are also present. While the histone deacetylation domain is conserved across Class IIa HDACs, the distinct catalytic activity of this class remains incompletely comprehended [48]. Unlike Class I and IIb HDACs, IIa HDACs feature a conserved histidine residue instead of a tyrosine residue at their active site, leading to limited deacetylation activity [49,50]. However, this constraint does not hinder their role as transcriptional repressors. In this regard, IIa HDACs contribute to epigenetic functions not only through deacetylation but also by recruiting Class I HDACs and interacting with transcription factors via their N-terminal binding site [51]. On the other hand, HDAC6, belonging to Class IIb, is mainly observed in the cytoplasm and comprises distinct structural domains such as NLS (localization signal region), NES1 and NES2 (leucine-rich nuclear export signal regions), DD1 and DD2 (tandem deacetylation catalytic regions), SE14 (serine–glutamate-containing tetradecapeptide repeat region), and ZnF-UBP (ubiquitin-binding zinc finger structure) [52]. Despite containing a nuclear localization signal, HDAC6’s cytoplasmic localization is primarily governed by NES and SE14, facilitating its translocation and anchoring in the cytoplasm [53,54].
The role of hadC6 within the cytoplasm remained unknown for a considerable period. However, in 2002, its characterization as a major histone deacetylase and its diverse array of non-histone substrates, including α-microtubulin, cortactin, Ku70, and HSP90, were elucidated [55]. Tubulin stands out as the principal substrate of HDAC6, influencing cytoskeletal dynamics, intracellular transport, and cellular motility. By regulating microtubule assembly and the localization of microtubule motor complexes, HDAC6 influences microfilament-based cell motility and the interaction of cortical actin with microfilaments [56]. Thus, its inhibition results in the hyperacetylation of microtubule proteins, enhancing intracellular vesicular transport, a process associated with neurological disorders like Parkinson’s and Huntington’s disease [57]. Additionally, HDAC6 participates in crucial intracellular signaling pathways, underscoring its cellular significance. Conversely, HDAC10 operates as a transcriptional repressor, capable of shuttling between the nucleus and cytoplasm. It harbors two conserved deacetylation catalytic domains at the N-terminal end. While the C-terminal region shares sequence similarity with the N-terminal end, it lacks deacetylase activity. The cytoplasmic localization is dictated by the leucine-rich domain situated at the C-terminal end [58]. HDAC10 exhibits interactions with HDAC3 akin to Type a HDACs, yet it possesses a distinct capability to function independently as a deacetylase.
Figure 4.
A 3D structure available for human Class IIa and IIb HDAC. HDAC4 catalytic domain bound to a hydroxamic acid inhibitor (PDB code 2VQM) [59]; HDAC6 with catalytic domain 2 in complex with Trichostatin A (PDB code 5EDU) [60]; HDAC7 catalytic domain in complex with Trichostatin A (PDB code 3C10) [61].
Figure 4.
A 3D structure available for human Class IIa and IIb HDAC. HDAC4 catalytic domain bound to a hydroxamic acid inhibitor (PDB code 2VQM) [59]; HDAC6 with catalytic domain 2 in complex with Trichostatin A (PDB code 5EDU) [60]; HDAC7 catalytic domain in complex with Trichostatin A (PDB code 3C10) [61].

HDAC11 is the only member of Class IV HDAC, which has fewer similarities to Class I and II HDACs. Among the identified HDACs, HDAC11 is the shortest, and is primarily comprised of the core catalytic domain that exhibits exclusive deacetylase activity [62]. This protein can be found in both the nucleus and cytoplasm of cells and has tissue-specific expression, with notable abundance in the kidney, heart, brain, skeletal muscle, testis, and other tissues [63]. In vivo, HDAC11 can also form complexes with HDAC6 [64]. Despite being the most recently discovered isoenzyme, HDAC11 remains one of the least studied and understood proteins within the HDAC family, with its 3D structure currently unavailable.
Class III HDACs are a group of NAD+-dependent deacetylases, which are capable of catalyzing the deacetylation of histone and non-histone substrates [65]. This class of HDACs belongs to the Sirtuin protein family, which has seven members in humans and are known as SIRT1–7. In yeast, this family is represented by the Sir2 protein [66]. For human Sirtuin proteins SIRT1, 2, 3, 5, and 6, the crystal structures of their catalytic core domains have been successfully resolved (Figure 5). These structures exhibit a well-organized comprehensive configuration, which can be ascribed to the selective evolutionary process and the preservation of the catalytically active region’s sequence. Specifically, their catalytic structural domain shows an elliptical shape and comprises two large and two small structural domains, each encompassing approximately 270 amino acid residues. The relatively conserved large structural domain adopts a typical Rossmann fold structure, featuring a central β-sheet and six β-strands. Additionally, this domain boasts several α-helices forming pockets for NAD+ accommodation and binding. On the other hand, the smaller domain is more variable and comprises two modules extending from the large structural domain, including a conserved Zn2+ binding element and a region of α-helices with relatively high variability [67]. The zinc finger structural domain is made up of three reverse parallel β-strands and one α-helix [68]. The highly conserved four-loop region connecting the structural domains forms the substrate binding pocket crucial for catalytic activity. Notably, the largest loop, i.e., a β1-α2 loop or cofactor binding loop, is a part of the NAD+ binding site and has a highly dynamic structure, which is critical for catalytic reactions.
Research has highlighted that SIRT1–3 display significant deacetylating activity, differing from the lower activity observed in SIRT5–7 [69]. According to several studies, different SIRTs may exhibit higher activity toward novel acylations. Notably, SIRT4 does not show any significant deacetylation activity. On the other hand, SIRT1–2 demonstrate significant activity against various acylations. This suggests that different SIRTs may have distinct functions and roles within cells, contributing to various biological processes [66]. Moreover, SIRT2 has the ability to facilitate the removal of benzoyl groups from histone lysine both in laboratory experiments and within living organisms [70]. SIRT5, a Sirtuin protein, offers an array of enzymatic activities, including debenzoylation, which can modify specific amino acid residues on target proteins. Furthermore, SIRT5 can act on different acyl-CoA derivatives, such as malonyl, butanoyl, and glutaryl groups, providing a wide range of regulatory functions. By contrast, SIRT4 and SIRT6 exhibit ADP ribosyltransferase activity, a post-translational modification that can alter protein function and localization. Notably, SIRT6 also exhibits debenzoylation activity on long-chain fatty acids, which can further diversify its functional range of activity [71]. SIRT7’s deacetylation activity is triggered by double-stranded DNA, resulting in histone H3 lysine 18 (H3K18) deacetylation within chromatin. Moreover, rRNA can enhance SIRT7’s long-chain fatty acylation activity, potentially exceeding its deacetylation activity [72]. The intracellular localization of the Sirtuin family deacetylases is clearly established. SIRT1, which shares close resemblance with yeast Sir2, has been the object of extensive research. SIRT3 is distributed in both the nucleus and mitochondria [73], while SIRT4 and SIRT5 are predominantly mitochondrial [74]. SIRT6 exclusively resides in the nucleus, whereas SIRT7 is specifically localized in the nucleolus [75]. In summary, the roles of SIRT1–7 in cells exhibit complexity and diversity [76].
Figure 5.
A 3D structure available for human Class III HDAC (SIRT-7). SIRT1 catalytic domain bound to NAD and an EX527 analog (PDB code 4I5I) [77]; SIRT2 apoform (PDB code 3ZGO) [78]; SIRT3 in complex with ADP-ribose (PDB code 4BN4) [79]; SIRT5 in complex with diazirine inhibitor 9 [PDB code 7X3P] [80]; SIRT6 in complex with ADP-ribose [81].
Figure 5.
A 3D structure available for human Class III HDAC (SIRT-7). SIRT1 catalytic domain bound to NAD and an EX527 analog (PDB code 4I5I) [77]; SIRT2 apoform (PDB code 3ZGO) [78]; SIRT3 in complex with ADP-ribose (PDB code 4BN4) [79]; SIRT5 in complex with diazirine inhibitor 9 [PDB code 7X3P] [80]; SIRT6 in complex with ADP-ribose [81].

HDACs are implicated in a range of diseases, spanning cancer, neurodegenerative disorders, and inflammatory conditions, underscoring their potential as therapeutic targets (Table 1) [82,83,84,85,86]. Particularly in cancer biology, their different functions are evident through their commonly altered expression across a variety of tumor types, emphasizing their significance as potential targets for cancer treatment (Table 2) [6,82,87,88,89].
As researchers delve deeper into the structural complexities of HDACs, they are gradually uncovering distinctions among various subgroups and subtypes. This knowledge offers a rational explanation for the selectivity of some HDAC inhibitors. For example, a series of novel o-phenylenediamine HDAC inhibitors were found to selectively inhibit HDAC1 and HDAC2 [90]. After conducting a detailed analysis of homologously modeled HDAC1 and HDAC3, researchers have discovered a structural difference in an amino acid located in the cavity at the bottom of the catalytically active center. This difference involves the substitution of the Ser113 residue of HDAC1 with the Tyr96 residue of HDAC3, which may serve as the structural basis for selectivity. Additionally, the lack of isoform selectivity in most current HDAC inhibitors is due to the highly conserved amino acid sequence of the catalytic active center of Zn2+-dependent HDAC [91]. On the other hand, the amino acid sequence surrounding the active site entrance on the protein surface remarkably differs among the isoforms. HDAC8, for example, has a shorter L1 loop near the active center entrance than other Class I members, resulting in a more expansive entrance and a more flexible protein surface. To develop inhibitors selective toward specific subtypes, researchers are employing a successful strategy that takes into account the structural differences around the entrance to the active center of each HDAC8 subtype. A comprehensive understanding of the structural differences of each HDAC isoform will also be invaluable in creating potent and specific inhibitors (Table 3).
Interestingly, as extensively documented in the scientific literature, the functions of most HDAC isoforms undergo significant regulation through various post-translational modifications (PTMs), such as phosphorylations, acetylations, sumoylations and ubiquitinations. Additionally, HDACs can form complexes with other proteins (Table 3), thereby modulating their deacetylase activity [97]. In this context, multiple HDAC isoforms may participate in these complexes, where one isoform can influence the activity of another. For instance, research on human HDAC1 and HDAC2 has uncovered their coexistence in at least three distinct multi-protein complexes (Sin3, NuRD/NRD/Mi2 and CoREST), exerting mutual effects on their activities [98]. Furthermore, Fischle et al. demonstrated that the enzymatic activity of HDAC4, 5 and 7 relies on their association with the HDAC3/SMRT/N-CoR complex [99]. Specifically, the C-terminal zinc-binding domain of HDAC4 plays a critical role in substrate recognition and its association with the HDAC3–NCoR corepressor complex [100].
1.3. Mutation Effects on HDACs Biology
The dysregulation of HDACs, either through excessive or reduced activity, contributes to tumorigenesis by affecting apoptosis, differentiation and angiogenesis [6]. In particular, specific mutations can significantly alter the deacetylase activity of HDACs, thereby driving tumorigenesis and promoting cancer development [101]. Indeed, these mutations alter the dynamic regulation of histone acetylation and deacetylation processes catalyzed by lysine acetyltransferases (KATs) and HDACs [101]. Notably, mutations in HDACs, like HDAC2, HDAC4 and HDAC9, have been identified in various cancers (colon, breast, and prostate cancer) [102,103,104]. For instance, the recurrent frameshift mutation in exon1 of HDAC2 is particularly common in colon cancer and leads to a loss of measurable HDAC2 expression in mutant tumors [103]. This mutation confers resistance to HDAC inhibitors and alters gene expression to promote tumorigenesis [105]. In this regard, in vitro experiments demonstrated that HDAC2-deficient cells were unresponsive to HDAC inhibitors, such as Trichostatin A. These cells did not exhibit increased acetylation of histones H3 and H4, and their proliferation was not reduced compared to cells expressing wild-type HDAC2 [103]. On the other hand, mutations in other proteins, such as AT-Rich Interaction Domain 1A (ARID1A), can also influence HDAC activity and therapeutic responses [106,107]. ARID1A mutations are common in ovarian clear cell carcinomas (OCCCs) and endometrioid carcinomas (OECs), leading to loss of ARID1A protein expression and driving ovarian cancer progression [106,107]. Together, elevated HDAC2 expression is associated with poor outcomes in ovarian cancer [108]. Thus, given that EZH2 inhibition is synthetically lethal with ARID1A mutation, and the EZH2-containing PRC2 complex interacts with HDAC2, attempts were made to determine whether ARID1A regulates the interaction between EZH2 and HDAC2. Co-immunoprecipitation (coIP) analysis demonstrated an interaction between EZH2 and HDAC2 in ARID1A-mutated Ovarian Tumor-derived Cell Line 21G (TOV21G), and the restoration of wild-type ARID1A disrupted this interaction, suggesting that EZH2 did not interact with HDAC2 in ARID1A wild-type cells. Furthermore, ARID1A knockout amplified the growth inhibition caused by HDAC2 knockdown in ARID1A wild-type RMG1 cells while restoring wild-type ARID1A in ARID1A-mutated cells, reducing sensitivity to HDAC2 knockdown [109]. Notably, the observed growth inhibition induced by HDAC2 knockdown could be rescued by a short hairpin RNA (shRNA)-resistant wild-type HDAC2, but not by a catalytically inactive mutant HDAC2 H142A [110]. Given the involvement of the catalytic site, the sensitivity of HDAC2 to SAHA was evaluated in preclinical models of ARID1A-mutated ovarian cancers. ARID1A-mutated cells exhibited significantly lower half-maximal inhibitory concentration (IC50) of SAHA than ARID1A wild-type cells. Furthermore, SAHA treatment effectively inhibited the growth of xenografted ARID1A-mutated tumors and improved survival in mice bearing orthotopically transplanted ARID1A-mutated tumors, suggesting potential for achieving selectivity with pan-HDAC inhibitors in ARID1A-inactivated cells [109].
1.4. HDACs, HDACIs, Metabolism and Emerging Technologies like Omics
Histone acetylation and deacetylation processes are highly sensitive to changes in metabolite levels, which can impact the effectiveness of histone deacetylase inhibitors (HDACis) and the intrinsic HDAC activity. Additionally, HDACs have demonstrated regulatory effects on proteins beyond histones, including enzymes participating in metabolic pathways. There are several examples of metabolic pathways influenced by HDAC activity [111]. For instance, in several cancers, such as hepatocellular carcinoma (HCC), heightened aerobic glycolysis contributes to increased tumor growth, a phenomenon known as the Warburg effect [112]. The gluconeogenesis pathway suppresses aerobic glycolysis; hence, the inhibition of gluconeogenesis can further increase cancer cell growth. Yang et al. identified elevated levels of HDAC1 and HDAC2 in HCC tissues. HDAC1 and HDAC2 inhibit Fructose-1,6-bisphosphate (FBP1) expression, the key enzyme in the gluconeogenesis pathway, through histone H3K27 deacetylation at the FBP1 enhancer. This repression of gluconeogenesis promotes aerobic glycolysis and cancer progression. Knockdown of HDAC1 and HDAC2 resulted in increased FBP1 expression and reduced cell growth in HCC cell lines [113]. Thus, these deacetylases are not only implicated in epigenetic modifications, but also in metabolic or, for example, immune modulation [113]. Therefore, understanding the impact of HDACs on cancers through metabolic or other processes can reveal new potential targets [111]. Consequently, computational -omics techniques encompassing genomics, transcriptomics, proteomics, metabolomics and epigenomics can play crucial roles in elucidating the HDACs functionality and the inhibitory activity of HDACis, especially in the context of specific mutations [111]. For instance, transcriptomics can identify gene expression changes upon HDAC inhibition, while metabolomics can reveal metabolic alterations influenced by HDAC activity. Moreover, epigenomics techniques, like Chromatin Immunoprecipitation Sequencing (ChIP-seq), can elucidate the genomic regions targeted by HDACs [114].
Various -omics approaches have begun to elucidate the underlying mechanisms of therapeutic or toxic effects associated with HDACis. These inhibitors modulate the expression of genes involved in diverse biological pathways, including cell cycle regulation, cell death, metabolism, and stress responses in cancer cells [114]. Beyond epigenomic and transcriptomic profiling, recent advancements in proteomics, metabolomics and chemoproteomics have provided datasets relevant to HDACis (Table 4) [114].
For example, Zhu et al. conducted multi-omics analyses involving bulk RNA sequencing (RNA-seq), transposase accessible chromatin sequencing (ATAC-seq), and H3K27ac-targeted cleavage under targets and tagmentation sequencing (CUT&Tag-seq) in HDACi-treated CAR-T cells, revealing comprehensive epigenetic remodeling and functional alterations, including changes in chromatin accessibility, transcription factor interaction networks and regulation of T cell differentiation. Also, specific HDAC inhibitors, like M344 and Chidamide (selective class I inhibitors), notably suppressed HDAC1 expression in CD19-28ζ CAR-T cells [115]. Regarding metabolism, inhibition of HDAC activity by HDACis can affect various metabolic processes. Amoedo et al. demonstrated that sodium butyrate (NaB) and Trichostatin A (TSA) treatment of lung cancer cells led to increased oxygen consumption coupled with ATP synthesis, activation of the pentose phosphate pathway (PPP), and enhanced mitochondria-bound hexokinase activity, promoting glycolysis [116]. Also, a study conducted by Wardell et al. examined the impact of HDACis valproate (VPA) and suberoylanilide hydroxamic acid (SAHA) on metabolism within the context of multiple myeloma. These HDACis elicited various metabolic changes in the cells, including reduced levels of acetyl-CoA, a decreased expression of glucose transporter type 1 (GLUT1) and the inhibition of hexokinase 1 (HXK1) activity [116]. The latter two effects are associated with diminished glucose uptake and glycolysis, processes essential for energy production in cancer cells [117]. On the other hand, some metabolites can regulate HDAC and HDACi activity. For instance, trapoxin (TPX) acts as an irreversible inhibitor of HDAC1 and HDAC4 by covalently binding to these targets [118,119]. Additionally, metabolism can enhance the activity of specific HDACis, such as the reduction of the disulfide bond in depsipeptide to create an active compound like romidepsin, which exhibits potent anticancer effects in leukemias and lymphomas [120,121]. In summary, employing systems technologies like proteomics and metabolomics is instrumental in fully understanding the mechanism of action of HDAC inhibitors and evaluating their therapeutic activity [111].
2. HDAC Inhibitors
HDAC inhibitors (HDACis) are a class of pharmacological agents that have shown great potential in the treatment of cancer. These compounds primarily function by altering the acetylation status of histones, which play a crucial role in regulating gene expression. By modifying histone acetylation, HDACis can induce changes in chromatin structure, leading to the activation of silenced genes and the inhibition of genes that promote cell proliferation. HDACis have been shown to exert their anticancer effects through a variety of mechanisms, including apoptosis, cell cycle arrest and autophagy [122,123]. Clinical trials have primarily focused on developing targeted Class I/II HDAC inhibitors, such as isohydroxamic acids like SAHA and cyclic peptides. These inhibitors have shown the most promising activity in inhibiting HDACs. However, they face several challenges, such as low bioavailability, rapid metabolism, irreversible differentiation, and lack of selectivity towards cancer cells. Therefore, it is crucial to explore the various functions of different HDACs to develop more effective and selective HDAC inhibitors. Additionally, HDAC inhibitors have exhibited effects on various cells and genes, indicating multiple antitumor mechanisms [124], including apoptosis and autophagy induction [125], tumor cell cycle arrest [126], and the inhibition of tumor cell angiogenesis [125]. For cell death in many cancer cells, HDACis can activate either the extrinsic pathway, influencing the receptor death pathway, or the intrinsic pathway, affecting the mitochondrial pathway [127]. Over the past few years, many HDAC inhibitors have been created either synthetically or by extracting them from natural sources [128]. The first natural hydroxamic acid known to inhibit HDACs was Trichostatin A (TSA). Vorinostat (suberoylanilide hydroxamic acid, SAHA), structurally similar to TSA, was the first FDA-approved HDAC inhibitor for the treatment of refractory cutaneous T-cell lymphoma (CTCL) [129]. At present, the US Food and Drug Administration (FDA) has given its approval to four HDACis for the treatment of various hematologic tumors as well as certain solid tumors (Figure 6) [130,131]. Moreover, Tucidinostat was approved in 2015 by the China Food and Drug Administration (CFDA) in the treatment of certain cancers [132].
Over the past two decades, there has been a significant expansion in the compound library of Zn2+-dependent HDAC inhibitors (HDACis). Despite the various structures of these compounds, whether synthesized or naturally occurring, a shared pharmacophore model predominates among most Zn2+ HDACis (Figure 7). This model comprises three essential elements:
- (1)
- the cap structure (Surface Recognition Domain), which typically constitutes a hydrophobic aromatic moiety that interfaces with the enzyme surface;
- (2)
- a Zn2+ binding group (ZBG), such as isohydroxamic acid, carboxylic acid, or benzamide, coordinating the Zn2+ ion at the enzyme catalytic center;
- (3)
Studies on co-crystalized complexes of isohydroxamic acid HDACis and HDACs have elucidated the interaction between the cap structure and the amino acids proximal to the enzyme catalytic site. Meanwhile, the ZBG structure binds to the metal ion at the bottom of the active site to form the complex [45]. The linker is a crucial component that plays a significant role in positioning the ZBG group within the active region of HDACs. The optimal length of the linker is of utmost importance, as it helps the ZBG group to chelate with Zn2+ and to establish hydrogen bonds with amino acids like histidylic acid and tyrosine. The long linker chain interacts with amino acid residues, occupying the active site, through forces like van der Waals interactions. On the other hand, the cap structure functions as a cover for the entrance to the enzyme active site [45]. HDACis work by competitively inhibiting the binding of acetyl-lysine residues of the substrate to the enzyme active site. However, alterations in any of the three components of the HDACis pharmacophore can influence their activity or selectivity, which can affect their therapeutic efficacy and safety. Isohydroxamic acid, benzamide, carboxylic acid, sulfhydryl groups, ketones, and epoxides are typical groups for ZBG. A comparative analysis of clinical HDACis SAHA, entinostat (MS275), and valproic acid, utilizing isohydroxamic acid, benzamide, and carboxylic acid as chelating groups, respectively, underscores significant differences in their inhibitory activity against HDACs. Notably, isohydroxamic acid exhibits the most potent zinc ion chelation capability [135]. Linker structures can differ in terms of their structural characteristics, such as lengths, saturation, unsaturation, linearity, cyclicality, and modifications. Scientists have found that modifying the linker can significantly affect the activity of an HDACi, making it more or less effective. Some of the most commonly used types of linkers include aliphatic chains, aromatic rings and vinyl–aromatic rings [136]. Docking and energy-optimized pharmacophore localization studies have revealed that a higher affinity for the target can be obtained with inhibitors containing at least one aromatic ring in their linker region. Moreover, the most effective enzyme inhibitory activity was observed when the carbon number of the linker region (n) is six. Phenyl, naphthyl and thiophene groups in the cap groups enhance the hydrophobic and high capacity of compounds, leading to improved HDAC inhibition. Additionally, the presence of substituents with higher lipophilicity, such as trifluoromethyl, tend to result in stronger HDAC inhibition. The effect is even more pronounced when methoxy and trifluoromethyl substitutions occur in the cap group at the adjacent, inter-, and para-positions. Lipophilicity unequivocally amplifies the hydrophobic interaction between the HDAC active site and its inhibitor, resulting in a marked increase in the inhibitor activity [137]. It is important to improve our understanding of the HDACi inhibition mechanism to develop drug compounds more effectively. One way to achieve this is by combining the pharmacophore model of HDAC inhibitors with structural insights into the enzymatic active region. This widely accepted approach enables the rational design and optimization of HDAC inhibitors by modifying their structure based on the three components of the pharmacophore.
