
Transcriptome Analysis of Seed in Dormancy and Dormancy Release State of Epimedium koreanum Nakai
by
Yonggang Zhang
1,
Feng Wu
1,
Jingjing Yu
1,
Zhiqiang Zhang
2,
Xiangdi Huang
1,
Huiling Hou
1 and
Limin Yang
1,* 1
Cultivation Base of State Key Laboratory for Ecological Restoration and Ecosystem Management of Jilin Province and Ministry of Science and Technology, College of Chinese Medicinal Materials, Jilin Agricultural University, Changchun 130118, China
2
Jilin Ginseng Research Institute (Changbai Mountain Natural Medicine Research Institute, Jilin Province), Tonghua 134100, China
*
Author to whom correspondence should be addressed.
Agronomy 2024, 14(5), 1037; https://doi.org/10.3390/agronomy14051037
Submission received: 2 March 2024
/
Revised: 12 April 2024
/
Accepted: 29 April 2024
/
Published: 13 May 2024
(This article belongs to the Section Crop Breeding and Genetics)
Abstract
:Epimedium koreanum is a perennial herb of the Berberidaceae family, which is a traditional tonic in Chinese medicine. Seed germination of E. koreanum is difficult. Dormancy is an intrinsic factor that affects seed germination. Elucidating the molecular mechanism of seed dormancy and the lifting process of E. koreanum is of great significance for the breeding, conservation, and utilization of E. koreanum. Previous studies have concluded that E. koreanum seed dormancy breakage requires warm-temperature stratification followed by low-temperature stratification treatments. Therefore, we performed transcriptome sequencing using freshly harvested, untreated seeds (NS), seeds that developed a cotyledonary embryo after 90 d of constant-temperature stratification at 15 °C (CS), and seeds that broke dormancy by 90 d of stratification at 15 °C and 60 d of stratification at 5 °C (ND) in order to find the responsive genes and regulatory genes that regulate dormancy. A total of 92,867 genes with differential expression were identified. GO enrichment analysis highlighted redox processes, as well as structural components of the nucleus and ribosomes. KEGG enrichment analysis revealed a significant enrichment of phytohormone signaling pathways, which play a crucial role in seed dormancy release. Additionally, protein–protein interactions (PPIs) were predicted with starch and sucrose metabolic pathways. This study introduces a novel concept for a more profound comprehension of the molecular regulatory mechanism of E. koreanum and lays a theoretical foundation for the screening of E. koreanum candidate genes.
1. Introduction
Seed dormancy, described as the inability of an intact, feasible seed to fully germinate under favorable conditions, is a crucial part of plant fitness [1,2]. Based on the classification of Rosbakh et al. [3], Baskin and Baskin [4,5] categorized seed dormancy into five types, namely morphological, physiological, morphophysiological (combination of morphological and physiological), physical, and combinational (combination of physiological and physical) dormancy. Physiological dormancy is the most common type of dormancy in a variety of plants and is generally found in Arabidopsis thaliana L. [6], Oryza sativa L [7], and some crops. Research on the processes involved in seed physiological dormancy has revealed that the initiation and persistence of seed dormancy are influenced by a range of regulatory elements, including seed maturation, hormonal impacts, and chromatin modification [8]. Of the five seed dormancy types, morphophysiological dormancy is one of the most complex and involves both morphodormancy (MD) and physiological dormancy (PD), with different levels and types of dormancy [9]. Seeds with morphophysiological dormancy are detached from the plant with an underdeveloped embryo. It is common for seeds to require a period of moist, warm stratification so that the embryo can continue to develop. Once the embryo has fully developed, it requires a period of moist, cold stratification to break physiological dormancy. To date, the molecular mechanisms underlying physiological dormancy in seed morphology remain unknown. Stratification is a common technique used to break seed dormancy, but the optimal temperature varies with the dormancy period. Low-temperature stratification overcomes the inhibitory effect of the endosperm and promotes seed embryo growth [10]. While variable-temperature stratification improves seed germination during low-temperature stratification, its effect is proportional to the length of variable-temperature treatment [11]. A network of signaling pathways controlled by multiple hormones and the environment controls seed dormancy. Spruce (Picea glauca) seeds underwent significant changes in abscisic acid (ABA), gibberellic acid (GA), and indole-3-acetic acid (IAA) levels during wet, cold stratification, and the expression of the transport inhibitor response 1 (TIR1) transcript was significantly up-regulated. ARF4 and Aux/IAA are components of the auxin signaling pathway that mediate dormancy release from white spruce seeds [12].
E. koreanum is a perennial herbaceous plant of the Berberidaceae family mainly found in the understory of sparse forests, thickets, or semi-shaded environments at the edge of forests and distributed in the Changbai Mountain area of China, the coast of the Sea of Japan, and the northern part of North Korea. It is a traditional bulk medicinal herb in China that is efficacious in tonifying the kidney and yang, strengthening the tendons and bones, and enhancing immune function [13]. However, E. koreanum has a limited distribution and slow regeneration, and its sexual reproductive ability is weak. Research has shown that the average flowering and fruiting rates of E. koreanum are not more than 8% and 6%, respectively. Additionally, Epimedium seeds exhibit obvious dormancy, and seedlings may not emerge until approximately one year under natural conditions [14]. The dormancy of E. koreanum seeds can be overcome by subjecting them to warm temperatures followed by cold stratification. This dormancy is characterized as a deep and straightforward morphophysiological dormancy [15]. Researchers compared the effects of natural stratification with those of cold stratification and hormone treatments on the release of dormancy in E. koreanum seeds and found that natural stratification plus 400 mg/L GA treatment was the most effective, resulting in the highest embryo rate [16]. Although seed propagation of E. koreanum is difficult, it allows for the selection of a good germplasm with stable genetic traits and facilitates seed conservation and expanded propagation. Therefore, it is crucial to strengthen research efforts to meet production needs and ensure variety conservation in Epimedium. Previous studies have mainly focused on promoting germination from a macroscopic perspective; however, the molecular mechanism of seed dormancy has not been reported. The present study aimed to investigate the molecular mechanism of seed germination at the molecular level to lay a solid theoretical foundation to guide practice and improve the germination rate of Korean mushroom seeds. Meanwhile, the study of the molecular mechanism of its dormancy lifting may also provide new ideas for the breeding process of E. koreanum.
2. Materials and Methods
2.1. Plant Materials and Lamination
Mature E. koreanum seeds were collected in June 2022 in Tonghua County, Tonghua City, Jilin Province, and sterilized and washed with 0.1% HgCl2 solution. Full and pest-free seeds with 98.00% viability as tested by the 2,3,5-triphenyltetrazolium chloride (TTC) method were selected, quickly frozen in liquid nitrogen in cryopreservation tubes, and stored in the refrigerator at −80 °C as the original seed, which was recorded as NS (not-processed seed). Part of the seed subjected to 15 °C constant-temperature stratification for 90 d until embryo morphology had just reached the cotyledon embryo seed was recorded as CS (cotyledon embryo seed). Part of the seed was subjected to 15 °C constant-temperature stratification for 90 d, then 5 °C stratification for 60 d until the morphology of a dormancy-lifted seed was reached and recorded as ND (not dormancy).
Seeds were mixed with wet sand (moisture content of wet sand was 17%) at a ratio of 1:10 (v:v) in a nylon mesh bag, which was spread flat in a foam box that was perforated for ventilation while covered with wet sand. The stratification was carried out in a constant-temperature incubator.
