

Initial Despair and Current Hope of Identifying a Clinically Useful Treatment of Myocardial Reperfusion Injury: Insights Derived from Studies of Platelet P2Y12 Antagonists and Interference with Inflammation and NLRP3 Assembly

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Acute Myocardial Infarction Involves Thrombus Formation
2. The Advent of P2Y12 Antagonists
3. Reperfusion Injury—Is It Real and Can It Be Blocked?
4. Ischemic Preconditioning
5. Ischemic Postconditioning
6. Possible Explanation for the Different Results in Pre-Clinical and Clinical Investigations
7. Drugs Administered to STEMI Patients Could Interfere with the Success of Conditioning Interventions
7.1. P2Y12 Antagonists
7.2. ASA
8. Multiple Simultaneous Interventions
9. Inflammation Contributes to Reperfusion Injury
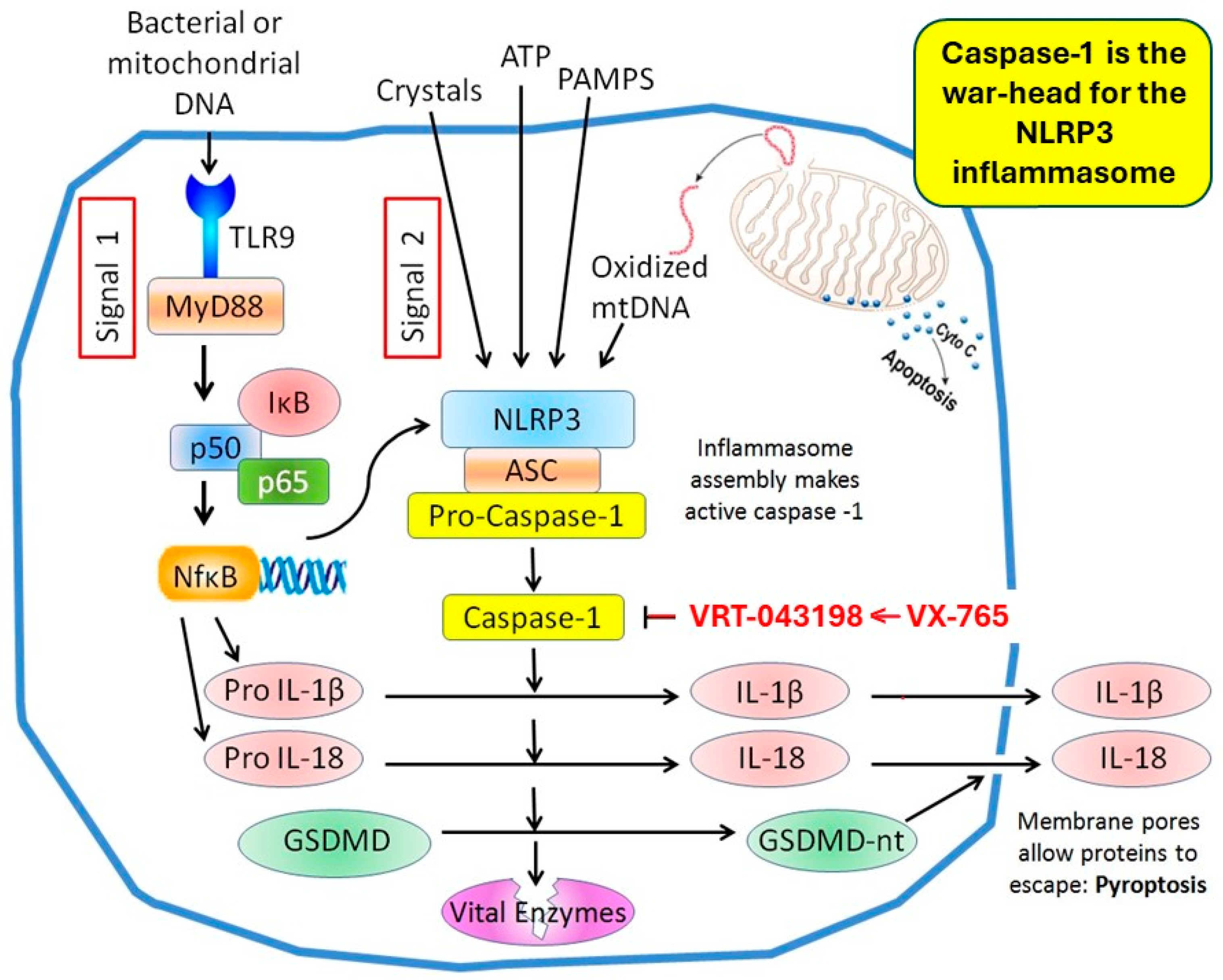
10. Gasdermin D and Caspase-1 Play Pivotal Roles in the Pyroptosis-Induced Death of Cardiomyocytes
11. Caspase-1 Blockers
12. Calpain
13. Gasdermin D Antagonism or Genetic Modification
14. Caspase-1 Inhibition Is Additive to That of IPC
15. Concluding Remarks
Funding
Conflicts of Interest
References
- Forrester, J.S.; Litvack, F.; Grundfest, W.; Hickey, A. A perspective of coronary disease seen through the arteries of living man. Circulation 1987, 75, 505–513. [Google Scholar] [CrossRef]
- DeWood, M.A.; Spores, J.; Notske, R.; Mouser, L.T.; Burroughs, R.; Golden, M.S.; Lang, H.T. Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. N. Engl. J. Med. 1980, 303, 897–902. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.-K. Reassessing the mechanisms of acute coronary syndromes: The “vulnerable plaque” and superficial erosion. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Stone, P.H.; Libby, P.; Boden, W.E. Fundamental pathobiology of coronary atherosclerosis and clinical implications for chronic ischemic heart disease management-the plaque hypothesis: A narrative review. JAMA Cardiol. 2023, 8, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.B.; Warner, T.D. Anti-platelet drugs and their necessary interaction with endothelial mediators and platelet cyclic nucleotides for therapeutic efficacy. Pharmacol. Ther. 2019, 193, 83–90. [Google Scholar] [CrossRef]
- Cairns, J.A.; Gent, M.; Singer, J.; Finnie, K.J.; Froggatt, G.M.; Holder, D.A.; Jablonsky, G.; Kostuk, W.J.; Melendez, L.J.; Myers, M.J.; et al. Aspirin, sulfinpyrazone, or both in unstable angina—Results of a Canadian multicenter trial. N. Engl. J. Med. 1985, 313, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Antiplatelet Trialists Collaboration. Secondary prevention of vascular disease by prolonged antiplatelet treatment. Br. Med. J. (Clin. Res. Ed.) 1988, 296, 320–331. [Google Scholar] [CrossRef]
- Antithrombotic Trialists Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002, 324, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Xu, J.; Qin, W.; Wang, Y.; Luo, S.; Wang, G. The role and molecular mechanism of P2Y12 receptors in the pathogenesis of atherosclerotic cardiovascular diseases. Appl. Sci. 2021, 11, 9078. [Google Scholar] [CrossRef]
- Cohen, M.V.; Downey, J.M. Combined cardioprotectant and antithrombotic actions of platelet P2Y12 receptor antagonists in acute coronary syndrome: Just what the doctor ordered. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 179–190. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Bliden, K.P.; Butler, K.; Tantry, U.S.; Gesheff, T.; Wei, C.; Teng, R.; Antonino, M.J.; Patil, S.B.; Karunakaranet, A.; et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: The ONSET/OFFSET study. Circulation 2009, 120, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Parodi, G.; Valenti, R.; Bellandi, B.; Migliorini, A.; Marcucci, R.; Comito, V.; Carrabba, N.; Santini, A.; Gensini, G.F.; Abbate, R.; et al. Comparison of prasugrel and ticagrelor loading doses in ST-segment elevation myocardial infarction patients: RAPID (Rapid Activity of Platelet Inhibitor Drugs) primary PCI study. J. Am. Coll. Cardiol. 2013, 61, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Reimer, K.A.; Lowe, J.E.; Rasmussen, M.M.; Jennings, R.B. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 1977, 56, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B. Commentary on selected aspects of cardioprotection. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Maroko, P.R.; Kjekshus, J.K.; Sobel, B.E.; Watanabe, T.; Covell, J.W.; Ross, J., Jr.; Braunwald, E. Factors influencing infarct size following experimental coronary artery occlusions. Circulation 1971, 43, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Maroko, P.R.; Libby, P.; Bloor, C.M.; Sobel, B.E.; Braunwald, E. Reduction by hyaluronidase of myocardial necrosis following coronary artery occlusion. Circulation 1972, 46, 430–437. [Google Scholar] [CrossRef]