The ZBG plays a pivotal role in the inhibitory activity of HDACis by binding to Zn2+ and its adjacent residues [46]. Using the type of ZBG to classify the six HDACIs, three primary categories are obtained (Table 5):
- (1)
- Isohydroxamic acids, which encompass SAHA, Belinostat (PXD101), and Panobinostat (LBH589);
- (2)
- Benzamide derivatives, exemplified by Mocetinostat (MGCD0103) and Chidamide;
- (3)
- Cyclic peptides, represented by romidepsin (FK228) [138].
Moreover, there are multiple HDACi drugs currently being studied in preclinical and clinical trials [139].
2.1. Isohydroxamic Acids
Isohydroxamic acid-based HDAC inhibitors have been extensively studied and are widely used due to their ability to inhibit nearly all Zn2+-dependent HDACs belonging to Classes I, II and IV. These inhibitors are known to be broad-spectrum inhibitors and are recognized as highly effective in regulating gene expression, cell differentiation and cell death. Unfortunately, this wide-ranging inhibition also gives rise to several adverse side effects. Despite these drawbacks, these drugs show potent antitumor effects and are frequently incorporated into combination therapies with various anticancer agents, amplifying their antitumor efficacy [140]. The hydrophobic channel of HDAC plays a crucial role in its physiological function by accommodating the acetyl-lysine side chain of the substrate. Zn2+ forms a five-tooth chelate at the bottom of the channel. However, when the isohydroxamic acid of HDAC inhibitors is present, the hydrophobic channel becomes occupied by the inhibitor’s hydrophobic linker competitively, while the ZBG of the inhibitor is chelated the zinc ion. Specifically, the isohydroxamic acid group, beyond forming a strong diphthong chelate with Zn2+, can be involved in oxygen bonds with His142, His143 and Tyr306. Thus, the isohydroxamic acid group acts as a ZBG and shows several advantages, such as unexacting synthesis, excellent in vitro stability, strong zinc binding and elevated solubility [141]. However, isohydroxamic acid groups show some negative aspects to consider. They act as a non-selective ZBG, also binding to other zinc-dependent enzymes, such as aminopeptidases, matrix metalloproteinases and carbonic anhydrases, with the appearance of unwanted side effects. Furthermore, isohydroxamic acid is susceptible to hydrolysis and glucuronidation, resulting in unfavorable pharmacokinetic characteristics and reduced in vivo efficacy [142]. Regarding the linker domain of isohydroxamic acid, either linear or cyclic structures, as well as saturated or unsaturated configurations, can be observed. Regarding the linear, straight-chain linker isohydroxamic acid HDAC inhibitors, the cap structure plays a pivotal role in compound modification and optimization, resulting in a diverse range of HDAC inhibitors. The flexibility of linear linkers enables them to interact more effectively with the surface amino acid residues, and this is crucial for HDAC activity. Due to this, in designing HDAC inhibitors, more complex cap structures such as branching caps are often employed. These cap structural domains usually consist of hydrophobic groups, specifically aromatic moieties. By incorporating these cap structures, the inhibitors can be optimized for their potency and selectivity towards HDAC enzymes [143].
In 2006, the US Food and Drug Administration (FDA) approved the clinical use of SAHA (Suberoylanilide Hydroxamic Acid) for the treatment of the cutaneous T-cell lymphoma. SAHA, also known as Vorinostat, was developed by Merck and represented the first approved HDACi [144]. This compound has shown potential in treating hematologic cancers, with a different efficacy in B-cell lymphomas, such as diffuse large B-cell lymphoma, follicular lymphoma and mantle cell lymphoma. For solid tumors like prostate and pancreatic cancers, SAHA has demonstrated the inhibition of the Akt/FOXO3a signaling pathway, which stimulates apoptosis in prostate tumor cells [145]. Additionally, SAHA plays a crucial role in inducing autophagy in tumor cells and preventing acute graft-versus-host disease. However, it is associated with significant toxicity, including fatigue, diarrhea, anorexia, bone marrow suppression and thrombocytopenia, due to its broad-spectrum inhibitory capacity [146]. Recent research has shown a promising potential in enhancing the selectivity of HDAC inhibitors, or developing new ones based on SAHA’s fundamental pharmacodynamic moiety. For instance, by replacing the hydrogen atom (H) at the C2 position of SAHA’s hydrophobic long chain with aliphatic or aromatic hydrocarbons, a new analog named C2-R-SAHA was created. This analog has shown the ability to enhance its selectivity for HDAC6 and 8 [147]. Thanks to the molecular docking approach, highly conserved active catalytic regions across all HDAC isoforms were observed, with class I HDAC exhibiting a narrower hydrophobic channel than HDAC6. Thus, by substituting aliphatic hydrocarbons on the hydrophobic chain of SAHA, it is possible to increase the barrier to the catalytic channel, which in turn impedes the catalysis of compounds HDAC1, 2 and 3. Similarly, by replacing unsaturated hydrocarbons on aromatic, cyclic, or adjacent isohydroxamic acid groups, it is possible to enhance the selectivity of HDAC6, as observed in tubastatin A [148]. Interestingly, Trichostatin A (TSA) shares its structure with SAHA, but displays significantly stronger HDAC inhibition activity. This increased activity is largely attributed to the bridging region of TSA, which includes a diene and an R-type methyl group. However, researchers have determined that these features alone do not fully explain the potency of TSA. The arylamine ring in the surface recognition region may also play a significant role in its effectiveness by interacting with amino acid residues in the enzyme capsule [149].
2.2. Benzamide Derivatives
Benzamide inhibitors are a new promising class of HDACis, characterized by enhanced selectivity and, thus, by reduced side effects. Specifically, they showed a higher selectivity towards HDAC1 and 2 than conventional isohydroxamic acid counterparts thanks to their unique N-(2-aminophenyl) benzamide pharmacodynamic group. Through a molecular docking study, Bass et al. suggested that benzamide inhibitors exhibit a distinct binding mode to histone deacetylase-like protein (HDLP), differing from isohydroxamic acid analogs [150]. Interestingly, their binding does not involve interactions with Zn2+. According to the docking findings, benzamides interact with the two benzene rings of Phe141 and Phe198 residues, narrowing the active pocket and obstructing the channel of the N-terminal Lys acetylation side chain of histone, the physiological substrate of HDAC. Such orientation allows for the formation of a hydrogen bond with either Tyr91 or Glu92, while the intermediate benzene ring adopts a sandwich structure with Phe141 and Phe198. This specific binding confers consistent selectivity to benzamide-based inhibitors compared to isohydroxamic acid inhibitors targeting Zn2+, thereby reducing toxicity.
The 3D structures of HDAC2 inhibitor complexes disclose the arrangement of the HDAC2 active site, comprising an approximately 8Å-long hydrophobic channel with the catalytic site containing the Zn2+ and an adjacent inner cavity termed the “foot pocket,” spanning approximately 14 Å. During inhibition, benzamide inhibitors deeply penetrate this cavity, with the o-amino group and carbonyl oxygen participating in Zn2+ chelation. Conversely, one side of the aromatic ring enters the catalytic “foot pocket,” prompting the repositioning of its residues to accommodate the aryl portion. In contrast, the structural characteristics of SAHA hinder its access to the catalytic foot pocket, explaining its lack of specificity in inhibiting HDAC2 [151]. Intramolecular hydrogen bonding can influence the effectiveness of benzamide inhibitors over time, unlike SAHA, which has a Zn2+ chelating group at the top of its molecule. This eliminates the need for extensive protein rearrangement or the breaking of internal ligand hydrogen bonds during the formation of drug-target complexes. The isohydroxamic acid of SAHA can directly bind to Zn2+ at the bottom of the hydrophobic channel and replace the bound water, resulting in rapid binding kinetics for ligands containing isohydroxamic acid esters. Benzamide inhibitors, however, must break their intramolecular hydrogen bonds before chelating with Zn2+. Furthermore, their large molecular size and curved hydrophobic channels limit their ability to rapidly bind the zinc ion [45].
Chidamide represents the pioneering oral inhibitor of histone deacetylase with subtype selectivity. It has been approved for clinical trials by the State Food and Drug Administration (China Food and Drug Administration, CFDA) [152]. Chidamide falls under the category of benzamide histone deacetylase subtype-selective inhibitors and it is characterized by its distinctive chemical structure. Known chemically as N-(2-amino-4-fluorophenyl)-4-[1]benzamide, Chidamide exhibits potent antitumor efficacy associated with low cytotoxicity relative to its counterparts. Its primary targets include subtypes 1, 2 and 3 of Class I HDACs and subtype 10 of Class IIb [153]. Moreover, Chidamide can induce the differentiation of tumor stem cells and reverse epithelial-mesenchymal phenotypic transformation (EMT) in tumor cells, thereby reinstating drug sensitivity in resistant tumor cells and impeding tumor metastasis and recurrence. This mechanism is attributed to its inhibition of relevant HDAC isoforms, elevation of chromatin histone acetylation levels, and initiation of chromatin remodeling, consequently inducing epigenetic alterations that disrupt the tumor cell cycle and promote apoptosis. Also, Chidamide demonstrates modulatory effects on cellular immunity, enhancing the activity of natural killer (NK) cells and antigen-specific cytotoxic T cells (CTLs) in mediating tumor cell elimination.
2.3. Cyclic Peptides
Cyclic peptides, the most structurally intricate class of HDAC inhibitors, can be categorized into two groups based on the presence of the 2-amino-8-oxo-9, 10-epoxy-decanoyl (Aoe) moiety. In the first group, including cyclic peptides with the Aoe moiety, trapoxin A, trapoxin B and WF-3161 can be mentioned. Apicidin and depsipeptide are examples of the second group, consisting of cyclic peptides without the Aoe moiety. Both peptide groups bind to HDACs similarly to isohydroxamic acids but with distinct mechanisms [154]. The spatial arrangement of Aoe-containing cyclic tetrapeptide macrocycles exhibits a configuration with D-amino acids and cycloamino acids, a spacer region adjacent to the amino acids, and numerous internal hydrogen bonds, resulting in a constricted 12-membered cyclic structure. Indeed, the presence of D-configured amino acids appears crucial for tight binding to the cap activation site edge. Some Aoe-containing inhibitors, for HDAC binding, require an epoxy keto group, a large cyclic peptide structure capable of binding to the duct entrance “groove,” a keto carbonyl group for interaction with Zn2+ and polar amino acid residues in the HDAC duct, and an epoxy group for alkylating the HDAC active site, thereby irreversibly inhibiting enzyme activity. Notably, substituting the epoxy keto group with isohydroxamic acid renders HDAC inhibition reversible [155]. Cyclic peptide HDAC inhibitors predominantly employ larger cyclic peptide structures as Cap groups. FK228, extensively studied, does not adhere to the classical pharmacophore model of HDAC. In vivo, it necessitates hydrolysis to release the sulfhydryl moiety of the zinc-chelating group, enabling effective chelation with zinc metal ions for enzyme inhibition. The larger cap group enhances interaction with amino acids at the active pocket periphery, thereby increasing target affinity [156]. Also, these inhibitors exhibit subtype selectivity for the HDAC family, showing potent inhibitory activity for Class I HDACs while poorly inhibiting Class IIb HDACs, particularly HDAC6. Thus, this selectivity offers insights into designing selective HDAC inhibitors. However, the design and synthesis of these inhibitors face challenges due to the complexity and poor drugability resulting from their large molecular skeleton and mass.
In 2012, the US FDA approved romidepsin (FK228), an atypical HDACi targeting Class I, for treating cutaneous T-cell lymphomas (CTCL) and peripheral T-cell lymphomas [157]. It derives from Gram-negative pigmented bacillus No. 968 and possesses a caged bicyclic phenolic peptide structure with rare disulfide bonds. These bonds are activated within human cells post-metabolism [158]. FK228, a precursor drug, exhibits greater stability than its reduced form, Red-FK228. The disulfide bond facilitates efficient diffusion across the cell membrane. Upon entering the cell, FK228 undergoes activation through glutathione reduction, allowing the Red-FK228 free sulfhydryl group to interact with the Zn2+ active site, thereby preventing HDAC from binding to the substrate.
2.4. Future Perspectives and PROTACs
Despite the considerable progress made, numerous unresolved issues in the exploration of HDAC inhibitors (HDACis) remain. Primarily, the majority of HDACis currently available are broad-spectrum inhibitors. In fact, these compounds compete for Zn2+ within the enzyme active site, lacking specificity towards distinct isoforms. By disrupting specific HDAC activities crucial for protein–protein interactions, a degree of selectivity for HDAC isoforms can be reached [159]. As previously highlighted, HDAC1, 2, and 3 are subunits within multiprotein complexes localized in the nucleus, and dislodging HDACs from these complexes significantly decreases enzyme activity. Thus, inhibiting the assembly of these complexes can partially block HDAC activity. Inositol phosphate is a well-conserved regulatory factor found within a complex of multiple proteins. It has a significant impact on enzyme activity. Indeed, by interacting with arginine residues close to the active site access, inositol phosphate is crucial for forming the complex and activating the enzyme. Therefore, the disruption of this interaction may intensify the inhibitory effect on HDAC1, 2 and 3, which can have negative consequences on enzyme activity.
Generally, HDAC inhibitors employ Zn2+-binding groups like isohydroxamic acid, thiol, carboxylic acid, ketone, or 2-aminoaniline. However, these functional groups may also strongly bind to other essential metalloenzymes, culminating in cytotoxicity and limiting the clinical utility of HDACis [160]. Combination therapy stands as a pivotal strategy to enhance efficacy, mitigate adverse effects, and neutralize drug resistance in tumor treatment. Extensive research has delved into combining HDACi with diverse drug categories, including antimetabolites, antimicrotubule drugs, topoisomerase II inhibitors, DNA cross-linking agents like cisplatin, HSP90 antagonists, and targeted therapies [161]. Also, HDACis have demonstrated synergy with the transcriptional regulator all-trans retinoic acid, DNA demethylating agents, and Bcr-Abl kinase inhibitors [162]. In summary, these results highlight the potential of HDACi combination therapy as an optimal therapeutic approach, exploiting synergistic effects to improve antitumor efficacy and overcome resistance to conventional treatments. Several combination drug regimens based on HDAC inhibitors (HDACi) are currently in clinical studies [163]. Thus, HDACis are progressively emerging as a promising novel therapeutic agent for tumors, showing broad potential in anticancer therapy.
In addition to isoform selectivity, HDAC1, HDAC2, and HDAC3 are present in vivo within seven distinct corepressor complexes, introducing an additional layer of complexity [164]. These different corepressor complexes play distinct physiological roles in cells, suggesting that targeting specific HDAC-containing corepressor complexes may be crucial for discovering new HDAC therapies with better efficacy and fewer side effects [165]. The future contributions of Proteolysis Targeting Chimeras (PROTACs) in this area are of particular interest. PROTACs, designed to degrade target proteins, generally include three components: a ligand to bind the protein of interest (POI), a ligand to trigger protein degradation (commonly an E3 ligand), and a linker covalently connecting these two ligands (Figure 8). Through the polyubiquitination of lysine amino acids on the POI, PROTACs facilitate eventual degradation by the proteasome. This ubiquitin transfer to the POI relies on the protein–protein interaction between the POI and E3 ligase mediated by the PROTAC [166].
For instance, the pan-BET inhibitor JQ1 functions broadly across BET proteins; however, when JQ1 is included in a PROTAC, it facilitates the specific degradation of BRD4 while sparing BRD2 and BRD3 [167]. Foretinib, a pan-kinase inhibitor, exhibits reduced kinase binding when functionalized into a PROTAC, indicating altered specificity [168]. Additionally, PROTACs’ ability to modify binding affinities and selectivity highlights their dynamic role in protein degradation, as evidenced by studies on p38 isoforms [169].
The remarkable ability of PROTAC-mediated degradation to alter protein selectivity has attracted researchers seeking to modulate HDAC activity. Notably, the first PROTAC targeting a histone deacetylase enzyme, NAD+-dependent SIRT2 [170], and the first zinc-dependent HDAC-targeting PROTAC for HDAC6 signify significant advancements in the field [171]. While approximately 20 PROTACs are progressing through or are already in clinical trials, none currently target HDACs; nevertheless, several studies have highlighted the great potential of this challenging approach. For instance, in a proteomics investigation by Xiong et al., HDAC degradation was explored with 48 PROTACs varying in HDAC ligand, linker length, and E3 ligand, revealing HDAC1, HDAC2, and HDAC9 as the least frequently degraded zinc-dependent HDAC isoforms, while HDAC6, HDAC8, and HDAC3 were most frequently degraded [172]. Thus, generating selective PROTACs for HDAC1 and HDAC2 posed challenges, given the susceptibility of the other Class I isoform (HDAC3) and HDAC6 to PROTAC-mediated degradation. Nevertheless, some PROTACs demonstrated selectivity in degrading specific HDAC isoforms, like HDAC3, exemplified by the synthesis reported by Xiao et al. of an HDAC3-selective degrader exhibiting significant potency in breast cancer cells [172,173,174,175]. Moreover, several studies have investigated the PROTAC-mediated degradation of HDAC8 [176,177,178,179,180], highlighting its propensity for degradation alongside HDAC3 and HDAC6, with Chotitumnavee et al. developing a selective HDAC8 PROTAC that outperformed its parent inhibitor in compromising cell viability [177].
Within the Class IIa enzymes, only HDAC4-selective PROTACs have been documented thus far, with Macabuag et al. pioneering the development of these PROTACs to explore the role of HDAC4 in Huntington’s Disease [181]. Their study introduced two sets of isoform-selective PROTACs, one incorporating a hydroxamic acid-based inhibitor linked to a VHL E3 ligase ligand via three different PEG lengths, and the other based on a trifluoromethyl oxadiazole HDAC inhibitor [182,183]. Both sets demonstrated dose-dependent degradation of HDAC4 in Jurkat E6-1 cells while sparing HDAC1, HDAC5, HDAC7, and HDAC9.
Meanwhile, among the eleven zinc-dependent HDAC enzymes, PROTACs targeting HDAC6 for degradation have been extensively reported, suggesting its particular susceptibility to proteasome-mediated degradation by PROTACs. This susceptibility might be attributed to the zinc finger ubiquitin-binding domain that characterizes this isoform [184].
Following K. Yang et al., who discovered selective HDAC6 degradation using a PROTAC incorporating a pan-HDAC inhibitor as the HDAC ligand [171], An et al. drew inspiration from the selective HDAC6 inhibitor Nexturastat A to design PROTACs targeting HDAC6 [185]. The PROTAC, incorporating Nexturastat A as the HDAC ligand and pomalidomide as the E3 ligand, demonstrated impressive efficacy in the B lymphoblast MM.1S cell line, with no degradation of HDAC1, HDAC2, or HDAC4. Additionally, Cao et al. devised a structurally unique HDAC6-targeting PROTAC based on the natural product indirubin as the ligand to engage HDAC6, revealing that shorter PEG linkers containing one PEG unit were notably more effective at HDAC6 degradation than longer PEG linkers [186].
Concerning PROTACs targeting Class III HDACs, J.Y. Hong et al. introduced a new SIRT2 inhibitor, incorporating it into two PROTACs [187], both effectively degrading SIRT2 within a concentration range of 0.5–10 mM in MCF7 and BT-549 cell lines. One of these PROTACs demonstrated the capability to reduce SIRT2 deacetylase and defatty-acylation activity in cells, whereas the SIRT2 inhibitor from which the PROTAC derives was not capable of reducing SIRT2 defatty-acylation activity, showcasing an advantage over the sole inhibition of SIRTs’ catalytic active site.
HDAC11, the sole HDAC isoenzyme in Class IV discovered in 2002, possesses significant fatty-acid deacylase activity and is recognized as a potential target for metabolic disorders [188,189,190]. Despite the existence of selective inhibitors for HDAC11 [191], no PROTACs targeting its degradation have been reported to date.
PROTACs exhibit the potent and selective degradation of individual HDAC isoforms, serving as valuable chemical probes for studying HDAC biology. By targeting HDACs for degradation via the proteasome, researchers can explore their biological roles beyond enzymatic function alone, potentially leading to novel therapeutic applications. While some HDAC-targeting PROTACs show enhanced efficacy in compromising cancer cell viability compared to HDAC inhibitors, others exhibit reverse effects, particularly against pan-HDAC inhibitors. However, future investigations should explore the therapeutic potential of HDAC-targeting PROTACs beyond cytotoxicity, considering their ability to enhance antitumor immune responses and overcome drug resistance in cancer immunotherapies, as evidenced by recent studies [192,193]. Ongoing clinical trials investigating combination therapies of HDAC inhibitors with immune checkpoint inhibitors suggest a promising avenue for the therapeutic application of PROTACs targeting HDACs [193].