2.2. RNA Extraction and cDNA Library Construction
The experimental procedure was strictly executed according to the standards provided by Illumina (San Diego, CA, USA), including library preparation and sequencing experiments. Using Trizol reagent (Invitrogen, Carlsbad, CA, USA), total RNA was extracted in accordance with the manufacturer’s instructions. An RNA 1000 Nano LabChip Kit (Agilent, Santa Clara, CA, USA) with an RIN number > 7.0 and a Bioanalyzer 2100 were used to analyze the total RNA amount and purity. Two rounds of purification were used to separate poly(A) RNA from total RNA (5 ug) using poly-T oligo-attached magnetic beads. After purification, divalent cations were used to break the mRNA into tiny fragments at a high temperature. Following the instructions for the mRNASeq sample preparation kit (Illumina, San Diego, CA, USA), the cleaved RNA fragments were reverse-transcribed to produce the final cDNA library. The average insert size for the paired-end libraries was 300 bp (±50 bp). We then performed the paired-end sequencing on an Illumina Novaseq™ 6000 at LC Sciences (Houston, TX, USA) following the vendor’s recommended protocol. After that, we followed the vendor’s suggested technique to complete the paired-end sequencing on an Illumina NovaseqTM 6000 at LC Sciences (Houston, TX, USA).
2.3. Gene Expression Analysis
First, in-house cutadapt [17] and perl scripts were used to remove the reads that contained adaptor contamination, low-quality bases, and undetermined bases. Then, sequence quality was verified using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/; accessed on 1 February 2023) [18], including the Q20, Q30, and GC content of the clean data. All downstream analyses were based on clean data of high quality. De novo assembly of the transcriptome was performed with Trinity 2.4.0 [19]. Trinity groups transcripts into clusters based on shared sequence content. Such a transcript cluster is very loosely referred to as a ‘gene’. The longest transcript in the cluster was chosen as the ‘gene’ sequence (aka unigene). Salmon [20] was used to perform expression level analysis for unigenes by calculating TPM [21]. The differentially expressed unigenes were selected with |log2 (fold change)| > 1 and with statistical significance (p value < 0.05) by R package edgeR (3.12.1) [22].
2.4. Enrichment and Interaction Analyses
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed using the Cluster Profiler R package (3.4.4) [23]. A corrected p-value ≤ 0.5 was used as the threshold condition for significant enrichment of differential genes in GO terms. KEGG enrichment was used as the reference data for Arabidopsis. Protein–protein interactions (PPIs) were analyzed using String (https://cn.string-db.org/; accessed on 8 February 2023), and the network was visualized using Cytoscape (3.9.0) (National Institute of General Medical Sciences, Bethesda, MD, USA).
2.5. Transcription Factor Family Analysis
E. koreanum has no reference genome, and the differential genes were compared to the Arabidopsis genome to annotate the genes to different transcription factor families. We then generated rose plots (https://www.chiplot.online/; accessed on 5 June 2023) and circle diagrams (https://www.omicstudio.cn/home; accessed on 5 June 2023).
2.6. Quantitative Real-Time PCR (qRT-PCR)
The total RNA of E. koreanum seeds at different time points was reverse-transcribed into cDNA using a SparkJade®SPARKscript (Shandong Sikejie Biotechnology Co., Ltd., Shandong, China). All-in-one RT SuperMix for qPCR (with gDNA eraser) kit, and the template was diluted 3-fold for the subsequent qRT-PCR reactions. Nine unigenes in the KEGG pathway were selected (TRINITY_DN108043_c2_g3, TRINITY_DN113468_c0_g1, TRINITY_DN118153_c2_g2, TRINITY_DN113427_c1_g3, TRINITY_DN107676_c1 g4, TRINITY_DN120537_c0_g3, TRINITY_DN91101_c0_g1, TRINITY_DN89441_c0_g2, and TRINITY_DN108287_c0_g7), the internal reference gene’s 18S rRNA was subjected to qRT-PCR, and the predicted CDS sequences were utilized to design specific primers with Primer3 plus(3.2.0) software. Specific primers were designed using the predicted CDS sequences and Primer3 plus software (Table 1), and the reaction program was based on the SparkJade® 2 × SYBR Green qPCR Mix (With ROX) kit. The relative gene expression was calculated by the 2−ΔΔCt method [24].
3. Results
3.1. Morphology of Seed Embryos at Different Times
As shown in Figure 1, the embryo is still very small and difficult to observe with the naked eye during the NS period. During the CS period it has developed into a cotyledonary embryo, and the structure is already obvious. During the ND period, the embryo’s morphological dormancy is basically lifted, and the embryo almost fills the seed.
3.2. RNA Sequencing Quality
A total of nine transcriptome libraries were obtained, with at least 3,947,626,264 clean reads, 5.43 G clean bases (Table 1), and a Q30 value of more than 93.19%.
3.3. Differentially Expressed Genes (DEGs)
A total of 92,867 differentially expressed genes were identified, as shown in Figure 2A, of which 62,988 were up-regulated and 29,879 were down-regulated. There were 49,511 and 43,356 genes differentially expressed in CS vs. NS and ND vs. NS, respectively. Moreover, more genes were up-regulated than down-regulated in each time node. Taking the intersecting genes in CS vs. NS and ND vs. NS, a total of 26,740 genes were co-expressed in the two comparison groups (Figure 2B).
3.4. Gene Ontology (GO) Enrichment Analysis
A total of 26,851 genes were annotated as significantly enriched in CS vs. NS according to the screening condition of a p-value ≤ 0.05. The oxidation/reduction process, the nucleus, and ATP binding were the major items (Figure 3A). A total of 24,738 genes were annotated as significantly enriched in ND vs. NS, with translation, the nucleus, and the structural constituents of the ribosome as the major items (Figure 3B). A total of 15,783 genes were annotated as significantly enriched in CS vs. NS and ND vs. NS intersecting genes, with the oxidation/reduction process, the nucleus, and the structural constituents of the ribosome as the major items (Figure 3C).
3.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
Based on the screening condition of a p-value ≤ 0.05, a total of 12,660 genes were annotated as significantly enriched in CS vs. NS, with 25 significantly enriched pathways. Plant hormone signal transduction (225 genes); pentose and glucuronate interconversions (176 genes); tropane, piperidine, and pyridine alkaloid biosynthesis (84 genes); and ascorbate and aldarate metabolism (134 genes) were the most enriched pathways (Figure 4A). A total of 11,874 ND vs. NS genes were annotated as significantly enriched, and there were seven significantly enriched pathways. Pentose and glucuronate interconversions (146 genes), porphyrin and chlorophyll metabolism (88 genes), steroid biosynthesis (106 genes), and plant hormone signal transduction (167 genes) were the most abundant pathways (Figure 4B). Among the CS vs. NS and ND vs. NS interconversions, a total of 7463 significantly enriched genes and 12 significantly enriched pathways were annotated. Plant hormone signal transduction (144 genes), pentose and glucuronate interconversions (123 genes), steroid biosynthesis (75 genes), and circadian rhythm (plant) (49 genes) were the most enriched pathways (Figure 4C).