- Hearse, D.J.; Humphrey, S.M.; Chain, E.B. Abrupt reoxygenation of the anoxic potassium-arrested perfused rat heart: A study of myocardial enzyme release. J. Mol. Cell. Cardiol. 1973, 5, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Jolly, S.R.; Kane, W.J.; Bailie, M.B.; Abrams, G.D.; Lucchesi, B.R. Canine myocardial reperfusion injury: Its reduction by the combined administration of superoxide dismutase and catalase. Circ. Res. 1984, 54, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Reimer, K.A.; Murry, C.E.; Richard, V.J. The role of neutrophils and free radicals in the ischemic-reperfused heart: Why the confusion and controversy? J. Mol. Cell. Cardiol. 1989, 21, 1225–1239. [Google Scholar] [CrossRef]
- Tissier, R.; Cohen, M.V.; Downey, J.M. Protecting the acutely ischemic myocardium beyond reperfusion therapies: Are we any closer to realizing the dream of infarct size elimination? Arch. Mal. Coeur Vaiss. 2007, 100, 794–802. [Google Scholar]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.J.; Auchampach, J.A. Blockade of ATP-sensitive potassium channels prevents myocardial preconditioning in dogs. Circ. Res. 1992, 70, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Downey, J.M. Ischemic preconditioning protects against infarction in rat heart. Am. J. Physiol. 1992, 263, H1107–H1112. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Alkhulaifi, A.M.; Browne, E.E.; Pugsley, W.B. Ischaemic preconditioning limits infarct size in the rat heart. Cardiovasc. Res. 1992, 26, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Schott, R.J.; Rohmann, S.; Braun, E.R.; Schaper, W. Ischemic preconditioning reduces infarct size in swine myocardium. Circ. Res. 1990, 66, 1133–1142. [Google Scholar] [CrossRef]
- Van Winkle, D.M.; Thornton, J.D.; Downey, D.M.; Downey, J.M. The natural history of preconditioning: Cardioprotection depends on duration of transient ischemia and time to subsequent ischemia. Coron. Artery Dis. 1991, 2, 613–619. [Google Scholar]
- Toombs, C.F.; Wiltse, A.L.; Shebuski, R.J. Ischemic preconditioning fails to limit infarct size in reserpinized rabbit myocardium. Implication of norepinephrine release in the preconditioning effect. Circulation 1993, 88, 2351–2358. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-M.; Liu, Y.; Liu, Y.; Tandon, N.; Kambayashi, J.; Downey, J.M.; Cohen, M.V. Attenuation of infarction in cynomolgus monkeys: Preconditioning and postconditioning. Basic Res. Cardiol. 2010, 105, 119–128. [Google Scholar] [CrossRef]
- Rischard, F.; McKean, T. Ischemia and ischemic preconditioning in the buffer-perfused pigeon heart. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1998, 119, 59–65. [Google Scholar] [CrossRef]
- Liu, G.S.; Thornton, J.; Van Winkle, D.M.; Stanley, A.W.H.; Olsson, R.A.; Downey, J.M. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation 1991, 84, 350–356. [Google Scholar] [CrossRef]
- Cohen, M.V.; Downey, J.M. Signalling pathways and mechanisms of protection in pre- and postconditioning: Historical perspective and lessons for the future. Br. J. Pharmacol. 2015, 172, 1913–1932. [Google Scholar] [CrossRef] [PubMed]
- Philipp, S.; Yang, X.-M.; Cui, L.; Davis, A.M.; Downey, J.M.; Cohen, M.V. Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc. Res. 2006, 70, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.; Wynne, A.; Duchen, M.; Yellon, D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation 2004, 109, 1714–1717. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Ong, S.-B.; Yellon, D.M. The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res. Cardiol. 2009, 104, 189–202. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. New directions for protecting the heart against ischaemia-reperfusion injury: Targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc. Res. 2004, 61, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Tsang, A.; Mocanu, M.M.; Yellon, D.M. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H971–H976. [Google Scholar] [CrossRef]
- Zhao, Z.-Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.-P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef]
- Skyschally, A.; van Caster, P.; Iliodromitis, E.K.; Schulz, R.; Kremastinos, D.T.; Heusch, G. Ischemic postconditioning: Experimental models and protocol algorithms. Basic Res. Cardiol. 2009, 104, 469–483. [Google Scholar] [CrossRef]