3. Animal Research and Clinical Trials with HDAC Inhibitors
Animal studies investigating HDAC inhibitors have provided valuable insights into their potential therapeutic applications across various diseases [194]. These studies often involve the administration of HDACis to animal models, such as mice or rats, to evaluate their efficacy, safety and mechanism of action [17,195]. By examining the effects of HDACis on disease progression, biomarkers and physiological parameters in these models, researchers can better understand their pharmacological properties and potential clinical benefits. Furthermore, animal studies allow for the exploration of optimal dosing regimens, routes of administration and combination therapies, which are essential for translating preclinical findings into successful clinical trials. Overall, animal studies play a crucial role in advancing our knowledge of HDACis and their therapeutic potential in treating a wide range of diseases [196,197,198,199]. On the other hand, clinical studies investigating HDACis in humans aim to assess their safety, efficacy and tolerability across various diseases, providing essential data for their potential use as therapeutic agents. The clinical trial information of some drugs is summarized in Table 5. The majority of pan-HDAC inhibitors are currently undergoing Phase II/III clinical trials, with several demonstrating promising outcomes [200,201,202]. A Phase II clinical study investigating Panobinostat revealed a substantial therapeutic benefit in treating multiple myeloma. This effect might be further enhanced by combining it with other related medications [200]. The next generation of selective inhibitors is expected to achieve heightened effectiveness in treating hematologic and solid tumors, broadening the range of diseases addressed by HDACis. With enhanced targeting mechanisms, these inhibitors will also help reduce side effects, bringing us closer to a future where cancer treatment is more effective and less harmful [203]. Beyond cancer, HDAC inhibitor drugs have demonstrated effectiveness in various other conditions. Vorinostat, for instance, is presently undergoing clinical trials for Alzheimer’s disease (NCT03056495). In neurodegenerative disorders, it is anticipated to be utilized for frontotemporal dementia resulting from progranulin deficiency, albeit with a need for further efficacy enhancement [204]. Significantly, a growing number of drugs have exhibited therapeutic potential in addressing HIV infection. This outcome could be linked to the cellular autophagy and immunomodulatory functions mediated by HDACs. With the expansive range of genetic regulatory functions attributed to HDACs, an escalating number of clinical trials for various diseases have progressed to the Phase II stage, offering further avenues for research in both clinical and preclinical settings. However, it is concerning that numerous adverse events persist in the clinical trials of marketed HDACis in different cancer types [205,206]. Despite the improved metabolic half-life and assured oral bioavailability of the latest clinical candidate, Pracinostat (Figure 4), the completion of a co-administration trial (NCT03848754) has revealed prevalent toxicities including nausea/vomiting (63%), anorexia (50%), hypokalemia (50%) and rhinorrhea accompanied by neutropenic symptoms [207]. Although present-day HDACis demonstrate potential in inhibiting cell proliferation in vitro, their lack of selectivity results in undesirable side effects, including off-targeting. This can lead to the harmful attack of healthy cells, causing significant toxic reactions. Furthermore, the increase in HDAC drug trials has raised worries about drug resistance [208,209,210]. This has led to the understanding that resistance to HDAC inhibitors involves many factors.
4. Computational Studies on HDACs
4.1. Molecular Modeling
Over the last two decades, several computational approaches have been employed to find HDAC inhibitors with enhanced potency and/or selectivity. The main purpose is to simplify the search process, reducing the search space and ensuring the identification of the most promising compounds with desired activities. Computational techniques, including ligand-based approaches (Figure 9) such as scaffold hopping, 3D-QSAR, and pharmacophore modeling, as well as structure-based methods like structure-based virtual screening/molecular docking (Figure 10) and fragment-based ligand design, have proven instrumental in developing HDAC inhibitors with targeted activity.
Thus, the potential hits identified undergo further validation through structure-based assessments, utilizing techniques such as molecular dynamics (MD) simulations coupled with MM-PBSA/MM-GBSA binding energy calculations (Figure 11). The MM/PBSA and MM/GBSA methodologies estimate the free energy of ligand binding to biological macromolecules, serving as intermediary tools bridging empirical scoring (e.g., docking and scoring) and rigorous alchemical perturbation (AP) methods [211,212]. The application of combined computational approaches in HDACi rational design allows us to significantly limit the risk of false positive hits. Moreover, such methodologies increase the possibility of retrieving specific inhibitors by employing different filters and scoring functions.
Scaffold hopping strategies, along with molecular docking, have been employed in various studies for the design of HDAC inhibitors. Usually, the majority of newly developed compounds with improved potency and/or desired selectivity are achieved through modifications in the three distinct structural regions of HDAC inhibitors. An exemplary demonstration of this strategy is seen in a series of quercetin-containing hydroxamic acid derivatives. These derivatives were synthesized by altering quercetin in both the cap and the linker regions, while their ability to bind HDAC was initially assessed in silico [213]. Given the recognized pan-HDAC inhibition activity of resveratrol, a combination of scaffold hopping, molecular docking and ADME prediction was utilized to create a set of resveratrol analogs. These prospective inhibitors were subsequently subjected to additional validation via MD simulations and in vitro enzyme inhibition assays targeting HDAC1 and HDAC2 [214,215]. The scaffold hopping strategy was also useful for the discovery of some hybrids bearing 1H-indazol-3-amine and benzohydroxamic acids with dual HDAC/EGFR1 inhibitory activity against breast cancer line MCF-7 [216], and of a novel Aminotetralin Class of HDAC6 and HDAC8 selective inhibitors with potent inhibitory activity against neuroblastoma BE(2)C cells [217]. Notably, a pair of HDAC6-selective inhibitors with 2-mercaptoquinazolinone as the cap moiety were drawn for the first time. This design foresaw manipulating the surface recognition group (quinazolinone core as a cap) and linker while retaining the hydroxamic acid side chains at the C-2 or N-3 position [218]. In a recent investigation, HDAC inhibitors featuring 4-acyl pyrrole caps were employed as a scaffold for developing potent hybrid inhibitors that target both bromodomain and extra-terminal (BET) proteins as well as HDACs [219]. Considering all these findings, the combined use of scaffold hopping with molecular docking emerges as a critical computational strategy for formulating HDAC inhibitors with the desired pharmacological characteristics
As previously stated, the conventional pharmacophore of HDAC inhibitors comprises three main components: a capping group (cap), a linker region, and a zinc-binding group [220]. Modifications in the cap and linker regions aim to achieve selectivity for specific HDAC isoforms, while variations in the zinc-binding groups aim for increased potency. Pharmacophore models can be classified as structure-based, developed from protein-ligand complexes, or ligand-based, utilizing known HDAC inhibitors’ structures. These models undergo validation to ensure their efficacy in distinguishing active and inactive compounds, often through methods like Receiver Operating Characteristic (ROC) analysis or inactive compounds (decoy) testing [221]. For instance, in the search of potential selective inhibitors targeting HDAC2, a screening process was conducted on 300,000 compounds sourced from Asinex, National Cancer Institute (NCI) and Maybridge databases using e-pharmacophore modeling [222]. Also, a 3D chemical feature-based QSAR pharmacophore model was developed to study the interaction between benzamide MS-275 and HDAC [220]. A potent HDAC3 inhibitor was identified by Kumbhar et al., by using a combined computational screening strategy involving ligand-based pharmacophore modeling, MD simulation, and MM-PBSA calculation methods [223]. It is noteworthy that combining MD simulations with energetically optimized structure-based pharmacophores (e-Pharmacophores) was useful in the rational design of potential HDAC inhibitors, as validated by MM-GBSA binding energy calculations [224]. Thanks to dynamic pharmacophore models, it became possible to address the issue concerning the flexibility within the protein’s active site. Specifically, this method was utilized to seek out potential inhibitors of HDAC8 by generating structure-based pharmacophore models from various conformations obtained through MD simulations [225].
Quinoline has been used as a cap group in the development of many HDAC inhibitors [226,227,228,229]. Among them, the quinoline-based HDAC inhibitor CHR3996 has completed a Phase I clinical study [230,231]. In 2017, Chen et al. designed several quinoline-based HDAC inhibitors whose binding mode was found to be the same as that of traditional HDACis. Interestingly, the authors observed that the eight positions of quinoline did not occupy the pocket, thus encouraging them to modify such positions in order to improve the activity and selectivity [232]. Therefore, in a more recent study, they designed and synthesized a new series of 8-substituted quinoline-2-carboxamide derivatives and identified a very potent compound (IC50 = 0.050 µM) that exhibited 3-fold greater HDAC inhibitory activity compared to the known HDAC inhibitor Vorinostat, with low toxicity against normal cells [233].
More recently, Gao and co-workers developed and synthesized novel HDAC inhibitors derived from the β-elemene scaffold [234]. β-elemene is specifically a sesquiterpene used in the treatment of lung cancer, pancreatic cancer, gastric cancer, breast cancer, bladder cancer, and malignant brain glioma [235,236,237,238,239,240]. Most of the prepared compounds, whose binding mode was fully investigated by means of molecular docking analyses, showed potent inhibitor activities against HDACs and significant inhibitory effects on the proliferation of K562 and MV4-11. Two derivatives demonstrated excellent in vitro antienzyme (IC50 values of 22 nM and 9 nM for HDAC1 and 8 nM and 14 nM for HDAC6, respectively) and broad spectrum in vitro antiproliferative activities (IC50 values ranging from 0.79 to 4.42 mM against K562, MV4-11, HEL, SU-DHL-2 and WSU-DLCL-2 cell lines) and, among them, one was found to induce cell apoptosis and to exhibit antitumor activity in the WSU-DLCL-2 xenograft mouse model, without significant toxicity.
Initial investigations into HDAC inhibitors utilized comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) for the design of novel HDAC inhibitors [241]. Afterwards, various QSAR analyses, including 3D-QSAR and multi-QSAR modeling, facilitated the discovery of potent HDAC inhibitors. As for indole amide analogs, a 3D-QSAR analysis was conducted on HDAC1 to identify the compounds with the highest predicted inhibitory activity [242,243]. Moreover, QSAR classification models, such as k-nearest neighbors (kNN) and neighborhood classifier (NEC), were employed to predict potential HDAC8 inhibitors [244]. Recent studies utilized 3D-QSAR analysis to design potential selective inhibitors of HDAC6 and explore selective HDAC8 inhibitors through QAAR studies [245,246]. A multi-QSAR modeling study successfully identified potent HDAC8 inhibitors [243]. Recent approaches involved 3D-QSAR analysis to design potential selective HDAC6 inhibitors [246]. QAAR studies, utilizing DFT-based calculation and molecular dynamic simulation, explored selective HDAC8 inhibitors [245]. Similarly, QAAR and molecular docking led to the discovery of selective HDAC8 inhibitors with antiproliferative activities [247]. Also, as for isoform 1, the 3D-QSAR model was utilized to predict potential HDAC1 inhibitors with high activity. Additionally, induced fit docking (IFD) optimized protein-ligand interactions and MD simulations with MM-GBSA calculations were employed [248]. In order to address the limitations of 3D-QSAR, researchers developed 4D-QSAR models incorporating molecular state ensemble averaging [248].
Due to the wealth of the crystal structures of HDACs, structure-based inhibitor design has become increasingly achievable. Currently, the crystal structures of various human HDAC classes are available on the Protein Data Bank (Figure 2, Figure 4 and Figure 5) (https://www.rcsb.org/), as previously described [249]. For other isoforms, homology models have been built to study their potential inhibitors. Notably Hsu et al. utilized homology modeling to construct human HDAC5 and 9 models. These models, along with crystal structures of HDAC4 and -7, were employed to perform structure-based/molecular docking-based virtual screening of the NCI compound library, aiming at identifying potential inhibitors of Class IIa HDACs. Subsequently, these compounds underwent evaluation against HeLa nuclear Class II HDACs to uncover Class IIa-selective inhibitors [250]. In 2019, Ibrahim Uba et al. applied a homology modeling study for human HDAC10, exploiting the crystal structure of HDAC10 derived from zebrafish (PDB code: 5TD7). Then, this theoretical model was submitted to structure-based virtual screening, MD simulations and ADMET prediction techniques, for identifying potential HDAC10 inhibitors [251]. In the study by Géraldy et al., an additional homology model of human HDAC10 was introduced, emphasizing the significance of a crucial hydrogen bond formation between a nitrogen atom in the cap group and the gatekeeper residue Glu272 in influencing HDAC10 binding [251]. Therefore, the accessibility of crystal structures for human HDACs has greatly improved the effectiveness of structure-based inhibitor design. Moreover, when biological activity data are unavailable, researchers have been using a method known as MM-GBSA/MM-PBSA ligand binding affinity calculations to determine the effectiveness of identified hits. For example, Sixto-López et al. used molecular docking, MD simulations and MM-GBSA energy calculations to design hydroxamic acid derivatives with potent inhibitory activity against HDAC1, HDAC6 and HDAC8. YSL-109 emerged as the most active compound against hepatocellular carcinoma, neuroblastoma and breast cancer [252]. Also, novel methods including e-pharmacophore modeling, structure-based virtual screening and MD simulations combined with MM-GBSA ligand binding energy calculations have been utilized in the quest to identify potent inhibitors of HDAC2 [222]. A thorough analysis was performed to discover potential inhibitors of Class IIa HDACs using a virtual screening process. This involved the usage of MD simulations and MM-PBSA to determine the ligand-free energy of binding. The compounds that exhibited optimal results based on these calculations were ultimately chosen as the final hits [253]. Similarly, a “multi-layer virtual screening workflow” was developed for identifying inhibitors selective for the HDAC6, demonstrating significant anticancer activity [254]. In a recent development, a compound demonstrating nanomolar activity against HDAC1, HDAC3 and HDAC6 was uncovered through a combined approach of pharmacophore modeling and structure-based virtual screening of an in-house database comprising 22,700 compounds. This compound displayed superior inhibitory effects compared to Vorinostat and exhibited potential in impeding the growth of solid cancers [255].
In summary, the computational methodologies mentioned above complement each other, providing multiple layers of filtration essential for successful hit discovery. Typically, pharmacophore modeling and/or 3D-QSAR modeling serve as the foundational models utilized in pharmacophore-based/ligand-based virtual screening. Following the identification of potential hits, structure-based virtual screening and molecular docking are utilized to anticipate binding poses and affinities. The most promising candidates are subjected to MD simulations to examine the stability of ligand binding modes. For a more precise determination of ligand binding affinity, MM-GBSA/MM-PBSA calculations are commonly integrated with MD simulations.
More recently, various studies have been focused on the elucidation of the simultaneous inhibition of two or more targets involved in critical pathways related to cancer progression. For example, Duan and co-workers applied computational and SAR approaches, and identified a series of pyridazinone-based PARP7/HDACs dual inhibitors whose in vitro and in vivo activities were evaluated. In particular, a hydroxyl propenamide derivative was reported as a potent and balanced dual inhibitor, with an excellent antitumor capability towards lung, B-myelomonocytic leukemia and histiocytic lymphoma cell lines, thus suggesting a relationship between anticancer immunity and HDAC inhibition [256].
Inspired by the synergistic effects of tubulin and HDAC inhibitors in dual targeting cancer therapy and the interaction between proteins, some o-aminobenzamide-based dual HDAC/tubulin inhibitors have been reported, which could target tumor tissues more accurately, thus enhancing their efficacy and improving the antitumor effects [257]. In 2022, Yao’s team reported a series of 2ME2 derivatives as dual HDAC/tubulin inhibitors by combining the pharmacophore of a HDAC inhibitor with the 2-methoxyestradiol (2ME2) skeleton [258]. Among them, a compound showed potent dual inhibitory activities on tubulin polymerization and HDAC (IC50 values were 0.06 and 0.12 μM of HDAC2 and HDAC6, respectively), as well as exhibited potent antiproliferative activities against MCF-7, MGC-803, HeLa, A549, HepG2 and U937 with IC50 values of 0.37–4.84 μM. In addition, the compound also exhibited potent in vitro and in vivo antitumor and antiangiogenic response. Its well-defined binding modes in tubulin (PDB code: 5LYJ) and HDAC2 (PDB code: 4LXZ) helped to explain in detail the high inhibitory potency on tubulin and HDAC2. More recently, novel dual tubulin/HDAC inhibitors were designed and synthesized based on the structure of natural product millepachine, which has been identified as a tubulin polymerization inhibitor [259]. A biological evaluation revealed that a derivative exhibited an impressive potency against PC-3 cells with the IC50 value of 16 nM, and effectively inhibited both microtubule polymerization and HDAC activity. Furthermore, the compound induced PC-3 cells apoptosis with a decrease in mitochondrial membrane potential and an elevation in reactive oxygen species levels in PC-3 cells. Additionally, it showed inhibitory effects on tumor cell migration and angiogenesis with favorable drug metabolism characteristics in vivo. Molecular docking analysis provided additional evidence supporting the binding of the identified dual inhibitor to tubulin and HDAC [260].
Also, the simultaneous inhibition of Class I phosphoinositide 3-kinases (PI3K) and HDAC has shown promise for treating various cancers [261]. Several PI3K/HDAC dual inhibitors have been disclosed, showing promising anticancer properties [262,263,264,265]. The first PI3K/HDAC dual inhibitor entering into the clinical trials, CUDC-907, was granted the fast-track designation by the FDA for treating relapsed or refractory diffuse large B-cell lymphoma [266]. Recently, Zhang et al. reported a novel series of 4-methylquinazoline based PI3K/HDAC dual inhibitors characterized by a hydroxamic acid moiety as a HDAC pharmacophore [267]. Despite favourable antiproliferative activities against a broad panel of cancer cell lines, these compounds only showed limited in vivo activities largely due to their poor pharmacokinetic properties. Therefore, the same research team incorporated the benzamide moiety as the zinc binding group and, by means of molecular docking and QSAR studies, obtained two potent PI3K/HDAC dual inhibitors for the treatment of acute myeloid leukemia with improved pharmacokinetic properties [268].
4.2. Machine Learning
Recently, machine learning has garnered significant attention in the initial phases of drug discovery studies [269]. Machine learning has opened up avenues to explore the vast chemical space beyond the limitations of conventional experimental techniques [270,271]. In medicinal chemistry, a range of machine learning models utilize algorithms including decision trees (DT), random forests (RF), support vector classifiers (SVC), k-nearest neighbors (kNN), Gaussian naive Bayes (GNB), and deep neural networks [271,272,273]. AlphaFold represents a promising machine learning approach, capable of accurately predicting the 3D structure of proteins, even in the absence of closely related structures. However, its models are generated without considering the presence of small molecules, ions, or cofactors, which complicates their direct application in drug design. In a recent study, Baselious and colleagues demonstrated the utility of an optimized AlphaFold model for virtual screening, specifically addressing HDAC subtype selectivity [274]. In the developed multistep screening process, various methodologies were employed, including structure-based pharmacophore screening to filter large databases, ligand docking, pose filtering, and prioritization. This stepwise virtual screening approach successfully identified a hit compound, subsequently validated using an in vitro enzymatic assay. The compound exhibited an IC50 value of 3.5 µM for HDAC11 and demonstrated the selective inhibition of HDAC11 over other HDAC subtypes at a concentration of 10 µM. Molecular dynamics simulations confirmed the stability of the initial binding mode, as evidenced by ligand RMSD, RMSF, bidentate chelation of the zinc ion, and interaction stability [275].
The absence of an experimental 3D model for HDAC10 poses a challenge to structure-based drug design for selective inhibitors. Consequently, various ligand-based modeling techniques offer a primary avenue to accelerate inhibitor design. In a recent investigation, Bhattacharya and collaborators employed diverse ligand-based modeling approaches on a wide array of HDAC10 inhibitors, leveraging machine learning models to screen for potential HDAC10 inhibitors within a vast chemical database. Furthermore, Bayesian classification and Recursive partitioning models were utilized to identify structural fingerprints governing HDAC10 inhibitory activity. Complementarily, a molecular docking analysis was conducted to elucidate the binding patterns of these identified structural fingerprints within the active site of HDAC10, providing invaluable insights for medicinal chemists in the design and development of effective HDAC10 inhibitors [276].
A recent study delved into HDAC8, a protein implicated in cancer progression. While many reported inhibitors targeting HDAC8 feature a hydroxamic acid group, known for its mutagenic potential, Nurani et al. turned to machine learning for drug screening, aiming to uncover alternative compounds devoid of hydroxamic acid while retaining HDAC8 inhibitory activity [277]. In this investigation, the authors devised a predictive model utilizing the random forest algorithm to screen for HDAC8 inhibitors, selected for its superior accuracy on the training dataset inclusive of data augmented by the synthetic minority oversampling technique (SMOTE). Employing the trained RF-SMOTE model, they successfully identified a selective non-hydroxamic acid derivative as an HDAC8 inhibitor, exhibiting an IC50 of 842 nM.
Furthermore, the study addressed HDAC1, another pivotal isoform implicated in numerous tumors. Li et al. compiled a dataset comprising 7318 HDAC1 inhibitors and computed four types of molecular fingerprints (MACCS, RDK, ECFP4, and TT fingerprints) to delineate molecular structural features. By calculating Tanimoto coefficients, they ensured a dataset rich in structural diversity, enabling the establishment of 80 classification models across four types of molecule fingerprints using five machine learning algorithms. Additionally, employing the DT algorithm, the authors dissected the structure–activity relationship of HDAC1 inhibitors, identifying certain substructures, such as N-(2-amino-phenyl)-benzamide, benzimidazole, hydroxamic acid with a middle-chain alkyl, and 4-aryl imidazole with a mid-chain alkyl featuring a chiral α carbon, as exerting a significant impact on high activity [278].
In contrast, HDAC6 has emerged as a potential therapeutic target associated with various diseases, notably cancer and neurological disorders, such as Rett syndrome, Alzheimer’s disease, and Huntington’s disease. In a recent study aimed at developing selective and potent HDAC6 inhibitors, Banerjee et al. investigated the structural determinants of quinazoline-cap-containing HDAC6 inhibitors through a combination of machine learning, conventional QSAR analysis, and MD simulation-based binding mode analysis. This integrated molecular modeling approach highlighted the critical role of the quinazoline moiety and its substitutions, as well as molecular properties such as the number of hydrogen bond donor–acceptor functions and the carbon–chlorine distance, in modulating the binding affinity of these inhibitors to HDAC6, thereby influencing their potency. Additionally, the study revealed that substitutions such as the chloroethyl group and bulky quinazolinyl cap group could impact the interaction of the cap function with amino acid residues near the catalytic site of HDAC6, potentially leading to both stabilization and destabilization of the cap function following the occupation of the hydrophobic catalytic site by the aryl hydroxamate linker–ZBG functions [279].
