
Molecular Characterization and Therapeutic Opportunities in KRAS Wildtype Pancreatic Ductal Adenocarcinoma
 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Sample Processing and NGS with Mutational Analysis
2.3. RNA Sequencing Analysis
2.4. Fusion Diagram
2.5. Subject Specimens
2.6. Patient-Derived Organoid (PDO) Generation from Biopsies
2.7. PDO Materials
2.8. PDO Drug Studies and Viability Assessment
3. Results
3.1. Subject Population
3.2. Genomic Characterization
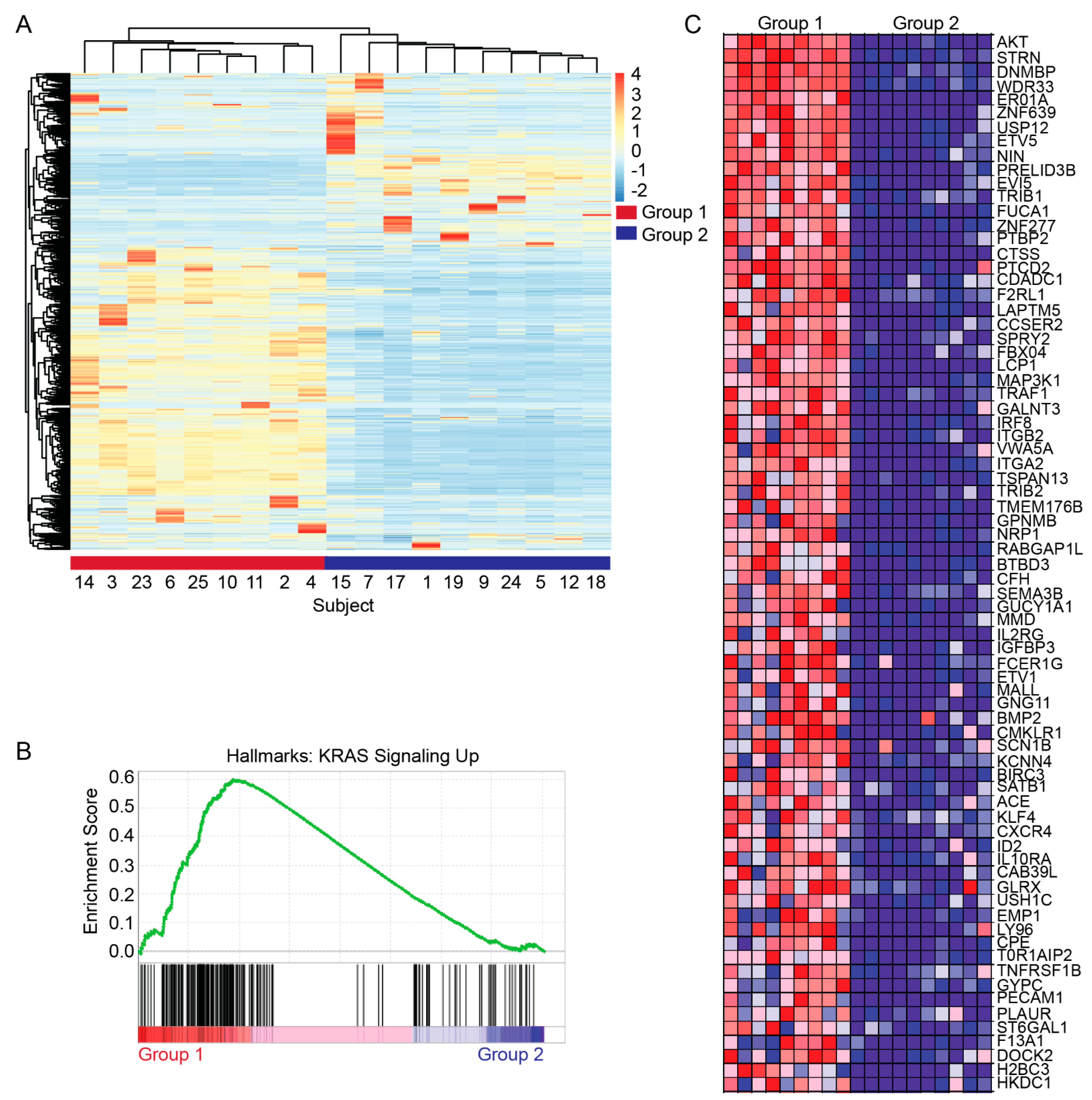
3.3. Transcriptomic Characterization
3.4. Translational Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. New Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Henley, S.J.; Ward, E.M.; Scott, S.; Ma, J.; Anderson, R.N.; Firth, A.U.; Thomas, C.C.; Islami, F.; Weir, H.K.; Lewis, D.R.; et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer 2020, 126, 2225–2249. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Hruban, R.H. Pathology and molecular genetics of pancreatic neoplasms. Cancer J. 2012, 18, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Downward, J. Exploiting the therapeutic implications of KRAS inhibition on tumor immunity. Cancer Cell 2024, 42, 338–357. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A., Jr.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Perkhofer, L.; Gout, J.; Roger, E.; Kude de Almeida, F.; Baptista Simões, C.; Wiesmüller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef]
- Golan, T.; Kindler, H.L.; Park, J.O.; Reni, M.; Macarulla, T.; Hammel, P.; Van Cutsem, E.; Arnold, D.; Hochhauser, D.; McGuinness, D.; et al. Geographic and Ethnic Heterogeneity of Germline BRCA1 or BRCA2 Mutation Prevalence Among Patients with Metastatic Pancreatic Cancer Screened for Entry into the POLO Trial. J. Clin. Oncol. 2020, 38, 1442–1454. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e2244. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Philip, P.A.; Azar, I.; Xiu, J.; Hall, M.J.; Hendifar, A.E.; Lou, E.; Hwang, J.J.; Gong, J.; Feldman, R.; Ellis, M. Molecular Characterization of KRAS Wild-type Tumors in Patients with Pancreatic Adenocarcinoma. Clin. Cancer Res. 2022, 28, 2704–2714. [Google Scholar] [CrossRef]