3.6. Gene Expression in Plant Hormone Signal Transduction
A total of 142 differential genes were screened in the phytohormone signaling pathways, with 20, 24, and 15 genes in the IAA (Figure 5A), CK (Figure 5B), and GA (Figure 5C) signaling pathways, respectively. Totals of 22, 21, and 11 genes were expressed in the ABA (Figure 5D), ETH (Figure 5E), and BR (Figure 5F) pathways, respectively. JA (Figure 5G) and SA (Figure 5H) had 5 and 24 genes expressed, respectively. In this study, the expression levels of genes regulating AUX1, TIR1, AUX/IAA, ARF, and SUR on the IAA signaling pathway showed a decreasing trend compared to NS, with the lowest value in the CS period. Transcriptional regulation of the IAA signaling pathway is dependent on auxin response factors (ARFs), which positively regulate IAA synthesis. Two genes regulating ARF were down-regulated during CS and ND compared to NS. On the cytokinin (CK) pathway, compared with NS, 14 genes were up-regulated and 10 genes were down-regulated in CS, and 17 genes were up-regulated and 7 genes were down-regulated in ND. B-ARR is a positive regulator of cytokinin production. Compared with NS, two genes regulating B-ARR were up-regulated, and four genes were down-regulated during CS; three genes regulating B-ARR were up-regulated, and three genes were down-regulated during ND. On the gibberellin (GA) pathway, compared to NS, CS had 1 gene up-regulated and 14 genes down-regulated, and ND had 3 genes up-regulated and 12 genes down-regulated. The DELLA protein family is a negative regulator in the GA signaling pathway. Compared with NS, all eight genes regulating DELLA were down-regulated during CS; seven genes regulating DELLA were down-regulated and one gene was up-regulated during ND. The expression levels of PYR/PYL, PP2C, SnRK2, and ABF on the abscisic acid (ABA) pathway showed a decreasing trend compared to NS, and the vast majority of genes were at their lowest values during the CS period. In the ethylene (ETH) pathway, 11 genes were up-regulated and 10 genes were down-regulated in CS, and 11 genes were up-regulated and 10 genes were down-regulated in ND compared with NS. CTR1 is a negative regulator of ETH. Compared with NS, two genes regulating CTR1 were up-regulated and one gene was down-regulated during CS, and two genes regulating CTR1 were up-regulated and one gene was down-regulated during ND. A total of 11 genes were down-regulated in all of CS, and 11 genes were down-regulated in all of ND compared with NS in the BR pathway. Five genes were down-regulated in all of CS, and five genes were down-regulated in all of ND compared with NS in the JA pathway. Five genes were up-regulated in all of CS, and five genes were down-regulated in all of ND compared with NS. Five genes were up-regulated in all of ND, and five genes were down-regulated in all of CS. Five genes were down-regulated in all of ND compared with NS in the SA pathway. A total of 5 genes were up-regulated, and 19 genes were down-regulated in the CS period. A total of 5 genes were up-regulated, and 19 genes were down-regulated in the ND period.
3.7. Gene Expression in Starch and Sucrose Metabolism
In the pathway related to starch and sucrose metabolism in E. koreanum, there are 8 genes regulating MGAM (maltase–glucoamylase), 1 gene regulating beta-fructofuranosidase, 6 genes regulating HK (hexokinase), 1 gene regulating FRK (fructokinase), 11 genes regulating GPI (glucose-6-phosphate isomerase), 2 genes regulating SUS (sucrose synthase), 3 genes regulating 1,3-beta-GS (1,3-beta-glucan synthase), 12 genes regulating glucan endo-1.3-beta-D-glucosidase, 8 genes regulate UGPase (UTP-glucose-1-phosphate uridylyltransferase), 2 genes regulating SS (starch synthase), 4 genes regulating alpha-amylase, and 4 genes regulating beta-amylase identified from the DEG of the transcriptome. Compared to the NS period, four of the MGAM, four of the HK, seven of the GPI, two of the 1,3-beta-GS, five of the UGPase, and two of the alpha-amylase genes were up-regulated, and the other genes were down-regulated during the CS period. In addition, four MGAM, one beta-fructofuranosidase, four HK, seven GPI, two SUS, two 1,3-beta-GS, two glucan endo-1,3-beta-D-glucosidase, five UGPase, two alpha-amylase, and one beta-amylase genes were up-regulated during ND, and other genes were down-regulated (Figure 6B).
3.8. Interaction Network Analysis
Protein–protein interaction (PPI) IDs of 132 proteins from the starch and sucrose metabolism pathway and 144 proteins from the plant hormone signal transduction pathway, which are involved in both pathways, were uploaded to the String online database. As shown in Figure 7A, 51 proteins in the starch and sucrose metabolic pathways and 71 proteins in the plant hormone signal transduction pathway formed an interaction network. In addition, as shown in Figure 7B, the top-ten proteins in terms of the degree value of each of the two pathways were selected for interactions, and we found that the SAG113 and ABI1 proteins on the plant hormone signal transduction pathway interacted with starch and sucrose metabolism pathway protein SUS3, protein ABI1 on the hormone signaling pathway interacted with starch and sucrose metabolism pathway protein SPS1-2, and the GAI protein on the hormone signaling pathway interacted with starch and sucrose metabolism pathway protein SPS3. SUS3 and ABI1 on the hormone signaling pathway interact with SPS1-2, and GAI on the hormone signaling pathway interacts with AMY2.
3.9. Transcription Factor Family Annotation
The intersecting genes of ND vs. NS and CS vs. NS were subjected to transcription factor annotation, and the top-15 transcription factor families were listed (Figure 8A). At the top of the list was the GRAS transcription family, followed by C3H, bHLH, and other transcription families. The top-ranked transcription factor families annotated in this study have been shown to functionally affect plant growth and development [25,26,27], and subsequent studies can focus on transcription factors with the aim of finding key transcription factors to break seed dormancy, accelerate seed development, and improve seed germination. Differential genes of the top-three transcription factor families were subjected to KEGG enrichment analysis (Figure 8B), which revealed that the top-twenty pathways of the GRAS family rank were enriched for the hormone-related pathway (K14494), bHLH was enriched for the hormone-related pathway (K14431), and C3H was not enriched.
3.10. Analysis of Data Reliability Using Quantitative Real-Time PCR
To validate the accuracy of RNA-seq data, the relative expression and log2-fold changes of nine genes were assessed using Ct values. Real-time quantitative fluorescence PCR (qRT-PCR) demonstrated a consistent up- or down-regulation trend, suggesting the reliability of RNA-seq data (Figure 9).
4. Discussion
In this study, RNA-Seq was used to sequence transcripts from different developmental stages of E. koreanum seeds, and DEGs were annotated by KEGG and GO functional annotations. The KEGG annotation results indicate that the hormone signaling pathway was significantly enriched in differential genes in the both the CS period and the ND period compared to untreated seeds. Seeds cannot grow and develop without the interconversion of energy, while the starch and sucrose metabolic pathways were investigated later. This research analyzed the key genes of hormone signaling, starch and sucrose metabolism, and transcription factors at different developmental stages of E. koreanum seeds.