4.3. Limitations of Computational Techniques
Advancements in computing technology and algorithmic sophistication have significantly enhanced the capability of executing computationally demanding biomolecular modeling tasks. Computer-aided drug design methods leverage these advancements to improve the accuracy and efficiency of molecular modeling predictions. Particularly noteworthy are the multilayered computational strategies employed in the quest for potential HDAC inhibitors, which harness the predictive capabilities of various scoring functions for effective filtering. For instance, empirical scoring functions are commonly utilized to predict ligand binding pose and affinity in structure-based drug design initiatives [280]. Yet, formulating active compounds or identifying them solely through computational modeling poses a formidable challenge. In certain cases, specific benchmarking techniques are required to manage extensive ligand datasets. Another consideration is the complete flexibility of the protein during molecular docking, which remains a concern even when induced fit and entropy effects do not significantly influence binding [281,282]. Contemporary trends in integrating MM-GBSA/MM-PBSA calculations of ligand binding affinity in the quest for potential HDAC inhibitors aim to enhance the precision of hit selection [223,283]. This is particularly significant considering that in certain studies employing multilayered approaches, the identified hits underwent no experimental validation. Overall, the utilization of combined computational methods has demonstrated efficacy in facilitating the design of both class- and isoform-selective inhibitors.
In LBVS, Support Vector Machines (SVMs) are commonly employed for binary property or activity predictions. For instance, they are utilized to differentiate between drugs and nondrugs [284,285], or between compounds with specific activity and those without [286,287]. They are also applied for predicting synthetic accessibility [288] or aqueous solubility. The scores generated by SVM classification have proven effective in ranking database compounds based on their decreasing likelihood of activity [289]. This ranking is often determined by the signed distance between a candidate compound and the hyperplane. Two studies have proposed specialized ranking functions for virtual screening to enhance SVM ranking accuracy, addressing the tendency of SVMs to prioritize classification performance over ranking optimization [290,291].
Various new kernel functions have been introduced for SVMs, including ligand and target kernels, which capture distinct information for a similarity assessment [292,293]. These kernels utilize different metrics such as graph or descriptor similarity for compounds and sequence, or binding site similarity for target proteins. For instance, graph kernels [294] facilitate the overall similarity computation between labeled graphs, eliminating the need to compute or store a vector representation of compounds. However, they entail high computational costs and require parameter tuning.
Decision tree (DT) models offer simplicity in understanding, interpreting, and validating predictions. However, they are prone to high variance, where even slight changes in the data can lead to different split sequences, complicating interpretation. This instability stems from the hierarchical process, where errors in higher splits cascade downward, affecting subsequent splits. Additionally, DT structure is sensitive to minor variations in the training data; small datasets can significantly impact the learning process, while large datasets may induce overfitting. To address these challenges, it is advisable to maintain a moderate training dataset size and a balanced tree structure with a reasonable number of levels. Heuristic approaches can be used to enhance classification accuracy by adjusting subtrees at lower levels. The performance of DTs also relies on selecting splitting attributes sorted by importance, ensuring that the most crucial attributes guide the splits at each level. To mitigate high variance, pruning techniques are commonly employed, utilizing either model complexity parameters or cross-validation. While individual DTs may not yield high-performance models, ensemble methods, such as bagging [294], boosting [295], and stacking [296], outperform individual learners by leveraging the variability among ensemble members, thereby capitalizing on the variance of DTs. In particular, Random Forest (RF) models have shown promise in enhancing the performance of individual DTs in ligand-based virtual screening (LBVS) and have applications in post-docking scoring functions and predicting protein-ligand binding affinity.
In ligand-based virtual screening (LBVS), Bayesian modeling methods are utilized to forecast the likelihood of a compound’s activity based on its descriptor vector. By leveraging known active (A) and inactive (Z) training compounds, these methods estimate conditional probability distributions P(B/A) and P(B/Z) given the representation B, respectively. Consequently, Bayesian classifiers excel in ranking compound databases according to their probability of activity. However, a significant drawback arises when there are substantial conditional dependencies between variables, rendering the naive Bayesian model unsuitable for such scenarios.
The kNN algorithm, a straightforward technique used to predict the class [297], property [298], or rank [299] of a molecule based on its nearest neighbors in the feature space, heavily relies on the local structure of the data. Hence, it is particularly effective for predicting properties with strong locality, such as protein function [300,301]. Despite its intuitive nature, the kNN method does have its limitations. Firstly, since it depends solely on the nearest k neighbors to predict a new compound, it is vulnerable to noisy data. A single misclassified training data point could lead to an incorrect prediction for a new molecule. Additionally, the inclusion of irrelevant descriptors may result in erroneous predictions. Moreover, the predicted value cannot exceed the maximum or minimum activity levels present in the training set.
In the field of medicinal chemistry, Artificial Neural Networks (ANNs) find applications in compound classification, QSAR studies, primary virtual screening of compounds, identification of potential drug targets, and the localization of structural and functional features of biopolymers [301,302]. Originally inspired by the structure and function of the brain, ANNs have evolved into versatile nonlinear regression models [303], offering flexibility in modeling complex relationships. However, one common challenge with ANN simulations is their ‘black box’ nature, where resulting classification models lack interpretability or explanation in physical or chemical terms. Nevertheless, ANNs excel at capturing and modeling nonlinear relationships, which is a significant advantage in many applications.
4.4. Achieving Selectivity for Each HDAC Isoform
The selectivity of HDAC inhibitors has emerged as a critical matter for their application in cancer therapy. First-generation HDAC inhibitors, such as Vorinostat, Belinostat and Panobinostat, target multiple isoforms, leading to cellular toxicities due to their hydroxamic acid functional group, as discussed elsewhere [304]. Seeking safer alternatives, the next generation of HDAC inhibitors predominantly aims for class or isoform selectivity, with various studies highlighting alternative zinc-binding groups (ZBGs) exhibiting high inhibitory activity along with selectivity [304]. For instance, imidazole thione-containing molecules have shown promise against HDAC8 [305]. Also, pyrimido[1,2-c][1,3]benzothiazin-6-imines have demonstrated high selectivity [306]. Further research, based on molecular docking and dynamic simulations, allowed for the identification of tropolone derivatives as selective HDAC2 inhibitors [307], while 3-hydroxypyridin-2-thione (3-HPT) was associated with the selective inhibition of HDAC6 [308]. Compounds with benzoylhydrazide as ZBG have exhibited potent inhibitory activity against Class I HDACs, particularly when possessing a 3-carbon-length β-nitrogen alkyl substituent chain [309]. Additionally, trifluoromethyloxadiazolyl moiety has shown selectivity for Class IIa HDACs [310], and 2-substituted benzamide has been used as a ZBG for selective inhibitors of HDAC3 [216]. Notably, an alternative approach proposed by Maolanon et al. focuses on disrupting protein–protein interactions essential for HDAC activity rather than chelating the active site zinc ion [311]. Employing a top-down combinatorial in silico approach, Ganai et al. explored strategies to selectively inhibit HDAC1 and HDAC2 (sequence identity: 94%), revealing distinct pharmacophore features for each isoform. According to the results of pharmacophore analysis, Dacinostat (LAQ824) showed a higher affinity towards HDAC1 when a positive ionizable group was present in the linker region. On the other hand, the ring in the linker region displayed a stronger interaction with HDAC2 [224]. Previously, Abdizadeh et al. used combined 3D-QSAR and molecular docking approaches to design biaryl benzamides as potent HDAC1 inhibitors [312]. Cao et al. conducted a review of different modifications carried out to the cap and linker groups to achieve selectivity for HDAC3. Meanwhile, Suzuki et al. identified specific compounds with phenyltriazole cap groups that exhibit greater selectivity for HDAC3 when compared to other Class I members [313,314].
HDAC6, although sharing structural similarities with HDAC10, presents slight distinctions compared to Class I HDACs, particularly in active site dimensions and surface residues. In the quest for HDAC6-selective inhibitors, a series of compounds incorporating 2-mercaptoquinazolinone as the cap moiety were developed. This effort focused on modifying the surface recognition group (utilizing the quinazolinone core as a cap) and linker, while preserving hydroxamic acid side chains at either the C-2 or N-3 position [218]. Recently, researchers have explored various avenues to design HDAC6-selective inhibitors. For instance, a 3D-QSAR analysis, leveraging the classical pharmacophore of HDAC inhibitors, was employed to devise potential HDAC6-selective compounds [315]. Pharmacophore models, notably HypoGen-based 3D QSAR, based on the crystal structure of HDAC6 (PDB ID: 5EDU), have been instrumental in identifying potent HDAC6 inhibitors and assessing their selectivity. Further assessment of these inhibitors involved MD simulations and analysis of protein–ligand interaction energy (PLIE) [283]. Lastly, in a separate initiative, Moi et al. utilized a cheminformatics approach to develop HDAC6 inhibitors, resulting in the surprising identification of aminotriazole with subnanomolar inhibitory potency and significant selectivity for Class I HDAC1 and HDAC8 [316].
Another successful virtual screening approach was applied to HDAC7 by our research group. Specifically, in line with our research, and to figure out novel anticancer strategies, an in-house chemical database of extracted molecules from both edible and non-edible mushrooms was adopted [317]. The SBVS pointed out the ibotenic acid as a promising HDAC7 inhibitor based on its theoretical binding affinity. In vivo studies demonstrated the capability of the ibotenic acid in decreasing the cellular viability on MCF-breast cancer cells, providing an interesting example of repurposing natural products to fight cancer disease.
What is highly interesting is, while single isoform inhibition has been extensively investigated, recent studies highlight the efficacy of dual/multi-targeting HDAC inhibitors in achieving synergistic and enhanced cancer therapy outcomes [318,319].
Therefore, in order to enhance antineoplastic activity and reduce undesirable effects, the world of research is focusing on the pursuit of dual binders capable of simultaneously inhibiting, for example, specific HDAC isoforms and tubulin, the Vascular Endothelial Growth Factor, Tyrosine kinase receptors, Hepatocyte growth factor receptor, Cyclin-dependent kinases, A2A adenosine receptor, Poly ADP-ribose polymerase and others [320].
5. In Vitro Validations Using Cell Lines
It is critical to ensure the accuracy and reliability of computational predictions by subjecting them to laboratory experiments for validation. This process verifies computational results through in vitro tests, which allow researchers to evaluate the robustness of computational methodologies and their applicability to real-world biological systems. In vitro validation also provides empirical evidence to support hypotheses and computational insights, thereby improving the credibility and reproducibility of computational studies. By identifying any discrepancies or limitations in computational models, experimental validation drives refinements and improvements for future analyses. Integrating in vitro validation with computational studies is critical in advancing our understanding of complex biological phenomena and facilitating the translation of computational results into practical applications.
Numerous in vitro validations utilizing cell lines have significantly contributed to our understanding of the molecular functions and regulatory roles of HDACs. These investigations typically involve the manipulation of HDAC expression levels or the application of HDAC inhibitors to cultured cell lines representing a spectrum of tissues and disease contexts [252,321,322]. Techniques such as immunoblotting, chromatin immunoprecipitation assays, and quantitative PCR analyses are commonly employed to dissect the intricate mechanisms underlying HDAC-mediated transcriptional regulation, cell cycle control, apoptosis, and cellular differentiation [111,323]. Moreover, cellular models have been instrumental in elucidating the crosstalk between HDACs and other signaling pathways, shedding light on their involvement in various physiological and pathological processes [111,324]. Additionally, the development and optimization of cell-based assays have facilitated the screening and validation of novel HDAC inhibitors, guiding the identification of promising candidates for therapeutic intervention [321,325].
One obstacle in identifying isoform-selective inhibitors stems from the limitations in current compound-screening technologies. The most commonly used in vitro deacetylation assay relies on monitoring the fluorescent signal resulting from HDAC-mediated degradation of the Fluor-De-Lys peptide substrate (Enzo Life Sciences). While this assay is robust and straightforward, screening for selectivity typically involves using purified, recombinant HDAC isoforms from baculovirus overexpression systems [326]. However, testing mammalian-cell derived HDAC proteins via immunoprecipitation of overexpressed isoforms from cell extracts introduces a typical error rate ranging from 30% to 60% [326]. Due to the significant error and labor-intensive nature of immunoprecipitation, many selective compounds have been exclusively tested against baculovirus-derived isoforms, such as tubastatin [327]. Yet, discrepancies arise when comparing inhibition data obtained from baculovirus- and mammalian cell–derived HDAC isoforms. For instance, apicidin exhibited potent activity against mammalian cell–derived HDAC1 (IC50 = 23 nM) [328], yet displayed variable potency against baculovirus-expressed HDAC1 (IC50 >10,000 nM or 0.7 nM in different reports) [326,329]. Although technical differences may account for these disparities, the similar assay formats suggest that the source of the HDAC isoforms can also impact screening results. Addressing this issue, Padige et al. introduced an enzyme-linked immunosorbent assay (ELISA)–based HDAC activity assay, aimed at utilizing mammalian cell–derived HDAC isoforms. Screening several known HDACis with diverse selectivity profiles validated the utility of this assay for inhibitor screening, suggesting its potential as a valuable tool for characterizing isoform-selective HDAC inhibitors against mammalian cell–derived HDAC isoforms [330].
Overall, the wealth of information garnered from in vitro studies using cell lines has been instrumental in advancing our knowledge of HDAC biology and in informing the development of targeted therapeutic strategies for a wide array of diseases.
6. Conclusions
In conclusion, undertaking a thorough examination of the conformational relationship of HDACis will facilitate the rational design of drugs and the development of effective and innovative treatments. Approved HDACis demonstrate efficacy against hematological malignancies, while numerous HDACis under evaluation show promise in various stages of clinical trials. In this scenario, computational techniques, including structure- and ligand-based virtual screening, pharmacophore modeling 3D-QSAR, molecular docking, dynamics simulations, and MM-PBSA/MM-GBSA ligand binding affinity calculations, have promoted and favoured the design of HDACis, by improving potency and/or selectivity. In particular, in this review it has been highlighted that combined computational strategies, through the application of multiple tools/force fields or scoring functions, stimulated the discovery of new lead compounds to optimize, leading to promising anticancer drugs within few years.
Although HDACis are still in the early stages of study, they have the potential to revolutionize our approach in fighting tumors, offering an attractive avenue for treating this complex pathology. Through the continued exploration of HDAC inhibition using computational methods, we establish the groundwork for revolutionary strides in cancer treatments, emphasizing the profound clinical and societal impact inherent in this field of inquiry.
Author Contributions
Conceptualization, A.C. and R.R.; writing—original draft preparation, A.A. and R.R.; writing—review and editing, S.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zhang, Q.; Dai, Y.; Cai, Z.; Mou, L. HDAC Inhibitors: Novel Immunosuppressants for Allo- and Xeno-Transplantation. Chem. Sel. 2018, 3, 176–187. [Google Scholar] [CrossRef]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, W.G. Targeting histone deacetylases for cancer therapy: From molecular mechanisms to clinical implications. Int. J. Biol. Sci. 2014, 10, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef]
- Brindisi, M.; Senger, J.; Cavella, C.; Grillo, A.; Chemi, G.; Gemma, S.; Cucinella, D.M.; Lamponi, S.; Sarno, F.; Iside, C.; et al. Novel spiroindoline HDAC inhibitors: Synthesis, molecular modelling and biological studies. Eur. J. Med. Chem. 2018, 157, 127–138. [Google Scholar] [CrossRef]
- Nian, H.; Bisson, W.H.; Dashwood, W.M.; Pinto, J.T.; Dashwood, R.H. Alpha-keto acid metabolites of organoselenium compounds inhibit histone deacetylase activity in human colon cancer cells. Carcinogenesis 2009, 30, 1416–1423. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Melesina, J.; Kolbinger, F.R.; Oehme, I.; Senger, J.; Witt, O.; Sippl, W.; Jung, M. Targeting histone deacetylase 8 as a therapeutic approach to cancer and neurodegenerative diseases. Future Med. Chem. 2016, 8, 1609–1634. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Pflum, M.K. LSD1 Substrate Binding and Gene Expression Are Affected by HDAC1-Mediated Deacetylation. ACS Chem. Biol. 2017, 12, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Pan, H.; Montgomery, R.L.; Olson, E.N.; Schultz, R.M. Compensatory functions of histone deacetylase 1 (HDAC1) and HDAC2 regulate transcription and apoptosis during mouse oocyte development. Proc. Natl. Acad. Sci. USA 2012, 109, E481–E489. [Google Scholar] [CrossRef] [PubMed]
- Segré, C.V.; Chiocca, S. Regulating the regulators: The post-translational code of class I HDAC1 and HDAC2. J. Biomed. Biotechnol. 2011, 2011, 690848. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. Pathological Role of HDAC8: Cancer and Beyond. Cells 2022, 11, 3161. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kundu, S.; Singh, A.; Singh, S. Understanding the Role of Histone Deacetylase and their Inhibitors in Neurodegenerative Disorders: Current Targets and Future Perspective. Curr. Neuropharmacol. 2022, 20, 158–178. [Google Scholar] [CrossRef] [PubMed]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar] [CrossRef]
- Verdin, E.; Dequiedt, F.; Kasler, H. HDAC7 regulates apoptosis in developing thymocytes. Novartis Found. Symp. 2004, 259, 115–129; discussion 129–131, 163–169. [Google Scholar]
- Das, S.; Natarajan, R. HDAC9: An Inflammatory Link in Atherosclerosis. Circ. Res. 2020, 127, 824–826. [Google Scholar] [CrossRef]
- LoPresti, P. HDAC6 in Diseases of Cognition and of Neurons. Cells 2020, 10, 12. [Google Scholar] [CrossRef]
