
QTL Mapping of Yield-Related Traits in Tetraploid Wheat Based on Wheat55K SNP Array

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Differences and Analysis
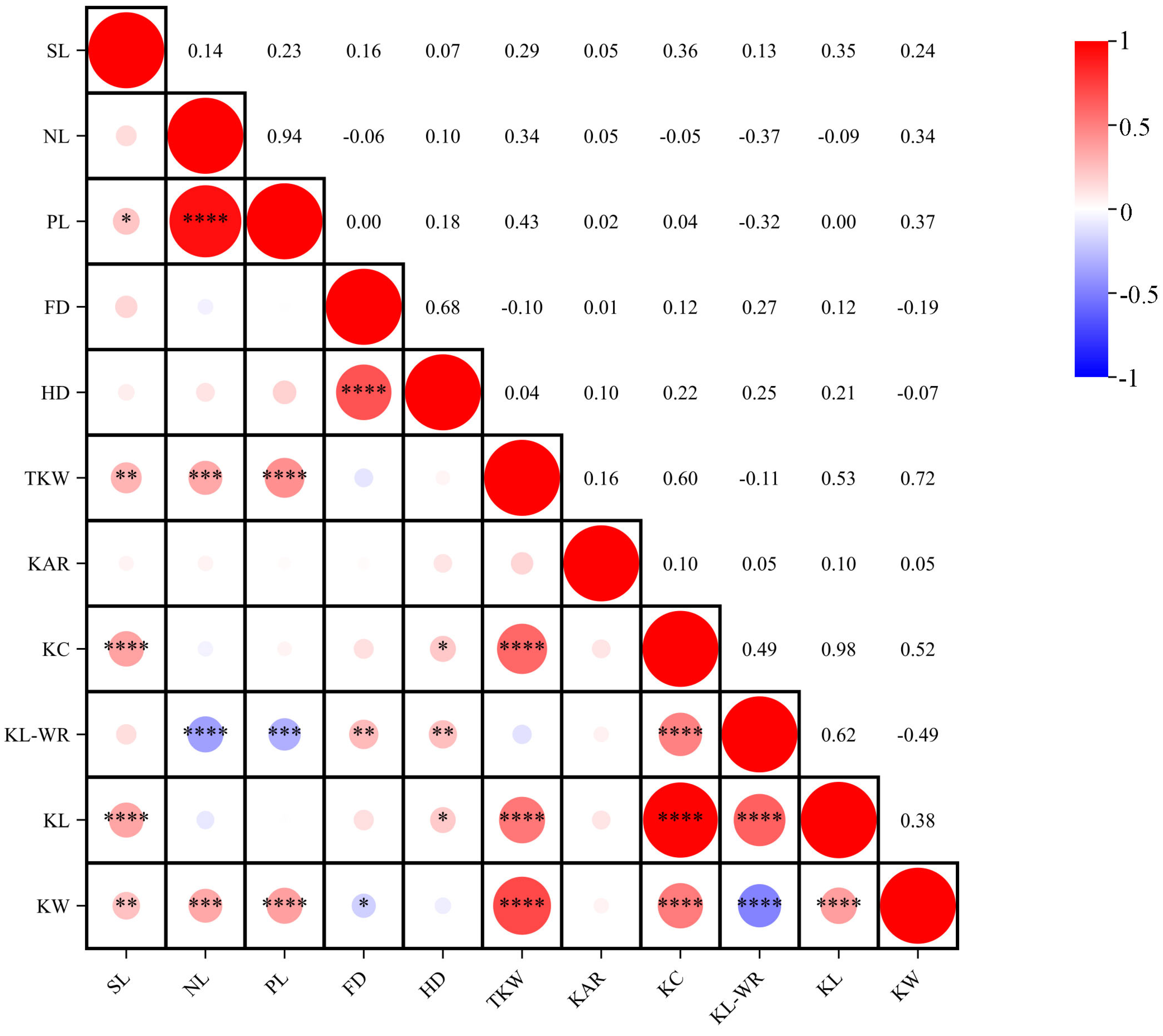
2.2. Correlation Analyses among Different Traits
2.3. Genetic Map Construction
2.4. QTL Mapping Analysis
2.5. Combined QTL–Environment Interaction Analysis
2.6. Analysis of the Effects of Stable Major Loci
2.7. Candidate Gene Prediction Analysis
3. Discussion
3.1. Analysis and Discussion of Enrichment of QTL Clusters in Agronomic Traits and Correlation among Agronomic Traits
3.2. Exploration of the Application of Different Types of Arrays in Wheat Materials with Different Ploidies
3.3. Candidate Gene Prediction Analysis within the QTL Mapping Interval
4. Materials and Methods
4.1. Plant Materials and Field Trials
4.2. Phenotypic Evaluation and Statistical Analysis
4.3. Genotyping and Genetic Map Construction
4.4. Quantitative Trait Loci Mapping
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Slafer, G. Genetic basis of yield as viewed from a crop physiologist’s perspective. Ann. Appl. Biol. 2003, 142, 117–128. [Google Scholar] [CrossRef]
- Kim, J.; Song, M.; Lee, J.; Ahn, S.; Ku, J. QTL Analysis of Seed and Growth Traits using RIL Population in Soybean. Cancer 2008, 83, 98–102. [Google Scholar]
- Zhang, N.; Fan, X.; Cui, F.; Zhao, C.; Zhang, W.; Zhao, X.; Yang, L.; Pan, R.; Chen, M.; Han, J.; et al. Characterization of the temporal and spatial expression of wheat (Triticum aestivum L.) plant height at the QTL level and their influence on yield-related traits. Theor. Appl. Genet. 2017, 130, 1235–1252. [Google Scholar] [CrossRef]
- Dixon, L.; Pasquariello, M.; Boden, S. TEOSINTE BRANCHED1 regulates height and stem internode length in bread wheat. J. Exp. Bot. 2020, 71, 4742–4750. [Google Scholar] [CrossRef]
- Dai, L.; Liu, C. Relationship between relative length of head neck and grain weight per spike of wheat. Agric. Technol. Commun. 1988, 11, 6. [Google Scholar]
- Bridgemohan, P.; Bridgemohan, R. Evaluation of anti-lodging plant growth regulators on the growth and development of rice (Oryza sativa). Acad J. 2014, 5, 12–16. [Google Scholar]
- Li, F.; Wen, W.; Liu, J.; Zhang, Y.; Cao, S.; He, Z.; Rasheed, A.; Jin, H.; Zhang, C.; Yan, J.; et al. Genetic architecture of grain yield in bread wheat based on genome-wide association studies. BMC Plant Biol. 2019, 19, 168. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Luo, W.; Zhang, X.; Mu, Y.; Deng, M.; Liu, Y.; Jiang, Q.; Chen, G.; Qi, P.; Pu, Z.; et al. A novel, validated, and plant height-independent QTL for spike extension length is associated with yield-related traits in wheat. Theor. Appl. Genet. 2020, 133, 3381–3393. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, P.; Lai, X.; Wang, X.; Ji, W.; Xu, S. The selection and application of peduncle length QTL QPL_6D.1 in modern wheat (Triticum aestivum L.) breeding. Theor. Appl. Genet. 2023, 136, 32. [Google Scholar] [CrossRef]