- Yang, X.-M.; Proctor, J.B.; Cui, L.; Krieg, T.; Downey, J.M.; Cohen, M.V. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J. Am. Coll. Cardiol. 2004, 44, 1103–1110. [Google Scholar] [CrossRef]
- Yang, X.-M.; Philipp, S.; Downey, J.M.; Cohen, M.V. Postconditioning’s protection is not dependent on circulating blood factors or cells but involves adenosine receptors and requires PI3-kinase and guanylyl cyclase activation. Basic Res. Cardiol. 2005, 100, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Argaud, L.; Gateau-Roesch, O.; Raisky, O.; Loufouat, J.; Robert, D.; Ovize, M. Postconditioning inhibits mitochondrial permeability transition. Circulation 2005, 111, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Gateau-Roesch, O.; Argaud, L.; Ovize, M. Mitochondrial permeability transition pore and postconditioning. Cardiovasc. Res. 2006, 70, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.V.; Yang, X.-M.; Downey, J.M. The pH hypothesis of postconditioning: Staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation 2007, 115, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.V.; Yang, X.-M.; Downey, J.M. Acidosis, oxygen, and interference with mitochondrial permeability transition pore formation in the early minutes of reperfusion are critical to postconditioning’s success. Basic Res. Cardiol. 2008, 103, 464–471. [Google Scholar] [CrossRef]
- Inserte, J.; Barba, I.; Poncelas-Nozal, M.; Hernando, V.; Agulló, L.; Ruiz-Meana, M.; Garcia-Dorado, D. cGMP/PKG pathway mediates myocardial postconditioning protection in rat hearts by delaying normalization of intracellular acidosis during reperfusion. J. Mol. Cell. Cardiol. 2011, 50, 903–909. [Google Scholar] [CrossRef]
- Philipp, S.; Yang, X.-M.; Willenbrock, R.; Downey, J.M.; Cohen, M.V. Postconditioning must be initiated in less than 1 minute following reperfusion and is dependent on adenosine 2b receptors and PI3-kinase. Z. Kardiol. 2005, 94 (Suppl. S1), V803. [Google Scholar]
- Mahaffey, K.W.; Puma, J.A.; Barbagelata, N.A.; DiCarli, M.F.; Leesar, M.A.; Browne, K.F.; Eisenberg, P.R.; Bolli, R.; Casas, A.C.; Molina-Viamonte, V.; et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction. Results of a multicenter, randomized, placebo-controlled trial: The Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J. Am. Coll. Cardiol. 1999, 34, 1711–1720. [Google Scholar] [CrossRef]
- Ross, A.M.; Gibbons, R.J.; Stone, G.W.; Kloner, R.A.; Alexander, R.W. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J. Am. Coll. Cardiol. 2005, 45, 1775–1780. [Google Scholar] [CrossRef]
- Yang, X.-M.; Philipp, S.; Downey, J.M.; Cohen, M.V. Atrial natriuretic peptide administered just prior to reperfusion limits infarction in rabbit hearts. Basic Res. Cardiol. 2006, 101, 311–318. [Google Scholar] [CrossRef]
- Kitakaze, M.; Asakura, M.; Kim, J.; Shintani, Y.; Asanuma, H.; Hamasaki, T.; Seguchi, O.; Myoishi, M.; Minamino, T.; Ohara, T.; et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): Two randomised trials. Lancet 2007, 370, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Théroux, P.; Chaitman, B.R.; Danchin, N.; Erhardt, L.; Meinertz, T.; Schroeder, J.S.; Tognoni, G.; White, H.D.; Willerson, J.T.; Jessel, A. Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations. Main results of the GUARDIAN trial. Circulation 2000, 102, 3032–3038. [Google Scholar] [CrossRef]
- Mentzer, R.M., Jr.; Bartels, C.; Bolli, R.; Boyce, S.; Buckberg, G.D.; Chaitman, B.; Haverich, A.; Knight, J.; Menasché, P.; Lee Myers, M.; et al. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: Results of the EXPEDITION study. Ann. Thorac. Surg. 2008, 85, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Zeymer, U.; Suryapranata, H.; Monassier, J.P.; Opolski, G.; Davies, J.; Rasmanis, G.; Linssen, G.; Tebbe, U.; Schröder, R.; Tiemann, R.; et al. The Na+/H+ exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the Evaluation of the Safety and Cardioprotective effects of eniporide in Acute Myocardial Infarction (ESCAMI) trial. J. Am. Coll. Cardiol. 2001, 38, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Ogawa, T.; Suzuki, K.; Goto, M.; Shimamoto, K. Infarct size limitation by a new Na+-H+ exchange inhibitor, Hoe 642: Difference from preconditioning in the role of protein kinase C. J. Am. Coll. Cardiol. 1997, 29, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Staat, P.; Rioufol, G.; Piot, C.; Cottin, Y.; Cung, T.T.; L’Huillier, I.; Aupetit, J.-F.; Bonnefoy, E.; Finet, G.; André-Fouët, X.; et al. Postconditioning the human heart. Circulation 2005, 112, 2143–2148. [Google Scholar] [CrossRef] [PubMed]