- Cheng, F.; Zheng, B.; Wang, J.; Zhao, G.; Yao, Z.; Niu, Z.; He, W. Histone deacetylase 10, a potential epigenetic target for therapy. Biosci. Rep. 2021, 41, BSR20210462. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xie, C.; Chen, Q.; Zhuang, S. HDAC11, an emerging therapeutic target for metabolic disorders. Front. Endocrinol. 2022, 13, 989305. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, D.; Feng, Y.; Li, B.; Cui, Y.; Chen, G.; Li, N. Association of sirtuins (SIRT1-7) with lung and intestinal diseases. Mol. Cell. Biochem. 2022, 477, 2539–2552. [Google Scholar] [CrossRef] [PubMed]
- Wątroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, S.; Zhou, N.; Cao, Z.; Zhang, Y. A proton-shuttle reaction mechanism for histone deacetylase 8 and the catalytic role of metal ions. J. Am. Chem. Soc. 2010, 132, 9471–9479. [Google Scholar] [CrossRef] [PubMed]
- Sauve, A.A.; Wolberger, C.; Schramm, V.L.; Boeke, J.D. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006, 75, 435–465. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Denu, J.M. Sirtuin catalysis and regulation. J. Biol. Chem. 2012, 287, 42419–42427. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, J.; Cai, H.; Li, Y.; Zhang, Y.; Zhang, X.; Zhao, D.; Li, Z.; Ma, H.; Wang, J.; et al. HDAC as a therapeutic target for treatment of endometrial cancers. Curr. Pharm. Des. 2014, 20, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Pflum, M.K.; Tong, J.K.; Lane, W.S.; Schreiber, S.L. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J. Biol. Chem. 2001, 276, 47733–47741. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, R.; Banerjee, S.; Amin, S.A.; Adhikari, N.; Jha, T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: A review. Eur. J. Med. Chem. 2020, 192, 112171. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.M.; Tsai, S.C.; Wen, Y.D.; Fejer, G.; Seto, E. Functional domains of histone deacetylase-3. J. Biol. Chem. 2002, 277, 9447–9454. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- De Souza, C.; Chatterji, B.P. HDAC Inhibitors as Novel Anti-Cancer Therapeutics. Recent. Pat. Anticancer Drug Discov. 2015, 10, 145–162. [Google Scholar] [CrossRef]
- Li, J.; Lu, L.; Liu, L.; Ren, X.; Chen, J.; Yin, X.; Xiao, Y.; Wei, G.; Huang, H.; Wei, W.; et al. HDAC1/2/3 are major histone desuccinylases critical for promoter desuccinylation. Cell Discov. 2023, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Adhikari, N.; Jha, T. Structure-activity relationships of HDAC8 inhibitors: Non-hydroxamates as anticancer agents. Pharmacol. Res. 2018, 131, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Celardo, I.; Gordiyenko, Y.; Cowley, S.M.; Robinson, C.V.; Fairall, L.; Schwabe, J.W. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell 2013, 51, 57–67. [Google Scholar] [CrossRef]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef]
- Hsu, K.C.; Liu, C.Y.; Lin, T.E.; Hsieh, J.H.; Sung, T.Y.; Tseng, H.J.; Yang, J.M.; Huang, W.J. Novel Class IIa-Selective Histone Deacetylase Inhibitors Discovered Using an in Silico Virtual Screening Approach. Sci. Rep. 2017, 7, 3228. [Google Scholar] [CrossRef]
- Liu, L.; Dong, L.; Bourguet, E.; Fairlie, D.P. Targeting Class IIa HDACs: Insights from Phenotypes and Inhibitors. Curr. Med. Chem. 2021, 28, 8628–8672. [Google Scholar] [CrossRef]
- Wright, L.H.; Menick, D.R. A class of their own: Exploring the nondeacetylase roles of class IIa HDACs in cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H199–H206. [Google Scholar] [CrossRef]
- Hess, L.; Moos, V.; Lauber, A.A.; Reiter, W.; Schuster, M.; Hartl, N.; Lackner, D.; Boenke, T.; Koren, A.; Guzzardo, P.M.; et al. A toolbox for class I HDACs reveals isoform specific roles in gene regulation and protein acetylation. PLoS Genet. 2022, 18, e1010376. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, G.S.; Hwang, H.J.; Nam, T.H.; Park, H.S.; Song, J.; Jang, T.H.; Lee, Y.C.; Kim, J.S. Structural basis of the specific interaction of SMRT corepressor with histone deacetylase 4. Nucleic Acids Res. 2018, 46, 11776–11788. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xiao, J.; Wang, Y.; Song, X.; Huang, L.; Ren, Z.; Kitazato, K. Posttranslational modification and beyond: Interplay between histone deacetylase 6 and heat-shock protein 90. Mol. Med. 2021, 27, 110. [Google Scholar] [CrossRef] [PubMed]
- Yue, K.; Qin, M.; Huang, C.; James Chou, C.; Jiang, Y.; Li, X. Comparison of three zinc binding groups for HDAC inhibitors—A potency, selectivity and enzymatic kinetics study. Bioorg Med. Chem. Lett. 2022, 70, 128797. [Google Scholar] [CrossRef] [PubMed]
- Bertos, N.R.; Gilquin, B.; Chan, G.K.; Yen, T.J.; Khochbin, S.; Yang, X.J. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J. Biol. Chem. 2004, 279, 48246–48254. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Zhang, F.; Maguire, A.; Byrne, T.; Weiner-Gorzel, K.; Bridgett, S.; O’Toole, S.; O’Leary, J.; Beggan, C.; Fitzpatrick, P.; et al. HDAC6 Degradation Inhibits the Growth of High-Grade Serous Ovarian Cancer Cells. Cancers 2020, 12, 3734. [Google Scholar] [CrossRef] [PubMed]
- Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. HDAC6-an Emerging Target Against Chronic Myeloid Leukemia? Cancers 2020, 12, 318. [Google Scholar] [CrossRef] [PubMed]
- English, K.; Barton, M.C. HDAC6: A Key Link Between Mitochondria and Development of Peripheral Neuropathy. Front. Mol. Neurosci. 2021, 14, 684714. [Google Scholar] [CrossRef]
- Herp, D.; Ridinger, J.; Robaa, D.; Shinsky, S.A.; Schmidtkunz, K.; Yesiloglu, T.Z.; Bayer, T.; Steimbach, R.R.; Herbst-Gervasoni, C.J.; Merz, A.; et al. First Fluorescent Acetylspermidine Deacetylation Assay for HDAC10 Identifies Selective Inhibitors with Cellular Target Engagement. ChemBioChem 2022, 23, e202200180. [Google Scholar] [CrossRef]
- Bottomley, M.J.; Lo Surdo, P.; Di Giovine, P.; Cirillo, A.; Scarpelli, R.; Ferrigno, F.; Jones, P.; Neddermann, P.; De Francesco, R.; Steinkühler, C.; et al. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J. Biol. Chem. 2008, 283, 26694–26704. [Google Scholar] [CrossRef]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef]
- Schuetz, A.; Min, J.; Allali-Hassani, A.; Schapira, M.; Shuen, M.; Loppnau, P.; Mazitschek, R.; Kwiatkowski, N.P.; Lewis, T.A.; Maglathin, R.L.; et al. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 2008, 283, 11355–11363. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Wu, F.; Jin, Y.M.; Chang, W.Q.; Xu, T.M. HDAC11: A rising star in epigenetics. Biomed. Pharmacother. 2020, 131, 110607. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tong, X.; Hu, W.; Chen, D. HDAC11: A novel target for improved cancer therapy. Biomed. Pharmacother. 2023, 166, 115418. [Google Scholar] [CrossRef] [PubMed]
- Yanginlar, C.; Logie, C. HDAC11 is a regulator of diverse immune functions. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef] [PubMed]
- Villalba, J.M.; Alcaín, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, M.; Mazzola, F.; Cuccioloni, M.; Sorci, L.; Audrito, V.; Zamporlini, F.; Fortunato, C.; Amici, A.; Cianci, M.; Deaglio, S.; et al. Molecular insights into the interaction between human nicotinamide phosphoribosyltransferase and Toll-like receptor 4. J. Biol. Chem. 2022, 298, 101669. [Google Scholar] [CrossRef]
- Simó-Mirabet, P.; Bermejo-Nogales, A.; Calduch-Giner, J.A.; Pérez-Sánchez, J. Tissue-specific gene expression and fasting regulation of sirtuin family in gilthead sea bream (Sparus aurata). J. Comp. Physiol. B 2017, 187, 153–163. [Google Scholar] [CrossRef]
- Chen, D.; Guarente, L. SIR2: A potential target for calorie restriction mimetics. Trends Mol. Med. 2007, 13, 64–71. [Google Scholar] [CrossRef]
- Arellano-Ballestero, H.; Sabry, M.; Lowdell, M.W. A Killer Disarmed: Natural Killer Cell Impairment in Myelodysplastic Syndrome. Cells 2023, 12, 633. [Google Scholar] [CrossRef] [PubMed]
- Lucatelli, P.; De Rubeis, G.; Trobiani, C.; Ungania, S.; Rocco, B.; De Gyurgyokai, S.Z.; Masi, M.; Pecorella, I.; Cappelli, F.; Lai, Q.; et al. In Vivo Comparison of Micro-Balloon Interventions (MBI) Advantage: A Retrospective Cohort Study of DEB-TACE Versus b-TACE and of SIRT Versus b-SIRT. Cardiovasc. Intervent Radiol. 2022, 45, 306–314. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef] [PubMed]
- Di Emidio, G.; Falone, S.; Artini, P.G.; Amicarelli, F.; D’Alessandro, A.M.; Tatone, C. Mitochondrial Sirtuins in Reproduction. Antioxidants 2021, 10, 1047. [Google Scholar] [CrossRef]
- Kida, Y.; Goligorsky, M.S. Sirtuins, Cell Senescence, and Vascular Aging. Can. J. Cardiol. 2016, 32, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Beegum, F.; Anuranjana, P.V.; George, K.T.; KP, D.; Begum, F.; Krishnadas, N.; Shenoy, R.R. Sirtuins as therapeutic targets for improving delayed wound healing in diabetes. J. Drug Target. 2022, 30, 911–926. [Google Scholar] [CrossRef]
- Zhao, X.; Allison, D.; Condon, B.; Zhang, F.; Gheyi, T.; Zhang, A.; Ashok, S.; Russell, M.; MacEwan, I.; Qian, Y.; et al. The 2.5 Å crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J. Med. Chem. 2013, 56, 963–969. [Google Scholar] [CrossRef]
- Moniot, S.; Schutkowski, M.; Steegborn, C. Crystal structure analysis of human Sirt2 and its ADP-ribose complex. J. Struct. Biol. 2013, 182, 136–143. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Schaefer, S.; Gertz, M.; Weyand, M.; Steegborn, C. Structures of human sirtuin 3 complexes with ADP-ribose and with carba-NAD+ and SRT1720: Binding details and inhibition mechanism. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1423–1432. [Google Scholar] [CrossRef]
- Li, G.-B.; Deng, J. Crystal Structure of Human SIRT5 in Complex with Diazidine Inhibitor 9. 2023. [Google Scholar] [CrossRef]
- Rajabi, N.; Auth, M.; Troelsen, K.R.; Pannek, M.; Bhatt, D.P.; Fontenas, M.; Hirschey, M.D.; Steegborn, C.; Madsen, A.S.; Olsen, C.A. Mechanism-Based Inhibitors of the Human Sirtuin 5 Deacylase: Structure-Activity Relationship, Biostructural, and Kinetic Insight. Angew. Chem. Int. Ed. Engl. 2017, 56, 14836–14841. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ge, J.; Li, H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 96–115. [Google Scholar] [CrossRef]
- Kulthinee, S.; Yano, N.; Zhuang, S.; Wang, L.; Zhao, T.C. Critical Functions of Histone Deacetylases (HDACs) in Modulating Inflammation Associated with Cardiovascular Diseases. Pathophysiology 2022, 29, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Nebbioso, A.; Altucci, L. Sirtuins and disease: The road ahead. Front. Pharmacol. 2012, 3, 4. [Google Scholar] [CrossRef]
- Zeng, L.S.; Yang, X.Z.; Wen, Y.F.; Mail, S.J.; Wang, M.H.; Zhang, M.Y.; Zheng, X.F.; Wang, H.Y. Overexpressed HDAC4 is associated with poor survival and promotes tumor progression in esophageal carcinoma. Aging 2016, 8, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, Y.; Wang, Z.; Wang, H.T.; Duan, B.; Ye, D.; Wang, C.; Jing, R.; Leng, Y.; Xi, J.; et al. HDAC10 promotes lung cancer proliferation via AKT phosphorylation. Oncotarget 2016, 7, 59388–59401. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, X.; Zhu, S.; Dejene, E.A.; Peng, W.; Sepulveda, A.; Seto, E. HDAC10 Regulates Cancer Stem-Like Cell Properties in KRAS-Driven Lung Adenocarcinoma. Cancer Res. 2020, 80, 3265–3278. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Han, S.; Cheng, W.; Miao, R.; Li, S.; Tian, X.; Kan, Q. Design, Synthesis, and Biological Evaluation of 2-Anilino-4-Triazolpyrimidine Derivatives as CDK4/HDACs Inhibitors. Drug Des. Dev. Ther. 2022, 16, 1083–1097. [Google Scholar] [CrossRef]
- Morse, J.S.; Sheng, Y.J.; Hampton, J.T.; Sylvain, L.D.; Das, S.; Alugubelli, Y.R.; Chen, P.C.; Yang, K.S.; Xu, S.; Fierke, C.A.; et al. Phage-assisted, active site-directed ligand evolution of a potent and selective histone deacetylase 8 inhibitor. Protein Sci. 2022, 31, e4512. [Google Scholar] [CrossRef]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef] [PubMed]
- Murayama, A.; Ohmori, K.; Fujimura, A.; Minami, H.; Yasuzawa-Tanaka, K.; Kuroda, T.; Oie, S.; Daitoku, H.; Okuwaki, M.; Nagata, K.; et al. Epigenetic control of rDNA loci in response to intracellular energy status. Cell 2008, 133, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Peserico, A.; Chiacchiera, F.; Grossi, V.; Matrone, A.; Latorre, D.; Simonatto, M.; Fusella, A.; Ryall, J.G.; Finley, L.W.; Haigis, M.C.; et al. A novel AMPK-dependent FoxO3A-SIRT3 intramitochondrial complex sensing glucose levels. Cell. Mol. Life Sci. 2013, 70, 2015–2029. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, U.; Ho, A.D.; Letzel, S.; Voelter-Mahlknecht, S. Assignment of the NAD-dependent deacetylase sirtuin 5 gene (SIRT5) to human chromosome band 6p23 by in situ hybridization. Cytogenet. Genome Res. 2006, 112, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Voelter-Mahlknecht, S.; Letzel, S.; Mahlknecht, U. Fluorescence in situ hybridization and chromosomal organization of the human Sirtuin 7 gene. Int. J. Oncol. 2006, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Guise, A.J.; Cristea, I.M. Post-translational modifications regulate class IIa histone deacetylase (HDAC) function in health and disease. Mol. Cell. Proteom. 2015, 14, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, N.; Seto, E. Regulation of histone deacetylase activities. J. Cell. Biochem. 2004, 93, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qin, G.; Zhao, T.C. HDAC4: Mechanism of regulation and biological functions. Epigenomics 2014, 6, 139–150. [Google Scholar] [CrossRef]
- Roy, D.M.; Walsh, L.A.; Chan, T.A. Driver mutations of cancer epigenomes. Protein Cell 2014, 5, 265–296. [Google Scholar] [CrossRef]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Fraga, M.F.; Ballestar, E.; Hamelin, R.; Yamamoto, H.; Boix-Chornet, M.; Caballero, R.; Alaminos, M.; Setien, F.; Paz, M.F.; et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 2006, 38, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Ropero, S.; Ballestar, E.; Alaminos, M.; Arango, D.; Schwartz, S.; Esteller, M. Transforming pathways unleashed by a HDAC2 mutation in human cancer. Oncogene 2008, 27, 4008–4012. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Shih, I.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Denkert, C.; Noske, A.; Darb-Esfahani, S.; Dietel, M.; Kalloger, S.E.; Huntsman, D.G.; Köbel, M. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia 2008, 10, 1021–1027. [Google Scholar] [CrossRef]
- Fukumoto, T.; Park, P.H.; Wu, S.; Fatkhutdinov, N.; Karakashev, S.; Nacarelli, T.; Kossenkov, A.V.; Speicher, D.W.; Jean, S.; Zhang, L.; et al. Repurposing Pan-HDAC Inhibitors for ARID1A-Mutated Ovarian Cancer. Cell Rep. 2018, 22, 3393–3400. [Google Scholar] [CrossRef]
- Kobayashi, T.; Nakazono, K.; Tokuda, M.; Mashima, Y.; Dynlacht, B.D.; Itoh, H. HDAC2 promotes loss of primary cilia in pancreatic ductal adenocarcinoma. EMBO Rep. 2017, 18, 334–343. [Google Scholar] [CrossRef]
- King, J.; Patel, M.; Chandrasekaran, S. Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective. Metabolites 2021, 11, 792. [Google Scholar] [CrossRef]
- Annibaldi, A.; Widmann, C. Glucose metabolism in cancer cells. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jin, X.; Yan, Y.; Shao, Y.; Pan, Y.; Roberts, L.R.; Zhang, J.; Huang, H.; Jiang, J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 2017, 7, 43864. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, Z. Mechanism of Action for HDAC Inhibitors-Insights from Omics Approaches. Int. J. Mol. Sci. 2019, 20, 1616. [Google Scholar] [CrossRef]
- Zhu, M.; Han, Y.; Gu, T.; Wang, R.; Si, X.; Kong, D.; Zhao, P.; Wang, X.; Li, J.; Zhai, X.; et al. Class I HDAC inhibitors enhance antitumor efficacy and persistence of CAR-T cells by activation of the Wnt pathway. Cell Rep. 2024, 43, 114065. [Google Scholar] [CrossRef] [PubMed]
- Amoêdo, N.D.; Rodrigues, M.F.; Pezzuto, P.; Galina, A.; da Costa, R.M.; de Almeida, F.C.; El-Bacha, T.; Rumjanek, F.D. Energy metabolism in H460 lung cancer cells: Effects of histone deacetylase inhibitors. PLoS ONE 2011, 6, e22264. [Google Scholar] [CrossRef] [PubMed]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Furumai, R.; Komatsu, Y.; Nishino, N.; Khochbin, S.; Yoshida, M.; Horinouchi, S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc. Natl. Acad. Sci. USA 2001, 98, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Kijima, M.; Yoshida, M.; Sugita, K.; Horinouchi, S.; Beppu, T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993, 268, 22429–22435. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Williams, D.E.; Ho, E.; Dashwood, R.H. Metabolism as a key to histone deacetylase inhibition. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 181–199. [Google Scholar] [CrossRef]
- Desai, D.; Salli, U.; Vrana, K.E.; Amin, S. SelSA, selenium analogs of SAHA as potent histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 2044–2047. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F. Histone Deacetylases and Histone Deacetylase Inhibitors: Molecular Mechanisms of Action in Various Cancers. Adv. Biomed. Res. 2019, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; D’Orazi, G.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell. Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hao, C.; Wang, L.; Liu, P.; Zhao, L.; Zhu, C.; Tian, X. Inhibition of leukemic cells by valproic acid, an HDAC inhibitor, in xenograft tumors. OncoTargets Ther. 2013, 6, 733–740. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Furumai, R.; Nishiyama, M.; Komatsu, Y.; Nishino, N.; Horinouchi, S. Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother. Pharmacol. 2001, 48 (Suppl. S1), S20–S26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, X.; Zhang, E.; Yang, L.; Yuan, H.; Tu, W.; Zhang, H.; Yin, Z.; Shen, W.; Chen, X.; et al. An epigenetic bioactive composite scaffold with well-aligned nanofibers for functional tendon tissue engineering. Acta Biomater. 2018, 66, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; O’Connor, O.A.; Krug, L.M.; Chiao, J.H.; Heaney, M.; Curley, T.; MacGregore-Cortelli, B.; Tong, W.; Secrist, J.P.; Schwartz, L.; et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 3923–3931. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Heaney, M.L.; Schwartz, L.; Richardson, S.; Willim, R.; MacGregor-Cortelli, B.; Curly, T.; Moskowitz, C.; Portlock, C.; Horwitz, S.; et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J. Clin. Oncol. 2006, 24, 166–173. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Lu, Z.; Cao, Z.; Zhang, Y. Zinc chelation with hydroxamate in histone deacetylases modulated by water access to the linker binding channel. J. Am. Chem. Soc. 2011, 133, 6110–6113. [Google Scholar] [CrossRef] [PubMed]
- Rajak, H.; Singh, A.; Raghuwanshi, K.; Kumar, R.; Dewangan, P.K.; Veerasamy, R.; Sharma, P.C.; Dixit, A.; Mishra, P. A structural insight into hydroxamic acid based histone deacetylase inhibitors for the presence of anticancer activity. Curr. Med. Chem. 2014, 21, 2642–2664. [Google Scholar] [CrossRef] [PubMed]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef]
- Heers, H.; Stanislaw, J.; Harrelson, J.; Lee, M.W. Valproic acid as an adjunctive therapeutic agent for the treatment of breast cancer. Eur. J. Pharmacol. 2018, 835, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Manal, M.; Chandrasekar, M.J.; Gomathi Priya, J.; Nanjan, M.J. Inhibitors of histone deacetylase as antitumor agents: A critical review. Bioorg. Chem. 2016, 67, 18–42. [Google Scholar] [CrossRef]
- Zhang, L.; Han, Y.; Jiang, Q.; Wang, C.; Chen, X.; Li, X.; Xu, F.; Jiang, Y.; Wang, Q.; Xu, W. Trend of histone deacetylase inhibitors in cancer therapy: Isoform selectivity or multitargeted strategy. Med. Res. Rev. 2015, 35, 63–84. [Google Scholar] [CrossRef]
- Giannini, G.; Cabri, W.; Fattorusso, C.; Rodriquez, M. Histone deacetylase inhibitors in the treatment of cancer: Overview and perspectives. Future Med. Chem. 2012, 4, 1439–1460. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.Y.; Ding, W.; Li, T.Q.; Zhang, Y.X.; Zhao, S.C. Histone Deacetylase (HDAC) Inhibitor, Suberoylanilide Hydroxamic Acid (SAHA), Induces Apoptosis in Prostate Cancer Cell Lines via the Akt/FOXO3a Signaling Pathway. Med. Sci. Monit. 2017, 23, 5793–5802. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. Histone Deacetylase Inhibitor-Induced Autophagy in Tumor Cells: Implications for p53. Int. J. Mol. Sci. 2017, 18, 1883. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhao, N.; Hu, Y.; Jiang, C.S.; Zhang, H. The Development Process: From SAHA to Hydroxamate HDAC Inhibitors with Branched CAP Region and Linear Linker. Chem. Biodivers. 2020, 17, e1900427. [Google Scholar] [CrossRef] [PubMed]