Hormone signaling, including gibberellin (GA), cytokinin (CK), brassinolide (BR), indole-3-acetic acid (IAA), ethylene (ET), abscisic acid (ABA), jasmonic acid (JA), and salicylic acid (SA), plays an essential role in seed development [28]. The DELLA protein is an important regulatory element in the gibberellin signaling pathway and negatively regulates GA signaling. When GA is absent, DELLA proteins are stable and inhibit the GA response; when GA is present, GID1 (gibberellin-insensitive dwarf 1) binds to GA. This binding promotes the formation of the GID1-GA-DELLA complex, which sequentially promotes its binding to SLY1 (SLEEPY1)/GID2 F-box proteins and polyubiquitinates DELLA, leading to targeted degradation of DELLA via the 26S proteasome. This relieves GA-responsive DELLA inhibition [29,30,31]. Currently, ABA-promoted seed dormancy in plants is mainly regulated in the ABA signaling pathway, where ABA signaling starts with ABA binding to ABA receptors (PY1/PYL/RCAR), and PP2C-like phosphatases (e.g., ABI1 and ABI2) and the SnRK protein kinases are involved, while ABA signaling terminates in ABT (ABA signaling terminator) proteins, which are able to interfere with the interaction of PYR1/PYL4 and ABI1/ABI2, hindering the inhibition of ABI1/ABI2 by ABA-bound PY-R1/PYL4 and, thus, regulating a series of physiological processes in seed dormancy [32]. The receptor for cytokinin (CK) is CRE1, and B-ARR is a class of response regulators that act to positively regulate cytokinin production. A-ARR proteins act as negative feedback regulators of CK signaling; however, unlike B-type RRs, they lack the typical output structural domain (output domain) of transcriptional regulation [33]. There are many studies on how auxin regulates plant growth and development, such as a Lemus chinensis seed germination test that showed low concentrations of auxin-promoted seed germination and high concentrations of auxin-inhibited seed germination [34]. Under high-salt conditions, Arabidopsis seeds were treated with different concentrations of auxin, and the higher the concentration of auxin, the more pronounced the inhibition effect on seed germination was [35,36]. In a study of transgenic Arabidopsis iaaM-OX, it was found that seed germination was strongly inhibited by the large amount of auxin produced by the transgenic Arabidopsis thaliana [37]. Thus, it can be concluded that auxin has a dual role in the regulation of seed germination and seed dormancy. Low concentrations of auxin inhibit seed dormancy and promote seed germination, while high concentrations of auxin inhibit seed germination and promote seed dormancy.
In this study, the expression of genes regulating DELLA proteins was down-regulated in both CS and ND compared to NS, and seed dormancy was gradually lifted. This study suggests that down-regulation of the gene encoding DELLA is closely related to the lifting of seed dormancy. In this study, the expression of genes that synthesize ABA receptors (PYR/PYL) was decreased, and the synthesis of PP2C phosphatase proteins was down-regulated, which suggests that there is a decreasing trend in the expression of ABA during the lifting of dormancy in seeds compared with the control. With dormancy lifting, the expression of most of the regulated CRE1 genes showed an upward trend, and the genes synthesizing A-ARR were not enriched in this study, which means that the synthesis of cytokinins was judged to be increased compared with NS, and it is also believed that cytokinins promoted dormancy lifting. In this paper, with the release of dormancy, the expression of genes regulating growth hormone synthesis showed a down-regulation trend compared with NS, and it was concluded in this study that auxin might not be a key hormone for promoting seed germination. The expression of genes regulating brassinolide (BR), jasmonic acid (JA), and salicylic acid (SA) showed a downward trend compared with the control, which means that these three types of hormones may not be the key hormones for the release of dormancy in the seeds of E. koreanum or they may be the hormones that regulate the germination of the seeds in a negative feedback manner.
Starch and sucrose are two of the most common carbohydrates in plants, and they play an essential role in plant growth and development. The metabolism of starch and sucrose involves a series of enzyme-catalyzed reactions that require the involvement of ATP, NADPH, and hydrolases. The content and distribution of starch and sucrose change with and are closely related to plant growth and development. Starch is primarily stored in the endosperm of seeds in the form of starch granules of various sizes, serving as a crucial storage material in seeds. The key regulatory enzymes involved in starch biosynthesis mainly include UDP-glucose pyrophosphorylase (UGPase), sucrose synthase (SUS), ADP-glucose pyrophosphorylase (AGPase), starch synthase (SS), granule-bound starch synthase (GBSS), and starch-branching enzyme (SBE) [38]. The first step of starch synthesis is catalyzed by SUS, which breaks down sucrose into fructose and UDP glucose (UDPG). UGPase catalyzes the production of glucose-1-P from UDPG. Glucose-1-P is then catalyzed by AGPase to produce ADP glucose, which serves as a substrate for amylosynthase and is involved in the synthesis of both straight-chain and branched-chain amylopectin [39]. This research identified two SUSs, eight UGPases, four AGPases, and two SSs that are differentially expressed during the development of E. koreanum seeds. These results suggest that the identified starch biosynthesis-related genes may play a significant role in starch biosynthesis in E. koreanum seeds. They also lay the groundwork for further functional analysis.
This research found that the SAG113 and ABI1 proteins on the phytohormone signaling pathway interact with starch and sucrose metabolism pathway protein SUS3, the ABI1 protein on the hormone signaling pathway interacts with starch and sucrose metabolism pathway SPS1-2, and the GAI protein on the hormone signaling pathway interacts with starch and sucrose metabolism pathway protein AMY2. Zhang et al. found that SAG113 is 47% similar to ABA-associated transcription factor ABI1, which may be a negative regulatory component of ABA regulation [40]. ABI1 is a member of the PP2C-A phosphatase, which is a negative regulatory component of the ABA signaling pathway and inhibits abscisic acid production by binding to and inhibiting Sn RK2, which blocks the ABA signaling pathway [41]. Sucrose catabolism is mediated by sucrose synthase (sucrose synthase, SUS, EC 2.4.1.13) or sucrose-converting enzyme (invertase, INV, EC 3.2.1.26) catalyzes [42]. INV hydrolyzes sucrose to glucose (Glu) and fructose (Fru), while SUS catalyzes sucrose into UDP-glucose (UDPG) and fructose [43]. Compared with INV, SUS, a UDP-glycosyltransferase widely found in plants, is the key enzyme catalyzing the metabolism of sucrose, catalyzes its degradation at a much faster rate, and plays a role in the development of the seeds and fruits [44]. The sucrose-phosphate synthase (SPS) enzyme is a key enzyme in the metabolism of Suc. (EC 2.4.1.14), a key rate-limiting enzyme in the regulation of sucrose synthesis and a soluble enzyme in the cytoplasm, where fructose 6-phosphate and UDPG (Uridine Diphosphate Glucose) are catalyzed by SPS to produce sucrose 6-phosphate (F6P) and uridine diphosphate (UDP), which is then hydrolyzed by sucrose phosphate phosphatase (SPP) to form sucrose [45]. This research extrapolated that the synthesis and breakdown of sucrose are regulated by abscisic acid. GAI, as one of the important DELLA family members, has been reported in a variety of plants. For example, Arabidopsis (Arabidopsis thaliana) has five DELLA family members, among which GAI mainly regulates stalk growth and leaf spreading [46,47]. Through the study of rice, Xue Hongwei’s team found that the regulation of GA metabolism in seeds can affect α-amylase activity and soluble sugar content, then synergize with ABA signaling to regulate seed dormancy, which can contribute to the development of gibberellins. Their study provided clues as to how gibberellin coordinates embryonic development and seed germination [48]. Overall, the interaction between gibberellin and amylase during seed germination is important for understanding the physiological process of seed germination.