- Yao, H.; Xie, Q.; Xue, S.; Luo, J.; Ma, Z. HL2 on chromosome 7D of wheat (Triticum aestivum L.) regulates both head length and spikelet number. Theor. Appl. Genet. 2019, 132, 1789–1797. [Google Scholar] [CrossRef]
- Xiong, H.; Li, Y.; Guo, H.; Xie, Y.; Liu, L. Genetic Mapping by Integration of 55K SNP Array and KASP Markers Reveals Candidate Genes for Important Agronomic Traits in Hexaploid Wheat. Front. Plant Sci. 2021, 12, 628478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiong, H.; Guo, H.; Li, Y.; Xie, X.; Xie, Y.; Zhao, L.; Gu, J.; Zhao, S.; Ding, Y. Identification of the Q Gene Playing a Role in Spike Morphology Variation in Wheat Mutants and Its Regulatory Network. Front. Plant Sci. 2022, 12, 807731. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Müller, A. Flowering time control and applications in plant breeding. Trends Plant Sci. 2009, 14, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, G.; Chuan, L.; Xia, T.; Zhao, J. Fine Mapping of a Novel Heading Date Gene, TaHdm605, in Hexaploid Wheat. Front. Plant Sci. 2018, 9, 1059. [Google Scholar] [CrossRef]
- Fu, D.; Szucs, P.; Yan, L.; Helguera, M.; Skinner, J.; von Zitzewitz, J.; Hayes, P.; Dubcovsky, J. Large deletions within the first intron in VRN-1 are associated with spring growth habit in barley and wheat. Mol. Genet. Genom. 2005, 273, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Kippes, N.; Zhu, J.; Chen, A.; Vanzetti, L.; Lukaszewski, A.; Nishida, H.; Kato, K.; Dvorak, J.; Dubcovsky, J. Fine mapping and epistatic interactions of the vernalization gene VRN-D4 in hexaploid wheat. Mol. Genet. Genom. 2014, 289, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Loukoianov, A.; Blechl, A.; Tranquilli, C.; Ramakrishna, W.; Sanmiguel, P.; Bennetzen, J.L.; Echenique, V.; Dubcovsky, J. The Wheat VRN2 Gene Is a Flowering Repressor Down-Regulated by Vernalization. Science 2004, 303, 1640–1644. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Nishida, H.; Zhu, J.; Nitcher, R.; Distelfeld, A.; Akashi, Y.; Kato, K.; Dubcovsky, J. Vrn-D4 is a vernalization gene located on the centromeric region of chromosome 5D in hexaploid wheat. Theor. Appl. Genet. 2010, 120, 543–552. [Google Scholar] [CrossRef]
- Murai, K.; Miyamae, M.; Kato, H.; Takumi, S.; Ogihara, Y. WAP1, a wheat APETALA1 homolog, plays a central role in the phase transition from vegetative to reproductive growth. Plant Cell Physiol. 2003, 44, 1255–1265. [Google Scholar] [CrossRef]