- Brightman, A.O.; Wang, J.; Miu, R.K.; Sun, I.L.; Barr, R.; Crane, F.L.; Morré, D.J. A growth factor- and hormone-stimulated NADH oxidase from rat liver plasma membrane. Biochim. Biophys. Acta 1992, 1105, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Wahan, S.K.; Vishakha, S.; Kurmi, B.D.; Gupta, G.D.; Rajak, H.; Asati, V. Recent Progress in Histone Deacetylase (HDAC) 1 Inhibitors as Anticancer Agent. Curr. Cancer Drug Targets 2022, 23, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Bass, A.K.A.; El-Zoghbi, M.S.; Nageeb, E.M.; Mohamed, M.F.A.; Badr, M.; Abuo-Rahma, G.E.A. Comprehensive review for anticancer hybridized multitargeting HDAC inhibitors. Eur. J. Med. Chem. 2021, 209, 112904. [Google Scholar] [CrossRef]
- Bressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.; Melkus, R.; De Jong, R.; O’Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. [Google Scholar] [CrossRef]
- Dong, M.; Ning, Z.Q.; Xing, P.Y.; Xu, J.L.; Cao, H.X.; Dou, G.F.; Meng, Z.Y.; Shi, Y.K.; Lu, X.P.; Feng, F.Y. Phase I study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemother. Pharmacol. 2012, 69, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, T.; Geng, C.; Zhang, Y.; Zhang, J.; Ning, Z.; Jiang, Z. Exploratory clinical study of chidamide, an oral subtype-selective histone deacetylase inhibitor, in combination with exemestane in hormone receptor-positive advanced breast cancer. Chin. J. Cancer Res. 2018, 30, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Buckton, L.K.; Rahimi, M.N.; McAlpine, S.R. Cyclic Peptides as Drugs for Intracellular Targets: The Next Frontier in Peptide Therapeutic Development. Chemistry 2021, 27, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Ramadhani, D.; Maharani, R.; Gazzali, A.M.; Muchtaridi, M. Cyclic Peptides for the Treatment of Cancers: A Review. Molecules 2022, 27, 4428. [Google Scholar] [CrossRef] [PubMed]
- Pojani, E.; Barlocco, D. Romidepsin (FK228), A Histone Deacetylase Inhibitor and its Analogues in Cancer Chemotherapy. Curr. Med. Chem. 2021, 28, 1290–1303. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Esposito, E.; Raj, V.P.; Muzi, L.; Zunino, F.; Zuco, V.; Cominetti, D.; Penco, S.; Dal Pozzo, A. New macrocyclic analogs of the natural histone deacetylase inhibitor FK228; design, synthesis and preliminary biological evaluation. Bioorg. Med. Chem. 2015, 23, 6785–6793. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Eckel, R.H.; Kahn, R.; Robertson, R.M.; Rizza, R.A. Preventing cardiovascular disease and diabetes: A call to action from the American Diabetes Association and the American Heart Association. Circulation 2006, 113, 2943–2946. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Lichtenstein, A.; Van Horn, L.; Harris, W.; Kris-Etherton, P.; Winston, M. Soy protein, isoflavones, and cardiovascular health: A summary of a statement for professionals from the american heart association nutrition committee. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1689–1692. [Google Scholar] [CrossRef]
- Wanczyk, M.; Roszczenko, K.; Marcinkiewicz, K.; Bojarczuk, K.; Kowara, M.; Winiarska, M. HDACi--going through the mechanisms. Front. Biosci. 2011, 16, 340–359. [Google Scholar] [CrossRef]
- Yang, R.; Lu, G.; Lv, Z.; Jia, L.; Cui, J. Different treatment regimens in breast cancer visceral crisis: A retrospective cohort study. Front. Oncol. 2022, 12, 1048781. [Google Scholar] [CrossRef]
- Tasneem, S.; Alam, M.M.; Amir, M.; Akhter, M.; Parvez, S.; Verma, G.; Nainwal, L.M.; Equbal, A.; Anwer, T.; Shaquiquzzaman, M. Heterocyclic Moieties as HDAC Inhibitors: Role in Cancer Therapeutics. Mini Rev. Med. Chem. 2022, 22, 1648–1706. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.D.; Cowley, S.M. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87.e75. [Google Scholar] [CrossRef]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef]
- Xiong, Y.; Donovan, K.A.; Eleuteri, N.A.; Kirmani, N.; Yue, H.; Razov, A.; Krupnick, N.M.; Nowak, R.P.; Fischer, E.S. Chemo-proteomics exploration of HDAC degradability by small molecule degraders. Cell Chem. Biol. 2021, 28, 1514–1527.e1514. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.P.; Baker, I.M.; Pytel, W.A.; Lin, L.Y.; Bowman, K.J.; Schwabe, J.W.R.; Cowley, S.M.; Hodgkinson, J.T. Optimization of Class I Histone Deacetylase PROTACs Reveals that HDAC1/2 Degradation is Critical to Induce Apoptosis and Cell Arrest in Cancer Cells. J. Med. Chem. 2022, 65, 5642–5659. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; de Weerd, S.; Chen, D.; Zwinderman, M.R.H.; van der Wouden, P.E.; Dekker, F.J. Induced protein degradation of histone deacetylases 3 (HDAC3) by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem. 2020, 208, 112800. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Geng, A.; Wang, Z.; Wu, C. Efficacy and safety of apatinib combined with liposomal doxorubicin or paclitaxel versus liposomal doxorubicin or paclitaxel monotherapy in patients with recurrent platinum-resistant ovarian cancer. J. Obstet. Gynaecol. Res. 2023, 49, 1611–1619. [Google Scholar] [CrossRef]
- Chotitumnavee, J.; Yamashita, Y.; Takahashi, Y.; Takada, Y.; Iida, T.; Oba, M.; Itoh, Y.; Suzuki, T. Selective degradation of histone deacetylase 8 mediated by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2022, 58, 4635–4638. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Wang, Y.; Jia, X.; Xu, P.; Qin, L.; Feng, X.; Li, Z.; Qiu, Z. Dual-target Janus kinase (JAK) inhibitors: Comprehensive review on the JAK-based strategies for treating solid or hematological malignancies and immune-related diseases. Eur. J. Med. Chem. 2022, 239, 114551. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, J.; Xu, W.; Wu, Q.; Zeng, R.; Liu, Z.; Tao, W.; Chen, Q.; Wang, Y.; Zhu, W.G. Structure-Based Discovery of Selective Histone Deacetylase 8 Degraders with Potent Anticancer Activity. J. Med. Chem. 2023, 66, 1186–1209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, D.; Suo, F.; Setroikromo, R.; Quax, W.J.; Dekker, F.J. Discovery of highly potent HDAC8 PROTACs with anti-tumor activity. Bioorg Chem. 2023, 136, 106546. [Google Scholar] [CrossRef]
- Macabuag, N.; Esmieu, W.; Breccia, P.; Jarvis, R.; Blackaby, W.; Lazari, O.; Urbonas, L.; Eznarriaga, M.; Williams, R.; Strijbosch, A.; et al. Developing HDAC4-Selective Protein Degraders to Investigate the Role of HDAC4 in Huntington’s Disease Pathology. J. Med. Chem. 2022, 65, 12445–12459. [Google Scholar] [CrossRef]
- Luckhurst, C.A.; Aziz, O.; Beaumont, V.; Bürli, R.W.; Breccia, P.; Maillard, M.C.; Haughan, A.F.; Lamers, M.; Leonard, P.; Matthews, K.L.; et al. Development and characterization of a CNS-penetrant benzhydryl hydroxamic acid class IIa histone deacetylase inhibitor. Bioorg. Med. Chem. Lett. 2019, 29, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Stott, A.J.; Maillard, M.C.; Beaumont, V.; Allcock, D.; Aziz, O.; Borchers, A.H.; Blackaby, W.; Breccia, P.; Creighton-Gutteridge, G.; Haughan, A.F.; et al. Evaluation of 5-(Trifluoromethyl)-1,2,4-oxadiazole-Based Class IIa HDAC Inhibitors for Huntington’s Disease. ACS Med. Chem. Lett. 2021, 12, 380–388. [Google Scholar] [CrossRef]
- Hook, S.S.; Orian, A.; Cowley, S.M.; Eisenman, R.N. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 13425–13430. [Google Scholar] [CrossRef]
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Gu, Z.; Lin, S.; Chen, D.; Wang, J.; Zhao, Y.; Li, Y.; Liu, T.; Wang, Y.; Lin, H.; et al. Attenuation of NLRP3 Inflammasome Activation by Indirubin-Derived PROTAC Targeting HDAC6. ACS Chem. Biol. 2021, 16, 2746–2751. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Jing, H.; Price, I.R.; Cao, J.; Bai, J.J.; Lin, H. Simultaneous Inhibition of SIRT2 Deacetylase and Defatty-Acylase Activities via a PROTAC Strategy. ACS Med. Chem. Lett. 2020, 11, 2305–2311. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Kutil, Z.; Novakova, Z.; Meleshin, M.; Mikesova, J.; Schutkowski, M.; Barinka, C. Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem. Biol. 2018, 13, 685–693. [Google Scholar] [CrossRef]
- Bhaskara, S. Histone deacetylase 11 as a key regulator of metabolism and obesity. eBioMedicine 2018, 35, 27–28. [Google Scholar] [CrossRef]
- Son, S.I.; Cao, J.; Zhu, C.L.; Miller, S.P.; Lin, H. Activity-Guided Design of HDAC11-Specific Inhibitors. ACS Chem. Biol. 2019, 14, 1393–1397. [Google Scholar] [CrossRef]
- Banik, D.; Noonepalle, S.; Hadley, M.; Palmer, E.; Gracia-Hernandez, M.; Zevallos-Delgado, C.; Manhas, N.; Simonyan, H.; Young, C.N.; Popratiloff, A.; et al. HDAC6 Plays a Noncanonical Role in the Regulation of Antitumor Immune Responses, Dissemination, and Invasiveness of Breast Cancer. Cancer Res. 2020, 80, 3649–3662. [Google Scholar] [CrossRef] [PubMed]
- Borcoman, E.; Kamal, M.; Marret, G.; Dupain, C.; Castel-Ajgal, Z.; Le Tourneau, C. HDAC Inhibition to Prime Immune Checkpoint Inhibitors. Cancers 2021, 14, 66. [Google Scholar] [CrossRef] [PubMed]
- Glauben, R.; Sonnenberg, E.; Zeitz, M.; Siegmund, B. HDAC inhibitors in models of inflammation-related tumorigenesis. Cancer Lett. 2009, 280, 154–159. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.L.; Daniels, E.G.; Molenaars, M.; Houtkooper, R.H.; Janssens, G.E. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol. Med. 2019, 11, e9854. [Google Scholar] [CrossRef] [PubMed]
- Sanna, M.D.; Guandalini, L.; Romanelli, M.N.; Galeotti, N. The new HDAC1 inhibitor LG325 ameliorates neuropathic pain in a mouse model. Pharmacol. Biochem. Behav. 2017, 160, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.; Soragni, E.; Jenssen, K.; Burnett, R.; Herman, D.; Coppola, G.; Geschwind, D.H.; Gottesfeld, J.M.; Pandolfo, M. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE 2008, 3, e1958. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Pallos, J.; Jacques, V.; Lau, A.; Tang, B.; Cooper, A.; Syed, A.; Purcell, J.; Chen, Y.; Sharma, S.; et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol. Dis. 2012, 46, 351–361. [Google Scholar] [CrossRef]
- Varela, R.B.; Resende, W.R.; Dal-Pont, G.C.; Gava, F.F.; Tye, S.J.; Quevedo, J.; Valvassori, S.S. HDAC inhibitors reverse mania-like behavior and modulate epigenetic regulatory enzymes in an animal model of mania induced by Ouabain. Pharmacol. Biochem. Behav. 2020, 193, 172917. [Google Scholar] [CrossRef]
- Chari, A.; Cho, H.J.; Dhadwal, A.; Morgan, G.; La, L.; Zarychta, K.; Catamero, D.; Florendo, E.; Stevens, N.; Verina, D.; et al. A phase 2 study of panobinostat with lenalidomide and weekly dexamethasone in myeloma. Blood Adv. 2017, 1, 1575–1583. [Google Scholar] [CrossRef]
- Choi, S.W.; Braun, T.; Henig, I.; Gatza, E.; Magenau, J.; Parkin, B.; Pawarode, A.; Riwes, M.; Yanik, G.; Dinarello, C.A.; et al. Vorinostat plus tacrolimus/methotrexate to prevent GVHD after myeloablative conditioning, unrelated donor HCT. Blood 2017, 130, 1760–1767. [Google Scholar] [CrossRef]
- Thomas, A.; Rajan, A.; Szabo, E.; Tomita, Y.; Carter, C.A.; Scepura, B.; Lopez-Chavez, A.; Lee, M.J.; Redon, C.E.; Frosch, A.; et al. A phase I/II trial of belinostat in combination with cisplatin, doxorubicin, and cyclophosphamide in thymic epithelial tumors: A clinical and translational study. Clin. Cancer Res. 2014, 20, 5392–5402. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Sun, Z.; Kuang, P.; Chen, J. Recent progress on HDAC inhibitors with dual targeting capabilities for cancer treatment. Eur. J. Med. Chem. 2020, 208, 112831. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Gao, F.; Coppola, G.; Gao, F.B. Suberoylanilide hydroxamic acid increases progranulin production in iPSC-derived cortical neurons of frontotemporal dementia patients. Neurobiol. Aging 2016, 42, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Milde, T.; Deubzer, H.E.; Oehme, I.; Witt, R.; Kulozik, A.; Eisenmenger, A.; Abel, U.; Karapanagiotou-Schenkel, I. Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma or leukemia. Klin. Padiatr. 2012, 224, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Hase, T.; Shimizu, S.; Ando, M.; Hata, A.; Murakami, H.; Kawakami, T.; Nagase, K.; Yoshimura, K.; Fujiwara, T.; et al. Phase I study of vorinostat with gefitinib in BIM deletion polymorphism/epidermal growth factor receptor mutation double-positive lung cancer. Cancer Sci. 2020, 111, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Abedin, S.M.; Badar, T.; Gauger, K.; Michaelis, L.C.; Runaas, L.; Carlson, K.S.; Murthy, G.G.; Atallah, E. Safety and efficacy of pracinostat in combination with gemtuzumab ozogamicin (PraGO) in patients with relapsed/refractory acute myeloid leukemia. Leuk. Res. 2022, 123, 106984. [Google Scholar] [CrossRef]
- Xue, F.; Cheng, Y.; Xu, L.; Tian, C.; Jiao, H.; Wang, R.; Gao, X. LncRNA NEAT1/miR-129/Bcl-2 signaling axis contributes to HDAC inhibitor tolerance in nasopharyngeal cancer. Aging 2020, 12, 14174–14188. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Wang, J.; Zhou, Z.; Cao, S.; Zhang, J. Strategies of Targeting CK2 in Drug Discovery: Challenges, Opportunities, and Emerging Prospects. J. Med. Chem. 2023, 66, 2257–2281. [Google Scholar] [CrossRef] [PubMed]
- Frommel, T.O.; Coon, J.S.; Tsuruo, T.; Roninson, I.B. Variable effects of sodium butyrate on the expression and function of the MDR1 (P-glycoprotein) gene in colon carcinoma cell lines. Int. J. Cancer 1993, 55, 297–302. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Uba, A.I.; Yelekçi, K. Identification of potential isoform-selective histone deacetylase inhibitors for cancer therapy: A combined approach of structure-based virtual screening, ADMET prediction and molecular dynamics simulation assay. J. Biomol. Struct. Dyn. 2018, 36, 3231–3245. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, S.; Berger, A.; Böcker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Schleicher, S.; Mayer, M.; Weiss, T.S.; et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLoS ONE 2013, 8, e73097. [Google Scholar] [CrossRef] [PubMed]
- Urias, B.S.; Pavan, A.R.; Albuquerque, G.R.; Prokopczyk, I.M.; Alves, T.M.F.; de Melo, T.R.F.; Sartori, G.R.; da Silva, J.H.M.; Chin, C.M.; Santos, J.L.D. Optimization of Resveratrol Used as a Scaffold to Design Histone Deacetylase (HDAC-1 and HDAC-2) Inhibitors. Pharmaceuticals 2022, 15, 1260. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qian, C.; Zhu, Y.; Cai, J.; He, Y.; Li, J.; Wang, T.; Zhu, H.; Li, Z.; Li, W.; et al. Design, synthesis and evaluate of novel dual FGFR1 and HDAC inhibitors bearing an indazole scaffold. Bioorg. Med. Chem. 2018, 26, 747–757. [Google Scholar] [CrossRef]
- Tang, G.; Wong, J.C.; Zhang, W.; Wang, Z.; Zhang, N.; Peng, Z.; Zhang, Z.; Rong, Y.; Li, S.; Zhang, M.; et al. Identification of a novel aminotetralin class of HDAC6 and HDAC8 selective inhibitors. J. Med. Chem. 2014, 57, 8026–8034. [Google Scholar] [CrossRef] [PubMed]
- Bui, H.T.B.; Nguyen, P.H.; Pham, Q.M.; Tran, H.P.; Tran, Q.; Jung, H.; Hong, Q.V.; Nguyen, Q.C.; Nguyen, Q.P.; Le, H.T.; et al. Target Design of Novel Histone Deacetylase 6 Selective Inhibitors with 2-Mercaptoquinazolinone as the Cap Moiety. Molecules 2022, 27, 2204. [Google Scholar] [CrossRef] [PubMed]
- Schäker-Hübner, L.; Warstat, R.; Ahlert, H.; Mishra, P.; Kraft, F.B.; Schliehe-Diecks, J.; Schöler, A.; Borkhardt, A.; Breit, B.; Bhatia, S.; et al. 4-Acyl Pyrrole Capped HDAC Inhibitors: A New Scaffold for Hybrid Inhibitors of BET Proteins and Histone Deacetylases as Antileukemia Drug Leads. J. Med. Chem. 2021, 64, 14620–14646. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.D.; Jiang, Y.J.; Zhou, J.W.; Yu, Q.S.; You, Q.D. Identification of ligand features essential for HDACs inhibitors by pharmacophore modeling. J. Mol. Graph. Model. 2008, 26, 1160–1168. [Google Scholar] [CrossRef]
- Uba, A.I.; Zengin, G. In the quest for histone deacetylase inhibitors: Current trends in the application of multilayered computational methods. Amino Acids 2023, 55, 1709–1726. [Google Scholar] [CrossRef]
- Pai, P.; Kumar, A.; Shetty, M.G.; Kini, S.G.; Krishna, M.B.; Satyamoorthy, K.; Babitha, K.S. Identification of potent HDAC 2 inhibitors using E-pharmacophore modelling, structure-based virtual screening and molecular dynamic simulation. J. Mol. Model. 2022, 28, 119. [Google Scholar] [CrossRef] [PubMed]
- Kumbhar, N.; Nimal, S.; Barale, S.; Kamble, S.; Bavi, R.; Sonawane, K.; Gacche, R. Identification of novel leads as potent inhibitors of HDAC3 using ligand-based pharmacophore modeling and MD simulation. Sci. Rep. 2022, 12, 1712. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A.; Abdullah, E.; Rashid, R.; Altaf, M. Combinatorial. Front. Mol. Neurosci. 2017, 10, 357. [Google Scholar] [CrossRef]
- Thangapandian, S.; John, S.; Lee, Y.; Kim, S.; Lee, K.W. Dynamic structure-based pharmacophore model development: A new and effective addition in the histone deacetylase 8 (HDAC8) inhibitor discovery. Int. J. Mol. Sci. 2011, 12, 9440–9462. [Google Scholar] [CrossRef]
- Nencetti, S.; Cuffaro, D.; Nuti, E.; Ciccone, L.; Rossello, A.; Fabbi, M.; Ballante, F.; Ortore, G.; Carbotti, G.; Campelli, F.; et al. Identification of histone deacetylase inhibitors with (arylidene)aminoxy scaffold active in uveal melanoma cell lines. J. Enzyme Inhib. Med. Chem. 2021, 36, 34–47. [Google Scholar] [CrossRef]
- Mehndiratta, S.; Chen, M.C.; Chao, Y.H.; Lee, C.H.; Liou, J.P.; Lai, M.J.; Lee, H.Y. Effect of 3-subsitution of quinolinehydroxamic acids on selectivity of histone deacetylase isoforms. J. Enzyme Inhib. Med. Chem. 2021, 36, 74–84. [Google Scholar] [CrossRef]
- Relitti, N.; Saraswati, A.P.; Chemi, G.; Brindisi, M.; Brogi, S.; Herp, D.; Schmidtkunz, K.; Saccoccia, F.; Ruberti, G.; Ulivieri, C.; et al. Novel quinolone-based potent and selective HDAC6 inhibitors: Synthesis, molecular modeling studies and biological investigation. Eur. J. Med. Chem. 2021, 212, 112998. [Google Scholar] [CrossRef]
- Hu, H.; Chen, F.; Dong, Y.; Li, M.; Xu, S.; Qin, M.; Gong, P. Discovery of Novel c-Mesenchymal-Epithelia transition factor and histone deacetylase dual inhibitors. Eur. J. Med. Chem. 2020, 204, 112651. [Google Scholar] [CrossRef] [PubMed]
- Moffat, D.; Patel, S.; Day, F.; Belfield, A.; Donald, A.; Rowlands, M.; Wibawa, J.; Brotherton, D.; Stimson, L.; Clark, V.; et al. Discovery of 2-(6-{[(6-fluoroquinolin-2-yl)methyl]amino}bicyclo[3.1.0]hex-3-yl)-N-hydroxypyrimidine-5-carboxamide (CHR-3996), a class I selective orally active histone deacetylase inhibitor. J. Med. Chem. 2010, 53, 8663–8678. [Google Scholar] [CrossRef]
- Banerji, U.; van Doorn, L.; Papadatos-Pastos, D.; Kristeleit, R.; Debnam, P.; Tall, M.; Stewart, A.; Raynaud, F.; Garrett, M.D.; Toal, M.; et al. A phase I pharmacokinetic and pharmacodynamic study of CHR-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin. Cancer Res. 2012, 18, 2687–2694. [Google Scholar] [CrossRef]
- Chen, C.; Hou, X.; Wang, G.; Pan, W.; Yang, X.; Zhang, Y.; Fang, H. Design, synthesis and biological evaluation of quinoline derivatives as HDAC class I inhibitors. Eur. J. Med. Chem. 2017, 133, 11–23. [Google Scholar] [CrossRef]
- Zhao, Y.; Yao, Z.; Ren, W.; Yang, X.; Hou, X.; Cao, S.; Fang, H. Design, synthesis and bioactivity evaluations of 8-substituted-quinoline-2-carboxamide derivatives as novel histone deacetylase (HDAC) inhibitors. Bioorg. Med. Chem. 2023, 85, 117242. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Duan, J.; Dang, X.; Yuan, Y.; Wang, Y.; He, X.; Bai, R.; Ye, X.Y.; Xie, T. Design, synthesis and biological evaluation of novel histone deacetylase (HDAC) inhibitors derived from. J. Enzyme Inhib. Med. Chem. 2023, 38, 2195991. [Google Scholar] [CrossRef]
- Zheng, F.; Tang, Q.; Zheng, X.H.; Wu, J.; Huang, H.; Zhang, H.; Hann, S.S. Inactivation of Stat3 and crosstalk of miRNA155-5p and FOXO3a contribute to the induction of IGFBP1 expression by beta-elemene in human lung cancer. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Long, J.; Liu, Z.; Hui, L. Anti-tumor effect and mechanistic study of elemene on pancreatic carcinoma. BMC Complement. Altern. Med. 2019, 19, 133. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Guo, T.; Qu, X.; Hu, X.; Zhang, Y.; Che, X.; Song, H.; Gong, J.; Ma, R.; Li, C.; et al. β-elemene increases the sensitivity of gastric cancer cells to TRAIL by promoting the formation of DISC in lipid rafts. Cell Biol. Int. 2018, 42, 1377–1385. [Google Scholar] [CrossRef]
- Xie, L.; Zhang, J.; Yan, H.; Cai, Y.; Xu, L. β-elemene induced apoptosis and senescence of triple-negative breast cancer cells through IGF1/IGF1R pathway. Tissue Cell 2022, 79, 101914. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Ma, L.; Nong, S.; Wu, Y.; Guo, X.; Pu, J. β-elemene induced anticancer effect in bladder cancer through upregulation of PTEN and suppression of AKT phosphorylation. Oncol. Lett. 2018, 16, 6019–6025. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Chang, L.; Guo, L.; Zhao, D.; Liu, H.B.; Wang, Q.S.; Zhang, P.; Du, W.Z.; Liu, X.; Zhang, H.T.; et al. β-elemene induces caspase-dependent apoptosis in human glioma cells in vitro through the upregulation of Bax and Fas/FasL and downregulation of Bcl-2. Asian Pac. J. Cancer Prev. 2014, 15, 10407–10412. [Google Scholar] [CrossRef]