Transcription factors are a significant factor in controlling gene expression, with important roles in plant growth, stress response, and seed development. The GRAS gene family, which is specific to plants, is essential in regulating transcription and managing different signaling pathways involved in plant growth and development. Currently, there are 10 different subfamilies in the GRAS gene family, and each subfamily is associated with distinct biological functions. Some studies have shown that the DELLA subfamily is involved in the signaling and transcriptional regulation processes of GA, light, and JA, which regulate plant growth and development. The SCR and SHR subfamilies are involved in the regulation of radial growth and development of plant roots [49,50]. The SCL3 subfamily is involved in the integration of the GA signaling pathway with the SCR/SHR pathway during root development. The LISCL subfamily is involved in growth hormone response via transcriptional regulation in processes such as growth hormone response and stress response. The PAT1 subfamily is involved in phytochrome light signaling and plant defense-related signaling pathways. The LAS subfamily is important in the regulation of the of axillary bud meristem development in plants [51]. Finally, the HAM subfamily is essential for the maintenance of adventitious shoots in flowering plants [52]. The C3H transcription factor, a zinc-finger protein, has been shown to play a significant role in regulating plant growth, development, and responses to biotic and abiotic factors in Arabidopsis thaliana and rice. Researchers found that C3H zinc-finger protein PEI1 (At5g075000, AtC3H52) from Arabidopsis is involved in seed embryo formation [53], whereas HUA1 (At3g12680, AtC3H35) plays an important role in the regulation of floral development in this species [54,55]. Similarly, GDS1 (At3g51120) is a C3H zinc-finger nuclear-speckle-localized protein that is essential for normal growth and development in Arabidopsis [56]. Arabidopsis KHZ1 and KHZ2 (At3g12130 and At5g06770, respectively) are two novel, non-tandem CCCH zinc-finger and K-homologous structural domain proteins that have a role in the flowering and senescence regulation, with redundant roles [57]. A non-canonical CCCH-tandem zinc-finger protein (LOC_Os04g38520) inhibits secondary wall synthesis and controls mechanical strength in rice [58]. Numerous C3H zinc-finger proteins play a role in hormone signaling, impacting plant growth and development. One such protein, OsDOS (Os01g09620, OsC3H2), is the initial gene identified in rice that encodes a CCCH-type zinc-finger protein. When this gene is overexpressed, it delays leaf senescence and participates in the jasmonic acid signaling pathway [59]. Rice OsLIC proteins are negative regulators of the oleoresin steroid response, which affects oleoresin lactone biosynthesis and/or signaling, which then regulates plant morphogenesis [60]. Similarly, OsTZF1 (OsC3H35, Os05g1067) positively regulates the ABA response [61]. The bHLH transcription factors are the second largest family of transcription factors in plants. They are widely present in the genomes of various plants and play important regulatory roles in plant growth and development, secondary metabolism, and responses to abiotic adversity stress. Plant physiology is closely related to transcriptional regulation. The bHLHs are involved in many growth and developmental processes, including seed germination, floral organ development, and lateral root growth.
Epimedium spp. seeds have not been extensively studied at the molecular level. Li et al. [62] investigated Epimedium brevicornu Maxim and determined that the optimal cold stratification temperature for seeds was 3.9 °C. The study analyzed the phytohormone signaling pathway and fatty acid degradation pathway, revealing changes in key genes during the process of seed dormancy release. This provides a basis for the screening of seed dormancy-related genes. The study found a difference in treatment temperature, which may be attributed to the distinct geographical distribution and biological characteristics of the two species. E. koreanum is the only species found in northeast China and is geographically isolated. Both studies analyzed the phytohormone signaling pathway and highlighted its crucial role in Epimedium seed development, indicating a similarity between the two. Ma et al. [63] conducted a study on Epimedium pseudowushanense BL Guo and found that seed dormancy was alleviated, and the levels of gibberellin and abscisic acid were significantly altered. The highest content of ABA was observed during the seed dormancy stage, while the content of GA3 was lower. As the seeds entered the germination stage, the content of GA3 increased, while the content of ABA decreased significantly. Transcriptome sequencing analysis identified eight key genes related to the metabolism and signaling of ABA and GAs. It was inferred that SnRK2 and DELLA play key roles in abscisic acid and gibberellin, respectively. Although the plant hormone content was not determined, this study analyzed the plant hormone signal transduction pathway. However, this study has a deficiency in not determining the hormone content. This study concluded that DELLA plays a significant role in the gibberellin pathway, which is a point of similarity. This study bridges the gap in the molecule-level study of E. koreanum seeds, providing new ideas for the study of E. koreanum seeds.
5. Conclusions
In this study, we revealed that seed dormancy and hormones are closely related to each other and explained that hormones are a means to break dormancy at the molecular level. Meanwhile, we concluded that gibberellins, abscisic acid, and cytokinins are the key regulatory hormones for seed germination and screened-out some of the dormancy-related genes. We also revealed the roles of hormone signaling and sucrose and starch metabolism in the process of seed dormancy release in E. koreanum. Furthermore, protein interaction analysis revealed potential connections between hormone signaling and sucrose and starch metabolism. To enhance seed germination and improve its rate, future studies could explore the application of hormones and other methods, offering novel insights for the breeding process of E. koreanum.
Author Contributions
Y.Z.: conceptualization, resources, writing—review and editing, project administration, and funding acquisition; F.W.: methodology, formal analysis, and writing—original draft; J.Y.: software, formal analysis, and investigation; Z.Z.: validation, data curation, and formal analysis; X.H.: investigation, visualization, and formal analysis; H.H.: visualization, validation, and formal analysis; L.Y.: conceptualization, writing—review and editing, project administration, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Jilin Province Science and Technology Development Plan Project, China (20200404006YY).
Data Availability Statement
The datasets used during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
We thank LC-Bio Technologies (Hangzhou) Co., Ltd., for the technical support of this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bewley, J.D. Seed Germination and Dormancy. Plant Cell 1997, 9, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Foley, M.E. Genetic and Molecular Control of Seed Dormancy. Trends Plant Sci. 1997, 2, 384–389. [Google Scholar] [CrossRef]
- Rosbakh, S.; Baskin, C.C.; Baskin, J.M. Nikolaeva et al.’s Reference Book on Seed Dormancy and Germination. Ecology 2020, 101, e03049. [Google Scholar] [CrossRef] [PubMed]