- Prashant, R.; Kadoo, N.; Desale, C.; Kore, P.; Dhaliwal, H.S.; Chhuneja, P.; Gupta, V. Kernel morphometric traits in hexaploid wheat (Triticum aestivum L.) are modulated by intricate QTL×QTL and genotype×environment interactions. J. Cereal Sci. 2012, 56, 432–439. [Google Scholar] [CrossRef]
- Lizana, X.; Riegel, R.; Gomez, L.; Herrera, J.; Isla, A.; McQueen-Mason, S.J.; Calderini, D.F. Expansins expression is associated with grain size dynamics in wheat (Triticum aestivum L.). J. Exp. Bot. 2010, 61, 1147–1157. [Google Scholar] [CrossRef]
- Ma, C.; Liu, L.; Liu, T.; Jia, Y.; Jiang, Q.; Bai, H.; Ma, S.; Li, S.; Wang, Z. QTL Mapping for Important Agronomic Traits Using a Wheat55K SNP Array-Based Genetic Map in Tetraploid Wheat. Plants 2023, 12, 847. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Li, C.; Liu, H.; Liu, J.; Luo, W.; Xu, Q.; Tang, H.; Mu, Y.; Deng, M.; Pu, Z. Quick mapping and characterization of a co-located kernel length and thousand-kernel weight-related QTL in wheat. Theor. Appl. Genet. 2022, 8, 135. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Xie, X.; Wang, Y.; Yang, Z.; Peng, H.; Xin, M.; Yao, Y.; Hu, Z.; Liu, J.; et al. Dispersed emergence and protracted domestication of polyploid wheat uncovered by mosaic ancestral haploblock inference. Nat. Commun. 2022, 13, 3891. [Google Scholar] [CrossRef]
- Wan, H.; Yang, F.; Li, J.; Wang, Q.; Liu, Z.; Tang, Y.; Yang, W. Genetic Improvement and Application Practices of Synthetic Hexaploid Wheat. Genes 2023, 14, 283. [Google Scholar] [CrossRef]
- Giancaspro, A.; Giove, S.; Zacheo, S.; Blanco, A.; Gadaleta, A. Genetic Variation for Protein Content and Yield-Related Traits in a Durum Population Derived From an Inter-Specific Cross Between Hexaploid and Tetraploid Wheat Cultivars. Front. Plant Sci. 2019, 10, 1509. [Google Scholar] [CrossRef]
- Mo, Z.; Zhu, J.; Wei, J.; Zhou, J.; Xu, Q.; Tang, H.; Mu, Y.; Deng, M.; Jiang, Q.; Liu, Y.; et al. The 55K SNP-Based Exploration of QTLs for Spikelet Number Per Spike in a Tetraploid Wheat (Triticum turgidum L.) Population: Chinese Landrace “Ailanmai” × Wild Emmer. Front. Plant Sci. 2021, 12, 732837. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Luo, W.; Qin, N.; Ding, P.; Zhang, H.; Yang, C.; Mu, Y.; Tang, H.; Liu, Y.; Li, W.; et al. A 55 K SNP array-based genetic map and its utilization in QTL mapping for productive tiller number in common wheat. Theor. Appl. Genet. 2018, 131, 2439–2450. [Google Scholar] [CrossRef]
- de Sousa, T.; Ribeiro, M.; Sabença, C.; Igrejas, G. The 10,000-Year Success Story of Wheat! Foods 2021, 10, 2134. [Google Scholar] [CrossRef] [PubMed]
- Shewry, P.; Sandra, H. Do “ancient” wheat species differ from modern bread wheat in their contents of bioactive components? J. Cereal Sci. 2015, 65, 469–476. [Google Scholar] [CrossRef]
