

Chimeric Cell Therapies as a Novel Approach for Duchenne Muscular Dystrophy (DMD) and Muscle Regeneration

 , ,
, ,  and
and
Abstract
:1. Introduction
2. History of Chimerism and the Development of Hematopoietic Chimeric Cells
2.1. Chimerism Promotes Tolerance Induction
2.2. Bone Marrow-Based Chimeric Cells
2.3. Umbilical Cord Blood-Based Chimeric Cells
3. Chimeric Cells for Treatment of Duchenne Muscular Dystrophy
3.1. Creation of Murine Dystrophin-Expressing Chimeric (DEC) Cells
3.2. Creation of Human Dystrophin-Expressing Chimeric (DEC) Cells
3.3. Dystrophin-Expressing Chimeric (DEC) Cell Therapy in the First in-Human Study
4. Future Potential of Chimeric Cells and Other Stem Cell Therapies in Treatment of Different Muscular and Neuromuscular Disorders
4.1. Regenerative Properties of DEC Cells as the Potential Treatment of Other Rare Muscular Dystrophies
4.2. Immunomodulatory and Anti-Inflammatory Properties of MSCs and Myoblast-Based DEC Cells as the Potential Therapeutic Strategy for Autoimmune Disorders Involving Muscle Degeneration
4.3. Therapeutic Potential of Stem-Cell-Based Therapies and DEC Therapy as a Novel Approach for Metabolic Muscle Disorders
4.4. Unique Properties of Stem Cells and DEC Cells as the Supportive Therapy for Neuromuscular Disorders
4.5. Promising Prospects of Mesenchymal Muscle Stem Cells and DEC Therapy in Addressing Multifactorial Pathophysiology of Sarcopenia
5. Conclusions
Funding
Conflicts of Interest
References
- Bader, P. Documentation of Engraftment and Chimerism after HSCT. In The EBMT Handbook; Carreras, E., Dufour, C., Mohty, M., Kröger, N., Eds.; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Hivelin, M.; Klimczak, A.; Cwykiel, J.; Sonmez, E.; Nasir, S.; Gatherwright, J.; Siemionow, M. Immunomodulatory Effects of Different Cellular Therapies of Bone Marrow Origin on Chimerism Induction and Maintenance across MHC Barriers in a Face Allotransplantation Model. Arch. Immunol. Ther. Exp. 2016, 64, 299–310. [Google Scholar] [CrossRef]
- Silk, A.D.; Gast, C.E.; Davies, P.S.; Fakhari, F.D.; Vanderbeek, G.E.; Mori, M.; Wong, M.H. Fusion between hematopoietic and epithelial cells in adult human intestine. PLoS ONE 2013, 8, e55572. [Google Scholar] [CrossRef]
- Mousavi, S.A.; Javadimoghadam, M.; Ghavamzadeh, A.; Alimoghaddam, K.; Sayarifard, A.; Ghaffari, S.H.; Chahardouli, B.; Basi, A. The Relationship between STR-PCR Chimerism Analysis and Chronic GvHD Following Hematopoietic Stem Cell Transplantation. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 24–29. [Google Scholar]
- Vassilopoulos, G.; Wang, P.R.; Russell, D.W. Transplanted bone marrow regenerates liver by cell fusion. Nature 2003, 422, 901–904. [Google Scholar] [CrossRef]
- Nygren, J.M.; Jovinge, S.; Breitbach, M.; Säwén, P.; Röll, W.; Hescheler, J.; Taneera, J.; Fleischmann, B.K.; Jacobsen, S.E. Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation. Nat. Med. 2004, 10, 494–501. [Google Scholar] [CrossRef]
- Rizvi, A.Z.; Swain, J.R.; Davies, P.S.; Bailey, A.S.; Decker, A.D.; Willenbring, H.; Grompe, M.; Fleming, W.H.; Wong, M.H. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 6321–6325. [Google Scholar] [CrossRef]
- Cwykiel, J.; Madajka-Niemeyer, M.; Siemionow, M. Development of Donor Recipient Chimeric Cells of bone marrow origin as a novel approach for tolerance induction in transplantation. Stem Cell Investig. 2021, 8, 8. [Google Scholar]
- Lee, K.W.; Park, J.B.; Park, H.; Kwon, Y.; Lee, J.S.; Kim, K.S.; Chung, Y.J.; Rju, J.S.; Choi, S.; Kwon, G.Y.; et al. Inducing Transient Mixed Chimerism for Allograft Survival Without Maintenance Immunosuppression With Combined Kidney and Bone Marrow Transplantation: Protocol Optimization. Transplantation 2020, 104, 1472–1482. [Google Scholar] [CrossRef]
- Sykes, M.; Sachs, D.H. Mixed allogeneic chimerism as an approach to transplantation tolerance. Immunol. Today 1988, 9, 23–27. [Google Scholar] [CrossRef]
- Mengrelis, K.; Muckenhuber, M.; Wekerle, T. Chimerism-based Tolerance Induction in Clinical Transplantation: Its Foundations and Mechanisms. Transplantation 2023, 107, 2473–2485. [Google Scholar] [CrossRef]
- Sachs, D.H.; Kawai, T.; Sykes, M. Induction of Tolerance through Mixed Chimerism. Cold Spring Harb. Perspect. Med. 2014, 4, a015529. [Google Scholar]
- Tolar, J. Regenerative Solutions for Inherited Diseases. Clin. Pharmacol. Ther. 2018, 103, 763–766. [Google Scholar]
- Nasir, S.; Bozkurt, M.; Krokowicz, L.; Klimczak, A.; Siemionow, M. Correlation of chimerism with graft size and revascularization in vascularized and nonvascularized skin allografts. Ann. Plast. Surg. 2009, 62, 430–438. [Google Scholar] [CrossRef]
- Ulusal, B.G.; Ulusal, A.E.; Ozmen, S.; Zins, J.E.; Siemionow, M.Z. A new composite facial and scalp transplantation model in rats. Plast. Reconstr. Surg. 2003, 112, 1302–1311. [Google Scholar] [CrossRef]
- Cwykiel, J.; Jundzill, A.; Klimczak, A.; Madajka-Niemeyer, M.; Siemionow, M. Donor Recipient Chimeric Cells Induce Chimerism and Extend Survival of Vascularized Composite Allografts. Arch. Immunol. Ther. Exp. 2021, 69, 13. [Google Scholar] [CrossRef]