- Juvale, D.C.; Kulkarni, V.V.; Deokar, H.S.; Wagh, N.K.; Padhye, S.B.; Kulkarni, V.M. 3D-QSAR of histone deacetylase inhibitors: Hydroxamate analogues. Org. Biomol. Chem. 2006, 4, 2858–2868. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, J.; Guo, Z.; Chu, F.; Cheng, Y.; Wu, S. Exploration of a binding mode of indole amide analogues as potent histone deacetylase inhibitors and 3D-QSAR analyses. Bioorg. Med. Chem. 2005, 13, 5424–5434. [Google Scholar] [CrossRef]
- Banerjee, S.; Adhikari, N.; Amin, S.A.; Jha, T. Structural exploration of tetrahydroisoquinoline derivatives as HDAC8 inhibitors through multi-QSAR modeling study. J. Biomol. Struct. Dyn. 2020, 38, 1551–1564. [Google Scholar] [CrossRef]
- Cao, G.P.; Arooj, M.; Thangapandian, S.; Park, C.; Arulalapperumal, V.; Kim, Y.; Kwon, Y.J.; Kim, H.H.; Suh, J.K.; Lee, K.W. A lazy learning-based QSAR classification study for screening potential histone deacetylase 8 (HDAC8) inhibitors. SAR QSAR Environ. Res. 2015, 26, 397–420. [Google Scholar] [CrossRef] [PubMed]
- Abdul, A.S.; Janish, K.; Samima, K.; Sanjib, D.; Ahmed, Q.I.; Tarun, J.; Shovanlal, G. Binary quantitative activity-activity relationship (QAAR) studies to explore selective HDAC8 inhibitors: In light of mathematical models, DFT-based calculation and molecular dynamic simulation studies. J. Mol. Struct. 2022, 1260, 132833. [Google Scholar]
- Zhao, L.; Fu, L.; Li, G.; Yu, Y.; Wang, J.; Liang, H.; Shu, M.; Lin, Z.; Wang, Y. Three-dimensional quantitative structural-activity relationship and molecular dynamics study of multivariate substituted 4-oxyquinazoline HDAC6 inhibitors. Mol. Divers. 2023, 27, 1123–1140. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Trivedi, P.; Adhikari, N.; Routholla, G.; Vijayasarathi, D.; Das, S.; Ghosh, B.; Jha, T. Quantitative activity–activity relationship (QAAR) driven design to develop hydroxamate derivatives of pentanoic acids as selective HDAC8 inhibitors: Synthesis, biological evaluation and binding mode of interaction studies. New J. Chem. 2021, 45, 17149–17162. [Google Scholar] [CrossRef]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef] [PubMed]
- Velankar, S.; Burley, S.K.; Kurisu, G.; Hoch, J.C.; Markley, J.L. The Protein Data Bank Archive. Methods Mol. Biol. 2021, 2305, 3–21. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7, 41615. [Google Scholar] [CrossRef]
- Ibrahim Uba, A.; Yelekçi, K. Homology modeling of human histone deacetylase 10 and design of potential selective inhibitors. J. Biomol. Struct. Dyn. 2019, 37, 3627–3636. [Google Scholar] [CrossRef]
- Sixto-López, Y.; Gómez-Vidal, J.A.; de Pedro, N.; Bello, M.; Rosales-Hernández, M.C.; Correa-Basurto, J. Hydroxamic acid derivatives as HDAC1, HDAC6 and HDAC8 inhibitors with antiproliferative activity in cancer cell lines. Sci. Rep. 2020, 10, 10462. [Google Scholar] [CrossRef] [PubMed]
- Elmezayen, A.D.; Kemal, Y. Structure-based virtual screening for novel potential selective inhibitors of class IIa histone deacetylases for cancer treatment. Comput. Biol. Chem. 2021, 92, 107491. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yan, W.; Wang, S.; Lu, M.; Yang, H.; Chai, X.; Shi, H.; Zhang, Y.; Jia, Q. Discovery of selective HDAC6 inhibitors based on a multi-layer virtual screening strategy. Comput. Biol. Med. 2023, 160, 107036. [Google Scholar] [CrossRef]
- Peng, Z.; Zhao, Q.; Tian, X.; Lei, T.; Xiang, R.; Chen, L.; Yang, Y. Discovery of Potent and Isoform-selective Histone Deacetylase Inhibitors Using Structure-based Virtual Screening and Biological Evaluation. Mol. Inform. 2022, 41, e2100295. [Google Scholar] [CrossRef]
- Pang, H.; Wu, L.; Tang, Y.; Zhou, G.; Qu, C.; Duan, J.A. Chemical Analysis of the Herbal Medicine Salviae miltiorrhizae Radix et Rhizoma (Danshen). Molecules 2016, 21, 51. [Google Scholar] [CrossRef] [PubMed]
- Shuai, W.; Wang, G.; Zhang, Y.; Bu, F.; Zhang, S.; Miller, D.D.; Li, W.; Ouyang, L.; Wang, Y. Recent Progress on Tubulin Inhibitors with Dual Targeting Capabilities for Cancer Therapy. J. Med. Chem. 2021, 64, 7963–7990. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Qin, J.; Kang, Y.; Zhang, Y.; Ba, M.; Yang, H.; Duan, Y.; Yao, Y. 2-Methoxydiol derivatives as new tubulin and HDAC dual-targeting inhibitors, displaying antitumor and antiangiogenic response. Bioorg. Chem. 2022, 120, 105625. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, W.; Yu, Y.; Wang, Y.; Yang, T.; Xue, L.; Yuan, X.; Long, C.; Liu, Z.; Chen, X.; et al. The compound millepachine and its derivatives inhibit tubulin polymerization by irreversibly binding to the colchicine-binding site in β-tubulin. J. Biol. Chem. 2018, 293, 9461–9472. [Google Scholar] [CrossRef]
- Xie, S.; Leng, J.; Zhao, S.; Zhu, L.; Zhang, M.; Ning, M.; Zhao, B.; Kong, L.; Yin, Y. Design and biological evaluation of dual tubulin/HDAC inhibitors based on millepachine for treatment of prostate cancer. Eur. J. Med. Chem. 2024, 268, 116301. [Google Scholar] [CrossRef]
- Ferro, A.; Pantazaka, E.; Athanassopoulos, C.M.; Cuendet, M. Histone deacetylase-based dual targeted inhibition in multiple myeloma. Med. Res. Rev. 2023, 43, 2177–2236. [Google Scholar] [CrossRef]
- Fan, F.; Liu, P.; Bao, R.; Chen, J.; Zhou, M.; Mo, Z.; Ma, Y.; Liu, H.; Zhou, Y.; Cai, X.; et al. A Dual PI3K/HDAC Inhibitor Induces Immunogenic Ferroptosis to Potentiate Cancer Immune Checkpoint Therapy. Cancer Res. 2021, 81, 6233–6245. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Soh, C.K.; Goh, W.H.; Wang, H. Design, Synthesis, and Preclinical Evaluation of Fused Pyrimidine-Based Hydroxamates for the Treatment of Hepatocellular Carcinoma. J. Med. Chem. 2018, 61, 1552–1575. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Tawa, G.J.; Henderson, M.J.; Danchik, C.; Liu, S.; Shah, P.; Wang, A.Q.; Dunn, G.; Kabir, M.; Padilha, E.C.; et al. Design, Synthesis, and Biological Evaluation of Quinazolin-4-one-Based Hydroxamic Acids as Dual PI3K/HDAC Inhibitors. J. Med. Chem. 2020, 63, 4256–4292. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dai, W.; Chen, X.; Geng, A.; Chen, Y.; Lu, T.; Zhu, Y. Design, synthesis and biological evaluation of 2,3-dihydroimidazo [1,2-c] quinazoline derivatives as novel phosphatidylinositol 3-kinase and histone deacetylase dual inhibitors. RSC Adv. 2017, 7, 52180–52186. [Google Scholar] [CrossRef]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2020, 40, 190–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Lai, F.; Lin, S.; Ji, M.; Zhang, J.; Zhang, Y.; Jin, J.; Fu, R.; Wu, D.; Tian, H.; et al. Design, Synthesis, and Biological Evaluation of 4-Methyl Quinazoline Derivatives as Anticancer Agents Simultaneously Targeting Phosphoinositide 3-Kinases and Histone Deacetylases. J. Med. Chem. 2019, 62, 6992–7014. [Google Scholar] [CrossRef]
- Zhang, K.; Huang, R.; Ji, M.; Lin, S.; Lai, F.; Wu, D.; Tian, H.; Bi, J.; Peng, S.; Hu, J.; et al. Rational design and optimization of novel 4-methyl quinazoline derivatives as PI3K/HDAC dual inhibitors with benzamide as zinc binding moiety for the treatment of acute myeloid leukemia. Eur. J. Med. Chem. 2024, 264, 116015. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.J. Machine Learning for Molecular Modelling in Drug Design. Biomolecules 2019, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 181, 475–483. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Nalbat, E.; Atalay, V.; Martin, M.J.; Cetin-Atalay, R.; Doğan, T. DEEPScreen: High performance drug-target interaction prediction with convolutional neural networks using 2-D structural compound representations. Chem. Sci. 2020, 11, 2531–2557. [Google Scholar] [CrossRef]
- Shimizu, Y.; Yonezawa, T.; Bao, Y.; Sakamoto, J.; Yokogawa, M.; Furuya, T.; Osawa, M.; Ikeda, K. Applying deep learning to iterative screening of medium-sized molecules for protein-protein interaction-targeted drug discovery. Chem. Commun. 2023, 59, 6722–6725. [Google Scholar] [CrossRef] [PubMed]
- Melesina, J.; Simoben, C.V.; Praetorius, L.; Bülbül, E.F.; Robaa, D.; Sippl, W. Strategies To Design Selective Histone Deacetylase Inhibitors. ChemMedChem 2021, 16, 1336–1359. [Google Scholar] [CrossRef] [PubMed]
- Baselious, F.; Hilscher, S.; Robaa, D.; Barinka, C.; Schutkowski, M.; Sippl, W. Comparative Structure-Based Virtual Screening Utilizing Optimized AlphaFold Model Identifies Selective HDAC11 Inhibitor. Int. J. Mol. Sci. 2024, 25, 1358. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Amin, S.A.; Kumar, P.; Jha, T.; Gayen, S. Exploring structural requirements of HDAC10 inhibitors through comparative machine learning approaches. J. Mol. Graph. Model. 2023, 123, 108510. [Google Scholar] [CrossRef] [PubMed]
- Nurani, A.; Yamashita, Y.; Taki, Y.; Takada, Y.; Itoh, Y.; Suzuki, T. Identification of a Histone Deacetylase 8 Inhibitor through Drug Screenings Based on Machine Learning. Chem. Pharm. Bull. 2024, 72, 173–178. [Google Scholar] [CrossRef]
- Li, R.; Tian, Y.; Yang, Z.; Ji, Y.; Ding, J.; Yan, A. Classification models and SAR analysis on HDAC1 inhibitors using machine learning methods. Mol. Divers. 2023, 27, 1037–1051. [Google Scholar] [CrossRef]
- Banerjee, S.; Jana, S.; Jha, T.; Ghosh, B.; Adhikari, N. An assessment of crucial structural contributors of HDAC6 inhibitors through fragment-based non-linear pattern recognition and molecular dynamics simulation approaches. Comput. Biol. Chem. 2024, 110, 108051. [Google Scholar] [CrossRef]
- Sotriffer, C.A.; Sanschagrin, P.; Matter, H.; Klebe, G. SFCscore: Scoring functions for affinity prediction of protein-ligand complexes. Proteins 2008, 73, 395–419. [Google Scholar] [CrossRef]
- Smith, R.D.; Damm-Ganamet, K.L.; Dunbar, J.B.; Ahmed, A.; Chinnaswamy, K.; Delproposto, J.E.; Kubish, G.M.; Tinberg, C.E.; Khare, S.D.; Dou, J.; et al. CSAR Benchmark Exercise 2013: Evaluation of Results from a Combined Computational Protein Design, Docking, and Scoring/Ranking Challenge. J. Chem. Inf. Model. 2016, 56, 1022–1031. [Google Scholar] [CrossRef]
- Spyrakis, F.; Cavasotto, C.N. Open challenges in structure-based virtual screening: Receptor modeling, target flexibility consideration and active site water molecules description. Arch. Biochem. Biophys. 2015, 583, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Poonia, P.; Sharma, M.; Jha, P.; Chopra, M. Pharmacophore-based virtual screening of ZINC database, molecular modeling and designing new derivatives as potential HDAC6 inhibitors. Mol. Divers. 2023, 27, 2053–2071. [Google Scholar] [CrossRef] [PubMed]
- Byvatov, E.; Fechner, U.; Sadowski, J.; Schneider, G. Comparison of support vector machine and artificial neural network systems for drug/nondrug classification. J. Chem. Inf. Comput. Sci. 2003, 43, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Zernov, V.V.; Balakin, K.V.; Ivaschenko, A.A.; Savchuk, N.P.; Pletnev, I.V. Drug discovery using support vector machines. The case studies of drug-likeness, agrochemical-likeness, and enzyme inhibition predictions. J. Chem. Inf. Comput. Sci. 2003, 43, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Warmuth, M.K.; Liao, J.; Rätsch, G.; Mathieson, M.; Putta, S.; Lemmen, C. Active learning with support vector machines in the drug discovery process. J. Chem. Inf. Comput. Sci. 2003, 43, 667–673. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Gilson, M.K. Virtual screening of molecular databases using a support vector machine. J. Chem. Inf. Model. 2005, 45, 549–561. [Google Scholar] [CrossRef]
- Podolyan, Y.; Walters, M.A.; Karypis, G. Assessing synthetic accessibility of chemical compounds using machine learning methods. J. Chem. Inf. Model. 2010, 50, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Geppert, H.; Horváth, T.; Gärtner, T.; Wrobel, S.; Bajorath, J. Support-vector-machine-based ranking significantly improves the effectiveness of similarity searching using 2D fingerprints and multiple reference compounds. J. Chem. Inf. Model. 2008, 48, 742–746. [Google Scholar] [CrossRef]
- Agarwal, S.; Dugar, D.; Sengupta, S. Ranking chemical structures for drug discovery: A new machine learning approach. J. Chem. Inf. Model. 2010, 50, 716–731. [Google Scholar] [CrossRef]
- Rathke, F.; Hansen, K.; Brefeld, U.; Müller, K.R. StructRank: A new approach for ligand-based virtual screening. J. Chem. Inf. Model. 2011, 51, 83–92. [Google Scholar] [CrossRef]
- Jacob, L.; Hoffmann, B.; Stoven, V.; Vert, J.-P. Virtual screening of GPCRs: An in silico chemogenomics approach. BMC Bioinform. 2008, 9, 363. [Google Scholar] [CrossRef]
- Jacob, L.; Vert, J.P. Protein-ligand interaction prediction: An improved chemogenomics approach. Bioinformatics 2008, 24, 2149–2156. [Google Scholar] [CrossRef]
- Schölkopf, B.; Warmuth, M.K. Learning Theory and Kernel Machines: 16th Annual Conference on Computational Learning Theory and 7th Kernel Workshop, COLT/Kernel 2003, Washington, DC, USA, 24–27 August 2003, Proceedings; Springer: Berlin/Heidelberg, Germany, 2003; Volume 2777. [Google Scholar]
- Vitányi, P. Computational Learning Theory: Second European Conference, EuroCOLT’95, Barcelona, Spain, 13–15 March 1995. Proceedings; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1995; Volume 2. [Google Scholar]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Kauffman, G.W.; Jurs, P.C. QSAR and k-nearest neighbor classification analysis of selective cyclooxygenase-2 inhibitors using topologically-based numerical descriptors. J. Chem. Inf. Comput. Sci. 2001, 41, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Konovalov, D.A.; Coomans, D.; Deconinck, E.; Heyden, Y.V. Benchmarking of QSAR models for blood-brain barrier permeation. J. Chem. Inf. Model. 2007, 47, 1648–1656. [Google Scholar] [CrossRef]
- Votano, J.R.; Parham, M.; Hall, L.H.; Kier, L.B.; Oloff, S.; Tropsha, A.; Xie, Q.; Tong, W. Three new consensus QSAR models for the prediction of Ames genotoxicity. Mutagenesis 2004, 19, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Zhang, F.; Chu, Q.; Liu, Z. High-performance blob-based iterative three-dimensional reconstruction in electron tomography using multi-GPUs. BMC Bioinform. 2012, 13 (Suppl. S10), S4. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, L.; Aitken, S.; van Hemert, J.; Goryanin, I. EnzML: Multi-label prediction of enzyme classes using InterPro signatures. BMC Bioinform. 2012, 13, 61. [Google Scholar] [CrossRef]
- Soyguder, S. Intelligent control based on wavelet decomposition and neural network for predicting of human trajectories with a novel vision-based robotic. Expert Syst. Appl. 2011, 38, 13994–14000. [Google Scholar] [CrossRef]
- Haykin, S. Neural Networks: A Comprehensive Foundation; Prentice Hall PTR: Hoboken, NJ, USA, 1998. [Google Scholar]
- Zhang, L.; Zhang, J.; Jiang, Q.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Hu, E.; Dul, E.; Sung, C.M.; Chen, Z.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Liu, R.; Lago, A.; Hofmann, G.; et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 2003, 307, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Kleinschek, A.; Meyners, C.; Digiorgio, E.; Brancolini, C.; Meyer-Almes, F.J. Potent and Selective Non-hydroxamate Histone Deacetylase 8 Inhibitors. ChemMedChem 2016, 11, 2598–2606. [Google Scholar] [CrossRef]
- Ononye, S.N.; VanHeyst, M.D.; Oblak, E.Z.; Zhou, W.; Ammar, M.; Anderson, A.C.; Wright, D.L. Tropolones as lead-like natural products: The development of potent and selective histone deacetylase inhibitors. ACS Med. Chem. Lett. 2013, 4, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Sodji, Q.H.; Kornacki, J.R.; Mrksich, M.; Oyelere, A.K. 3-Hydroxypyridin-2-thione as novel zinc binding group for selective histone deacetylase inhibition. J. Med. Chem. 2013, 56, 3492–3506. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Stowe, R.L.; Pinello, C.E.; Tian, G.; Madoux, F.; Li, D.; Zhao, L.Y.; Li, J.L.; Ma, H.; Hodder, P.; et al. Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem. Biol. 2015, 22, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Maolanon, A.R.; Madsen, A.S.; Olsen, C.A. Innovative Strategies for Selective Inhibition of Histone Deacetylases. Cell Chem. Biol. 2016, 23, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Abdizadeh, T.; Ghodsi, R.; Hadizadeh, F. 3D-QSAR (CoMFA, CoMSIA) and Molecular Docking Studies on Histone Deacetylase 1 Selective Inhibitors. Recent. Pat. Anticancer. Drug Discov. 2017, 12, 365–383. [Google Scholar] [CrossRef]
- Cao, F.; Zwinderman, M.R.H.; Dekker, F.J. The Process and Strategy for Developing Selective Histone Deacetylase 3 Inhibitors. Molecules 2018, 23, 551. [Google Scholar] [CrossRef]
- Suzuki, T.; Kasuya, Y.; Itoh, Y.; Ota, Y.; Zhan, P.; Asamitsu, K.; Nakagawa, H.; Okamoto, T.; Miyata, N. Identification of highly selective and potent histone deacetylase 3 inhibitors using click chemistry-based combinatorial fragment assembly. PLoS ONE 2013, 8, e68669. [Google Scholar] [CrossRef]
- Ruzic, D.; Djokovic, N.; Nikolic, K. Fragment-Based Drug Design of Selective HDAC6 Inhibitors. Methods Mol. Biol. 2021, 2266, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Moi, D.; Citarella, A.; Bonanni, D.; Pinzi, L.; Passarella, D.; Silvani, A.; Giannini, C.; Rastelli, G. Synthesis of potent and selective HDAC6 inhibitors led to unexpected opening of a quinazoline ring. RSC Adv. 2022, 12, 11548–11556. [Google Scholar] [CrossRef] [PubMed]
- Maruca, A.; Moraca, F.; Rocca, R.; Molisani, F.; Alcaro, F.; Gidaro, M.C.; Alcaro, S.; Costa, G.; Ortuso, F. Chemoinformatic Database Building and in Silico Hit-Identification of Potential Multi-Targeting Bioactive Compounds Extracted from Mushroom Species. Molecules 2017, 22, 1571. [Google Scholar] [CrossRef] [PubMed]
- Barbaraci, C.; di Giacomo, V.; Maruca, A.; Patamia, V.; Rocca, R.; Dichiara, M.; Di Rienzo, A.; Cacciatore, I.; Cataldi, A.; Balaha, M.; et al. Discovery of first novel sigma/HDACi dual-ligands with a potent in vitro antiproliferative activity. Bioorg. Chem. 2023, 140, 106794. [Google Scholar] [CrossRef]
- Patel, V.K.; Shirbhate, E.; Tiwari, P.; Kore, R.; Veerasamy, R.; Mishra, A.; Rajak, H. Multi-targeted HDAC Inhibitors as Anticancer Agents: Current Status and Future Prospective. Curr. Med. Chem. 2023, 30, 2762–2795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.X.; Huang, J.; Tian, X.Y.; Liu, Y.H.; Jia, M.Q.; Wang, W.; Jin, C.Y.; Song, J.; Zhang, S.Y. A review of progress in o-aminobenzamide-based HDAC inhibitors with dual targeting capabilities for cancer therapy. Eur. J. Med. Chem. 2023, 259, 115673. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Shou, D.; Huang, R.; Khuc, T.; Dai, S.; Zheng, W.; Klumpp-Thomas, C.; Xia, M. Identification of HDAC Inhibitors Using a Cell-Based HDAC I/II Assay. J. Biomol. Screen. 2016, 21, 643–652. [Google Scholar] [CrossRef]
- Mehdi, O.; Françoise, S.; Sofia, C.L.; Urs, G.; Kevin, Z.; Bernard, S.; Igor, S.; Anabela, C.D.; Dominique, L.; Eric, M.; et al. HDAC gene expression in pancreatic tumor cell lines following treatment with the HDAC inhibitors panobinostat (LBH589) and trichostatine (TSA). Pancreatology 2012, 12, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Weiss, U.; Möller, M.; Husseini, S.A.; Manderscheid, C.; Häusler, J.; Geisslinger, G.; Niederberger, E. Inhibition of HDAC Enzymes Contributes to Differential Expression of Pro-Inflammatory Proteins in the TLR-4 Signaling Cascade. Int. J. Mol. Sci. 2020, 21, 8943. [Google Scholar] [CrossRef]
- Lopez, G.; Song, Y.; Lam, R.; Ruder, D.; Creighton, C.J.; Bid, H.K.; Bill, K.L.; Bolshakov, S.; Zhang, X.; Lev, D.; et al. HDAC Inhibition for the Treatment of Epithelioid Sarcoma: Novel Cross Talk Between Epigenetic Components. Mol. Cancer Res. 2016, 14, 35–43. [Google Scholar] [CrossRef]
- Yu, W.C.; Yeh, T.Y.; Ye, C.H.; Chong, P.C.T.; Ho, Y.H.; So, D.K.; Yap, K.Y.; Peng, G.R.; Shao, C.H.; Jagtap, A.D.; et al. Discovery of HDAC6, HDAC8, and 6/8 Inhibitors and Development of Cell-Based Drug Screening Models for the Treatment of TGF-β-Induced Idiopathic Pulmonary Fibrosis. J. Med. Chem. 2023, 66, 10528–10557. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Altamura, S.; De Francesco, R.; Gallinari, P.; Lahm, A.; Neddermann, P.; Rowley, M.; Serafini, S.; Steinkühler, C. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg. Med. Chem. Lett. 2008, 18, 1814–1819. [Google Scholar] [CrossRef] [PubMed]
- Estiu, G.; West, N.; Mazitschek, R.; Greenberg, E.; Bradner, J.E.; Wiest, O. On the inhibition of histone deacetylase 8. Bioorg. Med. Chem. 2010, 18, 4103–4110. [Google Scholar] [CrossRef]
- Padige, G.; Negmeldin, A.T.; Pflum, M.K. Development of an ELISA-Based HDAC Activity Assay for Characterization of Isoform-Selective Inhibitors. J. Biomol. Screen. 2015, 20, 1277–1285. [Google Scholar] [CrossRef]
Figure 1.