- Su, Q.; Zhang, X.; Zhang, W.; Zhang, N.; Song, L.; Liu, L.; Xue, X.; Liu, G.; Liu, J.; Meng, D.; et al. QTL Detection for Kernel Size and Weight in Bread Wheat (Triticum aestivum L.) Using a High-Density SNP and SSR-Based Linkage Map. Front. Plant Sci. 2018, 9, 1484. [Google Scholar] [CrossRef]
- Hina, A.; Cao, Y.; Song, S.; Li, S.; Sharmin, R.A.; Elattar, M.A.; Bhat, J.A.; Zhao, T. High-Resolution Mapping in Two RIL Populations Refines Major “QTL Hotspot” Regions for Seed Size and Shape in Soybean (Glycine max L.). Int. J. Mol. Sci. 2020, 21, 1040. [Google Scholar] [CrossRef]
- Roncallo, P.F.; Akkiraju, P.C.; Cervigni, G.L.; Echenique, V.C. QTL mapping and analysis of epistatic interactions for grain yield and yield-related traits in Triticum turgidum L. var. durum. Euphytica 2017, 213, 277. [Google Scholar] [CrossRef]
- Maccaferri, M.; Cane’, M.; Sanguineti, M.; Salvi, S.; Colalongo, M.; Massi, A.; Clarke, F.; Knox, R.; Pozniak, C.; Clarke, J.; et al. A consensus framework map of durum wheat (Triticum durum Desf.) suitable for linkage disequilibrium analysis and genome-wide association mapping. BMC Genom. 2014, 15, 873. [Google Scholar] [CrossRef]
- Sun, C.; Dong, Z.; Zhao, L.; Ren, Y.; Zhang, N.; Chen, F. The Wheat 660K SNP array demonstrates great potential for marker-assisted selection in polyploid wheat. Plant Biotechnol. J. 2020, 18, 1354–1360. [Google Scholar] [CrossRef]
- Ren, T.; Fan, T.; Chen, S.; Li, C.; Chen, Y.; Ou, X.; Jiang, Q.; Ren, Z.; Tan, F.; Luo, P. Utilization of a Wheat55K SNP array-derived high-density genetic map for high-resolution mapping of quantitative trait loci for important kernel-related traits in common wheat. Theor. Appl. Genet. 2021, 134, 873. [Google Scholar]
- Zhou, S.; Zhang, J.; Che, Y.; Liu, W.; Lu, Y.; Yang, X.; Li, X.; Jia, J.; Liu, X.; Li, L. Construction of Agropyron Gaertn. genetic linkage maps using a wheat 660K SNP array reveals a homoeologous relationship with the wheat genome. Plant Biotechnol. J. 2018, 16, 818–827. [Google Scholar] [CrossRef]
- Mengistu, D.K.; Kidane, Y.G.; Catellani, M.; Frascaroli, E.; Fadda, C.; Pè, M.; Dell, A. High-density molecular characterization and association mapping in Ethiopian durum wheat landraces reveals high diversity and potential for wheat breeding. Plant Biotechnol. J. 2016, 14, 1800–1812. [Google Scholar] [CrossRef]
- Graziani, M.; Maccaferri, M.; Royo, C.; Salvatorelli, F.; Tuberosa, R.J.C.; Science, P. QTL dissection of yield components and morpho-physiological traits in a durum wheat elite population tested in contrasting thermo-pluviometric conditions. Field Crops Res. 1998, 59, 163–173. [Google Scholar] [CrossRef]
- Wang, Y.; Du, F.; Wang, J.; Wang, K.; Tian, C.; Qi, X.; Lu, F.; Liu, X.; Ye, X.; Jiao, Y. Improving bread wheat yield through modulating an unselected AP2/ERF gene. Nat. Plants 2022, 8, 930–939. [Google Scholar] [CrossRef]