- Beaubier, N.; Tell, R.; Lau, D.; Parsons, J.R.; Bush, S.; Perera, J.; Sorrells, S.; Baker, T.; Chang, A.; Michuda, J.; et al. Clinical validation of the tempus xT next-generation targeted oncology sequencing assay. Oncotarget 2019, 10, 2384–2396. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Hogenson, T.L.; Xie, H.; Phillips, W.J.; Toruner, M.D.; Li, J.J.; Horn, I.P.; Kennedy, D.J.; Almada, L.L.; Marks, D.L.; Carr, R.M.; et al. Culture media composition influences patient-derived organoid ability to predict therapeutic responses in gastrointestinal cancers. JCI Insight 2022, 7, e158060. [Google Scholar] [CrossRef]
- Menzies, A.M.; Yeh, I.; Botton, T.; Bastian, B.C.; Scolyer, R.A.; Long, G.V. Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment. Cell Melanoma Res. 2015, 28, 607. [Google Scholar] [CrossRef]
- Wang, C.; Teer, J.; Yao, J.; Anadon, C.; Noyes, D.; Landin, A.; Yu, X.; Du, D.; Thompson, Z.; Fang, B. 280 Both tumor intrinsic and extrinsic factors contribute to TIL resistance in lung cancer patients. J. ImmunoTherapy Cancer 2020, 8, A1–A559. [Google Scholar]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Recondo, G.; Che, J.; Janne, P.A.; Awad, M.M. Targeting MET Dysregulation in Cancer. Cancer Discov. 2020, 10, 922–934. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Modica, C.; Tortarolo, D.; Comoglio, P.M.; Basilico, C.; Vigna, E. MET/HGF Co-Targeting in Pancreatic Cancer: A Tool to Provide Insight into the Tumor/Stroma Crosstalk. Int. J. Mol. Sci. 2018, 19, 3920. [Google Scholar] [CrossRef]
- Luchini, C.; Paolino, G.; Mattiolo, P.; Piredda, M.L.; Cavaliere, A.; Gaule, M.; Melisi, D.; Salvia, R.; Malleo, G.; Shin, J.I. KRAS wild-type pancreatic ductal adenocarcinoma: Molecular pathology and therapeutic opportunities. J. Exp. Clin. Cancer Res. 2020, 39, 227. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Jeong, W.J.; Ro, E.J.; Choi, K.Y. Interaction between Wnt/beta-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of beta-catenin and RAS by targeting the Wnt/beta-catenin pathway. NPJ Precis. Oncol. 2018, 2, 5. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef]
- Wagle, M.-C.; Kirouac, D.; Klijn, C.; Liu, B.; Mahajan, S.; Junttila, M.; Moffat, J.; Merchant, M.; Huw, L.; Wongchenko, M. A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis. Oncol. 2018, 2, 7. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab with or Without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Windon, A.L.; Loaiza-Bonilla, A.; Jensen, C.E.; Randall, M.; Morrissette, J.J.D.; Shroff, S.G. A KRAS wild type mutational status confers a survival advantage in pancreatic ductal adenocarcinoma. J. Gastrointest. Oncol. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Yamamoto, H.; Itoh, F.; Nakamura, H.; Fukushima, H.; Sasaki, S.; Perucho, M.; Imai, K. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001, 61, 3139–3144. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Median Age (Years) | 61 (Range 24 to 84) | |||||||
| Male Sex (%) | 68 | |||||||
| Stage IV at Diagnosis (%) | 42 | |||||||
| Development of Metastatic Disease | 15 | |||||||
| Median Follow-up (months) | 23 (Range 0.6 to 142.9) | |||||||
| MSI Status | ||||||||
| High (%) | 2/21 (9.5) | |||||||
| Low (%) | 19/21 (90.5) | |||||||
| TMB Status | ||||||||
| High (>10 mutations/MB) (%) | 2/21 (10.5) | |||||||
| Low (>10 mutations/MB) (%) | 19/21 (89.5) | |||||||
| Treatment | Patient | Sample Number | Baseline Measurement | Best Response | % Change | Best Response | Objective Response Rate | Median Overall Survival in Months |
| FOLFIRINOX | Subject 1 | 1 | 2.5 cm | 3.3 cm | +32% | PD | 25% | 17 |
| Subject 2 | 8 | 2 cm | 1 cm | −50% | PR | |||
| Subject 3 | 10 | 11.1 cm | 3.2 cm | −72% | PR | |||
| Subject 4 | 17 | 3.7 cm | 2.9 cm | −21.6% | SD | |||
| Gemcitabine and nab-paclitaxel | Subject 1 | - | 5.4 cm | 5.2 cm | −3.7% | SD | 0% | 15 |
| Subject 2 | 12 | 2.8 cm | 5 cm | +78% | PD | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desai, A.; Xiao, A.H.; Choi, D.; Toruner, M.D.; Walden, D.; Halfdanarson, T.R.; Alberts, S.; McWilliams, R.R.; Mahipal, A.; Ahn, D.; et al. Molecular Characterization and Therapeutic Opportunities in KRAS Wildtype Pancreatic Ductal Adenocarcinoma. Cancers 2024, 16, 1861. https://doi.org/10.3390/cancers16101861
Desai A, Xiao AH, Choi D, Toruner MD, Walden D, Halfdanarson TR, Alberts S, McWilliams RR, Mahipal A, Ahn D, et al. Molecular Characterization and Therapeutic Opportunities in KRAS Wildtype Pancreatic Ductal Adenocarcinoma. Cancers. 2024; 16(10):1861. https://doi.org/10.3390/cancers16101861
Chicago/Turabian StyleDesai, Aakash, Alexander H. Xiao, Daheui Choi, Merih D. Toruner, Daniel Walden, Thorvardur R. Halfdanarson, Steven Alberts, Robert R. McWilliams, Amit Mahipal, Daniel Ahn, and et al. 2024. "Molecular Characterization and Therapeutic Opportunities in KRAS Wildtype Pancreatic Ductal Adenocarcinoma" Cancers 16, no. 10: 1861. https://doi.org/10.3390/cancers16101861