- Baskin, J.M.; Baskin, C.C. Seed Germination Ecophysiology of Jeffersonia diphylla, a Perennial Herb of Mesic Deciduous Forests. Am. J. Bot. 1989, 76, 1073–1080. [Google Scholar] [CrossRef]
- Baskin, C.C.; Baskin, J.M.; Meyer, S.E. Seed Dormancy in the Colorado Plateau Shrub Mahonia fremontii (Berberidaceae) and Its Ecological and Evolutionary Implications. Southwest. Nat. 1993, 38, 91–99. [Google Scholar] [CrossRef]
- Buijs, G. A Perspective on Secondary Seed Dormancy in Arabidopsis thaliana. Plants 2020, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Studies on the Dormancy Characteristics of Rice (Oryza sativa L.) Seeds. Master’s Thesis, Hunan Normal University, Changsha, China, 2012. [Google Scholar]
- Graeber, K.; Nakabayashi, K.; Miatton, E.; Leubner-Metzger, G.; Soppe, W.J.J. Molecular Mechanisms of Seed Dormancy. Plant Cell Environ. 2012, 35, 1769–1786. [Google Scholar] [CrossRef] [PubMed]
- Baskin, J.M.; Baskin, C.C. The Great Diversity in Kinds of Seed Dormancy: A Revision of the Nikolaeva–Baskin Classification System for Primary Seed Dormancy. Seed Sci. Res. 2021, 31, 249–277. [Google Scholar] [CrossRef]
- Yu-Guo, H. Study on Embryonic Dormancy of Kalopanax septemlobus Seeds. J. Northeast. For. Univ. 1986, 1, 39–44. [Google Scholar]
- Wang, J.; Liu, P.; He, L.; Zeng, Z.; Lu, J. Factors influencing seed germimation of Epimedium koreanum Nakai. J. Shenyang Pharm. Univ. 2013, 30, 807–811. [Google Scholar] [CrossRef]
- Liu, Y.; Müller, K.; El-Kassaby, Y.A.; Kermode, A.R. Changes in Hormone Flux and Signaling in White Spruce (Picea glauca) Seeds during the Transition from Dormancy to Germination in Response to Temperature Cues. BMC Plant Biol. 2015, 15, 292. [Google Scholar] [CrossRef] [PubMed]
- National Pharmacopoeia Committee. Pharmacopoeia of the People’s Republic of China; China Medical Science and Technology Press: Beijing, China, 2020; Volume 1, p. 340. [Google Scholar]
- Yang, L.; Han, M.; Wu, J.; Han, Z.; Zhang, L. Population biomass and renewal potential of Epimedium koreanum Nakai in diffferent habitats, Linjiang, Northeast China. Acta Ecol. Sin. 2007, 27, 2251–2258. [Google Scholar]
- Rhie, Y.H.; Lee, S.Y. Seed Dormancy and Germination of Epimedium koreanum Nakai. Sci. Hortic. 2020, 272, 109600. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, L.; Wang, K.; Dong, R. Effects of Stratification Conditions and Hormone Treatments on Seed Post-Ripening of Epimedium koreanum Nakai. Mol. Plant Breed. 2024, 22, 2044–2051. [Google Scholar]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Khan, Y.; Xiong, Z.; Zhang, H.; Liu, S.; Yaseen, T.; Hui, T. Expression and Roles of GRAS Gene Family in Plant Growth, Signal Transduction, Biotic and Abiotic Stress Resistance and Symbiosis Formation—A Review. Plant Biol. 2022, 24, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ma, C.; Xu, Y.; Wei, Q.; Imtiaz, M.; Lan, H.; Gao, S.; Cheng, L.; Wang, M.; Fei, Z.; et al. A Zinc Finger Protein Regulates Flowering Time and Abiotic Stress Tolerance in Chrysanthemum by Modulating Gibberellin Biosynthesis. Plant Cell 2014, 26, 2038–2054. [Google Scholar] [CrossRef]
- Lu, R.; Zhang, J.; Wu, Y.-W.; Wang, Y.; Zhang, J.; Zheng, Y.; Li, Y.; Li, X.-B. bHLH Transcription Factors LP1 and LP2 Regulate Longitudinal Cell Elongation. Plant Physiol. 2021, 187, 2577–2591. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, R.; Li, Y. Molecular Networks of Seed Size Control in Plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef]
- Sohn, S.-I.; Pandian, S.; Kumar, T.S.; Zoclanclounon, Y.A.B.; Muthuramalingam, P.; Shilpha, J.; Satish, L.; Ramesh, M. Seed Dormancy and Pre-Harvest Sprouting in Rice—An Updated Overview. Int. J. Mol. Sci. 2021, 22, 11804. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.K.; Steber, C.M. Gibberellin Hormone Signal Perception: Down-Regulating DELLA Repressors of Plant Growth and Development. Annu. Plant Rev. 2016, 49, 153–188. [Google Scholar]
- Song, S.; Liu, J.; Huang, H.; Wu, X.; Xu, H.; Zhang, Q.; Li, X.; Liang, J. Gibberellin hormone signal perception: Down-regulating DELLA repressors of plant growth and development. Sci. Sin. Vitae 2020, 50, 599–615. [Google Scholar]
- Finkelstein, R. Abscisic Acid Synthesis and Response. Arab. Book 2013, 11, e0166. [Google Scholar] [CrossRef]
- Kieber, J.J.; Schaller, G.E. Cytokinin Signaling in Plant Development. Development 2018, 145, dev149344. [Google Scholar] [CrossRef]
- Ma, H.; Liang, Z.; Huang, L.; Yan, C.; Kong, X. Effects of four kinds of exogenous hormones on the germination and seedling growth of Leymus chinensis. Agric. Res. Arid Areas 2008, 26, 69–73. [Google Scholar]
- Park, J.; Kim, Y.-S.; Kim, S.-G.; Jung, J.-H.; Woo, J.-C.; Park, C.-M. Integration of Auxin and Salt Signals by the NAC Transcription Factor NTM2 during Seed Germination in Arabidopsis. Plant Physiol. 2011, 156, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Remington, D.L.; Vision, T.J.; Guilfoyle, T.J.; Reed, J.W. Contrasting Modes of Diversification in the Aux/IAA and ARF Gene Families. Plant Physiol. 2004, 135, 1738–1752. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin Biosynthesis by the YUCCA Flavin Monooxygenases Controls the Formation of Floral Organs and Vascular Tissues in Arabidopsis. Genes Dev. 2006, 20, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Nougué, O.; Corbi, J.; Ball, S.G.; Manicacci, D.; Tenaillon, M.I. Molecular Evolution Accompanying Functional Divergence of Duplicated Genes along the Plant Starch Biosynthesis Pathway. BMC Evol. Biol. 2014, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.L.; Wood, J.R.; Tyson, R.H.; Bridges, I.G. Starch Biosynthesis in Developing Wheat Grain: Evidence against the Direct Involvement of Triose Phosphates in the Metabolic Pathway. Plant Physiol. 1988, 87, 311–319. [Google Scholar] [CrossRef]
- Zhang, K.; Xia, X.; Zhang, Y.; Gan, S. An ABA-Regulated and Golgi-Localized Protein Phosphatase Controls Water Loss during Leaf Senescence in Arabidopsis. Plant J. Cell Mol. Biol. 2012, 69, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.; Merlot, S.; Giraudat, J. The Arabidopsis ABSCISIC ACID-INSENSITIVE2 (ABI2) and ABI1 Genes Encode Homologous Protein Phosphatases 2C Involved in Abscisic Acid Signal Transduction. Plant Cell 1997, 9, 759–771. [Google Scholar] [CrossRef]
- Ruan, Y.-L. Sucrose Metabolism: Gateway to Diverse Carbon Use and Sugar Signaling. Annu. Rev. Plant Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef]
- Coleman, H.D.; Yan, J.; Mansfield, S.D. Sucrose Synthase Affects Carbon Partitioning to Increase Cellulose Production and Altered Cell Wall Ultrastructure. Proc. Natl. Acad. Sci. USA 2009, 106, 13118–13123. [Google Scholar] [CrossRef]
- Xu, S.-M.; Brill, E.; Llewellyn, D.J.; Furbank, R.T.; Ruan, Y.-L. Overexpression of a Potato Sucrose Synthase Gene in Cotton Accelerates Leaf Expansion, Reduces Seed Abortion, and Enhances Fiber Production. Mol. Plant 2012, 5, 430–441. [Google Scholar] [CrossRef]
- Winter, H.; Huber, S.C. Regulation of Sucrose Metabolism in Higher Plants: Localization and Regulation of Activity of Key Enzymes. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 253–289. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.-K.; Chang, C. Arabidopsis RGL1 Encodes a Negative Regulator of Gibberellin Responses. Plant Cell 2002, 14, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.; Piskurewicz, U.; Turečková, V.; Strnad, M.; Lopez-Molina, L. A Seed Coat Bedding Assay Shows That RGL2-Dependent Release of Abscisic Acid by the Endosperm Controls Embryo Growth in Arabidopsis Dormant Seeds. Proc. Natl. Acad. Sci. USA 2010, 107, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.-Q.; Chen, S.-H.; Zhang, X.-F.; Xue, H.-W. Rice OsGA2ox9 Regulates Seed GA Metabolism and Dormancy. Plant Biotechnol. J. 2023, 21, 2411–2413. [Google Scholar] [CrossRef] [PubMed]