- Thibault, H.; Piot, C.; Staat, P.; Bontemps, L.; Sportouch, C.; Rioufol, G.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; Aupetit, J.-F.; et al. Long-term benefit of postconditioning. Circulation 2008, 117, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef]
- Sörensson, P.; Saleh, N.; Bouvier, F.; Böhm, F.; Settergren, M.; Caidahl, K.; Tornvall, P.; Arheden, H.; Rydén, L.; Pernow, J. Effect of postconditioning on infarct size in patients with ST elevation myocardial infarction. Heart 2010, 96, 1710–1715. [Google Scholar] [CrossRef]
- Freixa, X.; Bellera, N.; Ortiz-Pérez, J.T.; Jiménez, M.; Paré, C.; Bosch, X.; De Caralt, T.M.; Betriu, A.; Masotti, M. Ischaemic postconditioning revisited: Lack of effects on infarct size following primary percutaneous coronary intervention. Eur. Heart J. 2012, 33, 103–112. [Google Scholar] [CrossRef]
- Hahn, J.-Y.; Song, Y.B.; Kim, E.K.; Yu, C.W.; Bae, J.-W.; Chung, W.-Y.; Choi, S.-H.; Choi, J.-H.; Bae, J.-H.; An, K.J.; et al. Ischemic postconditioning during primary percutaneous coronary intervention: The effects of postconditioning on myocardial reperfusion in patients with ST-segment elevation myocardial infarction (POST) randomized trial. Circulation 2013, 128, 1889–1896. [Google Scholar] [CrossRef] [PubMed]
- Limalanathan, S.; Andersen, G.Ø.; Kløw, N.-E.; Abdelnoor, M.; Hoffmann, P.; Eritsland, J. Effect of ischemic postconditioning on infarct size in patients with ST-elevation myocardial infarction treated by primary PCI: Results of the POSTEMI (POstconditioning in ST-Elevation Myocardial Infarction) randomized trial. J. Am. Heart Assoc. 2014, 3, e000679. [Google Scholar] [CrossRef] [PubMed]
- Favaretto, E.; Roffi, M.; Frigo, A.C.; Lee, M.S.; Marra, M.P.; Napodano, M.; Tarantini, G. Meta-analysis of randomized trials of postconditioning in ST-elevation myocardial infarction. Am. J. Cardiol. 2014, 114, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Boston-Griffiths, E.A.; Yellon, D.M. Cyclosporin A and cardioprotection: From investigative tool to therapeutic agent. Br. J. Pharmacol. 2012, 165, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Cung, T.-T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in patients with acute myocardial infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; et al. Cyclosporine A in reperfused myocardial infarction: The multicenter, controlled, open-label CYCLE trial. J. Am. Coll. Cardiol. 2016, 67, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Barrabes, J.A.; Bøtker, H.E.; Davidson, S.M.; Di Lisa, F.; Downey, J.; Engstrom, T.; Ferdinandy, P.; Carbrera-Fuentes, H.A.; Heusch, G.; et al. Ischaemic conditioning and targeting reperfusion injury: A 30 year voyage of discovery. Basic Res. Cardiol. 2016, 111, 70. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Bøtker, H.E.; Ovize, M.; Hausenloy, D.J.; Heusch, G. Co-morbidities and co-medications as confounders of cardioprotection-Does it matter in the clinical setting? Br. J. Pharmacol. 2020, 177, 5252–5269. [Google Scholar] [CrossRef]
- Lecour, S.; Andreadou, I.; Bøtker, H.E.; Davidson, S.M.; Heusch, G.; Ruiz-Meana, M.; Schulz, R.; Zuurbier, C.J.; Ferdinandy, P.; Hausenloy, D.J. IMproving Preclinical Assessment of Cardioprotective Therapies (IMPACT) criteria: Guidelines of the EU-CARDIOPROTECTION COST Action. Basic Res. Cardiol. 2021, 116, 52. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Andreadou, I.; Baxter, G.F.; Bøtker, H.E.; Davidson, S.M.; Dobrev, D.; Gersh, B.J.; Heusch, G.; Lecour, S.; Ruiz-Meana, M.; et al. Interaction of cardiovascular nonmodifiable risk factors, comorbidities and comedications with ischemia/reperfusion injury and cardioprotection by pharmacological treatments and ischemic conditioning. Pharmacol. Rev. 2023, 75, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-M.; Liu, Y.; Cui, L.; Yang, X.; Liu, Y.; Tandon, N.; Kambayashi, J.; Downey, J.M.; Cohen, M.V. Platelet P2Y12 blockers confer direct postconditioning-like protection in reperfused rabbit hearts. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-M.; Cui, L.; Alhammouri, A.; Downey, J.M.; Cohen, M.V. Triple therapy greatly increases myocardial salvage during ischemia/reperfusion in the in situ rat heart. Cardiovasc. Drugs Ther. 2013, 27, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-M.; Liu, Y.; Cui, L.; Yang, X.; Liu, Y.; Tandon, N.; Kambayashi, J.; Downey, J.M.; Cohen, M.V. Two classes of anti-platelet drugs reduce anatomical infarct size in monkey hearts. Cardiovasc. Drugs Ther. 2013, 27, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Sivaraman, V.; Kunuthur, S.P.; Cohen, M.V.; Downey, J.M.; Yellon, D.M. Cardioprotective properties of the platelet P2Y12 receptor inhibitor, cangrelor: Protective in diabetics and reliant upon the presence of blood. Cardiovasc. Drugs Ther. 2015, 29, 415–418. [Google Scholar] [CrossRef]