- Siemionow, M.; Cwykiel, J.; Heydemann, A.; Garcia-Martinez, J.; Siemionow, K.; Szilagyi, E. Creation of Dystrophin Expressing Chimeric Cells of Myoblast Origin as a Novel Stem Cell Based Therapy for Duchenne Muscular Dystrophy. Stem Cell Rev. Rep. 2018, 14, 189–199. [Google Scholar] [CrossRef]
- Siemionow, M.; Szilagyi, E.; Cwykiel, J.; Domaszewska-Szostek, A.; Heydemann, A.; Garcia-Martinez, J.; Siemionow, K. Transplantation of Dystrophin Expressing Chimeric Human Cells of Myoblast/Mesenchymal Stem Cell Origin Improves Function in Duchenne Muscular Dystrophy Model. Stem Cells Dev. 2021, 30, 190–202. [Google Scholar] [CrossRef]
- Siemionow, M.; Brodowska, S.; Różczka, K.; Roesler, C. Creation of human hematopoietic chimeric cell (HHCC) line as a novel strategy for tolerance induction in transplantation. Stem Cell Investig. 2022, 9, 11. [Google Scholar]
- Siemionow, M.; Cwykiel, J.; Chambily, L.; Gacek, S.; Brodowska, S. Novel Human Umbilical Di-Chimeric (HUDC) cell therapy for transplantation without life-long immunosuppression. Stem Cell Investig. 2023, 10, 16. [Google Scholar] [CrossRef]
- Siemionow, M.; Cwykiel, J.; Heydemann, A.; Garcia, J.; Marchese, E.; Siemionow, K.; Szilagyi, E. Dystrophin Expressing Chimeric (DEC) Human Cells Provide a Potential Therapy for Duchenne Muscular Dystrophy. Stem Cell Rev. Rep. 2018, 14, 370–384. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F. Muscular dystrophies. Lancet 2013, 381, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Sharawat, I.K.; Ramachandran, A.; Panda, P.K.; Elwadhi, A.; Tomar, A. Development and Validation of an Outpatient Clinical Predictive Score for the Diagnosis of Duchenne Muscular Dystrophy/Becker Muscular Dystrophy in Children Aged 2-18 Years. Ann. Indian Acad. Neurol. 2023, 26, 453–460. [Google Scholar] [PubMed]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.J.; Kunkel, L.M. The Pathogenesis and Therapy of Muscular Dystrophies. Annu. Rev. Genom. Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- Mercuri, E.; Pane, M.; Cicala, G.; Brogna, C.; Ciafaloni, E. Detecting early signs in Duchenne muscular dystrophy: Comprehensive review and diagnostic implications. Front. Pediatr. 2023, 11, 1276144. [Google Scholar] [CrossRef]
- Van Ruiten, H.J.; Marini Bettolo, C.; Cheetham, T.; Eagle, M.; Lochmuller, H.; Straub, V.; Bushby, K.; Guglieri, M. Why are some patients with Duchenne muscular dystrophy dying young: An analysis of causes of death in North East England. Eur. J. Paediatr. Neurol. 2016, 20, 904–909. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; de Kock, S.; Butt, T.; Jain, M.; Kleijen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef]
- Sienkiewicz, D.; Kulak, W.; Okurowska-Zawada, B.; Paszko-Patej, G.; Kawnik, K. Duchenne muscular dystrophy: Current cell therapies. Ther. Adv. Neurol. Disord. 2015, 8, 166–177. [Google Scholar] [CrossRef]
- Skuk, D.; Tremblay, J.P. Cell therapy in muscular dystrophies: Many promises in mice and dogs, few facts in patients. Expert Opin. Biol. Ther. 2015, 15, 1307–1319. [Google Scholar] [CrossRef]
- Sitzia, C.; Farini, A.; Jardim, L.; Razini, P.; Belicchi, M.; Cassinelli, L.; Villa, C.; Erratico, S.; Parolini, D.; Bella, P.; et al. Adaptive Immune Response Impairs the Efficacy of Autologous Transplantation of Engineered Stem Cells in Dystrophic Dogs. Mol. Ther. 2016, 24, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, Y.; Li, Y.; Cao, J.; Zhang, H.; Chen, M.; Wang, L.; Zhang, C. Long-term engraftment of myogenic progenitors from adipose-derived stem cells and muscle regeneration in dystrophic mice. Hum. Mol. Genet. 2015, 24, 6029–6040. [Google Scholar] [CrossRef]
- Meregalli, N.; Farini, A.; Belicchi, M.; Parolini, D.; Cassinelli, L.; Razini, P.; Sitizia, C.; Torrente, Y. Perspectives of stem cell therapy in Duchenne muscular dystrophy. FEBS J. 2013, 280, 4251–4262. [Google Scholar] [CrossRef] [PubMed]
- Kharfan-Dabaja, M.A.; Kumar, A.; Ayala, E.; Aljurf, M.; Nishihori, T.; Marsh, R.; Burroughs, L.M.; Majhail, N.; Al-Homsi, A.S.; Al-Kadhimi, Z.S.; et al. Standardizing Definitions of Hematopoietic Recovery, Graft Rejection, Graft Failure, Poor Graft Function, and Donor Chimerism in Allogeneic Hematopoietic Cell Transplantation: A Report on Behalf of the American Society for Transplantation and Cellular Therapy. Transpl. Cell Ther. 2021, 27, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Yunis, E.J.; Zuniga, J.; Romero, V.; Yunis, E.J. Chimerism and tetragametic chimerism in humans: Implications in autoimmunity, allorecognition and tolerance. Immunol. Res. 2007, 38, 213–236. [Google Scholar] [CrossRef] [PubMed]
- Kinder, J.M.; Stelzer, I.A.; Arck, P.C.; Way, S.S. Immunological implications of pregnancy-induced microchimerism. Nat. Rev. Immunol. 2017, 17, 483–494. [Google Scholar] [PubMed]
- Madan, K. Natural human chimeras: A review. Eur. J. Med. Genet. 2020, 63, 103971. [Google Scholar] [CrossRef] [PubMed]
- Billingham, R.E.; Brent, L.; Medawar, P.B. Actively acquired tolerance of foreign cells. Nature 1953, 172, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Billingham, R.E.; Bren, L. Quantitative studies on tissue transplantation immunity IV. Induction of tolerance in newborn mice and studies on the phenomenon of runt disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1959, 242, 439–477. [Google Scholar] [CrossRef]