- Hou, M. Cloning, Transformation and Functional Analysis of GRAS Transcription Factor Family Genes in Rice. Master’s Thesis, Jilin University, Changchun, China, 2013. [Google Scholar]
- Liu, Y.; Li, S.; Wang, Y.; Yang, J.; Zhou, W.; Shen, Y. Research progress on the functions of GRAS transcription factors in medical plants. J. Zhejiang A&F Univ. 2019, 36, 1233–1240. [Google Scholar]
- Sun, X.; Jones, W.T.; Rikkerink, E.H.A. GRAS Proteins: The Versatile Roles of Intrinsically Disordered Proteins in Plant Signalling. Biochem. J. 2012, 442, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, E.M. HAM Proteins Promote Organ Indeterminacy. Plant Signal. Behav. 2012, 7, 227–234. [Google Scholar] [CrossRef]
- Li, Z.; Thomas, T.L. PEI1, an Embryo-Specific Zinc Finger Protein Gene Required for Heart-Stage Embryo Formation in Arabidopsis. Plant Cell 1998, 10, 383–398. [Google Scholar] [CrossRef]
- Li, J.; Jia, D.; Chen, X. HUA1, a Regulator of Stamen and Carpel Identities in Arabidopsis, Codes for a Nuclear RNA Binding Protein. Plant Cell 2001, 13, 2269–2281. [Google Scholar] [CrossRef]
- Cheng, Y.; Kato, N.; Wang, W.; Li, J.; Chen, X. Two RNA Binding Proteins, HEN4 and HUA1, Act in the Processing of AGAMOUS Pre-mRNA in Arabidopsis thaliana. Dev. Cell 2003, 4, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Jeon, S.J.; Hwang, S.M.; Hong, J.C.; Bahk, J.D. The C3H-Type Zinc Finger Protein GDS1/C3H42 Is a Nuclear-Speckle-Localized Protein That Is Essential for Normal Growth and Development in Arabidopsis. Plant Sci. 2016, 250, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Jia, J.; Yan, X.; Shi, H.; Han, Y. Arabidopsis KHZ1 and KHZ2, Two Novel Non-Tandem CCCH Zinc-Finger and K-Homolog Domain Proteins, Have Redundant Roles in the Regulation of Flowering and Senescence. Plant Mol. Biol. 2017, 95, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xu, Z.; Cao, S.; Chen, K.; Li, S.; Liu, X.; Gao, C.; Zhang, B.; Zhou, Y. An Uncanonical CCCH-Tandem Zinc-Finger Protein Represses Secondary Wall Synthesis and Controls Mechanical Strength in Rice. Mol. Plant 2018, 11, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Li, M.; Yang, W.; Xu, W.; Xue, Y. A Novel Nuclear-Localized CCCH-Type Zinc Finger Protein, OsDOS, Is Involved in Delaying Leaf Senescence in Rice. Plant Physiol. 2006, 141, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, Y.; Zhang, C.; Ma, Q.; Joo, S.-H.; Kim, S.-K.; Xu, Z.; Chong, K. OsLIC, a Novel CCCH-Type Zinc Finger Protein with Transcription Activation, Mediates Rice Architecture via Brassinosteroids Signaling. PLoS ONE 2008, 3, e3521. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, F.; Zhou, J.; Fan, Z.; Chen, F.; Ma, H.; Xie, X. Overexpression of a Phytochrome-Regulated Tandem Zinc Finger Protein Gene, OsTZF1, Confers Hypersensitivity to ABA and Hyposensitivity to Red Light and Far-Red Light in Rice Seedlings. Plant Cell Rep. 2012, 31, 1333–1343. [Google Scholar] [CrossRef]
- Li, P.; Xiang, Q.; Dong, X.; Wang, Y. Characterizing Seed Dormancy in Epimedium brevicornu Maxim.: Development of Novel Chill Models and Determination of Dormancy Release Mechanisms by Transcriptomics. BMC Plant Biol. 2023. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, X.; Guo, B. Identification of Genes Involved in Metabolism and Signalling of Abscisic Acid and Gibberellins during Epimedium pseudowushanense B.L. Guo Seed Morphophysiological Dormancy. Plant Cell Rep. 2018, 37, 1061–1075. [Google Scholar] [CrossRef]
Figure 1.
Longitudinal sections of Epimedium koreanum seeds showing the cotyledon (Cot), embryo (Em), and endosperm (En). (A) NS (not-processed seed); (B) CS (cotyledon embryo seed); (C) ND (not dormancy).
Figure 1.
Longitudinal sections of Epimedium koreanum seeds showing the cotyledon (Cot), embryo (Em), and endosperm (En). (A) NS (not-processed seed); (B) CS (cotyledon embryo seed); (C) ND (not dormancy).

Figure 2.
(A) Numbers of DEGs in each time node; (B) Venn diagrams for CS vs. NS and ND vs. NS.

Figure 3.
Gene ontology (GO) enrichment analysis in the following groups: (A) CS vs. NS; (B) ND vs. NS; (C) CS vs. NS and ND vs. NS.
Figure 3.
Gene ontology (GO) enrichment analysis in the following groups: (A) CS vs. NS; (B) ND vs. NS; (C) CS vs. NS and ND vs. NS.

Figure 4.
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis in the following groups: (A) CS vs. NS; (B) ND vs. NS; (C) CS vs. NS and ND vs. NS.
Figure 4.
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis in the following groups: (A) CS vs. NS; (B) ND vs. NS; (C) CS vs. NS and ND vs. NS.

Figure 5.
Heatmap of annotated genes in the plant hormone signal transduction pathway. Solid lines and arrows indicate the signal transduction process and direction, respectively. (A) Indole-3–acetic acid (IAA) signaling pathway; (B) cytokinin (CK) signaling pathway; (C) gibberellin (GA) signaling pathway; (D) abscisic acid (ABA) signaling pathway; (E) ethylene (ETH) signaling pathway; (F) brassinosteroid (BR) signaling pathway; (G) jasmonic acid (JA) signaling pathway; (H) salicylic acid (SA) signaling pathway.
Figure 5.
Heatmap of annotated genes in the plant hormone signal transduction pathway. Solid lines and arrows indicate the signal transduction process and direction, respectively. (A) Indole-3–acetic acid (IAA) signaling pathway; (B) cytokinin (CK) signaling pathway; (C) gibberellin (GA) signaling pathway; (D) abscisic acid (ABA) signaling pathway; (E) ethylene (ETH) signaling pathway; (F) brassinosteroid (BR) signaling pathway; (G) jasmonic acid (JA) signaling pathway; (H) salicylic acid (SA) signaling pathway.


Figure 6.
(A) Starch and sucrose metabolic pathway diagrams; (B) heatmap of annotated genes in starch and sucrose metabolism pathway.
Figure 6.
(A) Starch and sucrose metabolic pathway diagrams; (B) heatmap of annotated genes in starch and sucrose metabolism pathway.

Figure 7.
(A) Protein–protein interaction (PPI) network among the plant hormone signal transduction pathway and the starch and sucrose metabolism pathway. The blue and orange nodes represent the plant hormone signal transduction pathway and the sucrose metabolism pathway, respectively. The diameters of the nodes represent the interaction frequency; the larger the circle and the more centrally located, the larger the value of its degree and the stronger the interaction. (B) Interaction network diagram of the top-ten degree values among proteins in the plant hormone signaling pathway (left) and the sucrose and starch metabolism pathway (right).
Figure 7.
(A) Protein–protein interaction (PPI) network among the plant hormone signal transduction pathway and the starch and sucrose metabolism pathway. The blue and orange nodes represent the plant hormone signal transduction pathway and the sucrose metabolism pathway, respectively. The diameters of the nodes represent the interaction frequency; the larger the circle and the more centrally located, the larger the value of its degree and the stronger the interaction. (B) Interaction network diagram of the top-ten degree values among proteins in the plant hormone signaling pathway (left) and the sucrose and starch metabolism pathway (right).