- Yang, X.-M.; Gadde, S.; Audia, J.P.; Alvarez, D.F.; Downey, J.M.; Cohen, M.V. Ticagrelor does not protect isolated rat hearts, thus clouding its proposed cardioprotective role through ENT 1 in heart tissue. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 371–376. [Google Scholar] [CrossRef]
- Liu, G.S.; Stanley, A.W.H.; Downey, J.M. Ischaemic preconditioning is not dependent on neutrophils or glycolytic substrate at reperfusion in rabbit heart. Cardiovasc. Res. 1992, 26, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.V.; Yang, X.-M.; White, J.; Yellon, D.M.; Bell, R.M.; Downey, J.M. Cangrelor-mediated cardioprotection requires platelets and sphingosine phosphorylation. Cardiovasc. Drugs Ther. 2016, 30, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Andreadou, I.; Vilahur, G. The platelet paradox of injury versus protection in myocardial infarction-has it been overlooked? Basic Res. Cardiol. 2021, 116, 37. [Google Scholar] [CrossRef]
- Penna, C.; Aragno, M.; Cento, A.S.; Femminò, S.; Russo, I.; Bello, F.D.; Chiazza, F.; Collotta, D.; Alves, G.V.; Bertinaria, M.; et al. Ticagrelor conditioning effects are not additive to cardioprotection induced by direct NLRP3 inflammasome inhibition: Role of RISK, NLRP3, and redox cascades. Oxid. Med. Cell. Longev. 2020, 2020, 9219825. [Google Scholar] [CrossRef]
- Roubille, F.; Lairez, O.; Mewton, N.; Rioufol, G.; Ranc, S.; Sanchez, I.; Cung, T.T.; Elbaz, M.; Piot, C.; Ovize, M. Cardioprotection by clopidogrel in acute ST-elevated myocardial infarction patients: A retrospective analysis. Basic Res. Cardiol. 2012, 107, 275. [Google Scholar] [CrossRef]
- Patti, G.; Bárczi, G.; Orlic, D.; Mangiacapra, F.; Colonna, G.; Pasceri, V.; Barbato, E.; Merkely, B.; Édes, I.; Ostojic, M.; et al. Outcome comparison of 600- and 300-mg loading doses of clopidogrel in patients undergoing primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: Results from the ARMYDA-6 MI (Antiplatelet therapy for Reducton of MYocardial Damage during Angioplasty-Myocardial Infarction) randomized study. J. Am. Coll. Cardiol. 2011, 58, 1592–1599. [Google Scholar]
- Ye, R.; Jneid, H.; Alam, M.; Uretsky, B.F.; Atar, D.; Kitakaze, M.; Davidson, S.M.; Yellon, D.M.; Birnbaum, Y. Do we really need aspirin loading for STEMI? Cardiovasc. Drugs Ther. 2022, 36, 1221–1238. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, Y.; Ye, R.; Ye, Y. Aspirin blocks the infarct-size limiting effect of ischemic postconditioning in the rat. Cardiovasc. Drugs Ther. 2023, 37, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Nanhwan, M.K.; Ling, S.; Kodakandla, M.; Nylander, S.; Ye, Y.; Birnbaum, Y. Chronic treatment with ticagrelor limits myocardial infarct size: An adenosine and cyclooxygenase-2-dependent effect. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2078–2085. [Google Scholar] [CrossRef] [PubMed]
- Montalescot, G.; van’t Hof, A.W.; Lapostolle, F.; Silvain, J.; Lassen, J.F.; Bolognese, L.; Cantor, W.J.; Cequier, Á.; Chettibi, M.; Goodman, S.G.; et al. Prehospital ticagrelor in ST-segment elevation myocardial infarction. N. Engl. J. Med. 2014, 371, 1016–1027. [Google Scholar] [CrossRef]
- Ubaid, S.; Ford, T.J.; Berry, C.; Murray, H.M.; Wrigley, B.; Khan, N.; Thomas, M.R.; Armesilla, A.L.; Townend, J.N.; Khogali, S.S.; et al. Cangrelor versus ticagrelor in patients treated with primary percutaneous coronary intervention: Impact on platelet activity, myocardial microvascular function and infarct size: A randomized controlled trial. Thromb. Haemost. 2019, 119, 1171–1181. [Google Scholar] [CrossRef]
- Mishra, P.K.; Adameova, A.; Hill, J.A.; Baines, C.P.; Kang, P.M.; Downey, J.M.; Narula, J.; Takahashi, M.; Abbate, A.; Piristine, H.C.; et al. Guidelines for evaluating myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H891–H922. [Google Scholar] [CrossRef]
- Vilela, E.M.; Fontes-Carvalho, R. Inflammation and ischemic heart disease: The next therapeutic target? Rev. Port. Cardiol. (Engl. Ed.) 2021, 40, 785–796. [Google Scholar] [CrossRef]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef]