- Ildstad, S.T.; Sachs, D.H. Reconstitution with syngeneic plus allogeneic or xenogeneic bone marrow leads to specific acceptance of allografts or xenografts. Nature 1984, 307, 168–170. [Google Scholar] [CrossRef]
- Sachs, D.H.; Sykes, M.; Kawai, T.; Cosimi, A.B. Immuno-intervention for the induction of transplantation tolerance through mixed chimerism. Semin. Immunol. 2011, 23, 165–173. [Google Scholar] [PubMed]
- Hartner, W.C.; De Fazio, S.R.; Maki, T.; Markees, T.G.; Monaco, A.P.; Gozzo, J.J. Prolongation of renal allograft survival in antilymphocyte-serum-treated dogs by postoperative injection of density-gradient-fractionated donor bone marrow. Transplantation 1986, 42, 593–597. [Google Scholar] [CrossRef]
- Thomas, F.T.; Carver, F.M.; Foil, M.B.; Pryor, W.H.; Larkin, E.W.; Hall, W.R.; Haisch, C.E.; Thomas, J.M. Long-term incompatible kidney survival in outbred higher primates without chronic immunosuppression. Ann. Surg. 1983, 198, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Clark, A.W.; Wood, M.L.; Sahyoun, A.I.; Codish, S.D.; Brown, R.W. Possible active enhancement of a human cadaver renal allograft with antilymphocyte serum (ALS) and donor bone marrow: Case report of an initial attempt. Surgery 1976, 79, 384–392. [Google Scholar] [PubMed]
- Calne, R.Y.; Williams, R. Orthotopic liver transplantation: The first 60 patients. Br. Med. J. 1977, 1, 471–476. [Google Scholar] [CrossRef]
- Scandling, J.D.; Busque, S.; Dejbakhsh-Jones, S.; Benike, C.; Millan, M.T.; Shizuru, J.A.; Hoppe, R.T.; Lowsky, R.; Engleman, E.G.; Strober, S. Tolerance and chimerism after renal and hematopoietic-cell transplantation. N. Engl. J. Med. 2008, 358, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Scandling, J.D.; Busque, S.; Shizuru, J.A.; Engleman, E.G.; Strober, S. Induced immune tolerance for kidney transplantation. N. Engl. J. Med. 2011, 365, 1359–1360. [Google Scholar] [CrossRef] [PubMed]
- Starzl, T.E.; Demetris, A.J.; Trucco, M.; Zeevi, A.; Ramos, H.; Terasaki, P.; Rudert, W.A.; Kocova, M.; Ricordi, C.; Ildstad, S.; et al. Chimerism and donor-specific nonreactivity 27 to 29 years after kidney allotransplantation. Transplantation 1993, 55, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Demetris, A.J.; Murase, N.; Rao, A.S.; Fung, J.J.; Starzl, T.E. The dichotomous functions of passenger leukocytes in solid-organ transplantation. Adv. Nephrol. Necker Hosp. 1995, 24, 341–354. [Google Scholar]
- Cwykiel, J.; Siemionow, M.Z. In vivo chimera model: Creation of primary and secondary chimera. In Plastic and Reconstructive Surgery: Experimental Models and Research Designs; Springer: London, UK, 2015; Volume 581, p. 591. [Google Scholar] [CrossRef]
- Eisenberg, L.M.; Eisenberg, C.A. Stem cell plasticity, cell fusion, and transdifferentiation. Birth Defects Res. C Embryo Today 2003, 69, 209–218. [Google Scholar] [CrossRef]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Immunol. 2003, 4, 815. [Google Scholar] [CrossRef] [PubMed]
- LeMaoult, J.; Caumartin, J.; Carosella, E.D. Exchanges of membrane patches (trogocytosis) split theoretical and actual functions of immune cells. Hum. Immunol. 2007, 68, 240–243. [Google Scholar] [CrossRef] [PubMed]
- LeMaoult, J.; Caumartin, J.; Daouya, M.; Favier, B.; Le Rond, S.; Gonzalez, A.; Carosella, E.D. Immune regulation by pretenders: Cell-to-cell transfers of HLA-G make effector T cells act as regulatory cells. Blood 2007, 109, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Ford McIntyre, M.S.; Young, K.J.; Gao, J.; Joe, B.; Zhang, L. Cutting edge: In vivo trogocytosis as a mechanism of double negative regulatory T cell-mediated antigen-specific suppression. J. Immunol. 2008, 181, 2271–2275. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.H.; Olson, E.N. Unveiling the mechanisms of cell-cell fusion. Science 2005, 308, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Schwann, T. Mikroskopische Untersuchungen uber die Ubereinstimmung in der Struktur und dem Wachsten der Thiere und Pflanzen; Saunderschen Buchhandlung: Berlin, Germany, 1839. [Google Scholar]
- Barski, G.; Sorieul, S.; Cornefert, F. “Hybrid” type cells in combined cultures of two different mammalian cell strains. J. Natl. Cancer Inst. 1961, 26, 1269–1291. [Google Scholar] [PubMed]
- Terada, N.; Hamazaki, T.; Oka, M.; Hoki, M.; Mastalerz, D.M.; Nakano, Y.; Meyer, E.M.; Morel, L.; Petersen, B.E.; Scott, E.W. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 2002, 416, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Ying, Q.L.; Nichols, J.; Evans, E.P.; Smith, A.G. Changing potency by spontaneous fusion. Nature 2002, 416, 545–548. [Google Scholar] [CrossRef]
- Mitra, S.; Tomar, P.C. Hybridoma technology; advancements, clinical significance, and future aspects. J. Genet. Eng. Biotechnol. 2021, 19, 159. [Google Scholar]
- LaBarge, M.A.; Blau, H.M. Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell 2002, 111, 589–601. [Google Scholar] [CrossRef]
- Wang, X.; Willenbring, H.; Akkari, Y.; Torimaru, Y.; Foster, M.; Al-Dhalimy, M.; Lagasse, E.; Finegold, M.; Olson, S.; Grompe, M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 2003, 422, 897–901. [Google Scholar] [CrossRef]