Schematic classification of HDAC family.

Figure 2.
A 3D structure available for human Class I HDAC. HDAC1 in complex with the dimeric ELM2-SANT domain of MTA1 from the NuRD complex (PDB code 4BKX) [44]; HDAC2 in complex with SAHA (Vorinostat) (PDB code 4LXZ) [45]; HDAC3 bound to corepressor and inositol tetraphosphate (PDB code 4A69) [43]; HDAC8 complexed with Trichostatin A (PDB code 1T64) [43].
Figure 2.
A 3D structure available for human Class I HDAC. HDAC1 in complex with the dimeric ELM2-SANT domain of MTA1 from the NuRD complex (PDB code 4BKX) [44]; HDAC2 in complex with SAHA (Vorinostat) (PDB code 4LXZ) [45]; HDAC3 bound to corepressor and inositol tetraphosphate (PDB code 4A69) [43]; HDAC8 complexed with Trichostatin A (PDB code 1T64) [43].

Figure 3.
Dynamics and catalytic mechanisms of zinc ion-dependent HDACs during the deacylation process. The hydrolyzing portion is highlighted in red.
Figure 3.
Dynamics and catalytic mechanisms of zinc ion-dependent HDACs during the deacylation process. The hydrolyzing portion is highlighted in red.
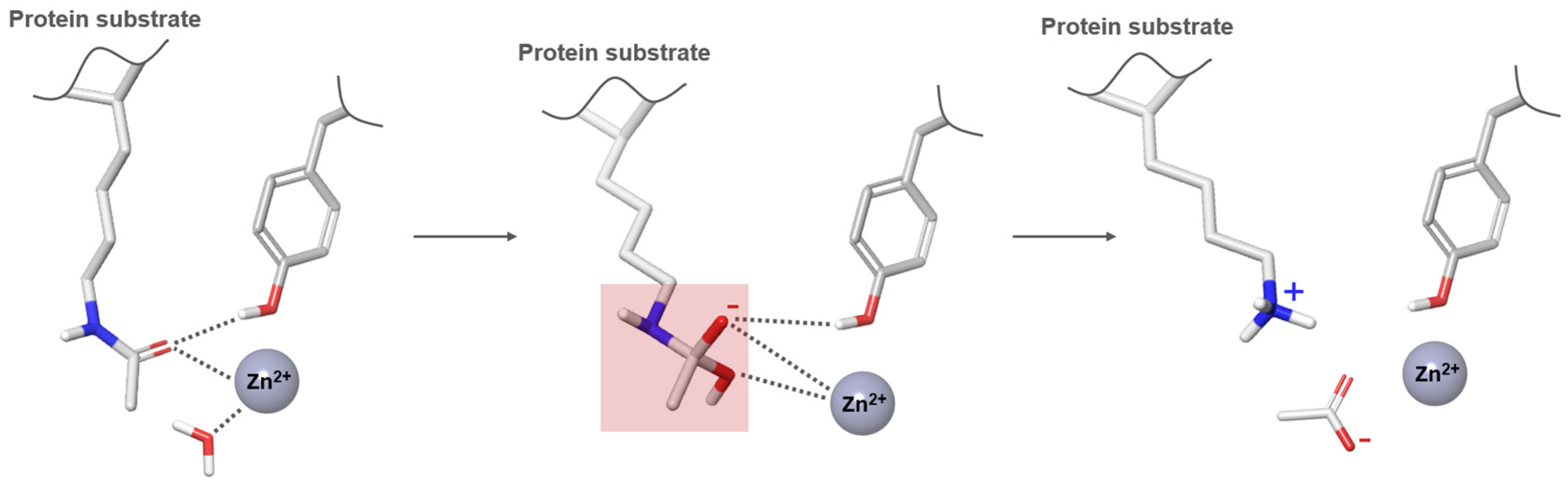
Figure 6.
A 2D structure of the four HDACis (Vorinostat, Panobinostat, Romidepsin and Belinostat) approved by FDA for the treatment of various cancers and of Tucidinostat, the only HDACi approved by CFDA.
Figure 6.
A 2D structure of the four HDACis (Vorinostat, Panobinostat, Romidepsin and Belinostat) approved by FDA for the treatment of various cancers and of Tucidinostat, the only HDACi approved by CFDA.

Figure 7.
A 2D structure of Vorinostat showing the HDACis shared pharmacophore model.

Figure 8.
Schematic diagram of a PROTAC, as an innovative approach to improve selectivity for a specific HDAC isoform.
Figure 8.
Schematic diagram of a PROTAC, as an innovative approach to improve selectivity for a specific HDAC isoform.

Figure 9.
Schematic representation of some ligand-based (LB) approaches for the virtual screening of large databases to discover new HDACis. The features of the ligand-based pharmacophore based on Vorinostat are illustrated as follows: the aromatic ring is represented by an orange ring, the hydrophobic feature is depicted as green spheres, and H-bond acceptors and donors are shown as red and light-blue spheres associated with arrows, respectively. In the scaffold hopping analysis, blue and yellow spheres denote the H-bond donor/acceptor moiety of Vorinostat. Specifically, blue spheres are designated as optional matches.
Figure 9.
Schematic representation of some ligand-based (LB) approaches for the virtual screening of large databases to discover new HDACis. The features of the ligand-based pharmacophore based on Vorinostat are illustrated as follows: the aromatic ring is represented by an orange ring, the hydrophobic feature is depicted as green spheres, and H-bond acceptors and donors are shown as red and light-blue spheres associated with arrows, respectively. In the scaffold hopping analysis, blue and yellow spheres denote the H-bond donor/acceptor moiety of Vorinostat. Specifically, blue spheres are designated as optional matches.

Figure 10.
Schematic representation of some structure-based (SB) approaches for the virtual screening of large databases to discover new HDACis. HDAC8 is represented as violet surface and ribbons, while the main residues of the catalytic site are shown as violet thin tube.
Figure 10.
Schematic representation of some structure-based (SB) approaches for the virtual screening of large databases to discover new HDACis. HDAC8 is represented as violet surface and ribbons, while the main residues of the catalytic site are shown as violet thin tube.
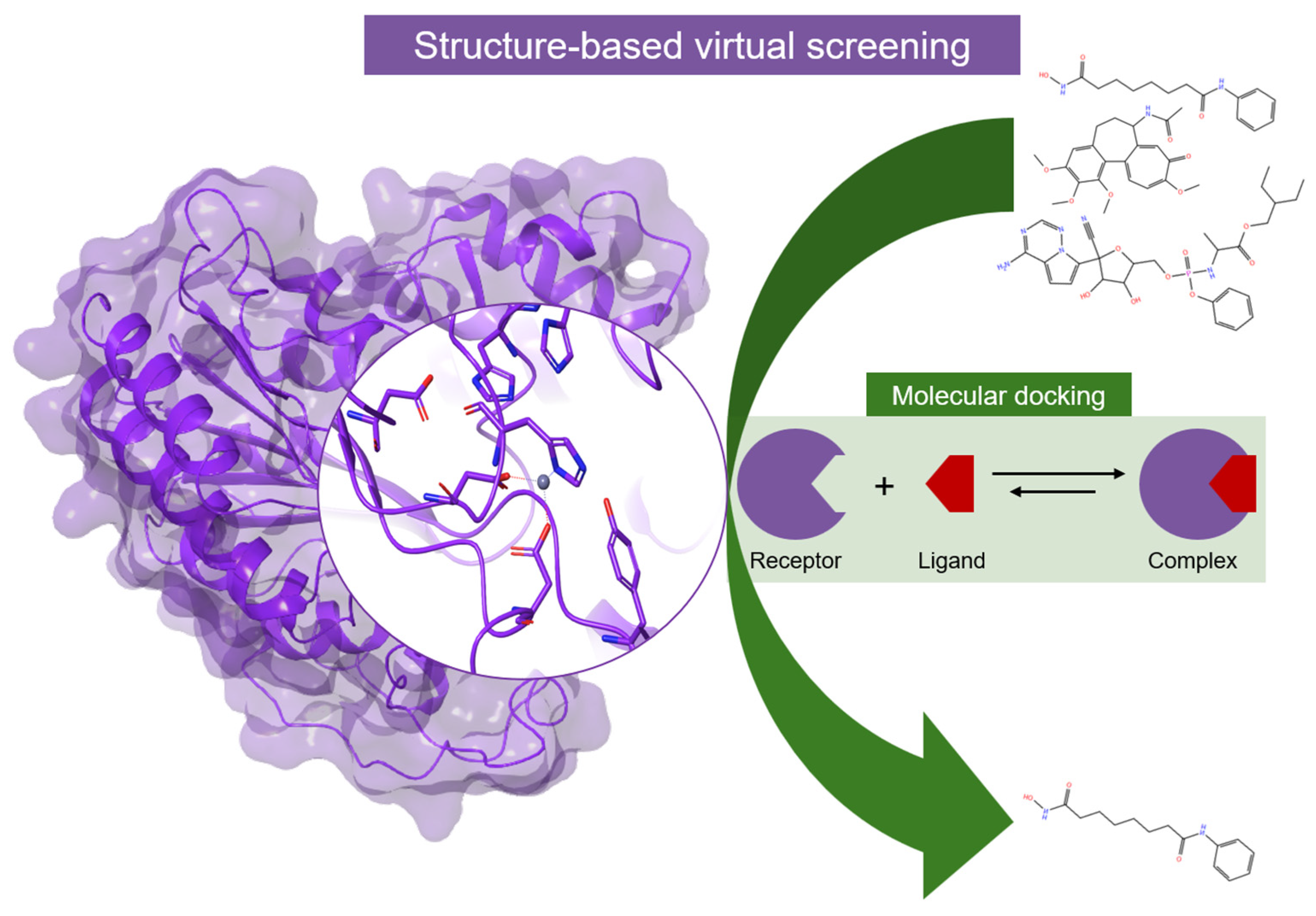
Figure 11.
Schematic representation of ADME prediction, Molecular Dynamics and MM-PBSA/MM-GBSA binding energy calculations. HDAC8 structure is depicted as violet surface and ribbons situated within a water box.
Figure 11.
Schematic representation of ADME prediction, Molecular Dynamics and MM-PBSA/MM-GBSA binding energy calculations. HDAC8 structure is depicted as violet surface and ribbons situated within a water box.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Diseases associated with each HDAC isoform.
| Class I | Zn2+-Dependent | |
|---|---|---|
| Isoform | Disease | |
| HDAC1, HDAC2, HDAC3, HDAC8 | Cancer (prostate, gastric, colorectal, Hodgkin lymphoma, lung, liver, acute lymphoblastic leukemia, breast, neuroblastoma); Neurological diseases (Huntington’s disease, Amyotrophic Lateral Sclerosis, Alzheimer’s Disease); Metabolic diseases (Diabetes, obesity); Cardiovascular diseases | |
| Class II | Zn2+-Dependent | |
| Isoform | Disease | |
| HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, HDAC10 | Cancer (esophageal, colon, Hodgkin lymphoma, lung, acute lymphoblastic leukemia, breast, medulloblastoma); Neurological diseases (Huntington’s disease, Parkinson’s disease, Amyotrophic Lateral Sclerosis, Alzheimer’s Disease); Cardiovascular diseases | |
| Class IV | Zn2+-Dependent | |
| Isoform | Disease | |
| HDAC11 | Cancer (breast, renal, liver); Neurological diseases (Multiple Sclerosis); Cardiovascular diseases | |
| Class III | NAD+-dependent | |
| Isoform | Disease | |
| SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, SIRT7 | Cancer (breast, pancreatic, colon, glioma); Neurological disease (Multiple Sclerosis); Metabolic diseases (Diabetes, obesity); Cardiovascular diseases | |
Table 2.
Description of the intricate role of each HDAC isoform in cancer biology and their distinct expression patterns across diverse tumor types.
Table 2.
Description of the intricate role of each HDAC isoform in cancer biology and their distinct expression patterns across diverse tumor types.
| Class I | Zn2+-Dependent | |
|---|---|---|
| Isoform | Role in Cancer Biology | Expression in Cancer |
| HDAC1 | (−) Apoptosis, (+) proliferation | Overexpressed in prostate (hormone-refractory), gastric A, colorectal, breast B, Hodgkin lymphoma, lung A, and liver A cancer |
| HDAC2 | (−) Apoptosis, (+) proliferation, (+) aneuploidy | Overexpressed in colorectal A, gastric A, prostvate A, Hodgkin lymphoma, acute lymphoblastic leukemia |
| HDAC3 | (−) Differentation, (+) proliferation | Overexpressed in lung, gastric A, breast AB, Hodgkin lymphoma, acute lymphoblastic leukemia Downregulated in liver cancer |
| HDAC8 | (−) Differentation, (+) proliferation | Overexpressed in neuroblastoma |
| Class II | Zn2+-Dependent | |
| Isoform | Role in Cancer Biology | Expression in Cancer |
| HDAC4 | (−) Differentation, (+) Angiogenesis | Overexpressed in esophageal cancer |
| HDAC5 | (−) Differentation, (−) Migration | Overexpressed in medulloblastoma Downregulated in lung, colon cancer and acute myeloid leukaemia |
| HDAC6 | (+) Migration | Overexpressed in breast cancer Downregulated in lung cancer |
| HDAC7 | (+) Angiogenesis | Overexpressed in acute lymphoblastic leukemia Downregulated in lung cancer |
| HDAC9 | (+) Angiogenesis | Overexpressed in medulloblastoma |
| HDAC10 | (+) Angiogenesis | Overexpressed in lung cancer |
| Class IV | Zn2+-Dependent | |
| Isoform | Role in Cancer Biology | Expression in Cancer |
| HDAC11 | Overexpressed in breast, renal and liver cancer | |
(+) Increased activity; (−) Reduced activity; A Independent prognosis indicator; B Associated with enhanced prognosis.
Table 3.
Size (number of amino acids), residues involved in the Zn2+ coordination, cellular distribution, corresponding complex and chromosome location for each HDAC isoform [92,93,94,95,96].
| Class I | Zn2+-Dependent | ||||
|---|---|---|---|---|---|
| Isoform | Size (aa) | Zn Coordination | Cellular Distribution | Complex | Chromosome Location |
| HDAC1 | 482 | His140 His141 Asp176 Asp264 | Nuclear | Sin3, NURD | 1p34 |
| HDAC2 | 488 | His141 His142 Asp177 Asp265 | Nuclear | Sin3, NURD | 6q21 |
| HDAC3 | 428 | His134 His135 Asp170 Asp259 | Nuclear | NCOR1/NCOR2-GPS2-TBL1X | 5q31 |
| HDAC8 | 377 | His142 His143 Asp178 Asp267 | Nuclear | Xq13 | |
| Class II | Zn2+-Dependent | ||||
| Isoform | Size (aa) | Zn Coordination | Cellular Distribution | Complex | Chromosome Location |
| HDAC4 | 1084 | His802 His803 Asp840 Asp934 | Nuclear, cytoplasm | NCOR1/NCOR2 | q37.2 |
| HDAC5 | 1122 | Nuclear, cytoplasm | 17q21 | ||
| HDAC6 | 1215 | H610 H611 Asp649 Asp742 | Nuclear, cytoplasm | Xp11.22–23 | |
| HDAC7 | 952 | His669 His670 Asp707 Asp801 | Nuclear, cytoplasm | NCOR1/NCOR2 | 12q13.1 |
| HDAC9 | 1011 | Nuclear, cytoplasm | p21–p15 | ||
| HDAC10 | 669 | Nuclear, cytoplasm | NCOR2 | 22q13.31 | |
| Class IV | Zn2+-Dependent | ||||
| Isoform | Size (aa) | Zn Coordination | Cellular Distribution | Complex | Chromosome Location |
| HDAC11 | 347 | Nuclear | - | - | |
| Class III (SIRT) | NAD+-Dependent | ||||
| Isoform | Size (aa) | Cellular Distribution | Complex | Chromosome Location | |
| SIRT1 | 747 | Nuclear, cytoplasm | eNoSC | ||
| SIRT2 | 389 | Nuclear, cytoplasm | |||
| SIRT3 | 399 | Mitochondria | FoxO3A | ||
| SIRT4 | 314 | Mitochondria | |||
| SIRT5 | 310 | Nuclear, cytoplasm, mitochondria | 6p23 | ||
| SIRT6 | 355 | Nucleas, endoplasmic reticulum | |||
| SIRT7 | 400 | Nuclear, cytoplasm | 17q25.3 | ||
Table 4.
Omics system technologies used to analyze HDACs and HDACis.
| Omics | Analysis | Detecting |
|---|---|---|
| Chemoproteome | MS, beads MS, MudPIT | Protein/HDACi interaction |
| Epigenome | ChIP-seq, ChIP-qPCR, ChIP-chip, DNase-seq, MNase-seq, ATAC-seq, MBD-seq, RNA-seq, NA-seq, HT-FAIRE | Histone modification and chromatin accessibility |
| Acetylome | Protein modification | |
| Transcriptome | Microarray, miRNA microarray, miRNA-seq, mRNA-seq, splicing-sensitive microarray, TempO-seq, GRO-seq, ChIP-seq | Gene expression |
| Proteome | LC-MS/MS, SILAC, HSMS, MS acetylome | Protein expression |
| Metabolome | MS metabolomics, NMR, LC/GC-MS/MS | Metabolic physiology |
MS (mass spectrometry); MudPIT (Multidimensional protein identification technology); ChIP (Chromatin immunoprecipitation); MNase (micrococcal nuclease); ATAC (assay for transposase-accessible chromatin); MBD (Methyl-CpG Binding Domain); FAIRE (formaldehyde-assisted isolation of regulatory elements); TempO (templated oligo assay); GRO (Global run-on); LC-MS/MS (liquid chromatography–tandem mass spectrometry); SILAC (Stable isotope labeling by amino acids in cell culture); HSMS (High-Resolution Mass Spectrometry); NMR (nuclear magnetic resonance); GC (gas chromatography) [114].
Table 5.
A 2D chemical structure, related disease and selectivity of current HDAC inhibitors divided by chemical class (hydroxamic acids, benzamides, short-chain fatty acids, and cyclic tetrapeptides).
Table 5.
A 2D chemical structure, related disease and selectivity of current HDAC inhibitors divided by chemical class (hydroxamic acids, benzamides, short-chain fatty acids, and cyclic tetrapeptides).
| Class | Inhibitor | 2D Structures | Selectivity | Disease |
|---|---|---|---|---|
| Hydroxamic acids | Vorinostat |  | Pan | Cutaneous T-cell lymphoma (Approved) Alzheimer’s disease (Phase I) |
| Trichostatin A |  | Pan | Preclinical use | |
| Belinostat |  | Pan | Peripheral T-cell lymphoma (Approved) | |
| Panobinostat |  | Pan | Multiple myeloma (Approved) | |
| Givinostat |  | Pan | Relapsed leukemia Multiple myeloma (Phase II) | |
| Resminostat |  | Pan | Hepatocellular carcinoma (Phase I and II) | |
| Abexinostat |  | Pan | B-cell lymphoma (Phase II) | |
| Quisinostat |  | Pan | Multiple myeloma (Phase I) | |
| Rocilinostat |  | II | Multiple myeloma (Phase I) | |
| Pracinostat |  | I, II, IV | Prostate cancer (Phase II) | |
| CHR-3996 |  | I | Advanced/metastatic solid tumors refractory to standard therapy (Phase I) | |
| Benzamides | Entinostat |  | I | Breast cancer, Hodgkin’s lymphoma, non-small cell lung cancer (Phase II and Phase III) |
| Tacedinaline |  | I | Hormone receptor-positive breast cancer, Non-small cell lung cancer and pancreatic cancer (Phase III) | |
| 4SC202 |  | I | Advanced hematological malignancies (Phase I) | |
| Mocetinostat | 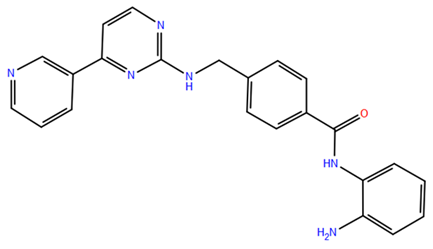 | I, IV | Hodgkin’s lymphoma (Phase II) | |
| Short-chain fatty acids | Valproic acid |  | I, IIa | Epilepsia, bipolar disorders, and migraine (Approved) |
| Butyric acid |  | I, II | Multiple conditions (Phase II) | |
| Phenylbutyric acid |  | I, II | Multiple conditions (Phase I) | |
| Cyclic tetrapeptides | Romidepsin |  | I | Cutaneous T-cell lymphoma (Approved) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Curcio, A.; Rocca, R.; Alcaro, S.; Artese, A. The Histone Deacetylase Family: Structural Features and Application of Combined Computational Methods. Pharmaceuticals 2024, 17, 620. https://doi.org/10.3390/ph17050620
AMA Style
Curcio A, Rocca R, Alcaro S, Artese A. The Histone Deacetylase Family: Structural Features and Application of Combined Computational Methods. Pharmaceuticals. 2024; 17(5):620. https://doi.org/10.3390/ph17050620
Chicago/Turabian StyleCurcio, Antonio, Roberta Rocca, Stefano Alcaro, and Anna Artese. 2024. "The Histone Deacetylase Family: Structural Features and Application of Combined Computational Methods" Pharmaceuticals 17, no. 5: 620. https://doi.org/10.3390/ph17050620
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.