


Genes 2024, 15(5), 618; https://doi.org/10.3390/genes15050618 (registering DOI) - 13 May 2024
Abstract
When stroke occurs in pediatric age, it might be mistakenly interpreted as non-accidental head injury (NAHI). In these situations, a multidisciplinary approach is fundamental, including a thorough personal and familial history, along with accurate physical examination and additional investigations. Especially when the clinical
[...] Read more.
When stroke occurs in pediatric age, it might be mistakenly interpreted as non-accidental head injury (NAHI). In these situations, a multidisciplinary approach is fundamental, including a thorough personal and familial history, along with accurate physical examination and additional investigations. Especially when the clinical picture is uncertain, it is important to remember that certain genetic conditions can cause bleeding inside the brain, which may resemble NAHI. Pediatric strokes occurring around the time of birth can also be an initial sign of undiagnosed genetic disorders. Hence, it is crucial to conduct a thorough evaluation, including genetic testing, when there is a suspicion of NAHI but the symptoms are unclear. In these cases, a characteristic set of symptoms is often observed. This study aims to summarize some of the genetic causes of hemorrhagic stroke in the pediatric population, thus mimicking non-accidental head injury, considering elements that can be useful in characterizing pathologies. A systematic review of genetic disorders that may cause ICH in children was carried out according to the Preferred Reporting Item for Systematic Review (PRISMA) standards. We selected 10 articles regarding the main genetic diseases in stroke; we additionally selected 11 papers concerning patients with pediatric stroke and genetic diseases, or studies outlining the characteristics of stroke in these patients. The disorders we identified were Moyamoya disease (MMD), COL4A1, COL4A2 pathogenic variant, Ehlers–Danlos syndrome (E-D), neurofibromatosis type 1 (Nf1), sickle cell disease (SCD), cerebral cavernous malformations (CCM), hereditary hemorrhagic telangiectasia (HHT) and Marfan syndrome. In conclusion, this paper provides a comprehensive overview of the genetic disorders that could be tested in children when there is a suspicion of NAHI but an unclear picture.
Full article
(This article belongs to the Special Issue Stroke Genomics and Exit Strategies)
►
Show Figures

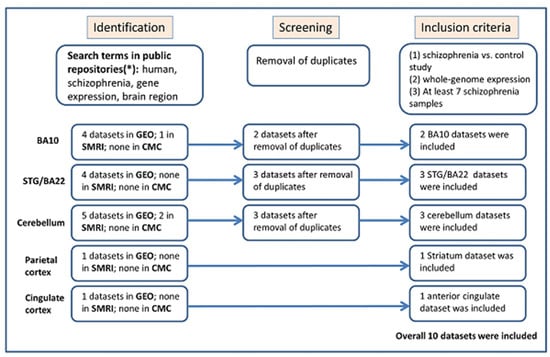
Figure 1















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}