- Camargo, F.D.; Finegold, M.; Goodell, M.A. Hematopoietic myelomonocytic cells are the major source of hepatocyte fusion partners. J. Clin. Investig. 2004, 113, 1266–1270. [Google Scholar] [CrossRef]
- Willenbring, H.; Bailey, A.S.; Foster, M.; Akkari, Y.; Dorrell, C.; Olson, S.; Finegold, M.; Fleming, W.H.; Grompe, M. Myelomonocytic cells are sufficient for therapeutic cell fusion in liver. Nat. Med. 2004, 10, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dolado, M.; Pardal, R.; Garcia-Verdugo, J.M.; Fike, J.R.; Lee, H.O.; Pfeffer, K.; Lois, C.; Morrison, S.J.; Alvarez-Buylla, A. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature 2003, 425, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.; Dazzi, F. Bone Marrow Transplantation 1957–2019. Front. Immunol. 2019, 10, 1246. [Google Scholar] [CrossRef] [PubMed]
- Berglund, S.; Magalhaes, I.; Gaballa, A.; Vanherberghen, B.; Uhlin, M. Advances in umbilical cord blood cell therapy: The present and the future. Expert Opin. Biol. Ther. 2017, 17, 691–699. [Google Scholar] [CrossRef]
- Orlando, N.; Pellegrino, C.; Valentini, C.G.; Bianchi, M.; Barbagallo, O.; Sparnacci, S.; Forni, F.; Fontana, T.M.; Teofili, L. Umbilical cord blood: Current uses for transfusion and regenerative medicine. Transfus. Apher. Sci. 2020, 59, 102952. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Tang, B.; Sun, Z. Umbilical cord blood transplantation: Still growing and improving. Stem Cells Transl. Med. 2021, 10, S62–S74. [Google Scholar] [CrossRef] [PubMed]
- Siemionow, M.; Cwykiel, J.; Brodowska, S.; Chambily, L. Human Multi-Chimeric Cell (HMCC) Therapy as a Novel Approach for Tolerance Induction in Transplantation. Stem Cell Rev. Rep. 2023, 19, 2741–2755. [Google Scholar] [CrossRef]
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Soslow, J.H.; Xu, M.; Slaughter, J.C.; Crum, K.; Kaslow, J.A.; George-Durrett, K.; Raucci, F.J., Jr.; Wilkinson, J.D.; Cripe, L.H.; Hor, K.N.; et al. Cardiovascular Measures of All-Cause Mortality in Duchenne Muscular Dystrophy. Circ. Heart Fail. 2023, 16, e010040. [Google Scholar] [CrossRef] [PubMed]
- Kieny, P.; Chollet, S.; Delalande, P.; Le Fort, M.; Magot, A.; Pereon, Y.; Perrouin Verbe, B. Evolution of life expectancy of patients with Duchenne muscular dystrophy at AFM Yolaine de Kepper centre between 1981 and 2011. Ann. Phys. Rehabil. Med. 2013, 56, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Clemens, P.R.; Hoffman, E.P. Exon-Skipping in Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, S343–S358. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.; Conner, H.; Groneman, M.; Blavo, C.; Parmar, M.S. Duchenne muscular dystrophy: Current treatment and emerging exon skipping and gene therapy approach. Eur. J. Pharmacol. 2023, 947, 175675. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Liu, J.; Janssen, J.M.; Chen, X.; Gonçalves, M.A.F.V. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci. Rep. 2016, 6, 37051. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Happi Mbakam, C.; Lamothe, G.; Tremblay, G.; Tremblay, J.P. CRISPR-Cas9 Gene Therapy for Duchenne Muscular Dystrophy. Neurotherapeutics 2022, 19, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Kornegay, J.N.; Li, J.; Bogan, J.R.; Bogan, D.J.; Chen, C.; Zheng, H.; Wang, B.; Qiao, C.; Howard, J.F., Jr.; Xiao, X. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol. Ther. 2010, 18, 1501–1508. [Google Scholar] [CrossRef]
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes 2020, 11, 837. [Google Scholar] [CrossRef]
- Bajek, A.; Porowinska, D.; Kloskowski, T.; Brzoska, E.; Ciemerych, M.A.; Drewa, T. Cell therapy in Duchenne muscular dystrophy treatment: Clinical trials overview. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Previtali, S.C.; Napolitano, S.; Cicalese, M.P.; Tedesco, F.S.; Nicastro, F.; Noviello, M.; Roostalu, U.; Natali Sora, M.G.; Scarlato, M.; et al. Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol. Med. 2015, 7, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Govoni, A.; Magri, F.; Brajkovic, S.; Zanetta, C.; Faravelli, I.; Corti, S.; Bresolin, N.; Comi, G.P. Ongoing therapeutic trials and outcome measures for Duchenne muscular dystrophy. Cell. Mol. Life Sci. 2013, 70, 4585–4602. [Google Scholar] [CrossRef] [PubMed]
- Rajput, B.S.; Chakrabarti, S.K.; Dongare, V.S.; Ramirez, C.M.; Deb, K.D. Human Umbilical Cord Mesenchymal Stem Cells in the Treatment of Duchenne Muscular Dystrophy: Safety and Feasibility Study in India. J. Stem Cells 2015, 10, 141–156. [Google Scholar] [PubMed]
- Torrente, Y.; Belicchi, M.; Marchesi, C.; D’antona, G.; Cogiamanian, F.; Pisati, F.; Gavina, M.; Giordano, R.; Tonlorenzi, R.; Fagiolari, G.; et al. Autologous Transplantation of Muscle-Derived CD133+ Stem Cells in Duchenne Muscle Patients. Cell Transplant. 2007, 16, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Muir, L.A.; Murry, C.E.; Chamberlain, J.S. Prosurvival Factors Improve Functional Engraftment of Myogenically Converted Dermal Cells into Dystrophic Skeletal Muscle. Stem Cells Dev. 2016, 25, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Sato, M.; Kuwahara, T.; Ebata, T.; Tabata, Y.; Sakurai, H. Transplantation of human iPSC-derived muscle stem cells in the diaphragm of Duchenne muscular dystrophy model mice. PLoS ONE 2022, 17, e0266391. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Serra, C.; Lee, G.; Wagner, K.R. Stem cell-based therapies for Duchenne muscular dystrophy. Exp. Neurol. 2020, 323, 113086. [Google Scholar] [CrossRef]