- Kloner, R.A. Treating acute myocardial infarctions with anti-inflammatory agents. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 73673–73678. [Google Scholar] [CrossRef] [PubMed]
- Buckley, L.F.; Libby, P. Inhibiting NLRP3 inflammasome activity in acute myocardial infarction: A review of pharmacologic agents and clinical outcomes. J. Cardiovasc. Pharmacol. 2019, 74, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.C.; Quinn, S.; Nasis, A.; Hiew, C.; Roberts-Thomson, P.; Adams, H.; Sriamareswaran, E.; Htun, N.M.; Wilson, W.; Stub, D.; et al. Colchicine in patients with acute coronary syndrome: The Australian COPS randomized clinical trial. Circulation 2020, 142, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Trankle, C.R.; Buckley, L.F.; Lipinski, M.J.; Appleton, D.; Kadariya, D.; Canada, J.M.; Carbone, S.; Roberts, C.S.; Abouzaki, N.; et al. Interleukin-1 blockade inhibits the acute inflammatory response in patients With ST-segment-elevation myocardial infarction. J. Am. Heart Assoc. 2020, 9, e014941. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.C.; Rothman, A.M.K.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, A.S.; Fox, K.; Foley, C.; Banya, W.; et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study. Eur. Heart J. 2015, 36, 377–384. [Google Scholar] [CrossRef]
- Broch, K.; Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Tøllefsen, I.M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damås, J.K.; et al. Randomized trial of interleukin-6 receptor inhibition in patients with acute ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Mauro, A.G.; Bonaventura, A.; Mezzaroma, E.; Quader, M.; Toldo, S. NLRP3 Inflammasome in acute myocardial infarction. J. Cardiovasc. Pharmacol. 2019, 74, 175–187. [Google Scholar] [CrossRef]
- Ma, Q. Pharmacological inhibition of the NLRP3 inflammasome: Structure, molecular activation, and inhibitor-NLRP3 interaction. Pharmacol. Rev. 2023, 75, 487–520. [Google Scholar] [CrossRef] [PubMed]
- Vande Walle, L.; Lamkanfi, M. Drugging the NLRP3 inflammasome: From signalling mechanisms to therapeutic targets. Nat. Rev. Drug Discov. 2024, 23, 43–66. [Google Scholar] [CrossRef]
- Fu, J.; Wu, H. Structural mechanisms of NLRP3 inflammasome assembly and activation. Annu. Rev. Immunol. 2023, 41, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Makoni, N.J.; Nichols, M.R. The intricate biophysical puzzle of caspase-1 activation. Arch. Biochem. Biophys. 2021, 699, 108753. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Wang, L.; Hauenstein, A.V. The NLRP3 inflammasome: Mechanism of action, role in disease and therapies. Mol. Aspects Med. 2020, 76, 100889. [Google Scholar] [CrossRef]
- Martin-Sánchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.J.D.; Mortellaro, A.; et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef]
- Liu, X.; Xia, S.; Zhang, Z.; Wu, H.; Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nat. Rev. Drug Discov. 2021, 20, 384–405. [Google Scholar] [CrossRef]
- Popov, S.V.; Maslov, L.N.; Naryzhnaya, N.V.; Mukhomezyanov, A.V.; Krylatov, A.V.; Tsibulnikov, S.Y.; Ryabov, V.V.; Cohen, M.V.; Downey, J.M. The role of pyroptosis in ischemic and reperfusion injury of the heart. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Neuhof, C.; Neuhof, H. Calpain system and its involvement in myocardial ischemia and reperfusion injury. World J. Cardiol. 2014, 6, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-T.; Feng, R.-Q.; Tang, J.-K.; Zhou, J.-J.; Gao, F.; Ren, J. CaMKII/calpain interaction mediates ischemia/reperfusion injury in isolated rat hearts. Cell Death Dis. 2020, 11, 388. [Google Scholar] [CrossRef] [PubMed]
- Hernando, V.; Inserte, J.; Sartório, C.L.; Parra, V.M.; Poncelas-Nozal, M.; Garcia-Dorado, D. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J. Mol. Cell Cardiol. 2010, 49, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Hernando, V.; Garcia-Dorado, D. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2012, 96, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rong, H.; Zhang, F.X.; Wu, K.; Mu, L.; Meng, J.; Xiao, B.; Zamponi, G.W.; Shi, Y. A membrane potential- and calpain-dependent reversal of caspase-1 inhibition regulates canonical NLRP3 inflammasome. Cell Rep. 2018, 24, 2356–2369.e5. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannana, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef]