- Reyes, F.; Buono, R.; Roschzttardtz, H.; Di Rubbo, S.; Yeun, L.; Russinova, E.; Otegui, M. A novel endosomal sorting complex required for transport (ESCRT) component in Arabidopsis thaliana controls cell expansion and development. J. Biol. Chem. 2014, 289, 4980–4988. [Google Scholar] [CrossRef]
- Steffens, A.; Jakoby, M.; Hülskamp, M. Physical, Functional and Genetic Interactions between the BEACH Domain Protein SPIRRIG and LIP5 and SKD1 and Its Role in Endosomal Trafficking to the Vacuole in Arabidopsis. Front. Plant Sci. 2017, 8, 1969. [Google Scholar] [CrossRef]
- Giunta, F.; De, P.; Mastrangelo, A.; Sanna, G.; Motzo, R. Environmental and Genetic Variation for Yield-Related Traits of Durum Wheat as Affected by Development. Front. Plant Sci. 2018, 18, 9. [Google Scholar] [CrossRef]
- Milner, S.; Maccaferri, M.; Huang, B.; Mantovani, P.; Massi, A.; Frascaroli, E.; Tuberosa, R.; Salvi, S. A multiparental cross population for mapping QTL for agronomic traits in durum wheat (Triticum turgidum ssp. durum). Plant Biotechnol. J. 2016, 14, 735–748. [Google Scholar] [CrossRef]
- Soriano, J.M.; Malosetti, M.; Roselló, M.; Sorrells, M.; Royo, C. Dissecting the old Mediterranean durum wheat genetic architecture for phenology, biomass and yield formation by association mapping and QTL meta-analysis. PLoS ONE 2017, 12, e0178290. [Google Scholar] [CrossRef]
- Mangini, G.; Gadaleta, A.; Colasuonno, P.; Marcotuli, I.; Signorile, A.; Simeone, R.; De, P.; Mastrangelo, A.; Laidò, G.; Pecchioni, N.; et al. Genetic dissection of the relationships between grain yield components by genome-wide association mapping in a collection of tetraploid wheats. PLoS ONE 2018, 13, e0190162. [Google Scholar] [CrossRef]
- Peleg, Z.; Fahima, T.; Krugman, T.; Abbo, S.; Saranga, Y. Genomic dissection of drought resistance in durum wheat x wild emmer wheat recombinant inbreed line population. Plant Cell Environ. 2009, 32, 758–779. [Google Scholar] [CrossRef] [PubMed]
- Peleg, Z.; Fahima, T.; Korol, A.B.; Abbo, S.; Saranga, Y. Genetic analysis of wheat domestication and evolution under domestication. J. Exp. Bot. 2011, 62, 5051–5061. [Google Scholar] [CrossRef]
- Zhao, Y.; Peng, T.; Sun, H.; Teotia, S.; Wen, H.; Du, Y.; Zhang, J.; Li, J.; Tang, G.; Xue, H.; et al. miR1432-OsACOT (Acyl-CoA thioesterase) module determines grain yield via enhancing grain filling rate in rice. Plant Biotechnol. J. 2019, 17, 712–723. [Google Scholar] [CrossRef]
- Dong, E.; Liu, C.; Bai, Y.; Mei, X.; Zhao, Z.; Zhu, S.; Zhang, J.; Cai, Y. A new network containing MYB109-ZmCesA5 is involved in kernel development. Biochem. Biophys. Res. Commun. 2022, 602, 15–20. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Year | Parental Lines | RIL Population | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QH-1 | DW | Min | Max | Mean | SD | CV% | SK | KU | H2 | ||