- Siemionow, M.; Langa, P.; Harasymczuk, M.; Cwykiel, J.; Sielewicz, M.; Smieszek, J.; Heydemann, A. Human dystrophin expressing chimeric (DEC) cell therapy ameliorates cardiac, respiratory, and skeletal muscle’s function in Duchenne muscular dystrophy. Stem Cells Transl. Med. 2021, 10, 1406–1418. [Google Scholar] [CrossRef]
- Siemionow, M.; Langa, P.; Brodowska, S.; Kozlowska, K.; Zalants, K.; Budzynska, K.; Heydemann, A. Long-Term Protective Effect of Human Dystrophin Expressing Chimeric (DEC) Cell Therapy on Amelioration of Function of Cardiac, Respiratory and Skeletal Muscles in Duchenne Muscular Dystrophy. Stem Cell Rev. Rep. 2022, 18, 2872–2892. [Google Scholar]
- Malik, M.; Siemionow, M.; Cwykiel, J.; Heydemann, A.; Garcia-Martinez, J.; Siemionow, K.; Szilagyi, E. Intraosseous transplant of dystrophin expressing chimeric (DEC) cells improves skeletal muscle function in mdx mouse model of Duchenne muscular dystrophy. Postepy Kardiol. Interwencyjnej 2022, 18, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Siemionow, M.; Malik, M.; Langa, P.; Cwykiel, J.; Brodowska, S.; Heydemann, A. Cardiac Protection after Systemic Transplant of Dystrophin Expressing Chimeric (DEC) Cells to the mdx Mouse Model of Duchenne Muscular Dystrophy. Stem Cell Rev. Rep. 2019, 15, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Siemionow, M.; Brodowska, S.; Langa, P.; Zalants, K.; Kozlowska, K.; Grau-Kazmierczak, W.; Heydemann, A. Long-Term Biodistribution and Safety of Human Dystrophin Expressing Chimeric Cell Therapy After Systemic-Intraosseous Administration to Duchenne Muscular Dystrophy Model. Arch. Immunol. Ther. Exp. 2022, 70, 20. [Google Scholar] [CrossRef] [PubMed]
- Siemionow, M.; Budzynska, K.; Zalants, K.; Langa, P.; Brodowska, S.; Siemionow, K.; Heydemann, A. Amelioration of Morphological Pathology in Cardiac, Respiratory, and Skeletal Muscles Following Intraosseous Administration of Human Dystrophin Expressing Chimeric (DEC) Cells in Duchenne Muscular Dystrophy Model. Biomedicines 2024, 12, 586. [Google Scholar] [CrossRef] [PubMed]
- Heydemann, A.; Bieganski, G.; Wachowiak, J.; Czarnota, J.; Niezgoda, A.; Siemionow, K.; Ziemiecka, A.; Sikorska, M.H.; Bozyk, K.; Tullius, S.G.; et al. Dystrophin Expressing Chimeric (DEC) Cell Therapy for Duchenne Muscular Dystrophy: A First-in-Human Study with Minimum 6 Months Follow-up. Stem Cell Rev. Rep. 2023, 19, 1340–1359. [Google Scholar] [CrossRef] [PubMed]
- Siemionow, M.; Biegański, G.; Niezgoda, A.; Wachowiak, J.; Czarnota, J.; Siemionow, K.; Ziemiecka, A.; Sikorska, M.H.; Bozyk, K.; Heydemann, A. Safety and Efficacy of DT-DEC01 Therapy in Duchenne Muscular Dystrophy Patients: A 12-Month Follow-Up Study After Systemic Intraosseous Administration. Stem Cell Rev. Rep. 2023, 19, 2724–2740. [Google Scholar] [CrossRef] [PubMed]
- Niezgoda, A.; Biegański, G.; Wachowiak, J.; Czarnota, J.; Siemionow, K.; Heydemann, A.; Ziemiecka, A.; Sikorska, M.H.; Bozyk, K.; Siemionow, M. Assessment of Motor Unit Potentials Duration as the Biomarker of DT-DEC01 Cell Therapy Efficacy in Duchenne Muscular Dystrophy Patients up to 12 Months After Systemic-Intraosseous Administration. Arch. Immunol. Ther. Exp. 2023, 71, 24. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Liu, G.; Halim, A.; Ju, Y.; Luo, Q.; Song, A.G. Mesenchymal Stem Cell Migration and Tissue Repair. Cells 2019, 8, 784. [Google Scholar] [CrossRef]
- Mazo, I.B.; Massber, S.; von Andrian, U.H. Hematopoietic stem and progenitor cell trafficking. Trends Immunol. 2011, 32, 493–503. [Google Scholar] [CrossRef]
- Straub, V.; Guglieri, M. An update on Becker muscular dystrophy. Curr. Opin. Neurol. 2023, 36, 450–454. [Google Scholar] [CrossRef]
- Nicolas, A.; Raguénès-Nicol, C.; Ben Yaou, R.; Ameziane-Le Hir, S.; Chéron, A.; Vié, V.; Claustres, M.; Leturcq, F.; Delalande, O.; Hubert, J.F.; et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet. 2015, 24, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Mul, K. Facioscapulohumeral Muscular Dystrophy. Continuum 2022, 28, 1735–1751. [Google Scholar] [CrossRef] [PubMed]
- Taheri, F.; Taghizadeh, E.; Pour, M.J.R.; Rostami, D.; Renani, P.G.; Rastgar-Moghadam, A.; Hayat, S.M.G. Limb-girdle Muscular Dystrophy and Therapy: Insights into Cell and Gene-based Approaches. Curr. Gene Ther. 2020, 19, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Falsaperla, R.; Praticò, A.D.; Ruggieri, M.; Parano, E.; Rizzo, R.; Corsello, G.; Vitaliti, G.; Pavone, P. Congenital muscular dystrophy: From muscle to brain. Ital. J. Pediatr. 2016, 42, 78. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, I.E.; Fujimoto, M.; Vencovsky, J.; Aggarwal, R.; Holmqvist, M.; Christopher-Stine, L.; Mammen, A.L.; Miller, F.W. Idiopathic inflammatory myopathies. Nat. Rev. Dis. Primers 2021, 7, 86. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Hohlfeld, R. Polymyositis and dermatomyositis. Lancet 2003, 362, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, K.; Lundberg, I.E. Polymyositis and dermatomyositis: Pathophysiology. Rheum. Dis. Clin. N. Am. 2011, 37, 159–171, v. [Google Scholar] [CrossRef] [PubMed]