- Xiao, H.; Li, H.; Wang, J.-J.; Zhang, J.-S.; Shen, J.; An, X.-B.; Zhang, C.-C.; Wu, J.-M.; Song, Y.; Wang, X.-Y.; et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon β-adrenergic insult. Eur. Heart J. 2018, 39, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Willeford, A.; Suetomi, T.; Nickle, A.; Hoffman, H.M.; Miyamoto, S.; Brown, J.H. CaMKIIδ-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight 2018, 3, e97054. [Google Scholar] [CrossRef]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca2+/calmodulin-dependent protein kinase II δ signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef]
- Suetomi, T.; Miyamoto, S.; Brown, J.H. Inflammation in nonischemic heart disease: Initiation by cardiomyocyte CaMKII and NLRP3 inflammasome signaling. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H877–H890. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lai, X.; Fan, X.; Ye, B.; Zhong, L.; Zhang, Y.; Shao, R.; Shi, S.; Huang, W.; Su, L.; et al. Oridonin protects against myocardial ischemia-reperfusion injury by inhibiting GSDMD-mediated pyroptosis. Genes 2022, 13, 2133. [Google Scholar] [CrossRef]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femminò, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways. Oxid. Med. Cell. Longev. 2016, 2016, 5271251. [Google Scholar] [CrossRef] [PubMed]
- Darwesh, A.M.; Jamieson, K.L.; Wang, C.; Samokhvalov, V.; Seubert, J.M. Cardioprotective effects of CYP-derived epoxy metabolites of docosahexaenoic acid involve limiting NLRP3 inflammasome activation. Can. J. Physiol. Pharmacol. 2019, 97, 544–556. [Google Scholar] [CrossRef]
- Pomerantz, B.J.; Reznikov, L.L.; Harken, A.H.; Dinarello, C.A. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1b. Proc. Natl. Acad. Sci. USA 2001, 98, 2871–2876. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Marchetti, C.; Chojnacki, J.; Toldo, S.; Mezzaroma, E.; Tranchida, N.; Rose, S.W.; Federici, M.; Van Tassell, B.W.; Zhang, S.; Abbate, A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 2014, 63, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Toldo, S.; Chojnacki, J.; Mezzaroma, E.; Liu, K.; Salloum, F.N.; Nordio, A.; Carbone, S.; Mauro, A.G.; Das, A.; et al. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. J. Cardiovasc. Pharmacol. 2015, 66, 1–8. [Google Scholar] [CrossRef]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A.; et al. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef]
- van Hout, G.P.J.; Bosch, L.; Ellenbroek, G.H.J.M.; de Haan, J.J.; van Solinge, W.W.; Cooper, M.A.; Arslan, F.; de Jager, S.C.A.; Robertson, A.A.B.; Pasterkamp, G.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef]
- Cohen, M.V.; Yang, X.-M.; Tuckey, A.N.; Audia, J.; Downey, J.M. Reperfusion injury is reduced by antagonism of NLRP3’s caspase-1. Circulation 2021, 144 (Suppl. S1), A13089. [Google Scholar] [CrossRef]
- Qiu, Z.; Lei, S.; Zhao, B.; Wu, Y.; Su, W.; Liu, M.; Meng, Q.; Zhou, B.; Leng, Y.; Xia, Z.-y. NLRP3 inflammasome activation-mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid. Med. Cell. Longev. 2017, 2017, 9743280. [Google Scholar] [CrossRef] [PubMed]
- Silvis, M.J.M.; Demkes, E.J.; Timmers, L.; Arslan, F.; de Jager, S.C.A.; Sluijter, J.P.G.; Mosterd, A.; de Kleijn, D.P.V.; Bosch, L.; van Hout, G.P.J. NLRP3-inflammasome inhibition with IZD334 does not reduce cardiac damage in a pig model of myocardial infarction. Biomedicines 2022, 10, 3056. [Google Scholar] [CrossRef] [PubMed]
- Sandanger, Ø.; Ranheim, T.; Vinge, L.E.; Bliksøen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Sandanger, Ø.; Gao, E.; Ranheim, T.; Bliksøen, M.; Kaasbøll, O.J.; Alfsnes, K.; Nymo, S.H.; Rashidi, A.; Ohm, I.K.; Attramadal, H.; et al. NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem. Biophys. Res. Commun. 2016, 469, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, H.; Yang, L.; Yong, H.; Qin, Q.; Tan, M.; Xu, L.; Liang, K.; Zong, J.; Qian, W. NLRP3 deficiency accelerates pressure overload-induced cardiac remodeling via increased TLR4 expression. J. Mol. Med. 2018, 96, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Jong, W.M.C.; Leemans, J.C.; Weber, N.C.; Juffermans, N.P.; Schultz, M.J.; Hollmann, M.W.; Zuurbier, C.J. Nlrp3 plays no role in acute cardiac infarction due to low cardiac expression. Int. J. Cardiol. 2014, 177, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Boxer, M.B.; Quinn, A.M.; Shen, M.; Jadhav, A.; Leister, W.; Simeonov, A.; Auld, D.S.; Thomas, C.J. A highly potent and selective caspase 1 inhibitor that utilizes a key 3-cyanopropanoic acid moiety. ChemMedChem. 2010, 5, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Wannamaker, W.; Davies, R.; Namchuk, M.; Pollard, J.; Ford, P.; Ku, G.; Decker, C.; Charifson, P.; Weber, P.; Germann, U.A.; et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1β and IL-18. J. Pharmacol. Exp. Ther. 2007, 321, 509–516. [Google Scholar]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; White, H.S. Progress report on new antiepileptic drugs: A summary of the Eleventh Eilat Conference (EILAT XI). Epilepsy Res. 2013, 103, 2–30. [Google Scholar] [CrossRef]