Figure 8.
(A) CS vs. NS and ND vs. NS gene transcription factor annotation results; (B) top-three transcription factor family KEGG enrichment results. (a) (GRAS); (b) (bHLH); (c) (C3H); circles are coloured from lighter to darker as the number of genes enriched ranges from few to many;the one in the red border is a node in the phytohormone signal transduction pathway.
Figure 8.
(A) CS vs. NS and ND vs. NS gene transcription factor annotation results; (B) top-three transcription factor family KEGG enrichment results. (a) (GRAS); (b) (bHLH); (c) (C3H); circles are coloured from lighter to darker as the number of genes enriched ranges from few to many;the one in the red border is a node in the phytohormone signal transduction pathway.
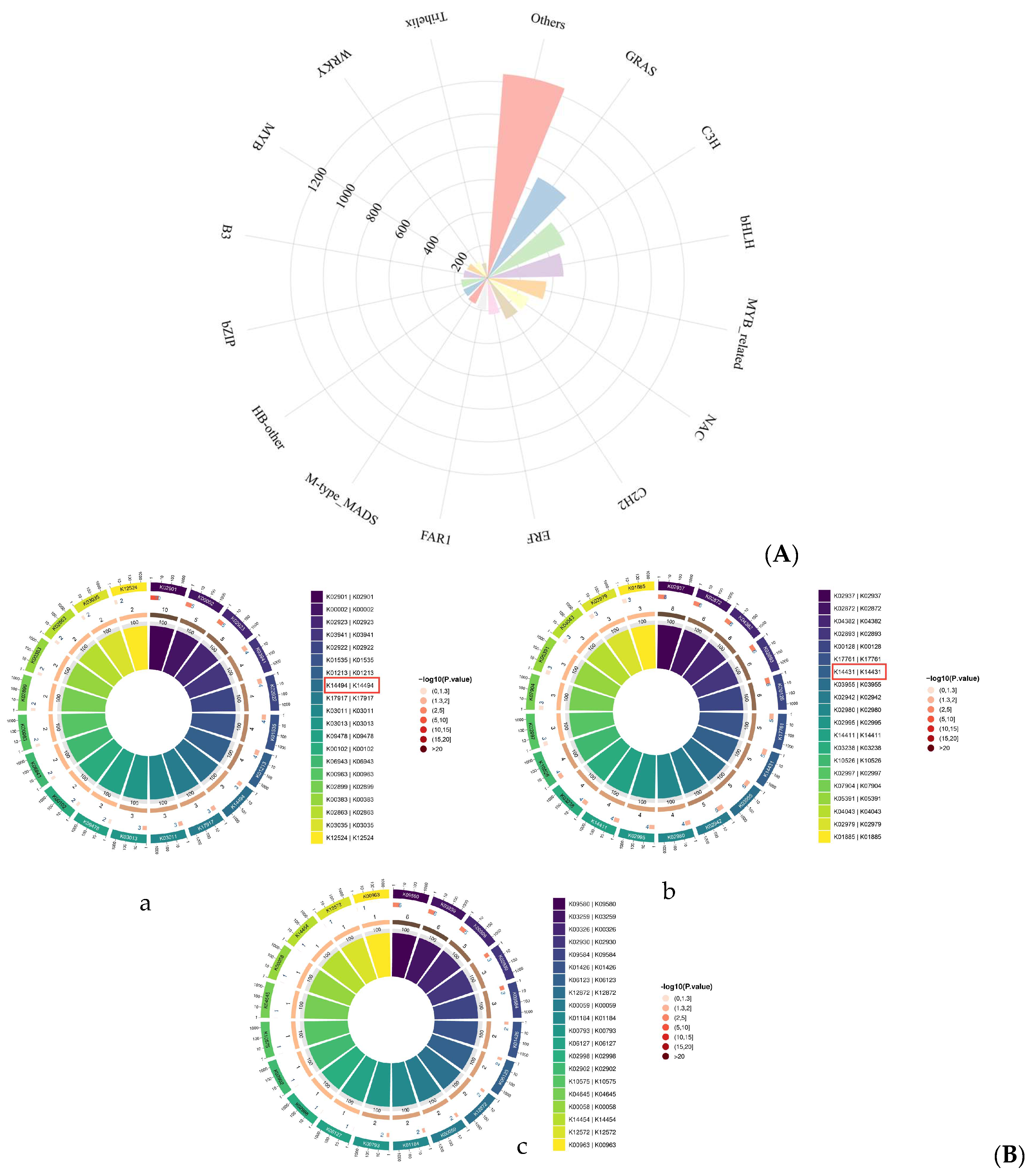
Figure 9.
Log2-fold changes of 9 genes in quantitative real-time PCR (qRT-PCR) and RNA-seq; error bars represent the standard deviation of three replicates.
Figure 9.
Log2-fold changes of 9 genes in quantitative real-time PCR (qRT-PCR) and RNA-seq; error bars represent the standard deviation of three replicates.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
RNA sequencing quality.
| Sample | Raw Reads | Raw Bases | Valid Reads | Valid Bases | Valid% | Q20% | Q30% | GC% |
|---|---|---|---|---|---|---|---|---|
| NS_1 | 41,957,680 | 6.29 G | 41,393,464 | 5.77 G | 98.66 | 97.74 | 93.16 | 47.41 |
| NS_2 | 40,797,168 | 6.12 G | 40,226,414 | 5.61 G | 98.60 | 97.76 | 93.21 | 47.46 |
| NS_3 | 40,803,704 | 6.12 G | 40,229,626 | 5.61 G | 98.59 | 97.75 | 93.19 | 47.42 |
| CS_1 | 40,468,880 | 6.07 G | 39,924,266 | 5.57 G | 98.65 | 97.79 | 93.46 | 55.40 |
| CS_2 | 39,476,264 | 5.92 G | 38,970,190 | 5.43 G | 98.72 | 97.86 | 93.65 | 55.45 |
| CS_3 | 41,336,382 | 6.20 G | 40,794,118 | 5.69 G | 98.69 | 97.85 | 93.63 | 55.52 |
| ND_1 | 43,005,770 | 6.45 G | 42,169,380 | 5.88 G | 98.06 | 97.91 | 93.63 | 52.44 |
| ND_2 | 40,732,900 | 6.11 G | 40,066,156 | 5.58 G | 98.36 | 97.79 | 93.36 | 50.43 |
| ND_3 | 44,456,686 | 6.67 G | 43,867,368 | 6.13 G | 98.67 | 98.10 | 94.14 | 52.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, Y.; Wu, F.; Yu, J.; Zhang, Z.; Huang, X.; Hou, H.; Yang, L. Transcriptome Analysis of Seed in Dormancy and Dormancy Release State of Epimedium koreanum Nakai. Agronomy 2024, 14, 1037. https://doi.org/10.3390/agronomy14051037
AMA Style
Zhang Y, Wu F, Yu J, Zhang Z, Huang X, Hou H, Yang L. Transcriptome Analysis of Seed in Dormancy and Dormancy Release State of Epimedium koreanum Nakai. Agronomy. 2024; 14(5):1037. https://doi.org/10.3390/agronomy14051037
Chicago/Turabian StyleZhang, Yonggang, Feng Wu, Jingjing Yu, Zhiqiang Zhang, Xiangdi Huang, Huiling Hou, and Limin Yang. 2024. "Transcriptome Analysis of Seed in Dormancy and Dormancy Release State of Epimedium koreanum Nakai" Agronomy 14, no. 5: 1037. https://doi.org/10.3390/agronomy14051037
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.