- Findlay, A.R.; Goyal, N.A.; Mozaffar, T. An overview of polymyositis and dermatomyositis. Muscle Nerve 2015, 51, 638–656. [Google Scholar] [CrossRef] [PubMed]
- Marvi, U.; Chung, L.; Fiorentino, D.F. Clinical presentation and evaluation of dermatomyositis. Indian J. Dermatol. 2012, 57, 375–381. [Google Scholar] [CrossRef]
- Connolly, A.; Gordon, P.A.; Hannah, J.; Creamer, D. The chameleon rash: A review of the polyphenotypic dermatoses of dermatomyositis. Clin. Exp. Dermatol. 2021, 46, 1016–1022. [Google Scholar] [CrossRef]
- Marie, I.; Mouthon, L. Therapy of polymyositis and dermatomyositis. Autoimmun. Rev. 2011, 11, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Kohsaka, H. Current diagnosis and treatment of polymyositis and dermatomyositis. Mod. Rheumatol. 2018, 28, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Tyndall, A.; van Laar, J.M. Stem cell transplantation and mesenchymal cells to treat autoimmune diseases. Presse Med. 2016, 45, e159–e169. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, H.; Cao, M.; Tang, Y.; Liang, J.; Feng, X.; Wang, H.; Hua, B.; Liu, B.; Sun, L. Efficacy of allogeneic mesenchymal stem cell transplantation in patients with drug-resistant polymyositis and dermatomyositis. Ann. Rheum. Dis. 2011, 70, 1285–1288. [Google Scholar] [CrossRef] [PubMed]
- Kok, K.; Zwiers, K.C.; Boot, R.G.; Overkleeft, H.S.; Aerts, J.M.F.G.; Artola, M. Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions. Biomolecules 2021, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Politei, J.M.; Bouhassira, D.; Germain, D.P.; Goizet, C.; Guerrero-Sola, A.; Hilz, M.J.; Hutton, E.J.; Karaa, A.; Liguori, R.; Üçeyler, N.; et al. Pain in Fabry Disease: Practical Recommendations for Diagnosis and Treatment. CNS Neurosci. Ther. 2016, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Padua, L.; Pazzaglia, C.; Morgante, E.; Centurion, C.; Antuzzi, D.; Russo, M.A.; Frustaci, A. Cardiac and skeletal myopathy in Fabry disease: A clinicopathologic correlative study. Hum. Pathol. 2012, 43, 1444–1452. [Google Scholar]
- Van der Tol, L.; Cassiman, D.; Houge, G.; Janssen, M.C.; Lachmann, R.H.; Linthorst, G.E.; Ramaswami, U.; Sommer, C.; Tondel, C.; West, M.L.; et al. Uncertain diagnosis of fabry disease in patients with neuropathic pain, angiokeratoma or cornea verticillata: Consensus on the approach to diagnosis and follow-up. JIMD Rep. 2014, 17, 83–90. [Google Scholar] [CrossRef]
- Perretta, F.; Jaurretche, S. Fabry Disease: Switch from Enzyme Replacement Therapy to Oral Chaperone Migalastat: What Do We Know Today? Healthcare 2023, 11, 449. [Google Scholar] [CrossRef]
- Taverna, S.; Cammarata, G.; Colomba, P.; Sciarrino, S.; Zizzo, C.; Francofonte, D.; Zora, M.; Scalia, S.; Brando, C.; Curto, A.L.; et al. Pompe disease: Pathogenesis, molecular genetics and diagnosis. Aging 2020, 12, 15856–15874. [Google Scholar] [CrossRef]
- Case, L.E.; Kishnani, P.S. Physical therapy management of Pompe disease. Genet. Med. 2006, 8, 318–327. [Google Scholar] [CrossRef] [PubMed]
- MENA Pompe Working Group; Al Jasmi, F.; Al Jumah, M.; Alqarni, F.; Al-Sanna’a, N.; Al-Sharif, F.; Bohlega, S.; Cupler, E.J.; Fathalla, W.; Hamdan, M.A.; et al. Diagnosis and treatment of late-onset Pompe disease in the Middle East and North Africa region: Consensus recommendations from an expert group. BMC Neurol. 2015, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Toscano, A.; Rodolico, C.; Musumeci, O. Multisystem late onset Pompe disease (LOPD): An update on clinical aspects. Ann. Transl. Med. 2019, 7, 284. [Google Scholar]
- Khan, A.; Barber, D.L.; Huang, J.; Rupar, C.A.; Rip, J.W.; Auray-Blais, C.; Boutin, M.; O’Hoski, P.; Gargulak, K.; McKillop, W.M.; et al. Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 2021, 12, 1178. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Fukuda, T.; Gilbert, A.L.; de Jong, D.; Thurberg, B.L.; Mattaliano, R.J.; Meikle, P.; Hopwood, J.J.; Nagashima, K.; Nagaraju, K.; et al. Replacing acid alpha-glucosidase in Pompe disease: Recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol. Ther. 2005, 11, 48–56. [Google Scholar] [CrossRef] [PubMed]
- van Til, N.P.; Stok, M.; Aerts Kaya, F.S.; de Waard, M.C.; Farahbakhshian, E.; Visser, T.P.; Kroos, M.A.; Jacobs, E.H.; Willart, M.A.; van der Wegen, P.; et al. Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood 2010, 115, 5329–5337. [Google Scholar] [CrossRef] [PubMed]
- Unnisa, Z.; Yoon, J.K.; Schindler, J.W.; Mason, C.; van Til, N.P. Gene Therapy Developments for Pompe Disease. Biomedicines 2022, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Domm, J.M.; Wootton, S.K.; Medin, J.A.; West, M.L. Gene therapy for Fabry disease: Progress, challenges, and outlooks on gene-editing. Mol. Genet. Metab. 2021, 134, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Santalla, A.; Nogales-Gadea, G.; Ørtenblad, N.; Brull, A.; de Luna, N.; Pinós, T.; Lucia, A. McArdle disease: A unique study model in sports medicine. Sports Med. 2014, 44, 1531–1544. [Google Scholar] [CrossRef]