| SL | 22LH | 11.17 | 6.25 | 6.00 | 37.50 | 9.04 | 3.03 | 33.49 | 8.12 | 76.37 | 0.48 |
| 22YL | 12.72 | 9.66 | 7.30 | 12.12 | 9.34 | 1.06 | 11.31 | 0.24 | −0.08 | ||
| 23LH | 11.17 | 6.93 | 6.33 | 11.80 | 8.50 | 1.06 | 12.43 | 0.62 | 0.18 | ||
| 23YL | 10.77 | 7.50 | 4.50 | 40.90 | 9.09 | 3.14 | 34.52 | 8.84 | 90.10 | ||
| BLUE | 11.46 | 7.58 | 6.33 | 19.75 | 9.01 | 1.51 | 16.80 | 3.98 | 24.76 | ||
| NL | 21YL | 21.33 | 16.17 | 4.50 | 35.17 | 17.02 | 5.88 | 34.51 | 0.64 | 0.35 | 0.86 |
| 22LH | 17.33 | 15.00 | 4.25 | 28.50 | 17.52 | 6.07 | 34.65 | −0.25 | −0.86 | ||
| 22YL | 20.18 | 19.14 | 3.50 | 36.94 | 22.00 | 7.18 | 32.63 | −0.27 | −1.02 | ||
| 23LH | 24.33 | 20.00 | 3.00 | 35.00 | 20.18 | 7.24 | 35.90 | 0.00 | −0.87 | ||
| 23YL | 31.97 | 16.80 | 9.03 | 46.67 | 24.83 | 11.46 | 46.14 | 3.38 | 24.61 | ||
| BLUE | 23.03 | 17.42 | 8.57 | 41.54 | 20.31 | 6.09 | 29.97 | 0.22 | −0.07 | ||
| PL | 21YL | 46.00 | 35.83 | 16.00 | 62.17 | 38.28 | 8.19 | 21.40 | 0.33 | 0.03 | 0.77 |
| 22LH | 39.33 | 31.00 | 18.25 | 50.00 | 36.39 | 7.91 | 21.73 | −0.37 | −0.88 | ||
| 22YL | 24.30 | 19.98 | 14.48 | 27.80 | 20.71 | 3.00 | 14.48 | 0.14 | −0.47 | ||
| 23LH | 45.63 | 37.00 | 21.00 | 59.00 | 38.73 | 9.17 | 23.67 | −0.03 | −0.97 | ||
| 23YL | 55.00 | 35.50 | 24.77 | 67.67 | 45.34 | 10.90 | 24.04 | −0.13 | −1.17 | ||
| BLUE | 42.17 | 31.98 | 22.74 | 51.62 | 36.01 | 6.70 | 18.60 | −0.12 | −0.89 | ||
| FD | 22YL | 5.4 | 4.25 | 4.24 | 5.90 | 4.41 | 0.39 | 8.87 | 2.41 | 4.36 | 0.19 |
| 23LH | 6.10 | 6.06 | 5.12 | 6.17 | 6.08 | 0.12 | 1.99 | −6.53 | 47.19 | ||
| 23YL | 4.26 | 4.21 | 4.17 | 5.08 | 4.28 | 0.16 | 3.81 | 4.35 | 18.22 | ||
| BLUE | 5.54 | 5.13 | 4.83 | 5.85 | 5.21 | 0.16 | 3.16 | 1.93 | 4.01 | ||
| HD | 21YL | 4.20 | 4.19 | 4.16 | 4.28 | 4.21 | 0.03 | 0.61 | 0.35 | −0.31 | 0.09 |
| 22YL | 4.30 | 4.20 | 4.18 | 5.60 | 4.28 | 0.24 | 5.60 | 4.31 | 17.63 | ||
| 23YL | 4.19 | 4.15 | 4.12 | 4.23 | 4.18 | 0.02 | 0.53 | 0.02 | 0.14 | ||
| BLUE | 4.24 | 4.19 | 4.17 | 4.70 | 4.24 | 0.08 | 2.00 | 4.02 | 15.95 | ||
| TKW | 21YL | 41.82 | 61.09 | 34.50 | 63.89 | 49.42 | 5.53 | 11.18 | −0.38 | 0.66 | 0.73 |
| 22LH | 46.80 | 50.02 | 31.07 | 71.06 | 47.99 | 4.90 | 10.21 | 1.04 | 5.36 | ||
| 22YL | 39.06 | 56.20 | 39.92 | 61.64 | 49.74 | 4.34 | 8.73 | 0.01 | 0.00 | ||
| 23LH | 48.79 | 52.95 | 38.92 | 59.67 | 48.47 | 4.11 | 8.48 | 0.06 | 0.12 | ||
| 23YL | 37.70 | 49.40 | 28.12 | 55.46 | 43.90 | 5.13 | 11.68 | −0.26 | 0.58 | ||
| BLUE | 42.83 | 53.93 | 39.20 | 56.88 | 47.90 | 3.27 | 6.83 | 0.22 | 0.34 | ||
| KAR | 21YL | 24.50 | 19.30 | 1.75 | 17.12 | 8.25 | 2.41 | 29.19 | 0.57 | 1.19 | 0.00 |