- Kudelova, J.; Fleischmannova, J.; Adamova, E.; Matalova, E. Pharmacological caspase inhibitors: Research towards therapeutic perspectives. J. Physiol. Pharmacol. 2015, 66, 473–482. [Google Scholar] [PubMed]
- Kaufman, R. Phase 2 Clinical Study in Psoriasis with Oral Investigational Drug VX-765; clinicaltrials.gov/ct2/show/NCT00205465; National Library of Medicine, National Institutes of Health: Bethesda, MD, USA, 2007. [Google Scholar]
- Audia, J.P.; Yang, X.-M.; Crockett, E.S.; Housley, N.; Ul Haq, E.; O’Donnell, K.; Cohen, M.V.; Downey, J.M.; Alvarez, D.F. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y12 receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Res. Cardiol. 2018, 113, 32. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-M.; Downey, J.M.; Cohen, M.V.; Housley, N.A.; Alvarez, D.F.; Audia, J.P. The highly selective caspase-1 inhibitor VX-765 provides additive protection against myocardial infarction in rat hearts when combined with a platelet inhibitor. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, H.; Arjun, S.; Petrucci, O.; Yellon, D.M.; Davidson, S.M. The caspase 1 inhibitor VX-765 protects the isolated rat heart via the RISK pathway. Cardiovasc. Drugs Ther. 2018, 32, 165–168. [Google Scholar] [CrossRef]
- Yang, X.-M.; Cohen, M.V.; Sayner, S.; Audia, J.P.; Downey, J.M. Lethal caspase-1/4-dependent injury occurs in the first minutes of coronary reperfusion and requires calpain activity. Int. J. Mol. Sci. 2023, 24, 3801. [Google Scholar] [CrossRef] [PubMed]
- Yue, R.-C.; Lu, S.-Z.; Luo, Y.; Wang, T.; Liang, H.; Zeng, J.; Liu, J.; Hu, H.-X. Calpain silencing alleviates myocardial ischemia-reperfusion injury through the NLRP3/ASC/caspase-1 axis in mice. Life Sci. 2019, 233, 116631. [Google Scholar] [CrossRef] [PubMed]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Han, J.; Fan, X.; Huang, Z.; Su, L.; Cai, X.; Lin, S.; Chen, X.; Huang, W.; Dai, S.; et al. Novel GSDMD inhibitor GI-Y1 protects heart against pyroptosis and ischemia/reperfusion injury by blocking pyroptotic pore formation. Basic Res. Cardiol. 2023, 118, 40. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Tu, Z.; Chen, K.; Xu, Y.; Chen, F.; Xu, S.; Shi, T.; Qian, J.; Shen, L.; Hwa, J.; et al. Gasdermin D inhibition confers antineutrophil-mediated cardioprotection in acute myocardial infarction. J. Clin. Investig. 2022, 132, e151268. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Zhang, Z.-G.; Chen, M.-W.; Yang, Y.; Li, R.J.; Xu, J.-J.; Yang, C.; Li, Y.-Y.; Chen, H.-W.; Liu, S.-X.; et al. Inhibition of GSDMD activates poly(ADP-ribosyl)ation and promotes myocardial ischemia-reperfusion injury. Oxid. Med. Cell Longev. 2022, 2022, 1115749. [Google Scholar] [CrossRef]
- Shi, H.; Gao, Y.; Dong, Z.; Yang, J.; Gao, R.; Li, X.; Zhang, S.; Ma, L.; Sun, X.; Wang, Z.; et al. GSDMD-mediated cardiomyocyte pyroptosis promotes myocardial I/R injury. Circ. Res. 2021, 129, 383–396. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohen, M.V.; Downey, J.M. Initial Despair and Current Hope of Identifying a Clinically Useful Treatment of Myocardial Reperfusion Injury: Insights Derived from Studies of Platelet P2Y12 Antagonists and Interference with Inflammation and NLRP3 Assembly. Int. J. Mol. Sci. 2024, 25, 5477. https://doi.org/10.3390/ijms25105477
Cohen MV, Downey JM. Initial Despair and Current Hope of Identifying a Clinically Useful Treatment of Myocardial Reperfusion Injury: Insights Derived from Studies of Platelet P2Y12 Antagonists and Interference with Inflammation and NLRP3 Assembly. International Journal of Molecular Sciences. 2024; 25(10):5477. https://doi.org/10.3390/ijms25105477
Chicago/Turabian StyleCohen, Michael V., and James M. Downey. 2024. "Initial Despair and Current Hope of Identifying a Clinically Useful Treatment of Myocardial Reperfusion Injury: Insights Derived from Studies of Platelet P2Y12 Antagonists and Interference with Inflammation and NLRP3 Assembly" International Journal of Molecular Sciences 25, no. 10: 5477. https://doi.org/10.3390/ijms25105477