- Llavero, F.; Arrazola Sastre, A.; Luque Montoro, M.; Gálvez, P.; Lacerda, H.M.; Parada, L.A.; Zugaza, J.L. McArdle Disease: New Insights into Its Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2019, 20, 5919. [Google Scholar] [CrossRef]
- Teles, J.S.; Ramos, C.T.; Almeida, B.M.; Sousa, A.V. A Case Report of McArdle Disease Diagnosed Following Statin-Induced Myositis. Cureus 2023, 15, e44701. [Google Scholar] [CrossRef]
- Quinlivan, R.; Beynon, R.J. Pharmacological and nutritional treatment for McArdle’s disease (Glycogen Storage Disease type V). Cochrane Database Syst. Rev. 2004, 11, CD003458. [Google Scholar] [CrossRef]
- D’souza, R.S.; Levandowski, C.; Slavov, D.; Graw, S.L.; Allen, L.A.; Adler, E.; Mestroni, L.; Taylor, M.R. Danon disease: Clinical features, evaluation, and management. Circ. Heart Fail. 2014, 7, 843–849. [Google Scholar] [CrossRef]
- Shalata, A.; Bar-Shai, M.; Hadid, Y.; Mahroum, M.; Mintz, H.; Shalata, Z.E.; Radzishevsky, E.; Genizi, J.; Lorber, A.; Ben-Yosef, T.; et al. Danon Disease: Entire LAMP2 Gene Deletion with Unusual Clinical Presentation-Case Report and Review of the Literature. Genes 2023, 14, 1539. [Google Scholar] [CrossRef]
- Boucek, D.; Jirikowic, J.; Taylor, M. Natural history of Danon disease. Genet. Med. 2011, 13, 563–568. [Google Scholar] [CrossRef]
- Csányi, B.; Popoiu, A.; Hategan, L.; Hegedűs, Z.; Nagy, V.; Rácz, K.; Hőgye, M.; Sághy, L.; Iványi, B.; Csanády, M.; et al. Identification of Two Novel LAMP2 Gene Mutations in Danon Disease. Can. J. Cardiol. 2016, 32, 1355.e23–1355.e30. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Fanin, M.; Angelini, C.; Sandri, M. Autophagy dysregulation in Danon disease. Cell Death Dis. 2017, 8, e2565. [Google Scholar] [CrossRef]
- Ortuño-Costela, M.D.C.; Cerrada, V.; Moreno-Izquierdo, A.; García-Consuegra, I.; Laberthonnière, C.; Delourme, M.; Garesse, R.; Arenas, J.; Fuster García, C.; García García, G.; et al. Generation of the First Human In Vitro Model for McArdle Disease Based on iPSC Technology. Int. J. Mol. Sci. 2022, 23, 13964. [Google Scholar] [CrossRef]
- Zhang, J.; Chou, O.H.; Tse, Y.L.; Ng, K.M.; Tse, H.F. Application of Patient-Specific iPSCs for Modelling and Treatment of X-Linked Cardiomyopathies. Int. J. Mol. Sci. 2021, 22, 8132. [Google Scholar] [CrossRef]
- Talib, S.; Shepard, K.A. Unleashing the cure: Overcoming persistent obstacles in the translation and expanded use of hematopoietic stem cell-based therapies. Stem Cells Transl. Med. 2020, 9, 420–426. [Google Scholar] [CrossRef]
- Cluskey, S.; Ramsden, D.B. Mechanisms of neurodegeneration in amyotrophic lateral sclerosis. Mol. Pathol. 2001, 54, 386–392. [Google Scholar] [PubMed]
- van den Bos, M.A.J.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int. J. Mol. Sci. 2019, 20, 2818. [Google Scholar] [CrossRef]
- Yoshida, M. Amyotrophic lateral sclerosis with dementia: The clinicopathological spectrum. Neuropathology 2004, 24, 87–102. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Fahrner-Scott, K.; Zapata, C.; O’Riordan, D.L.; Cohen, E.; Rosow, L.; Pantilat, S.Z.; Lomen-Hoerth, C.; Bischoff, K.E. Embedded Palliative Care for Amyotrophic Lateral Sclerosis: A Pilot Program and Lessons Learned. Neurol. Clin. Pract. 2022, 12, 68–75. [Google Scholar] [CrossRef]
- Shoesmith, C. Palliative care principles in ALS. Handb. Clin. Neurol. 2023, 191, 139–155. [Google Scholar] [CrossRef]
- Lefebvre, S.; Sarret, C. Pathogenesis and therapeutic targets in spinal muscular atrophy (SMA). Arch. Pediatr. 2020, 27, 7S3–7S8. [Google Scholar] [CrossRef]
- Keinath, M.C.; Prior, D.E.; Prior, T.W. Spinal Muscular Atrophy: Mutations, Testing, and Clinical Relevance. Appl. Clin. Genet. 2021, 14, 11–25. [Google Scholar] [CrossRef]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Qiu, J.; Wu, L.; Qu, R.; Jiang, T.; Bai, J.; Sheng, L.; Feng, P.; Sun, J. History of development of the life-saving drug “Nusinersen” in spinal muscular atrophy. Front. Cell. Neurosci. 2022, 16, 942976. [Google Scholar] [CrossRef]
- Fox, D.; To, T.M.; Seetasith, A.; Patel, A.M.; Iannaccone, S.T. Adherence and Persistence to Nusinersen for Spinal Muscular Atrophy: A US Claims-Based Analysis. Adv. Ther. 2023, 40, 903–919. [Google Scholar] [CrossRef]
- Okano, H.; Morimoto, S. iPSC-based disease modeling and drug discovery in cardinal neurodegenerative disorders. Cell Stem Cell 2022, 29, 189–208. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Therapeutic opportunities and challenges of induced pluripotent stem cells-derived motor neurons for treatment of amyotrophic lateral sclerosis and motor neuron disease. Neural Regen. Res. 2017, 12, 723–736. [Google Scholar] [CrossRef]
- De Filippis, L.; Zalfa, C.; Ferrari, D. Neural Stem Cells and Human Induced Pluripotent Stem Cells to Model Rare CNS Diseases. CNS Neurol. Disord. Drug Targets 2017, 16, 915–926. [Google Scholar] [CrossRef]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R. Molecular Mechanism and Pathogenesis of Sarcopenia: An Overview. Int. J. Mol. Sci. 2021, 22, 3032. [Google Scholar] [CrossRef]