| 22LH | 6.01 | 10.56 | 1.31 | 18.88 | 10.22 | 3.03 | 29.60 | 0.29 | 0.29 | ||
| 22YL | 7.89 | 5.52 | 4.02 | 15.12 | 8.40 | 2.71 | 32.34 | 0.44 | −0.80 | ||
| 23LH | 12.67 | 17.14 | 0.94 | 21.56 | 14.32 | 2.78 | 19.44 | −0.85 | 4.49 | ||
| 23YL | 10.32 | 9.71 | 7.81 | 21.30 | 13.88 | 2.60 | 18.70 | 0.27 | −0.06 | ||
| BLUE | 12.01 | 12.18 | 6.72 | 13.48 | 10.75 | 1.21 | 11.28 | −0.24 | 0.16 | ||
| KC | 21YL | 20.20 | 21.11 | 12.61 | 21.52 | 18.91 | 1.53 | 8.08 | −1.45 | 2.86 | 0.48 |
| 22LH | 17.81 | 17.69 | 16.65 | 19.98 | 18.11 | 0.70 | 3.87 | 0.24 | −0.41 | ||
| 22YL | 17.73 | 19.70 | 14.69 | 21.34 | 18.89 | 1.21 | 6.40 | −1.72 | 3.84 | ||
| 23LH | 19.64 | 19.42 | 17.34 | 20.61 | 18.70 | 0.67 | 3.58 | 0.45 | 0.40 | ||
| 23YL | 19.25 | 18.81 | 17.32 | 20.79 | 18.75 | 0.69 | 3.67 | 0.40 | 0.13 | ||
| BLUE | 18.89 | 19.31 | 16.95 | 20.56 | 18.64 | 0.63 | 3.36 | 0.08 | 0.23 | ||
| KL-WR | 21YL | 2.63 | 2.27 | 2.16 | 2.76 | 2.41 | 0.11 | 4.58 | 0.26 | −0.09 | 0.82 |
| 22LH | 2.47 | 2.28 | 2.07 | 2.67 | 2.31 | 0.11 | 4.90 | 0.40 | 0.28 | ||
| 22YL | 2.75 | 2.38 | 2.13 | 2.76 | 2.41 | 0.11 | 4.73 | 0.43 | 0.40 | ||
| 23LH | 2.50 | 2.33 | 2.04 | 2.68 | 2.30 | 0.12 | 5.32 | 0.21 | 0.11 | ||
| 23YL | 2.74 | 2.40 | 2.12 | 2.86 | 2.41 | 0.12 | 5.08 | 0.40 | 0.41 | ||
| BLUE | 2.61 | 2.32 | 2.13 | 2.60 | 2.36 | 0.09 | 3.89 | 0.21 | 0.03 | ||
| KL | 21YL | 7.94 | 8.07 | 5.14 | 8.41 | 7.45 | 0.58 | 7.72 | −1.30 | 2.57 | 0.63 |
| 22LH | 7.28 | 7.09 | 6.63 | 8.18 | 7.27 | 0.31 | 4.32 | 0.31 | −0.37 | ||
| 22YL | 7.47 | 7.97 | 6.03 | 8.79 | 7.71 | 0.51 | 6.57 | −1.36 | 2.81 | ||
| 23LH | 8.00 | 7.80 | 6.82 | 8.36 | 7.51 | 0.31 | 4.09 | 0.27 | 0.07 | ||
| 23YL | 7.97 | 7.67 | 6.94 | 8.60 | 7.60 | 0.30 | 3.96 | 0.35 | 0.39 | ||
| BLUE | 7.72 | 7.71 | 6.85 | 8.41 | 7.50 | 0.28 | 3.68 | 0.24 | 0.28 | ||
| KW | 21YL | 3.04 | 3.57 | 2.20 | 3.62 | 3.12 | 0.24 | 7.76 | −1.08 | 1.67 | 0.49 |
| 22LH | 2.98 | 3.13 | 2.88 | 3.54 | 3.17 | 0.13 | 4.00 | 0.04 | 0.56 | ||
| 22YL | 2.73 | 3.37 | 2.50 | 3.50 | 3.22 | 0.20 | 6.26 | −1.69 | 3.01 | ||
| 23LH | 3.22 | 3.38 | 2.92 | 3.62 | 3.28 | 0.13 | 3.91 | −0.04 | −0.04 | ||
| 23YL | 2.93 | 3.22 | 2.76 | 3.51 | 3.19 | 0.15 | 4.64 | −0.24 | −0.11 | ||
| BLUE | 2.98 | 3.33 | 2.90 | 3.46 | 3.20 | 0.10 | 3.22 | −0.06 | −0.02 | ||
| Traits | QTL | Env | Interval(cM) | Left Marker | Right Marker | LOD | PVE(%) | Add |
|---|---|---|---|---|---|---|---|---|
| SL | QSL.QD-1A.1 | 2022YL | 20.71–22.71 | AX-109410802 | AX-108955152 | 4.5567 | 13.5381 | 0.3519 |
| 2023LH | 20.71–22.71 | AX-109410802 | AX-108955152 | 9.2387 | 20.2021 | 0.4992 | ||
| NL | QNL.QD-4B.2 | 2021YL | 78.59–79.09 | AX-110031800 | AX-108848122 | 12.6214 | 34.7074 | 3.3629 |
| 2022LH | 78.59–79.09 | AX-110031800 | AX-108848122 | 6.6713 | 18.1964 | 2.4966 | ||