- Wei, L.; Gregorich, Z.R.; Lin, Z.; Cai, W.; Jin, Y.; McKiernan, S.H.; McIlwain, S.; Aiken, J.M.; Moss, R.L.; Diffe, G.M.; et al. Novel Sarcopenia-related Alterations in Sarcomeric Protein Post-translational Modifications (PTMs) in Skeletal Muscles Identified by Top-down Proteomics. Mol. Cell. Proteom. 2018, 17, 134–145. [Google Scholar] [CrossRef]
- Dowling, P.; Gargan, S.; Swandulla, D.; Ohlendieck, K. Fiber-Type Shifting in Sarcopenia of Old Age: Proteomic Profiling of the Contractile Apparatus of Skeletal Muscles. Int. J. Mol. Sci. 2023, 24, 2415. [Google Scholar] [CrossRef]
- Nishikawa, H.; Fukunishi, S.; Asai, A.; Yokohama, K.; Nishiguchi, S.; Higuchi, K. Pathophysiology and mechanisms of primary sarcopenia (Review). Int. J. Mol. Med. 2021, 48, 156. [Google Scholar] [CrossRef]
- Cho, M.R.; Lee, S.; Song, S.K. A Review of Sarcopenia Pathophysiology, Diagnosis, Treatment and Future Direction. J. Korean Med. Sci. 2022, 37, e146. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, D.; Yang, Y.; Xie, W.; He, M.; Yu, D.; Wu, Y.; Wang, X.; Xiao, W.; Li, Y. The role and therapeutic potential of stem cells in skeletal muscle in sarcopenia. Stem Cell Res. Ther. 2022, 13, 28. [Google Scholar] [CrossRef]
- Wong, R.S.Y.; Cheong, S.K. Therapeutic potential of mesenchymal stem cells and their derivatives in sarcopenia. Malays. J. Pathol. 2022, 44, 429–442. [Google Scholar] [PubMed]

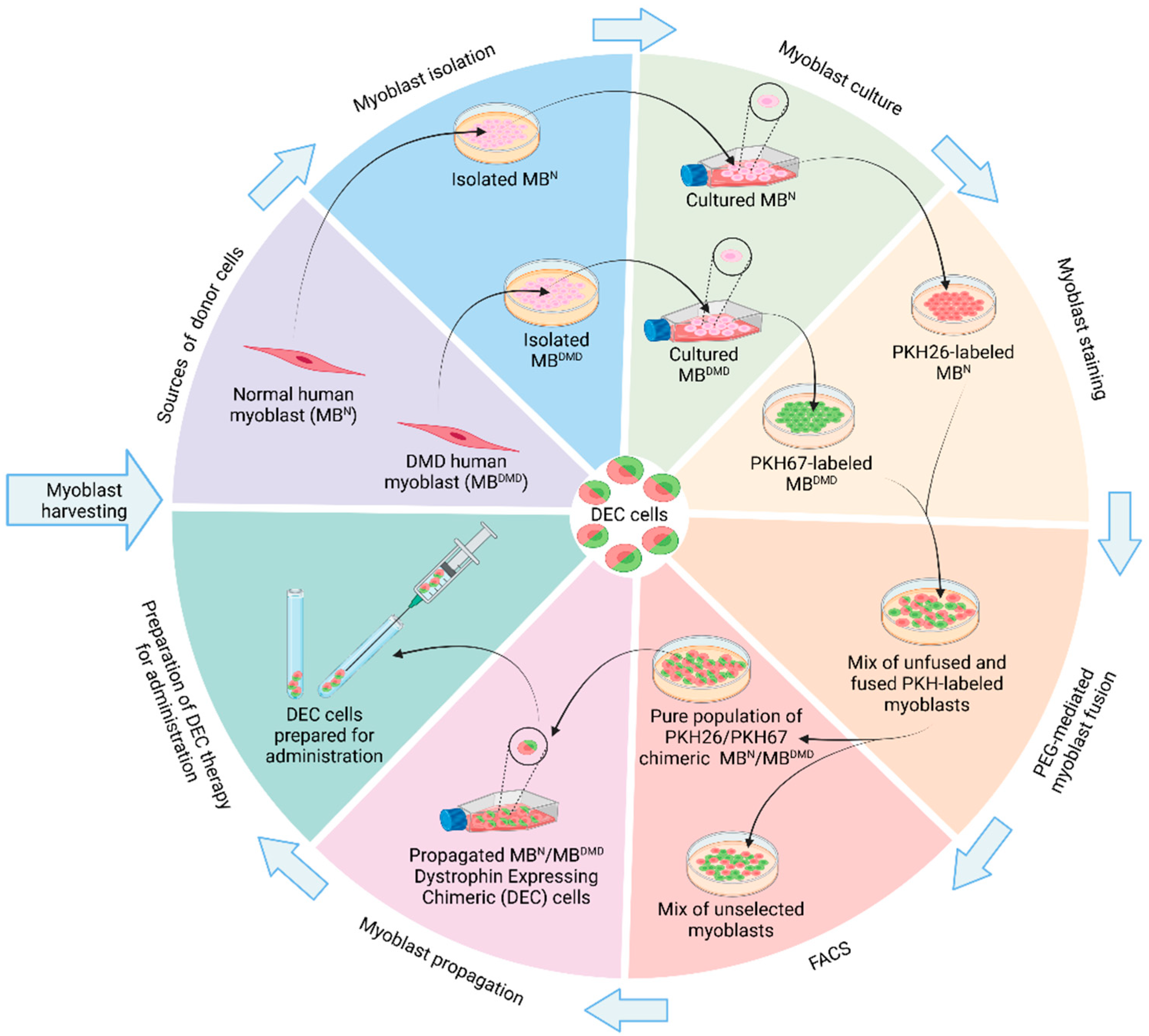

{kind=link}
{kind=link}
| A | ||||||
|---|---|---|---|---|---|---|
| Donor | Recipient | Cell Source | Name of the Therapy | Title of the Manuscript | Year | Refs |
| Mice | Mdx mouse | Myoblast | DEC | Creation of Dystrophin Expressing Chimeric Cells of Myoblast Origin as a Novel Stem Cell Based for Duchenne Muscular Dystrophy | 2018 | [17] |
| Mdx mouse | Myoblast and MSC | DEC | Cardiac Protection after Systemic Transplant of Dystrophin Expressing Chimeric (DEC) Cells to the mdx Mouse Model of Duchenne Muscular Dystrophy | 2019 | [95] | |
| Mdx mouse | Myoblast and MSC | DEC | Intraosseous Transplant of Dystrophin Expressing Chimeric (DEC) Cells Improves Skeletal Muscle Function in mdx Mouse Model of Duchenne Muscular Dystrophy | 2022 | [94] | |
| Human (Tissues and Cells Bank *) | Mdx/scid mouse | Myoblast | DEC | Dystrophin Expressing Chimeric (DEC) Human Cells Provide a Potential Therapy for Duchenne Muscular Dystrophy | 2018 | [21] |
| Mdx/scid mouse | Myoblast and MSC | DEC | Transplantation of Dystrophin Expressing Chimeric Human Cells of Myoblast/Mesenchymal Stem Cell origin Improves Function in Duchenne Muscular Dystrophy | 2021 | [18] | |
| Mdx/scid mouse | Myoblast | DEC | Human Dystrophin Expressing Chimeric (DEC) Cell Therapy Ameliorates Cardiac, Respiratory and Skeletal Muscle’s Function in Duchenne Muscular Dystrophy | 2021 | [92] | |
| Mdx/scid mouse | Myoblast | DEC | Long-Term Protective Effect of Human Dystrophin Expressing Chimeric (DEC) Cell Therapy on Amelioration of Function of Cardiac, Respiratory and Skeletal Muscles in Duchenne Muscular Dystrophy | 2022 | [93] | |
| Mdx/scid mouse | Myoblast | DEC | Long-Term Biodistribution and Safety of Human Dystrophin Expressing Chimeric Cell Therapy After Systemic-Intraosseous Administration to Duchenne Muscular Dystrophy Model | 2022 | [96] | |
| Mdx/scid mouse | Myoblast | DEC | Amelioration of Morphological Pathology in Cardiac, Respiratory, and Skeletal Muscles Following Intraosseous Administration of Human Dystrophin Expressing Chimeric (DEC) Cells in Duchenne Muscular Dystrophy Model | 2024 | [97] | |
| B | ||||||
| Donor | Recipient | Cell Source | Name of the Therapy | Title of the Manuscript | Year | DOI |
| Human (normal and DMD-affected donors) | Human | Myoblast | DT-DEC01 | Dystrophin Expressing Chimeric (DEC) Cell Therapy for Duchenne Muscular Dystrophy: A First-in-Human Study with Minimum 6 Months Follow-up | 2023 | [98] |
| Human | Myoblast | DT-DEC01 | Safety and Efficacy of DT-DEC01 Therapy in Duchenne Muscular Dystrophy Patients: A 12-Month Follow-Up Study After Systemic Intraosseous Administration | 2023 | [99] | |
| Human | Myoblast | DT-DEC01 | Assessment of Motor Unit Potentials Duration as the Biomarker of DT-DEC01 Cell Therapy Efficacy in Duchenne Muscular Dystrophy Patients up to 12 Months After Systemic-Intraosseous Administration | 2023 | [100] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budzynska, K.; Siemionow, M.; Stawarz, K.; Chambily, L.; Siemionow, K. Chimeric Cell Therapies as a Novel Approach for Duchenne Muscular Dystrophy (DMD) and Muscle Regeneration. Biomolecules 2024, 14, 575. https://doi.org/10.3390/biom14050575
Budzynska K, Siemionow M, Stawarz K, Chambily L, Siemionow K. Chimeric Cell Therapies as a Novel Approach for Duchenne Muscular Dystrophy (DMD) and Muscle Regeneration. Biomolecules. 2024; 14(5):575. https://doi.org/10.3390/biom14050575
Chicago/Turabian StyleBudzynska, Katarzyna, Maria Siemionow, Katarzyna Stawarz, Lucile Chambily, and Krzysztof Siemionow. 2024. "Chimeric Cell Therapies as a Novel Approach for Duchenne Muscular Dystrophy (DMD) and Muscle Regeneration" Biomolecules 14, no. 5: 575. https://doi.org/10.3390/biom14050575