| 2023LH | 78.59–79.09 | AX-110031800 | AX-108848122 | 20.4944 | 44.2068 | 4.7673 | ||
| 2023YL | 78.59–79.09 | AX-110031800 | AX-108848122 | 70.3906 | 27.2688 | 28.1164 | ||
| PL | QPL.QD-4B.1 | 2021YL | 78.59–79.09 | AX-110031800 | AX-108848122 | 20.3889 | 52.4851 | 5.7827 |
| 2022LH | 78.59–79.09 | AX-110031800 | AX-108848122 | 8.3522 | 31.331 | 3.9071 | ||
| 2022YL | 78.59–79.09 | AX-110031800 | AX-108848122 | 20.583 | 43.3116 | 2.0306 | ||
| 2023LH | 78.59–79.09 | AX-110031800 | AX-108848122 | 19.2727 | 44.2748 | 6.0188 | ||
| FD | QFD.QD-2B | 2022YL | 103.48–110.53 | AX-108817536 | AX-109927771 | 15.2566 | 29.4535 | 0.275 |
| 2023YL | 103.48–110.53 | AX-108817536 | AX-109927771 | 3.7689 | 3.8373 | 0.0701 | ||
| HD | QHD.QD-2B.1 | 2021YL | 25.99–26.47 | AX-89458348 | AX-111717978 | 4.3118 | 12.679 | −0.0086 |
| 2023YL | 25.99–26.47 | AX-89458348 | AX-111717978 | 4.2206 | 14.4486 | −0.0076 | ||
| QHD.QD-4B | 2021YL | 79.09–89.74 | AX-108848122 | AX-111068079 | 5.207 | 16.2552 | −0.0099 | |
| 2023YL | 79.09–89.74 | AX-108848122 | AX-111068079 | 3.9059 | 13.967 | −0.0075 | ||
| KC | QKC.QD-4B.2 | 2022LH | 64.88–69.63 | AX-108801712 | AX-109959423 | 7.8789 | 21.595 | −0.352 |
| 2022YL | 64.88–69.63 | AX-108801712 | AX-109959423 | 4.065 | 14.4427 | −0.4677 | ||
| 2023LH | 64.88–69.63 | AX-108801712 | AX-109959423 | 7.1755 | 16.32 | −0.2934 | ||
| 2023YL | 64.88–69.63 | AX-108801712 | AX-109959423 | 6.4894 | 16.8047 | −0.3074 | ||
| KL-WR | QKL-WR.QD-4B.6 | 2021YL | 72.85–74.24 | AX-86176450 | AX-109925267 | 8.9608 | 16.7457 | −0.0499 |
| 2023YL | 72.85–74.24 | AX-86176450 | AX-109925267 | 9.0497 | 6.2925 | −0.0545 | ||
| KL | QKL.QD-4B.2 | 2022LH | 64.88–69.63 | AX-108801712 | AX-109959423 | 11.234 | 27.3212 | −0.1853 |
| 2022YL | 64.88–69.63 | AX-108801712 | AX-109959423 | 4.6622 | 7.6794 | −0.2111 | ||
| 2023YL | 64.88–69.63 | AX-108801712 | AX-109959423 | 5.9072 | 16.453 | −0.1307 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Y.; Zhang, Y.; Sun, Y.; Ma, C.; Bai, Y.; Zhang, H.; Hou, J.; Wang, Y.; Ji, W.; Bai, H.; et al. QTL Mapping of Yield-Related Traits in Tetraploid Wheat Based on Wheat55K SNP Array. Plants 2024, 13, 1285. https://doi.org/10.3390/plants13101285
Jia Y, Zhang Y, Sun Y, Ma C, Bai Y, Zhang H, Hou J, Wang Y, Ji W, Bai H, et al. QTL Mapping of Yield-Related Traits in Tetraploid Wheat Based on Wheat55K SNP Array. Plants. 2024; 13(10):1285. https://doi.org/10.3390/plants13101285
Chicago/Turabian StyleJia, Yatao, Yifan Zhang, Yingkai Sun, Chao Ma, Yixiong Bai, Hanbing Zhang, Junbin Hou, Yong Wang, Wanquan Ji, Haibo Bai, and et al. 2024. "QTL Mapping of Yield-Related Traits in Tetraploid Wheat Based on Wheat55K SNP Array" Plants 13, no. 10: 1285. https://doi.org/10.3390/plants13101285